Introduction
There is accumulating evidence to suggest that the
inhalation of anaesthetics can induce nervous system injury which
is characterized by cognitive dysfunction, a lack of concentration
and an impaired ability to process information (1). Sevoflurane, for example, which is
one of the most commonly used inhaled anesthetics, has been
demonstrated to impair learning and memory abilities in aged rats
and to induce the apoptosis of rat hippocampal neurons (2). In addition, sevoflurane has been
demonstrated to induce apoptosis, to increase interleukin (IL)-6
expression and to induce the activation of the nuclear factor-κB
(NF-κB) signaling pathway in human neuroglioma cells (3). Therefore, more attention should be
paid to the use of inhaled anesthetics, such as sevoflurane in
elderly patients, while finding an agent that can counteract the
nervous system injury induced by the inhalation of anesthestics
would be of great significance for reducing the potential adverse
effects of anesthetics.
Minocycline, a second generation semi-synthetic
derivative of tetracycline antibiotics, is mainly used in the
treatment of infectious diseases in clinical practice. Minocycline
can easily pass through the blood-brain barrier (BBB) (4), and has been demonstrated to
attenuate neuronal cell death and improve the symptoms of
neurodegenerative disorders in animal models (5,6).
Recent studies have demonstrated that minocycline exerts
neuroprotective effects by suppressing inflammation (7,8)
and combating oxidative stress in the brain (9). Our group recently reported that
minocycline attenuated sevoflurane-induced cognitive impairment in
aged rats (10), suggesting that
minocycline may be a promising agent which may be used to
counteract anesthetic-induced neurotoxicity. Hence, in this study,
we aimed to validate the cytoprotective effects of minocycline and
to investigate the molecular mechanisms underlying the protective
effects of minocycline against anesthetic-induced cytotoxicity.
Hopefully, our study will shed some light on the discovery of more
neuroprotective agents for the prevention and treatment of
anesthetic-induced nervous system injury.
In our previous study, we identified that the NF-κB
signaling pathway was activated in sevoflurane-induced neuronal
injury in aged rats, and the excessive activation of NF-κB
signaling was inhibited by minocycline treatment (10). Yet, the exact mechanisms
underlying the sevoflurane-induced activation of NF-κB and its
inhibition by minocycline remain unclear. Nuclear factor E2-related
factor 2 (Nrf2) is an important stress response transcription
factor. In response to various stimulators, Nrf2 translocates from
the cytosol to the nucleus, where it binds to the antioxidant
response element (ARE) located in the promoter region of a spectrum
of oxidative stress-inducible genes (11). A crosstalk between the NF-κB and
Nrf2 pathways under conditions of oxidative stress and during
inflammation has been proposed in several studies (12–14). In this study, the involvement of
Nrf2 in the minocycline-implemented protective effects against
sevoflurane-induced cell injury was investigated. Furthermore, the
interaction between the Nrf2 and NF-κB pathways during this process
was characterized via a small interfering RNA (siRNA)-mediated
knockdown approach.
Materials and methods
Animal model of sevoflurane-induced
hippocampal neuronal injury and treatment with minocycline
Hippocampal neuronal injury in aged rats was induced
by the inhalation of sevoflurane as previously described (10). Briefly, 24 male, 20-month-old
Sprague-Dawley rats (n=6 per group) were purchased from the
Laboratory Animal Center of China Medical University (Shenyang,
China). The rats were either pre-treated or not with 45 mg/kg
minocycline (Dalian Meilun Biology Technology Co., Ltd., Dalian,
China) by intraperitoneal injection before being were subjected to
the inhalation of 2% sevoflurane (Jiangsu Hengrui Medicine Co.,
Ltd., Lianyungang, China) for 5 h, and the histology of the
hippocampus was analyzed 24 h later after the rats were sacrificed
and the brains were removed. All animal experiments and protocols
were approved by the Animal Ethics Committee of China Medical
University, Shenyang, China.
Terminal deoxynucleotidyl
transferase-mediated dUTP nick-end labeling (TUNEL) assay
The histology of the hippocampus and the apoptotic
status were examined by hematoxylin staining and by TUNEL assay
using the in situ Cell Death Detection kit (Roche
Diagnostics, Basel, Switzerland). Specifically, the brains were
fixed in 4% paraformaldehyde, paraffin-embedded and cut into 5
µm-thick sections. The sections were then dewaxed,
permeabilized with 0.1% Triton X-100 (Beyotime Institute of
Biotechnology, Haimen, China) and subjected to TUNEL assay
according to the manufacturer's instructions. Hematoxylin was used
to stain the nuclei. The sections were examined under an optical
microscopy at magnification, ×400.
Malondialdehyde (MDA) assay
The hippocampal tissue was excised from the mouse
brains, homogenized and centrifuged at 10,000 × g for 10 min at
4°C. The supernatant was collected, and the protein concentration
was determined using a BCA protein assay kit (Beyotime Institute of
Biotechnology). The level of MDA in the supernatant was measured
using the MDA assay kit (Jiancheng Bioengineering Institute,
Nanjing, China) according to the manufacturer's instructions.
Cell culture, transfection and
treatment
The H4 human neuroglioma cell line was obtained from
the Cell Bank of the Chinese Academy of Sciences (Shanghai, China).
The cells were cultured in RPMI-1640 (Gibco Life Technologies,
Grand Island, NY, USA) supplemented with 10% fetal bovine serum
(FBS; HyClone, Logan, UT, USA) in a humidified atmosphere of 5%
CO2 at 37°C.
The H4 cells were transfected with Nrf2 siRNA
(5′-GCACCUUAUAUCUCGAAGUTT-3′) or non-targeting scramble (NS)
oligonucleotides (5′-UUCUCCGAACGUGUCACGUTT-3′) (both were
synthesized by GenePharma, Shanghai, China) using Lipofectamine
2000 (Invitrogen, Carlsbad, CA, USA) according to the instructions
provided by the manufacturer. The cells were subjected to treatment
24 h after transfection.
The H4 cells were assigned to 4 experimental groups
as follows: i) the control group; ii) the minocycline group; iii)
the sevoflurane group; and iv) the minocycline + sevoflurane group.
Minocycline was dissolved in DMSO to yield a stock solution (40.5
mM), which was added to the culture medium to a final concentration
of 200 µM for treatment. An equal volume of DMSO was added
to the culture medium of the cells in the control and sevoflurane
groups. An ambient 4.1% sevoflurane in vitro, equivalent to
2 minimum alveolar concentration (MAC) in humans (15), has been previously shown to
increase IL-6 levels and to activate NF-κB signaling pathway in H4
cells (3). The cells were
incubated with or without 200 mM minocycline for 6 h in the
presence or absence of 4.1% sevoflurane in the atmosphere
consisting of 21% O2, 5% CO2 and the
remainder, N2.
Hoechst staining
The Hoechst staining kit (Beyotime Institute of
Biotechnology) was used to detect apoptotic cells according to the
manufacturer's instructions. The cells were seeded in 12-well
microplates and allowed to adhere. After treatment, the cells were
fixed, washed with PBS and stained with 0.5 ml Hoechst staining
solution for 5 min. Thereafter, the cells were washed, observed
under a fluorescence microscopy (BX53; Olympus, Tokyo, Japan) and
photographed at a magnification of ×400.
Apoptosis assay by flow cytometry
Cell apoptosis was determined by flow cytometry
using an Annexin V/propidium iodide (PI) apoptosis detection kit
(Beyotime Institute of Biotechnology). The cells were harvested and
resuspended in 500 µl binding buffer. Anti-Annexin V-FITC
antibody (5 µl) and PI (5 µl) were added to the cell
suspension, and the cells were then incubated for 15 min at room
temperature, followed by flow cytometric analysis on a FACSCalibur
flow cytometer (BD Biosciences, Franklin Lakes, NJ, USA).
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was extracted using a RNAsimple total RNA
kit (Tiangen Biotech Co., Ltd., Beijing, China), and reversed
transcribed into cDNA by Super M-MLV reverse transcriptase (BioTeke
Corporation, Beijing, China). Quantitative PCR (qPCR) was performed
using the cDNA template, SYBR-Green Master Mix (Solarbio, Beijing,
China) and the following primers: homo Nrf2 forward,
5′-CAGTCAGCGACGGAAAGAGTA-3′ and reverse,
5′-TGTGGGCAACCTGGGAGTAG-3′; homo β-actin forward,
5′-CTTAGTTGCGTTACACCCTTTCTTG-3′ and reverse,
5′-CTGTCACCTTCACCGTTCCAGTTT-3′; rat Nrf2 forward,
5′-CCCATTGAGGGCTGTGATCT-3′ and reverse, 5′-GTTGGCTGTGCTTTAGGTCC-3′;
rat β-actin forward, 5′-GGAGATTACTGCCCTGGCTCCTAGC-3′ and reverse,
5′-GGCCGGACTCATCGTACTCCTGCTT-3′. The fluorescence signals were
detected using the Exicycler™ 96 quantitative fluorescence analyzer
(Bioneer, Daejeon, Korea).
Western blot analysis
NP-40 cell lysis buffer (Beyotime Institute of
Biotechnology) was used to extract total proteins from the H4
cells, and RIPA lysis buffer (Beyotime Institute of Biotechnology)
was used to extract total proteins from the hippocampal tissues.
Nuclear and cytosolic proteins were extracted using the nuclear and
cytosolic protein extraction kit (Beyotime Institute of
Biotechnology) in accordance with the manufacturer's instructions.
BCA protein assays (Beyotime Institute of Biotechnology) were used
to determine protein concentrations. Proteins (40 µg) from
each sample were separated by SDS-PAGE and transferred onto a PVDF
membrane (Millipore, Bedford, MA, USA). After blocking with 5%
non-fat milk at room temperature for 1 h, the membrane was
incubated with the following specific primary antibodies:
anti-p-IκBα (1:500; Cat. no. bs-5515R; BIOSS, Beijing, China),
anti-Nrf2 (1:400; Cat. no. BA3790; Boster, Wuhan, China),
anti-caspase-3 (Cat. no. sc-56053), anti-Bax (Cat. no. sc-7480),
anti-Bcl-2 (Cat. no. sc-7382), anti-IL-6 (Cat. no. sc-130326) and
anti-NF-κB (Cat. no. sc-8008) (all 1:1000; all from Santa Cruz
Biotechnology, Inc., Dallas, TX, USA) overnight at 4°C.
Subsequently, the membrane was incubated with corresponding
horseradish peroxidase (HRP)-labeled secondary antibodies (Cat.
nos. A0208 and A0216; Beyotime Institute of Biotechnology) at 37°C
for 45 min. The signals of the proteins were visualized using the
ECL reagents (7SeaPharmTech, Shanghai, China). The membranes
blotted for p-IκBα was stripped and re-blotted for IκBα with an
anti-IκBα antibody (Cat. no. sc-1643, Santa Cruz Biotechnology,
Inc.). All membranes were stripped and re-probed with anti-β-actin
(Cat. no. sc-69879; Santa Cruz Biotechnology, Inc.), anti-histone
H3 (Cat. no. bsm-33042M; BIOSS) or anti-lamin A (Cat. no. sc-56137;
Santa Cruz Biotechnology, Inc.) to verify equal loading and
transfer of protein samples, wherein β-actin and lamin A/histone H3
were used as the corresponding antibodies (Santa Cruz
Biotechnology, Inc.) as the internal controls for cytosolic and
nuclear proteins, respectively. Following exposure, the films were
scanned and analyzed by Gel-Pro analyzer software to determine the
densitometric values of the target bands.
Enzyme-linked immunosorbent assay
(ELISA)
A human IL-6 ELISA kit (R&D Systems, Inc.,
Minneapolis, MN, USA) was used to detect the level of IL-6 in the
H4 culture medium. The cells were seeded in 12-well microplates and
cultured to 80% confluence. After the indicated treatments, the
culture medium was collected and tested for the level of IL-6. The
absorbance at 450 nm was measured using the ELX-800 absorbance
microplate reader (Bio-Tek Instruments, Inc., Winooski, VT, USA).
The concentration of IL-6 was calculated based on the standard
curve generated with the BSA standards in the kit.
Reactive oxygen species (ROS) assay
Intracellular ROS production was determined using
the ROS assay kit (Beyotime Institute of Biotechnology) according
to the manufacturer's instructions. After being subjected to the
indicated treatments, the cells were incubated in serum-free medium
containing 10 µM 2′,7′-dichlorodihydrofluorescein diacetate
(H2DCFDA) for 20 min at 37°C. The cells were then washed
with serumfree medium 3 times, resuspended in 500 µl PBS,
and analyzed by flow cytometry.
Immunofluorescence staining
The cells were fixed in 4% paraformaldehyde for 15
min, briefly rinsed with PBS, and permeabilized with 0.1% Triton
X-100. After blocking with goat serum for 15 min, the cells were
incubated with 1:100 diluted anti-NF-κB p65 antibody (1:100; Cat.
no. sc-8008; Santa Cruz Biotechnology) overnight at 4°C, and then
with 1:200 diluted FITC-labeled goat anti-rabbit secondary antibody
(Cat. no. A0562; Beyotime Institute of Biotechnology) for 60 min at
room temperature. Thereafter, the cell nuclei were counter-stained
with DAPI, and the cells were observed under a fluorescence
microscopy (BX53; Olympus) and photographed at a magnification of
×400.
Statistical analysis
GraphPad Prism 5 software was used for data
processing and plotting. Values are expressed as the means ±
standard deviation. Comparisons between multiple groups were
performed by one-way analysis of variance (ANOVA), followed by
Bonferroni's post-hoc test for comparisons between 2 groups. A
value of P<0.05 was considered to indicate a statistically
significant difference.
Results
Minocycline attenuates
sevoflurane-induced neuronal injury and oxidative stress in the rat
hippocampus
The effect of minocycline on hippocampal neuronal
injury in sevoflurane-exposed rats was assessed by TUNEL assay and
histological examination. As shown in Fig. 1A, sevoflurane induced the
degeneration of rat hippocampal neurons, as evidenced by
condensation of the cell nuclei, TUNEL-labeled chromosomal
fragmentation, as well as by the sparse and irregular arrangement
of the neurons. By contrast, minocycline significantly inhibited
sevoflurane-induced neuronal apoptosis and prevented neuronal
degeneration in the hippocampus.
ROS is known to be generated during cell metabolic
processes and can induce apoptosis through oxidative reactions
(16,17). Thus, in this study, we assessed
oxidative stress in the rat hippocampus by measuring the level of
MDA, a final product of lipid peroxidation. The results revealed
that sevoflurane markedly increased the MDA content in the
hippocampus, whereas minocycline significantly suppressed the
sevoflurane-induced elevation of MDA (Fig. 1B), suggesting that the
neuroprotective effects of minocycline are associated with its
antioxidant properties.
Minocycline inhibits the
sevoflurane-induced apoptosis of and ROS generation in H4
cells
To investigate the mechanisms underlying the
protective effects of minocycline against anesthesia-induced
toxicity, we employed an in vitro model using the H4 cell
line. Hoechst staining and flow cytometric analysis were performed
to assess the apoptotic status of the H4 cells subjected to the
different treatments (exposure to sevoflurane and/or treatment with
minocycline). The increased membrane permeability of apoptotic
cells allows the uptake of the Hoechst stain by the cells, such
that the Hoechst stain binds to the chromosomal DNA and emits
fluorescence under a fluorescence microscope. As shown in Fig. 2A, the H4 cells that were
co-treated with minocycline and sevoflurane exhibited lower numbers
of Hoechst-positive cells, as compared with those treated with
sevoflurane alone, suggesting that minocycline attenuated the
sevoflurane-induced apoptosis of H4 cells. Consistently, the
results of flow cytometric analysis revealed that the apoptotic
ratio in the sevoflurane-treated cells (19.14±1.97%) was
significantly increased compared with that in the control cells
(6.96±0.84%) (Fig. 2B and C;
P<0.001), whereas minocycline co-treatment markedly reduced the
apoptotic ratio (11.33±1.39%) compared with the sevoflurane-exposed
cells (Fig. 2B and C;
P<0.001). These results confirmed the protective effects of
minocycline against sevoflurane-induced cell apoptosis. In
addition, the results of western blot analysis indicated that
exposure to sevoflurane led to the downregulation of Bcl-2
(Fig. 2D and E; P<0.01), the
upregulation of Bax and to the elevated level of cleaved caspase-3,
as compared with the control cells (Fig. 2D, F and G; P<0.001 and
P<0.01, respectively). By contrast, the sevoflurane-induced
alterations in the protein levels of Bcl-2, Bax and cleaved
caspase-3 were reversed by co-treatment with minocycline. Taken
together, the above-mentioned results indicated that minocycline
inhibited the sevoflurane-induced apoptosis of H4 cells.
H2DCFDA assay was performed to
investigate whether sevoflurane-induced cell injury and apoptosis
are attributed to intracellular ROS production. The ratio of cells
generating moderate to high levels of ROS
(H2DCFDA-positive) was significantly increased in the
sevoflurane-exposed cells as compared with the control cells
(P<0.001), whereas minocycline reduced the number of
ROS-producing cells induced by sevoflurane (P<0.01) (Fig. 2H and I). Thus, minocycline
suppressed sevoflurane-induced ROS production in H4 cells.
Minocycline suppresses the
sevoflurane-induced upregulation of IL-6 expression and the
activation of the NF-κB signaling pathway
Our previous study demonstrated that minocycline
inhibited the sevoflurane-induced upregulation of IL-6 and the
activation of NF-κB in the rat hippocampus (10). In this study, in the H4 cells,
minocycline significantly reduced the sevoflurane-induced elevation
of secreted and intracellular IL-6 (Fig. 3A and B). In addition, compared
with the control cells, the levels of the proteolytic and
phosphorylated form of IκBα (i.e., p-IκBα) and nuclear NF-κB p65
were increased in the sevoflurane-exposed cells, resulting in
reduced levels of full-length IκBα and cytosolic NF-κB p65.
Sevoflurane-stimulated IκBα phosphorylation and NF-κB nuclear
translocation were significantly inhibited by minocycline
co-treatment (Fig. 3C–E).
Moreover, immunofluorescence staining also revealed the nuclear
translocation of NF-κB p65 upon exposure to sevoflurane, which was
blocked by minocycline (Fig. 3F).
These results indicated that minocycline inhibited the
sevoflurane-induced activation of NF-κB signaling and the
production of IL-6 in H4 cells.
Minocycline further augments the
upregulation and activation of Nrf2 in the hippocampus of
sevoflurane-treated rats and in H4 cells
In our rat model, the prolonged inhalation of
sevoflurane led to increased mRNA and protein levels of Nrf2 and
Nrf2 in the hippocampus (Fig. 4A and
B), implying that exposure to sevoflurane led to the
upregulated expression and activation/nuclear transclocation of
Nrf2. Of note, minocycline alone did not affect Nrf2 expression or
activation in the rat hippocampus, but further augmented Nrf2
expression and activation when used in conjunction with sevoflurane
(Fig. 4A and B). Similar
observations were noted in the H4 cells; minocycline further
enhanced the sevoflurane-induced upregulation and activation of
Nrf2 (Fig. 4C and D).
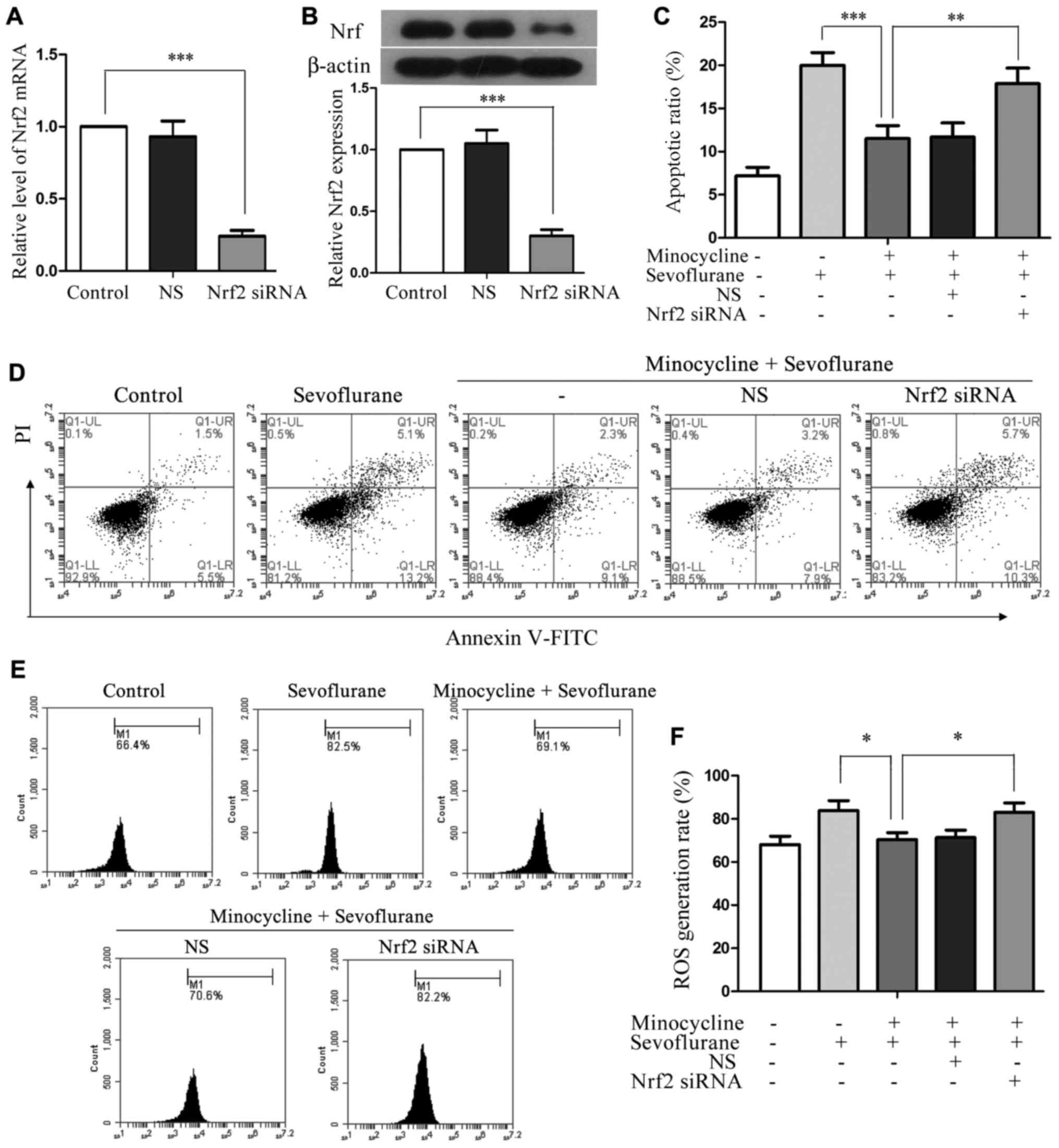
Silencing of Nrf2 attenuates the
cytoprotective effects of minocycline in sevoflurane-treated H4
cells
To investigate whether minocycline exerts its
cytoprotective effect via Nrf2, we used a siRNA-mediated knockdown
approach. Nrf2 siRNA was transfected into H4 cells and achieved
approximately 75% reduction in the expression of Nrf2 at both the
mRNA and protein level, whereas the NS control sequence did not
affect Nrf2 expression or activation (Fig. 5A and B). Compared with the cells
co-treated with sevoflurane and minocycline, the Nrf2
siRNA-transfected cells displayed a significantly increased
apoptotic ratio (P<0.01; Fig. 5C
and D) and an increased number of ROS-producing cells following
co-treatment (P<0.05; Fig. 5E and
F). These results demonstrated that the anti-apoptotic and
anti-oxidative effects of minocycline in sevoflurane-induced cell
injury were compromised when Nrf2 was silenced, suggesting that
minocycline may exert its cytoprotective effects via the activation
of Nrf2.
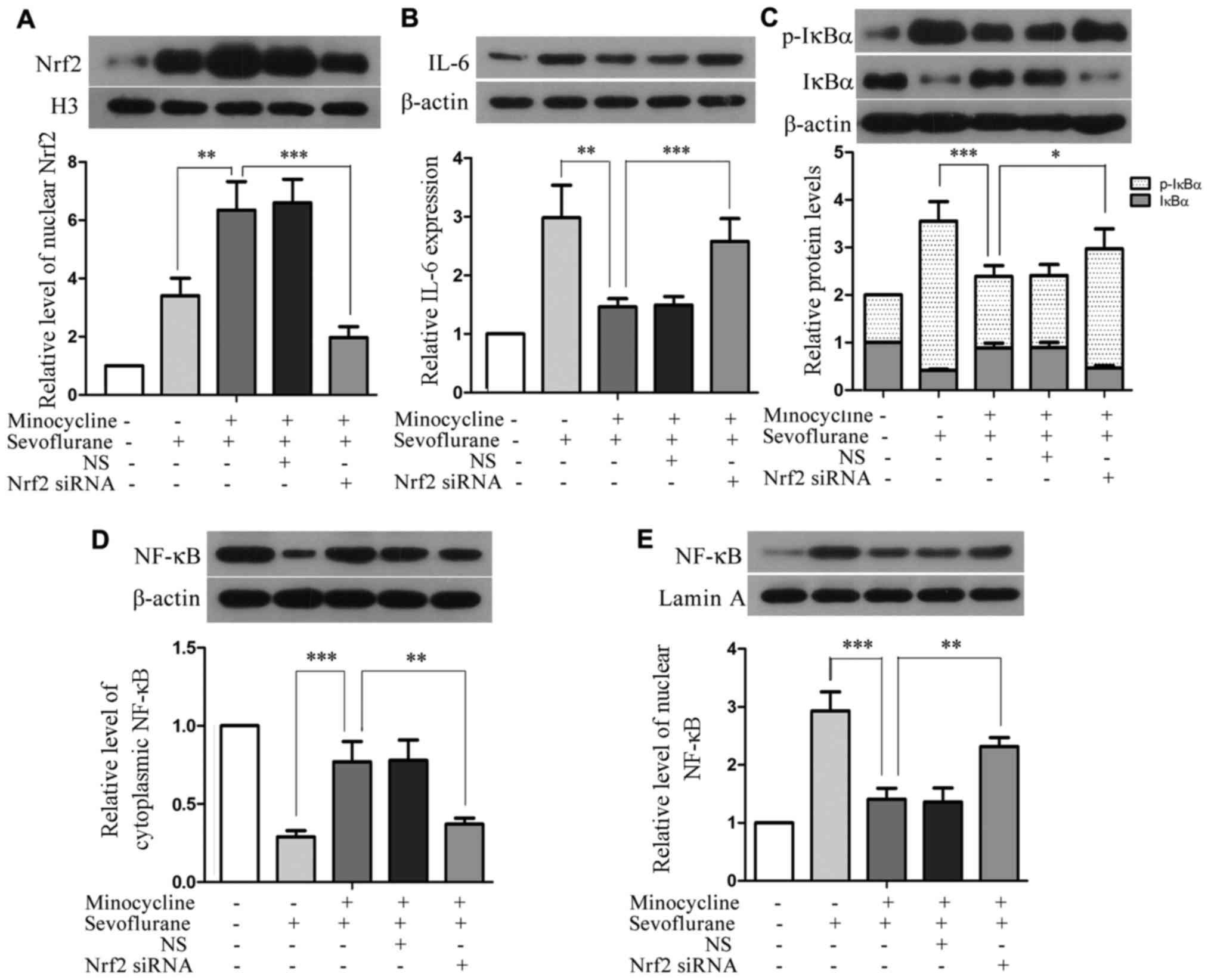
Silencing of Nrf2 attenuates the
inhibitory effects of minocycline on the sevoflurane-induced
upregulation of IL-6 and the activation of NF-κB
Compared with the H4 cells co-treated with
sevoflurane and minocycline, the level of nuclear Nrf2 was
profoundly reduced in the Nrf2 siRNA-transfected cells subjected to
co-treatment (P<0.01), although Nrf2 siRNA did not completely
abolish Nrf2 upregulation (Fig.
6A). Moreover, compared with the cells co-treated with
sevoflurane and minocycline, the silencing of Nrf2 resulted in
elevated levels of IL-6, p-IκBα and nuclear NF-κB, and reduced
levels of IκBα and cytosolic NF-κB in the cells subjected to
co-treatment (Fig. 6B–E),
implying that the minocycline-mediated inhibition of IL-6
upregulation and the activation of the NF-κB signaling pathway in
sevoflurane-induced cell injury were attenuated upon the silencing
of Nrf2. These results suggest that minocycline suppresses the
sevoflurane-induced upregulation of IL-6 and the activation of
NF-κB through Nrf2.
Discussion
Minocycline, a broad-spectrum tetracycline
antibiotic, has been shown to exhibit independent neuroprotective
properties and therapeutic effects in various experimental models
of neurodegenerative diseases (5–9);
yet the underlying molecular mechanisms responsible for its
neuroprotective function remain to be elucidated. Our group
previously reported the neuroprotective effect of minocycline in
counteracting sevoflurane-induced neurotoxicity in rats (10). In this study, we further
characterized the molecular mechanisms responsible for the
protective effects of minocycline against sevoflurane-induced
neuronal injury. Our results demonstrated that minocycline
effectively inhibited sevoflurane-induced cell apoptosis, ROS
production and the activation of the NF-κB pathway, and
simultaneously stimulated the activation of the Nrf2 antioxidant
pathway. Our study provides evidences of the potential clinical
value of minocycline in the prevention of anesthetic-induced
neurotoxicity.
Cell apoptosis is regulated by multiple factors,
including caspase family proteins, Bcl-2, Bax and NF-κB signaling
(18). Among these factors, Bax
which belongs to the Bcl-2 gene family and caspase-3 have been
demonstrated to accelerate apoptosis, while Bcl-2 plays an
anti-apoptotic role in cells (19). Previous studies have shown that
minocycline inhibits the activation of the caspase pathway
(20), and exerts an
anti-apoptotic effect by upregulating the apoptotic suppressor
Bcl-2 and downregulating pro-apoptotic caspase-1 and caspase-3
(19,21). In this study, we demonstrated that
minocycline counteracted the sevoflurane-induced downregulation of
Bcl-2, while it significantly reduced the levels of Bax and cleaved
caspase-3, which were elevated upon exposure to sevoflurane. These
results indicate that minocycline exerts an anti-apoptotic effect
in injured cells by suppressing alterations in apoptotic regulatory
proteins.
ROS generation, as part of the normal physiological
metabolic process, plays important roles in the regulation of cell
survival and apoptosis (16,17). Minocycline has been shown to play
a protective role by reducing the production of ROS in
6-OHDA-induced PC12 cell injury and in primary rat oligodendroglial
precursor cells upon oxygen-glucose deprivation (22,23). Consistently, our results also
demonstrated that minocycline suppressed the elevation of ROS
production in the injured H4 cells challenged with sevoflurane.
Nrf2 is known to be activated in response to oxidative stress, and
it in turn transactivates the expression of antioxidant response
genes (11). In this study, Nrf2
was activated upon sevoflurane-induced oxidative stress both in
vivo and in vitro. Intriguingly, minocycline alone had
no effect on Nrf2 activation or ROS generation, but it further
augmented the activation of Nrf2 when used in conjunction with
sevoflurane, and suppressed the sevoflurane-induced elevation of
ROS production. Furthermore, the antioxidant effects of minocycline
were markedly attenuated when Nrf2 expression was downregulated by
siRNA. These results suggest that minocycline exerts its
antioxidant effects by activating the antioxidant machinery via
Nrf2.
NF-κB exists as a dimer composing of polypeptide
chain p50 and p65. IκB, an inhibitor of NF-κB signaling, binds to
the NF-κB dimer to form a trimer in the cytoplasm, and undergoes
proteolysis and phosophorylation in response to the activating
stimuli of NF-κB signaling, resulting in the dissociation of the
trimer and NF-κB nuclear translocation. Previous studies have shown
that elevated cellular ROS levels can contribute to the prolonged
activation of c-Jun N-terminal kinase (JNK) and other kinases,
leading to the phosphorylation of IκB by IκB kinase (IKK) (24), and the nuclear translocation of
NF-κB. Hence, sevoflurane-induced ROS elevation and the
counteracting action by minocycline in this study is probably the
regulatory mechanism for the activation of the NF-κB signaling
pathway. In the nucleus, NF-κB activates the transcription of
downstream genes that are critical players in inflammation and
apoptosis, such as IL-6 (25).
Moreover, both Bcl-2 and Bax have been shown to be downstream
targets of NF-κB, and the transcriptional tuning of these two genes
by NF-κB for promoting or inhibiting apoptosis depends on the
stimulus and the cell type (26).
In addition, it is well recognized that the ROS-mediated crosstalk
between NF-κB and JNK signaling is critical for the regulation of
the Bcl-2/Bax ratio and apoptosis (27). In the present study, our results
suggested that the activation of NF-κB signaling was involved in
the sevoflurane-induced cytotoxicity and apoptosis. Moreover, the
silencing of Nrf2 inhibited the activation of the NF-κB signaling
pathway, and this may be mediated via Nrf2-controlled intracellular
oxidative stress. Therefore, alterations in cellular ROS levels,
modulated by the Nrf2 pathway, signal the NF-κB pathway and control
cell fate.
In conclusion, our findings demonstrate that
minocycline attenuates sevoflurane-induced cell injury by
inhibiting apoptosis, reducing ROS production and blocking the
activation of the NF-κB signaling pathway. Its antioxidant and
anti-apoptotic effects are likely to be mediated via the activation
of Nrf2. Our study suggests a protective property of minocycline
against anesthetic-induced cell injury, and provides insight into
the mechanisms underlying the cytoprotective properties of
minocycline. Thus, we propose further development and validation of
minocycline as a cytoprotective agent for its potential clinical
application in the prevention/treatment of anesthetic-induced
nervous system injury.
Acknowledgments
This study was supported by grants from the National
Natural Science Foundation of China (nos. 81473285, 81101402 and
81302534).
References
|
1
|
Bittner EA, Yue Y and Xie Z: Brief review:
anesthetic neurotoxicity in the elderly, cognitive dysfunction and
Alzheimer's disease. Can J Anaesth. 58:216–223. 2011. View Article : Google Scholar
|
|
2
|
Xiong WX, Zhou GX, Wang B, Xue ZG, Wang L,
Sun HC and Ge SJ: Impaired spatial learning and memory after
sevoflurane-nitrous oxide anesthesia in aged rats is associated
with down-regulated cAMP/CREB signaling. PLoS One. 8:e794082013.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Zhang L, Zhang J, Yang L, Dong Y, Zhang Y
and Xie Z: Isoflurane and sevoflurane increase interleukin-6 levels
through the nuclear factor-kappa B pathway in neuroglioma cells. Br
J Anaesth. 110(Suppl 1): i82–i91. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Yang LP, Zhu XA and Tso MO: Minocycline
and sulforaphane inhibited lipopolysaccharide-mediated retinal
microglial activation. Mol Vis. 13:1083–1093. 2007.PubMed/NCBI
|
|
5
|
Choi Y, Kim HS, Shin KY, Kim EM, Kim M,
Kim HS, Park CH, Jeong YH, Yoo J, Lee JP, et al: Minocycline
attenuates neuronal cell death and improves cognitive impairment in
Alzheimer's disease models. Neuropsychopharmacology. 32:2393–2404.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Yrjänheikki J, Tikka T, Keinänen R,
Goldsteins G, Chan PH and Koistinaho J: A tetracycline derivative,
minocycline, reduces inflammation and protects against focal
cerebral ischemia with a wide therapeutic window. Proc Natl Acad
Sci USA. 96:13496–13500. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Desjarlais M, Pratt J, Lounis A, Mounier
C, Haidara K and Annabi B: Tetracycline derivative minocycline
inhibits autophagy and inflammation in concanavalin-a-activated
human hepatoma cells. Gene Regul Syst Bio. 8:63–73. 2014.PubMed/NCBI
|
|
8
|
Yoon SY, Patel D and Dougherty PM:
Minocycline blocks lipopolysaccharide induced hyperalgesia by
suppression of microglia but not astrocytes. Neuroscience.
221:214–224. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kuang X, Scofield VL, Yan M, Stoica G, Liu
N and Wong PK: Attenuation of oxidative stress, inflammation and
apoptosis by minocycline prevents retrovirus-induced
neurodegeneration in mice. Brain Res. 1286:174–184. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Tian Y, Guo S, Wu X, Ma L and Zhao X:
Minocycline alleviates sevoflurane-induced cognitive impairment in
aged rats. Cell Mol Neurobiol. 35:585–594. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ishii T, Itoh K, Takahashi S, Sato H,
Yanagawa T, Katoh Y, Bannai S and Yamamoto M: Transcription factor
Nrf2 coordinately regulates a group of oxidative stress-inducible
genes in macrophages. J Biol Chem. 275:16023–16029. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lyu JH, Kim KH, Kim HW, Cho SI, Ha KT,
Choi JY, Han CW, Jeong HS, Lee HK, Ahn KS, et al: Dangkwisoo-san,
an herbal medicinal formula, ameliorates acute lung inflammation
via activation of Nrf2 and suppression of NF-kappaB. J
Ethnopharmacol. 140:107–116. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Cuadrado A, Martín-Moldes Z, Ye J and
Lastres-Becker I: Transcription factors NRF2 and NF-kappaB are
coordinated effectors of the Rho family, GTP-binding protein RAC1
during inflammation. J Biol Chem. 289:15244–15258. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Mahmoud-Awny M, Attia AS, Abd-Ellah MF and
El-Abhar HS: Mangiferin mitigates gastric ulcer in
ischemia/reperfused rats: involvement of PPAR-gamma, NF-kappaB and
Nrf2/HO-1 signaling pathways. PLoS One. 10:e01324972015. View Article : Google Scholar
|
|
15
|
Franks NP and Lieb WR: Temperature
dependence of the potency of volatile general anesthetics:
implications for in vitro experiments. Anesthesiology. 84:716–720.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Jacobson MD: Reactive oxygen species and
programmed cell death. Trends Biochem Sci. 21:83–86. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Simon HU, Haj-Yehia A and Levi-Schaffer F:
Role of reactive oxygen species (ROS) in apoptosis induction.
Apoptosis. 5:415–418. 2000. View Article : Google Scholar
|
|
18
|
Heo K, Cho YJ, Cho KJ, Kim HW, Kim HJ,
Shin HY, Lee BI and Kim GW: Minocycline inhibits caspase-dependent
and -independent cell death pathways and is neuroprotective against
hippocampal damage after treatment with kainic acid in mice.
Neurosci Lett. 398:195–200. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wang J, Wei Q, Wang CY, Hill WD, Hess DC
and Dong Z: Minocycline up-regulates Bcl-2 and protects against
cell death in mitochondria. J Biol Chem. 279:19948–19954. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Huang C, Li R, Zeng Q, Ding Y, Zou Y, Mao
X, Hu W, Xiong R and Li M: Effect of minocycline postconditioning
and ischemic postconditioning on myocardial ischemia-reperfusion
injury in atherosclerosis rabbits. J Huazhong Univ Sci Technolog
Med Sci. 32:524–529. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Chen M, Ona VO, Li M, Ferrante RJ, Fink
KB, Zhu S, Bian J, Guo L, Farrell LA, Hersch SM, et al: Minocycline
inhibits caspase-1 and caspase-3 expression and delays mortality in
a transgenic mouse model of Huntington disease. Nat Med. 6:797–801.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Jiang BP, Le L, Xu LJ and Xiao PG:
Minocycline inhibits ICAD degradation and the NF-kappaB activation
induced by 6-OHDA in PC12 cells. Brain Res. 1586:1–11. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Schmitz T, Endesfelder S, Chew LJ, Zaak I
and Bührer C: Minocycline protects oligodendroglial precursor cells
against injury caused by oxygen-glucose deprivation. J Neurosci
Res. 90:933–944. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Sakon S, Xue X, Takekawa M, Sasazuki T,
Okazaki T, Kojima Y, Piao JH, Yagita H, Okumura K, Doi T, et al:
NF-kappaB inhibits TNF-induced accumulation of ROS that mediate
prolonged MAPK activation and necrotic cell death. EMBO J.
22:3898–3909. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Xiao W, Hodge DR, Wang L, Yang X, Zhang X
and Farrar WL: Co-operative functions between nuclear factors
NF-kappaB and CCAT/enhancer-binding protein-beta (C/EBP-beta)
regulate the IL-6 promoter in autocrine human prostate cancer
cells. Prostate. 61:354–370. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Dutta J, Fan Y, Gupta N, Fan G and Gélinas
C: Current insights into the regulation of programmed cell death by
NF-kappaB. Oncogene. 25:6800–6816. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Nakano H, Nakajima A, Sakon-Komazawa S,
Piao JH, Xue X and Okumura K: Reactive oxygen species mediate
crosstalk between NF-kappaB and JNK. Cell Death Differ. 13:730–737.
2006. View Article : Google Scholar
|