Introduction
Hepatocellular carcinoma (HCC), the predominant form
of adult liver malignancies, is one of the leading causes of
cancer-related mortality worldwide (1,2).
According to a report by the World Health Organization (WHO)
('World Cancer Report 2014'), China now ranks first in the number
of new cancer cases worldwide, and in particular, it ranks first in
the number of new cases of HCC and related deaths worldwide.
Currently, the incidence of liver cancer is approximately
25.7/100,000, becoming the type of cancer with the third largest
mortality rate after gastric cancer and lung cancer. The detailed
molecular mechanisms of hepatocarcinogenesis are not yet fully
understood (3).
Histone acetylation occurs at the N-termini of the
protein octamers and neutralizes the basic charge of the affected
lysine (4). It is a reversible
process, mediated by either histone acetyltransferases (HATs) or
histone deacetylases (HDACs). It also plays a leading role in
several cell functions and regulates numerous processes, including
nucleosome assembly, chromatin condensation, folding,
heterochromatin silencing and gene transcription (5). In recent years, histone H3
acetylation has become one of the hotspots in epigenetic
regulation. It has been reported to be closely associated with the
occurrence and development of multiple types of cancer and to also
be related to the prognosis of multiple types of cancer, including
HCC (6–9). The major acetylation sites of
histone H3 tails are histone H3 lysine 9 (H3K9) and histone H3
lysine 14 (H3K14) and both of their modifications are associated
with the promoters and enhancers of actively transcribed genes
(10–12). The mechanisms responsible for the
interaction of H3K9ac and H3K14ac with ubiquitin-like with PHD and
ring finger domains 2 (UHRF2) remain largely unknown.
UHRF2 is a multi-domain E3 ubiquitin ligase, which
consists of an ubiquitin-like (UBL) domain, a tandem Tudor domain
(TTD), a plant homeodomain (PHD) finger domain, a SET and RING
associated YDG motif (SRA/YDG) domain, and a really interesting new
gene (RING) finger domain (13).
It has been reported that UHRF2 plays a very important role in cell
cycle regulation through its association with multiple cell
cycle-related proteins, including cyclins (A2, B1, D1 and E1),
cyclin-dependent kinases (CDK2, CDK4 and CDK6), retinoblastoma
protein (pRB), p53 and proliferating cell nuclear antigen (PCNA)
(13). It has also been reported
that UHRF2 is involved in epigenetic regulation, including DNA
methylation and histone modifications by interacting with
hemi-methylated DNA, DNA methyltransferases (DNMT1, DNMT3a and
DNMT3b), histone methyltransferase G9a, H3K9 methylation patterns
(H3K9me2/me3) and HDAC1 (14–17). Therefore, we hypothesized that
UHRF2 may be associated with H3K9ac and H3K14ac. In our previous
studies, we demonstrated that UHRF2 interacted with hepatitis B
virus (HBV) core protein and promoted its degradation (18), and we also demonstrated that UHRF2
inhibited the HBV replication cycle, not only through direct
interaction with HBc, but it also reduced the acetylation of HBV
cccDNA-bound H3 histones (19).
HBV is one of the major risk factors for HCC (1,3).
These data suggest that UHRF2 may be associated with acetylated H3
and may play an important role in HCC.
In the present study, we demonstrated that the
levels of H3K9ac and H3K14ac were higher in HCC tissues and HepG2
cancer cells compared with adjacent non-tumor tissues and L02
normal cells. We also confirmed that a higher expression of UHRF2
correlated with a lower expression of H3K9ac in HCC tissues (when
comparing HCC tissues) and that the overexpression of UHRF2
regulated the expression of H3K9ac in different cells. Moreover,
UHRF2 co-localized and interacted with H3K9ac in L02 and HepG2
cells, and UHRF2 interacted with H3K9ac through its PHD domain.
Overall, these data reveal the molecular mechanisms responsible for
the interaction of UHRF2 with acetylated H3 and provide new
moleculer targets for HCC therapeutics.
Materials and methods
Reagents, cell lines and plasmids
Polyclonal rabbit human anti-UHRF2/NIRF antibody
(ab28673), anti-H3 (ab1791) antibody and monoclonal rabbit human
antiH3K9ac (ab32129) antibody, H3K14ac (ab52946) were purchased
from Abcam (Cambridge, MA, USA). Monoclonal mouse anti-Flag
antibody (AF 519) and β-actin (AA128) were obtained from Beyotime
Institute of Biotechnology (Jiangsu, China). Trichostatin A (TSA;
T1952, 5 mM in DMSO) was purchased from Sigma-Aldrich (St. Louis,
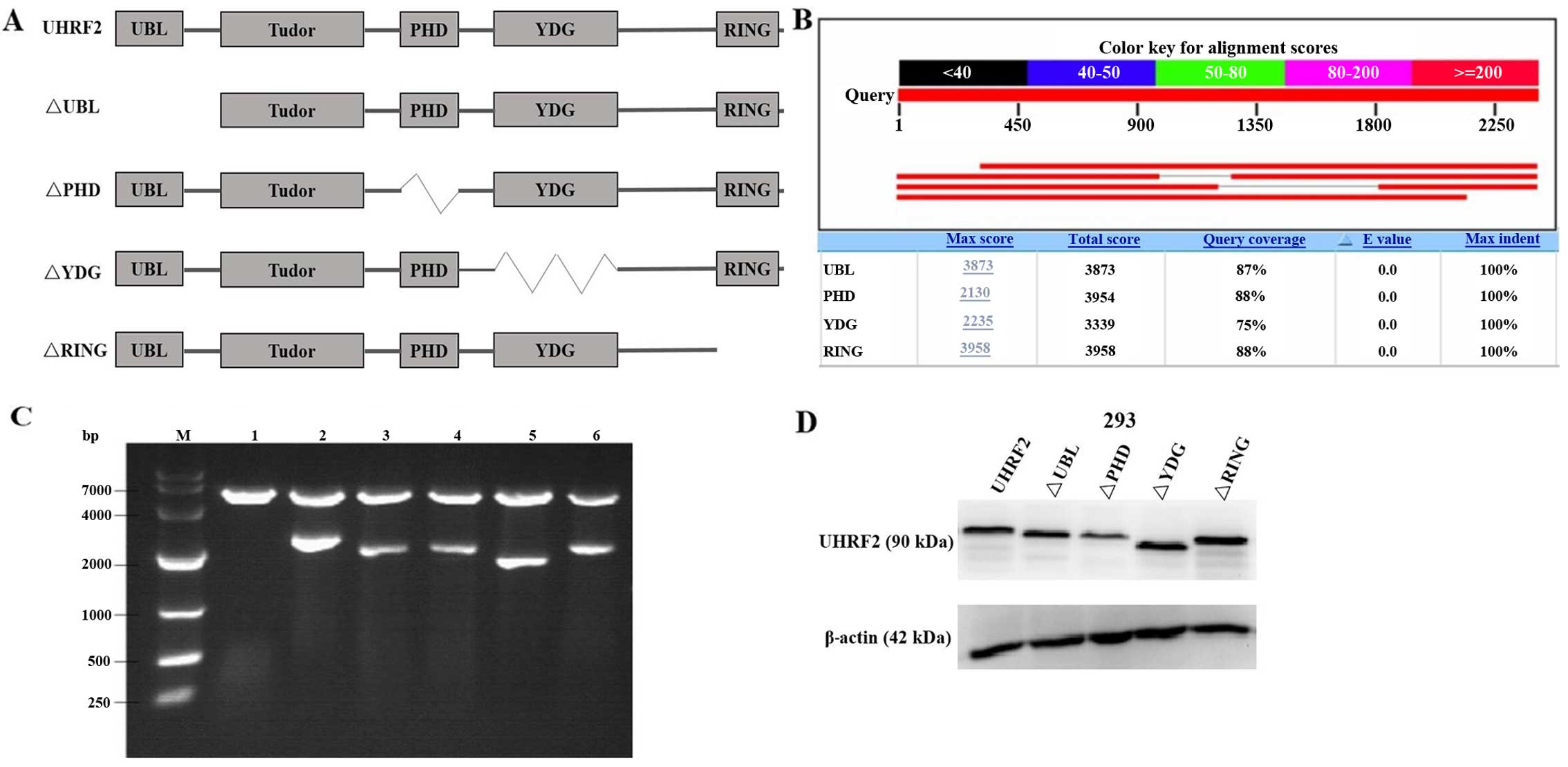
MO, USA). Full-length and mutants UHRF2 plasmids were obtained by
cloning the PCR fragment corresponding to UHRF2 into the
pCMV-3xFlag vector (Fig. 1). The
293 normal renal epithelial cells, L02 normal liver cells, HepG2
HCC cells and pCMV-Flag-UHRF2, pCMV-Flag and deletion mutants of
UHRF2 plasmids were stored by our laboratory.
Cell culture and transfection
The L02 cells were maintained in Roswell Park
Memorial Institute-1640 (RPMI-1640; Gibco, Carlsbad, CA, USA), the
293 cells and HepG2 cells were maintained in Dulbecco's modified
Eagle's medium (DMEM; HyClone Laboratories, Red Bank, NJ, USA).
Both cells were supplemented with 10% fetal bovine serum (FBS; TBD
Science Biotechnology, Tianjing, China) in a humidified atmosphere
containing 5% CO2 at 37°C. Cell transfection (with
pCMV-Flag-UHRF2 or pCMV-Flag) was carried out using Lipofectamine
2000 (Invitrogen, Carlsbad, CA, USA) according to the
manufacturer's instructions. TSA (400 nM) was added to the culture
medium after 4–6 h of transfection and harvested at the end of 24 h
if necessary. TSA is a HDACI and promotes the level of histone
acetylation (20,21). The cells were harvested at 24 h
post-transfection.
Human tumor and non-neoplastic
tissue
HCC tissues (n=5) and corresponding adjacent
non-tumor liver tissues (n=5) were obtained from patients at the
Department of Pathology of the First Affiliated Hospital of
Chongqing Medical University. Ethics approval for the study was
obtained from the First Affiliated Hospital of Chongqing Medical
University, and informed consent was obtained from each
patient.
Immunohistochemistry
The tissues were deparaffinized for 1 h at 60°C in
an oven and rehydrated by gradient alcohol. The antigen-retrieving
steps were carried out in citrate buffer for 10 min in a microwave
oven. The tissue sections were incubated overnight with primary
antibodies to UHRF2 (1:200 dilution), H3 (1:200 dilution), H3K9ac
(1:100 dilution) or H3K14ac (1:100 dilution) at 4°C, and then
incubated with the following secondary antibodies: biotinylated
goat anti-rabbit IgG (SP9001) and biotinylated goat anti-mouse IgG
(SP9002) (Zhongshan Goldenbridge Biotechnology, Corp., Beijing,
China) for 15 min at 37°C, using streptavidin/peroxidase (SP)
histostain™-Plus kits, and stained with DAB (Zhongshan Goldenbridge
Biotechnology Corp.). The nuclei were counterstained with
hematoxylin. The rest of the procedure was performed according to
manufacturer's instructions. Observations were performed using an
Olympus multifunction microscope (Olympus, Tokyo, Japan). The
staining intensity for UHRF2 was graded as 0 (no staining), 1 (mild
staining), 2 (moderate staining) and 3 (intense staining). The
staining extent was scored using the scale as follows: 0 (no
staining of cells), 1 (<10% of tissue stained positive), 2
(10–50% stained positive), 3 (>50% stained positive). The final
staining score was defined as the sum of the intensity and extent
scores. The specimens were divided into 3 groups according to their
overall scores as follows: 0–2, negative; 3–4, weak staining (lower
expression); 5–6, intense staining (higher expression).
Western blot analysis
For western blot analysis, the cells were washed 3
times with ice-cold phosphate-buffered saline (PBS) and lysed in
RIPA buffer containing protease inhibitor cocktail (Roche
Diagnostics Corp., Indianapolis, IN, USA) and phenylmethanesulfonyl
fluoride (PMSF). Following incubation at 4°C for 30 min, the lysate
was centrifuged at 14,000 × g for 30 min at 4°C. The protein
concentration was determined using the bicinchoninic acid (BCA)
protein assay kit (Beyotime Institute of Biotechnology) with BSA as
the standard. Samples were denatured by 5X SDS loading buffer for 5
min at 100°C and separated by sodium dodecyl sulfate-polyacrylamide
gel electrophoresis (SDS-PAGE). Following electrotransfer, the
membrane was probed with antibodies against UHRF2 (1:1,000
dilution), β-actin (1:1,000 dilution), H3 (1:1,000 dilution),
H3K9ac (1:1,000 dilution) or H3K14ac (1:500 dilution). The blots
were then incubated with the secondary antibodies: HRP-labeled goat
anti-mouse IgG (H+L) (A0192) and HRP-labeled goat anti-rabbit IgG
(H+L) (A0208) (from Beyotime Institute of Biotechnology). Detection
was carried out using the ECL chemiluminescent system (Merck
Millipore KGaA, Darmstadt, Germany) according to the manufacturer's
instructions.
Co-immunoprecipitation (Co-IP) assay
For Co-IP, the L02 and HepG2 cells were collected at
24 h post-transfection, washed 3 times with ice-cold PBS and lysed
with ice-cold NP-40-based lysis buffer containing protease
inhibitor cocktail and PMSF for 30 min, followed by centrifugation
at 15,800 × g for 15 min at 4°C. The protein concentration was
determined using the BCA protein assay kit. The protein extracts
were then incubated overnight with specific antibody against Flag
and protein G-agarose beads (Beyotime Institute of Biotechnology)
were then added for 4 h at 4°C. The immunoprecipitated complexes
were washed 5 times using ice-cold lysis buffer, and bound proteins
were released by boiling in 2X SDS loading buffer for 5 min at
100°C. The precipitated proteins were then examined by western blot
analysis. The input was used as a positive control.
Immunofluorescence staining
The L02 and HepG2 cells were grown on cover slips in
6-well plates for 24 h and transfected with pCMV-Flag-UHRF2. TSA
was added to the culture medium at 4–6 h of transfection, and the
cells were then incubated for a further 24 h. The cells were washed
3 times with PBS, fixed with 4% paraformaldehyde for 30 min,
permeabilized in PBS containing 0.1% Triton X-100, and then washed
and blocked with 5% goat serum albumin in PBS for 30 min at room
temperature to block non-specific antibodies and followed by
incubation with antibodies against Flag (1:800 dilution), H3 (1:250
dilution), H3K9ac (1:250 dilution) or H3K14ac (1:250 dilution)
overnight at 4°C. After being washed 3 times with PBS, the cells
were incubated with the secondary antibody, namely Alexa Fluor 488
goat anti-mouse IgG (ZF 0512; 1:100 dilution) or Alexa Fluor 594
goat anti-rabbit IgG (ZF 0516; 1:100 dilution) (Zhongshan
Goldenbridge Biotechnology, Corp.) for 1 h at 37°C in the dark and
then washed 3 times. The cell nuclei were stained with
4,6-diamidino-2-phenylindole (DAPI), washed 3 times again with PBS
and finally sealed with 50% glycerin. Observations were performed
under a laser scanning confocal microscope (Leica TCS-SP2, Leica,
Wetzler, German).
Statistical analysis
The results are expressed as the means ± standard
deviation (SD). All data represent the means ± SD of 3 separate
experiments. Student's t-tests were used for statistical analysis.
A value of P<0.05 was considered to indicate a statistically
significant difference.
Results
Endogenous expression of UHRF2, H3K9ac
and H3K14ac in different cells and tissues
In our previous study, we demonstrated that UHRF2
decreased the acetylation of HBV cccDNA-bound H3 histones (19). In order to explore the molecular
mechanisms responsible for the interaction between UHRF2 and
acetylated H3, we first performed immunohistochemistry and western
blot analysis to detect the endogenous expression levels of UHRF2,
H3K9ac and H3K14ac in HCC tissues and adjacent non-tumor tissues,
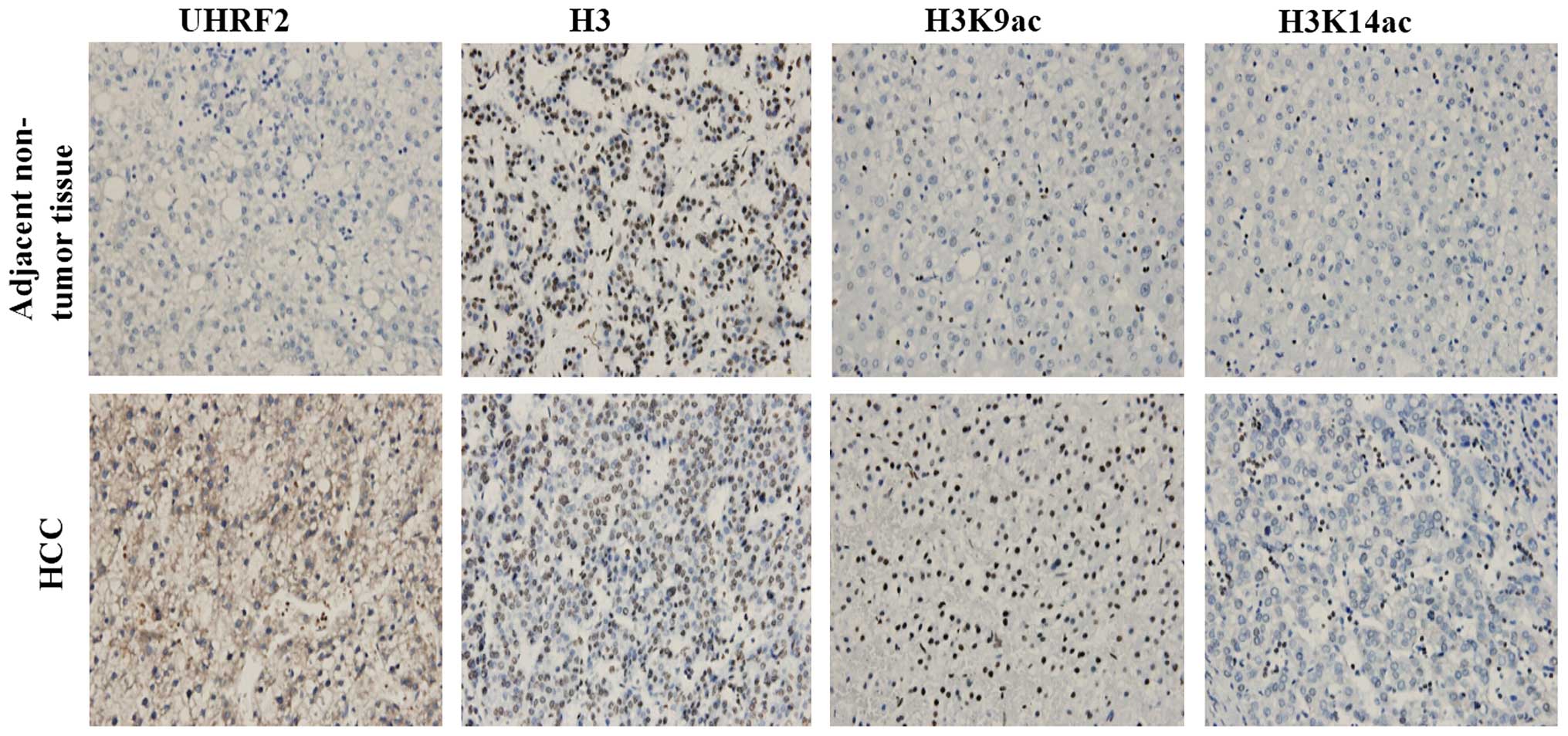
and in L02 and HepG2 cells, respectively. The results of
immunohistochemistry revealed the negative staining of UHRF2 in the
adjacent non-tumor tissues, and the positive staining of UHRF2 in
HCC tissues. In addition, the expression of H3K9ac and H3K14ac was
higher in the HCC tissues compared to the adjacent non-tumor
tissues (Fig. 2). The results of
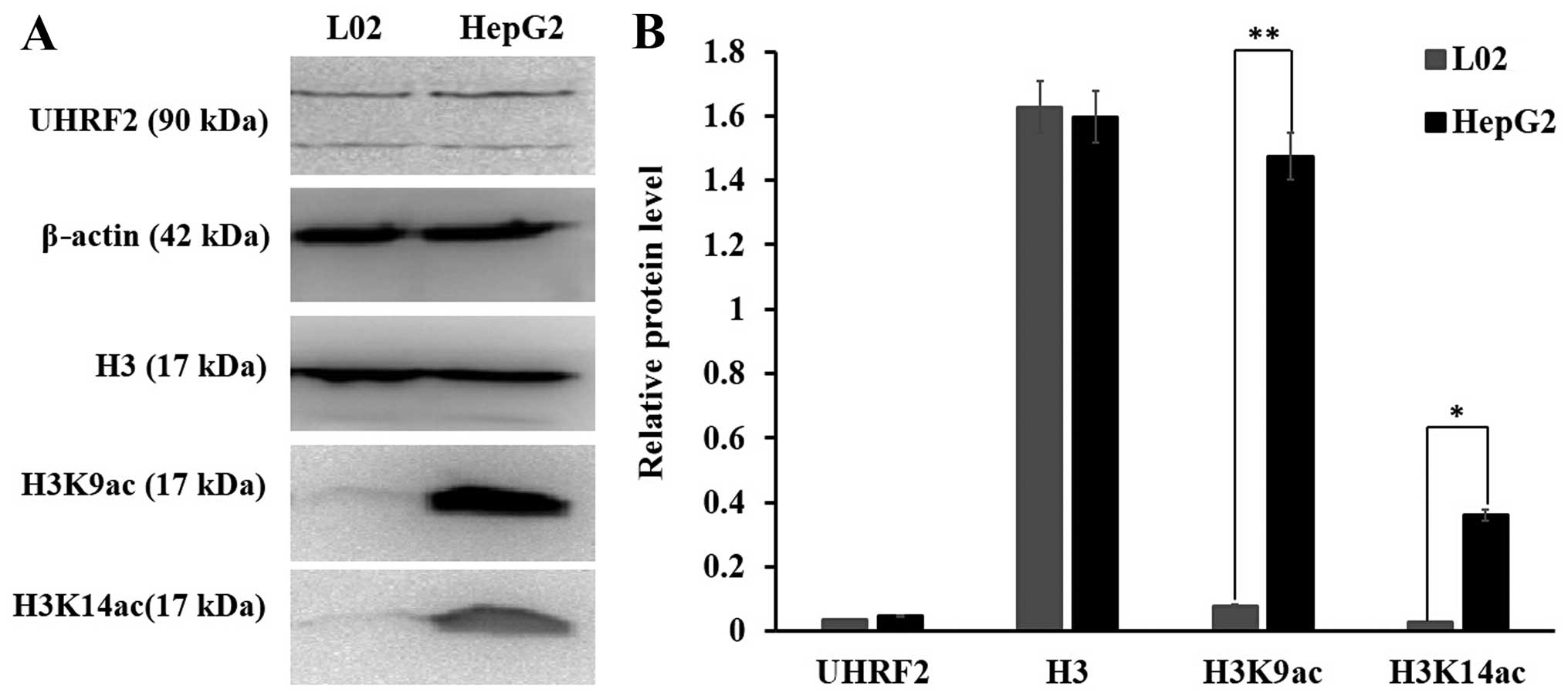
western blot analysis indicated that the protein level of UHRF2 did
not exhibit a significant difference between the L02 normal cells
and the HepG2 HCC cells; however, the protein levels of H3K9ac were
higher in the HepG2 cells than in the L02 cells (P<0.01) and the
protein levels of H3K14ac were also higher in the HepG2 cells than
in the L02 cells (P<0.05) (Fig.
3). These data indicated that the protein level of UHRF2 was
consistent with that of H3K9ac and H3K14ac in the HCC tissues; a
higher expression of UHRF2 was associated with higher levels of
H3K9ac and H3K14ac in HCC tissues when compared to adjacent
non-tumor tissues.
A higher level of UHRF2 is associated
with a lower level of H3K9ac in HCC tissues (comparison of
different HCC tissues only)
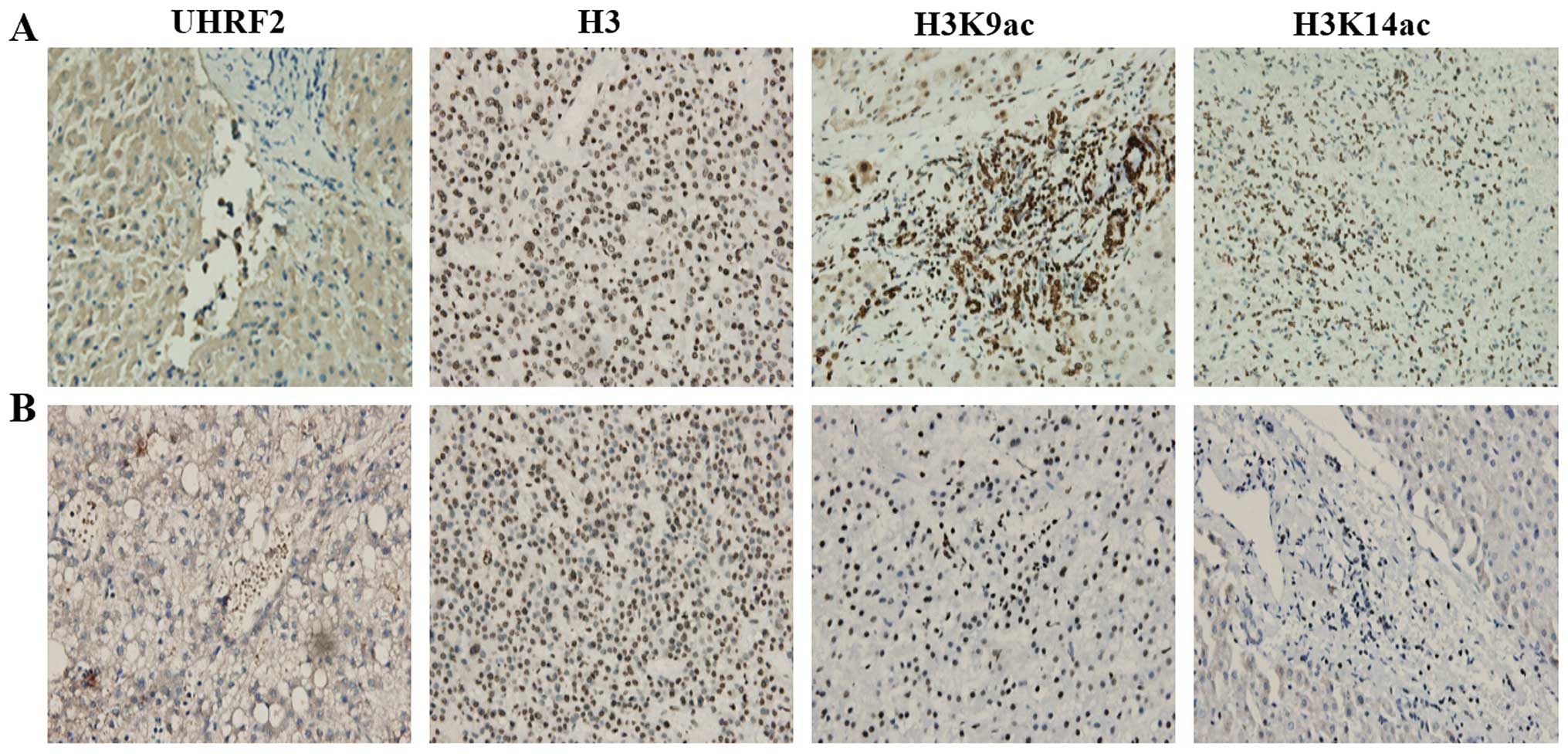
We then examined UHRF2, H3K9ac and H3K14ac
expression between different HCC tissues only. We observed some
interesting results regarding the expression of UHRF2 and H3K9ac in
HCC tissues (Fig. 4). We found
weak UHRF2 staining in some HCC tissues (lower expression of UHRF2;
Fig. 4A), as well as intense
UHRF2 staining in other HCC tissues (higher expression of UHRF2;
Fig. 4B). The results of
immunohistochemistry revealed that the expression of H3K9ac was
higher in the HCC tissues with a lower UHRF2 expression (Fig. 4A), but it was lower in the HCC
tissues with a higher UHRF2 expression (Fig. 4B). The expression of H3K9ac was
higher in the HCC tissues with a lower UHRF2 expression compared
with the HCC tissues with a higher UHRF2 expression. Thus, it
appeared that a higher expression of UHRF2 correlated with a lower
expression of H3K9ac when comparing the HCC tissues. H3K14ac
staining did not exhibit any obvious difference between the HCC
tissues (Fig. 4). These results
demonstrated that the expression of UHRF2 negatively correlated
with H3K9ac expression when comparing different HCC tissues.
Overexpression of UHRF2 effects on the
expression of H3K9ac and H3K14ac
We then explored the molecular mechanisms
responsible for the interaction between UHRF2 and acetylated H3 by
upregulating UHRF2 expression by transfecting the cells with
pCMV-Flag-UHRF2. Semi-quantitative analysis of the protein levels
of H3K9ac and H3K14ac was performed by western blot analysis. The
overexpression of UHRF2 increased the level of H3K9ac in the L02
cells compared to the cells transfected with the empty vector
(P<0.01; Fig. 5A and B);
however, it decreased the level of H3K9ac in the HepG2 cells
(P<0.05; Fig. 5C and D). In
addition, the overexpression of UHRF2 increased the level of
H3K14ac in the L02 cells, whereas it did not significantly alter
its level in the HepG2 cells (P<0.01) (Fig. 5). These results indicated that the
overexpression of UHRF2 increased the expression of H3K14ac in L02
normal cells, but it did not significantly alter the expression of
H3K14ac in HepG2 cells, whereas it had a differential effect on the
expression of H3K9ac in L02 normal cells as compared with HepG2 HCC
cells.
UHRF2 co-localizes with H3, H3K9ac and
H3K14ac
To investigate the regulatory effect of UHRF2 on the
expression of H3K9ac and H3K14ac, the subcellular localization of
UHRF2, H3, H3K9ac and H3K14ac was detected in the L02 and HepG2
cells by immunofluorescence staining with a laser scanning confocal
microscope. UHRF2 is an essential protein for the maintenance of
methylated histone H3 lysine 9 (H3K9me) (14,17). We hypothesized that UHRF2 may
responsible for recruiting H3K9ac and H3K14ac. The results of
immunofluorescence staining indicated that UHRF2 was localized in
the nucleus (green). H3, H3K9ac and H3K14ac exhibited nucelar
distribution (red) (Fig. 6). More
significantly, in the merged images in Fig. 6, a yellow area (Merge1) could be
observed, which indicated that there was a co-localization of UHRF2
with H3, H3K9ac and H3K14ac in both the L02 and HepG2 cells
(Fig. 6).
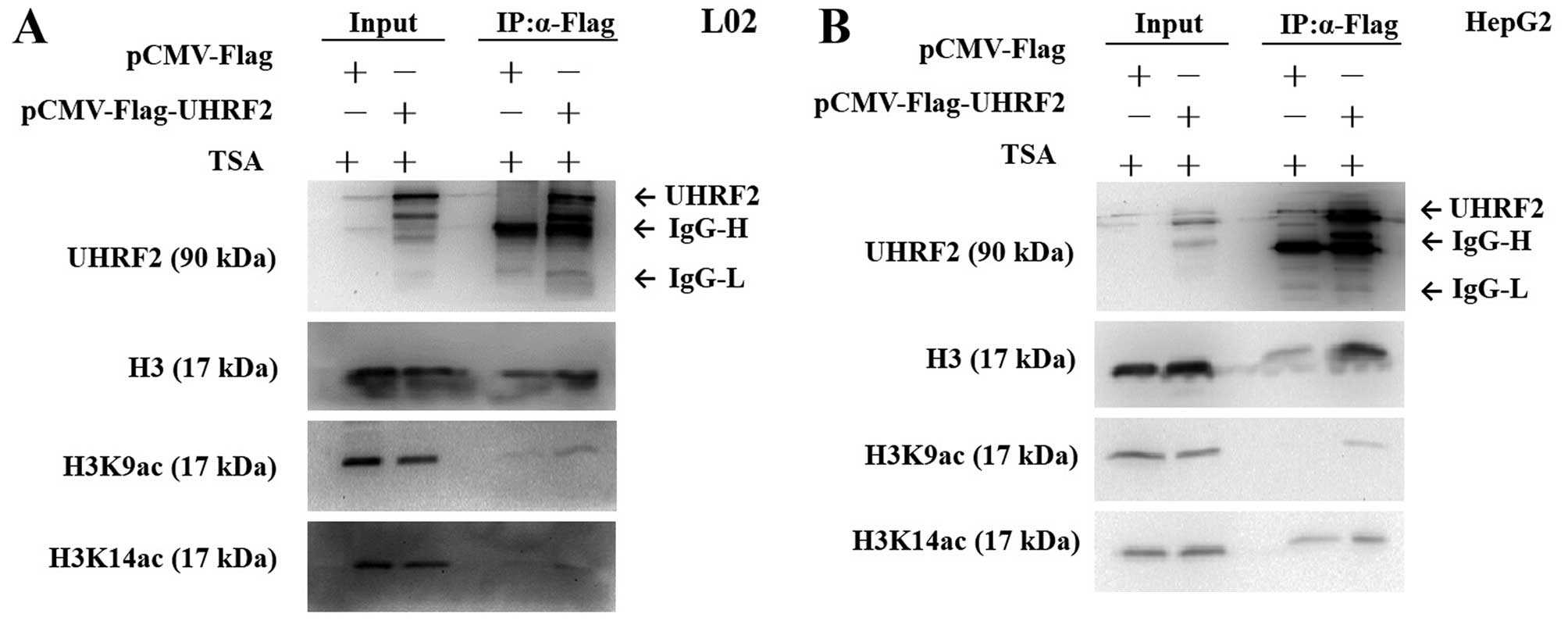
UHRF2 interacts with H3K9ac but not
H3K14ac
To further investigate the interaction between UHRF2
and acetylated H3, we performed Co-IP assay. The cells were
transfected with pCMV-Flag-UHRF2 or pCMV-Flag (mock vector). Flag
antibody and protein G-agarose beads were used to
co-immunoprecipitate H3, H3K9ac, H3K14ac protein from the L02 and
HepG2 cell extracts. The results revealed that only the
precipitation of Flag-UHRF2-interacting protein complexes from
cells transfected with pCMV-Flag-UHRF2 could detect the expression
of H3K9ac, indicating that H3K9ac was indeed efficiently
precipitated with UHRF2 (Fig. 7).
However, H3K14ac was not detected in the L02 cells (Fig. 7A), but it was detected in both the
pCMV-Flag-UHRF2- and pCMV-Flag-transfected HepG2 HCC cells,
indicating that H3K14ac could not be precipitated with UHRF2
(Fig. 7B).
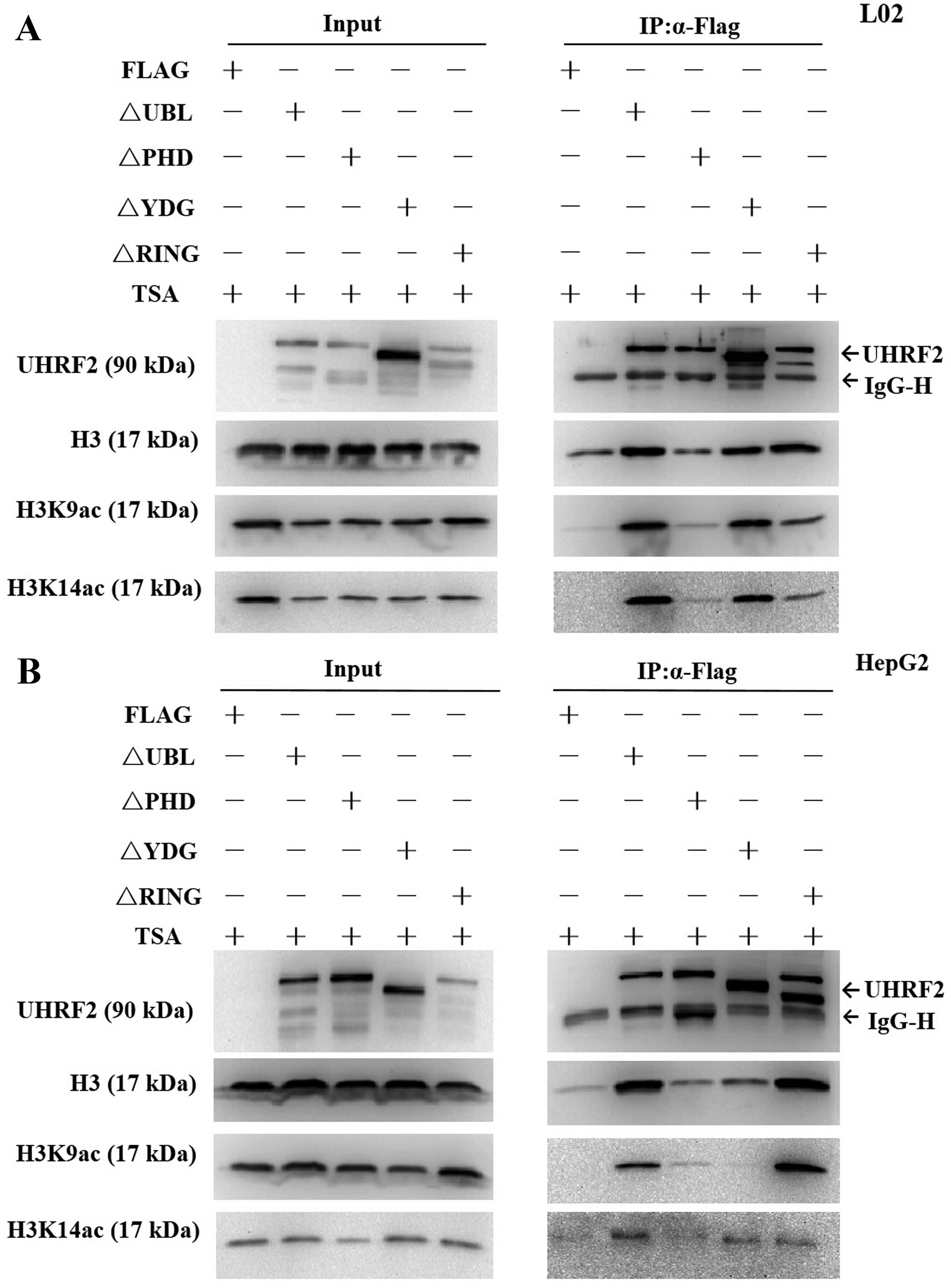
The PHD domain of UHRF2 is important for
H3K9ac binding
UHRF2 is a multi-domain protein (Fig. 1), its PHD domain has been showed
to read part of the histone code (16) and it is characterized as a
versatile epigenetic reader, linking post-translational
modifications (PTMs), particularly the histone H3 tail (22). In order to find the required
domains of UHRF2 interacting with H3K9ac, we performed IP assay of
the cells transfected with deletion mutants of UHRF2 plasmids or
pCMV-Flag. The results revealed that H3K9ac expression was
significantly lower compared with the input at the PHD deletion
mutant domain, indicating that the PHD domain was the key domain
for the interaction of UHRF2 with H3K9ac (Fig. 8). Of note, H3K9ac expression was
significantly lower compared with the input at both the PHD
deletion mutant domain and SRA/YDG deletion mutant domain in the
HepG2 HCC cells, indicating not only the PHD domain, but also the
SRA/YDG domain were important for the interaction of UHRF2 with
H3K9ac in HepG2 HCC cells (Fig.
8B).
Discussion
UHRF2 had been proposed as a potential tumor
suppressor in breast cancer cell lines and A549 lung cancer cells
(23,24), but has been shown to play an
oncongenic role in colorectal cancer and glioma cells (25–27). The expression UHRF2 differs in
different types of cancer, illustrating the heterogeneity of
cancer, and has also been suggested that UHRF2 plays an important
role in tumorigenesis. However, few studies have reported the role
of UHRF2 in HCC. In this study, demonstrated that the expression of
UHRF2 was higher in HCC tissues compared with adjacent non-tumor
tissues. Accumulating evidence has established that
tumor-associated epigenetic alterations, including DNA methylation
and histone modifications are important determinants in the
initiation and progression of HCC and represent promising
biomarkers and therapeutic targets (1,3,9,28).
A high expression of trimethylated histone H3 lysine 4 has been
shown to be associated with a poor prognosis in HCC, and the
increased acetylation of H3K9 has been associated with increased
H3K4 methylation (7,29). This study also demonstrated that
the levels of H3K9ac and H3K14ac were higher in HepG2 HCC cells and
HCC tissues compared with L02 normal cells and adjacent non-tumor
tissues. Moreover, the first evidence was provided that the
overexpression of UHRF2 downregulated the protein level of H3K9ac
in HepG2 cancer cells, but upregulated the protein level of H3K9ac
in L02 normal cells. A higher level of UHRF2 was associated with a
lower level of H3K9ac when comparing HCC tissues. Therefore, UHRF2
and H3K9ac together may serve as a biological marker for HCC
therapeutics.
UHRF2 is involved in several important epigenetic
modifications, including DNA methylation, histone methylation and
acetylation (14,17,26,30). In this study, we first reported
that UHRF2 co-localized with H3K9ac and H3K14ac. In addition, UHRF2
interacted with H3K9ac, but not H3K14ac, and this result indicated
site binding specificity when UHRF2 combined with acetylated H3.
When a specific site of acetylated histones combines with UHRF2,
this triggers a specific event downstream. UHRF1, another member of
the UHRF family of proteins, is highly similar to UHRF2 in both
sequence and structure, and interacts with methylated H3K9 through
the PHD domain (16). UHRF2
specifically binds to H3K9me2/3 at the TTD-PHD region (17). The crystal structure basically
displays that the PHD domain interacts with the H3 N-terminal tail
(22,31). In this study, we clarified that
the PHD domain of UHRF2 is the key region for its interaction with
H3K9ac. These results revealed the crosstalk and the mechanistic
link between UHRF2 and acetylated H3. Furthermore, it has been
reported that the SRA/YDG domain is essential for binding histone
H3 tails (32). We also found
that the SRA/YDG domain was also required in combining with H3K9ac
in HepG2 cells, unlike in L02 normal cells. Further studies are
required in order to determine the difference in the required
domain in normal and cancer cells, as regards UHRF2 binding to
H3K9ac.
In conclusion, our findings demonstrated that the
overexpression of UHRF2 decreased the protein level of H3K9ac in
HepG2 cancer cells, but it increased the protein level of H3K9ac in
L02 normal cells, and a higher level of UHRF2 expression was
associated with a lower level of H3K9ac expression in HCC tissues.
Moreover, our findings also demonstrated that UHRF2 interacted with
H3K9ac directly and the PHD domain was the key domain for the
binding of UHRF2 to H3K9ac. These results may provide a better
understanding of the crosstalk between UHRF2 and H3K9ac, and
highlight the possibilities that UHRF2 and H3K9ac together may be
therapeutic target in HCC.
References
|
1
|
Ma L, Chua MS, Andrisani O and So S:
Epigenetics in hepatocellular carcinoma: An update and future
therapy perspectives. World J Gastroenterol. 20:333–345. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Siegel R, Naishadham D and Jemal A: Cancer
statistics, 2012. CA Cancer J Clin. 62:10–29. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Puszyk WM, Trinh TL, Chapple SJ and Liu C:
Linking metabolism and epigenetic regulation in development of
hepatocellular carcinoma. Lab Invest. 93:983–990. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kratz A, Arner E, Saito R, Kubosaki A,
Kawai J, Suzuki H, Carninci P, Arakawa T, Tomita M, Hayashizaki Y
and Daub CO: Core promoter structure and genomic context reflect
histone 3 lysine 9 acetylation patterns. BMC Genomics. 11:2572010.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Shahbazian MD and Grunstein M: Functions
of site-specific histone acetylation and deacetylation. Annu Rev
Biochem. 76:75–100. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Elsheikh SE, Green AR, Rakha EA, Powe DG,
Ahmed RA, Collins HM, Soria D, Garibaldi JM, Paish CE, Ammar AA, et
al: Global histone modifications in breast cancer correlate with
tumor phenotypes, prognostic factors, and patient outcome. Cancer
Res. 69:3802–3809. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
He C, Xu J, Zhang J, Xie D, Ye H, Xiao Z,
Cai M, Xu K, Zeng Y, Li H and Wang J: High expression of
trimethylated histone H3 lysine 4 is associated with poor prognosis
in hepatocellular carcinoma. Hum Pathol. 43:1425–1435. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Neureiter D, Jäger T, Ocker M and
Kiesslich T: Epigenetics and pancreatic cancer: Pathophysiology and
novel treatment aspects. World J Gastroenterol. 20:7830–7848. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Hung SY, Lin HH, Yeh KT and Chang JG:
Histone-modifying genes as biomarkers in hepatocellular carcinoma.
Int J Clin Exp Pathol. 7:2496–2507. 2014.PubMed/NCBI
|
|
10
|
Karmodiya K, Krebs AR, Oulad-Abdelghani M,
Kimura H and Tora L: H3K9 and H3K14 acetylation co-occur at many
gene regulatory elements, while H3K14ac marks a subset of inactive
inducible promoters in mouse embryonic stem cells. BMC Genomics.
13:4242012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Pokholok DK, Harbison CT, Levine S, Cole
M, Hannett NM, Lee TI, Bell GW, Walker K, Rolfe PA, Herbolsheimer
E, et al: Genome-wide map of nucleosome acetylation and methylation
in yeast. Cell. 122:517–527. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wang Z, Zang C, Rosenfeld JA, Schones DE,
Barski A, Cuddapah S, Cui K, Roh TY, Peng W, Zhang MQ and Zhao K:
Combinatorial patterns of histone acetylations and methylations in
the human genome. Nat Genet. 40:897–903. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Mori T, Ikeda DD, Yamaguchi Y and Unoki M:
NIRF/UHRF2 occupies a central position in the cell cycle network
and allows coupling with the epigenetic landscape. FEBS Lett.
586:1570–1583. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zhang J, Gao Q, Li P, Liu X, Jia Y, Wu W,
Li J, Dong S, Koseki H and Wong J: S phase-dependent interaction
with DNMT1 dictates the role of UHRF1 but not UHRF2 in DNA
methylation maintenance. Cell Res. 21:1723–1739. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Bronner C, Achour M, Arima Y, Chataigneau
T, Saya H and Schini-Kerth VB: The UHRF family: Oncogenes that are
drugable targets for cancer therapy in the near future? Pharmacol
Ther. 115:419–434. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Karagianni P, Amazit L, Qin J and Wong J:
ICBP90, a novel methyl K9 H3 binding protein linking protein
ubiquitination with heterochromatin formation. Mol Cell Biol.
28:705–717. 2008. View Article : Google Scholar :
|
|
17
|
Pichler G, Wolf P, Schmidt CS, Meilinger
D, Schneider K, Frauer C, Fellinger K, Rottach A and Leonhardt H:
Cooperative DNA and histone binding by Uhrf2 links the two major
repressive epigenetic pathways. J Cell Biochem. 112:2585–2593.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Qian G, Jin F, Chang L, Yang Y, Peng H and
Duan C: NIRF, a novel ubiquitin ligase, interacts with hepatitis B
virus core protein and promotes its degradation. Biotechnol Lett.
34:29–36. 2012. View Article : Google Scholar
|
|
19
|
Qian G, Hu B, Zhou D, Xuan Y, Bai L and
Duan C: NIRF, a novel ubiquitin ligase, inhibits hepatitis B virus
replication through effect on HBV core protein and H3 histones. DNA
Cell Biol. 34:327–332. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lu JC, Chang YT, Wang CT, Lin YC, Lin CK
and Wu ZS: Trichostatin A modulates thiazolidinedione-mediated
suppression of tumor necrosis factor α-induced lipolysis in 3T3-L1
adipocytes. PLoS One. 8:e715172013. View Article : Google Scholar
|
|
21
|
Sailaja BS, Cohen-Carmon D, Zimmerman G,
Soreq H and Meshorer E: Stress-induced epigenetic transcriptional
memory of acetylcholinesterase by HDAC4. Proc Natl Acad Sci USA.
109:E3687–E3695. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Musselman CA and Kutateladze TG:
Handpicking epigenetic marks with PHD fingers. Nucleic Acids Res.
39:9061–9071. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
He X, Duan C, Chen J, Ou-Yang X, Zhang Z,
Li C and Peng H: Let-7a elevates p21(WAF1) levels by targeting of
NIRF and suppresses the growth of A549 lung cancer cells. FEBS
Lett. 583:3501–3507. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wu J, Liu S, Liu G, Dombkowski A, Abrams
J, Martin-Trevino R, Wicha MS, Ethier SP and Yang ZQ:
Identification and functional analysis of 9p24 amplified genes in
human breast cancer. Oncogene. 333–341. 2012. View Article : Google Scholar
|
|
25
|
Wang F, Zhang P, Ma Y, Yang J, Moyer MP,
Shi C, Peng J and Qin H: NIRF is frequently upregulated in
colorectal cancer and its oncogenicity can be suppressed by let-7a
microRNA. Cancer Lett. 314:223–231. 2012. View Article : Google Scholar
|
|
26
|
Lu S, Yan D, Wu Z, Jiang T, Chen J, Yuan
L, Lin J, Peng Z and Tang H: Ubiquitin-like with PHD and ring
finger domains 2 is a predictor of survival and a potential
therapeutic target in colon cancer. Oncol Rep. 31:1802–1810.
2014.PubMed/NCBI
|
|
27
|
Wu TF, Zhang W, Su ZP, Chen SS, Chen GL,
Wei YX, Sun T, Xie XS, Li B, Zhou YX, et al: UHRF2 mRNA expression
is low in malignant glioma but silencing inhibits the growth of
U251 glioma cells in vitro. Asian Pac J Cancer Prev. 13:5137–5142.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Venturelli S, Armeanu S, Pathil A, Hsieh
CJ, Weiss TS, Vonthein R, Wehrmann M, Gregor M, Lauer UM and Bitzer
M: Epigenetic combination therapy as a tumor-selective treatment
approach for hepatocellular carcinoma. Cancer. 109:2132–2141. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Nightingale KP, Gendreizig S, White DA,
Bradbury C, Hollfelder F and Turner BM: Cross-talk between histone
modifications in response to histone deacetylase inhibitors: MLL4
links histone H3 acetylation and histone H3K4 methylation. J Biol
Chem. 282:4408–4416. 2007. View Article : Google Scholar
|
|
30
|
Luo T, Cui S, Bian C and Yu X: Uhrf2 is
important for DNA damage response in vascular smooth muscle cells.
Biochem Biophys Res Commun. 441:65–70. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Rajakumara E, Wang Z, Ma H, Hu L, Chen H,
Lin Y, Guo R, Wu F, Li H, Lan F, et al: PHD finger recognition of
unmodified histone H3R2 links UHRF1 to regulation of euchromatic
gene expression. Mol Cell. 43:275–284. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Citterio E, Papait R, Nicassio F, Vecchi
M, Gomiero P, Mantovani R, Di Fiore PP and Bonapace IM: Np95 is a
histone-binding protein endowed with ubiquitin ligase activity. Mol
Cell Biol. 24:2526–2535. 2004. View Article : Google Scholar : PubMed/NCBI
|