Introduction
Hepatic fibrosis is caused by various types of
chronic liver injury. This progressive pathological process is
described as accumulation of extracellular matrix (ECM) proteins in
and around injured liver tissues (1). Cholestasis results in intrahepatic
accumulation of cytotoxic bile acids and hepatic inflammation,
which is then followed by biliary fibrosis, cirrhosis and finally
end-stage liver disease (2,3).
Cholestatic liver disease such as primary biliary cirrhosis and
primary sclerosing cholangitis is characterized by a progressive
destruction of biliary epithelial cells (BECs) and inflammatory and
autoimmune disorders (4,5).
Proliferating BECs have been shown to secrete
transforming growth factor-β1 (TGF-β1) and platelet-derived growth
factor (PDGF), which stimulate the activation and proliferation of
hepatic stellate cells (HSCs) and portal fibroblasts (6,7).
Activated HSCs and portal fibroblasts cause enhanced collagen
deposition and are the major cellular effectors in liver fibrosis
(1,8). This ultimately leads to excessive
generation of ECM and accelerates the progression of fibrosis
(9). Thus, the suppression of
proliferating BECs and activated HSCs has been considered to be a
therapeutic target for treating liver fibrosis.
Apamin is an 18 amino acid peptide neurotoxin found
in apitoxin (bee venom) (10). It
has long been known as a specifically selective blocker of
Ca2+-activated K+ (SK) channels (11). These channels play an important
role in mediating the increase in transepithelial secretion due to
increases in intracellular Ca2+ (12). Moreover, apamin has been
demonstrated to exhibit anti-inflammatory and anti-fibrotic
activity in various cell types and mouse models (13,14). A previous study carried out by our
group confirmed that apamin is an anti-fibrotic agent which acts
through suppression of TGF-β1-induced hepatocyte
epithelial-mesenchymal transition (13). However, the effects of apamin in
biliary cirrhosis and the molecular mechanism underlying HSC
proliferation have not been explored.
In the present study, we fed mice with
3,5-diethoxycarbonyl-1,4-dihydrocollidine (DDC), which induces
sclerosing cholangitis and biliary fibrosis. We demonstrated that
apamin inhibited DDC-induced liver fibrosis and mediated BEC
proliferation and ductular reaction, which are repair responses to
cholestatic injury. Moreover, apamin treatment caused the
suppression of activated HSCs through the TGF-β1/Smad signaling
pathway.
Materials and methods
Reagents
Apamin was purchased from Sigma (St. Louis, MO,
USA). TGF-β1 was purchased from R&D Systems (Minneapolis, MN,
USA) and dissolved in 4 mM HCl containing 0.1% bovine serum albumin
(BSA).
DDC-induced mouse model of biliary
fibrosis
For induction of liver injury, 8-week-old C57BL/6
male mice (20–25 g; Samtako, Osan, Korea) were selected. Male
C57BL/6 mice were fed a control diet or a DDC supplemented diet
(0.1%) for 4 weeks to induce advanced biliary fibrosis as
previously described (15). All
animal protocols were approved by the Institutional Animal Care and
Use Committee of Catholic University of Daegu (Daegu, Korea). The
mice received an intraperitoneal injection of apamin (0.1 mg/kg)
dissolved in saline twice a week. Mice were sacrificed after 4
weeks from the first DDC diet administration.
Cell culture
HSC-T6 cells, an immortalized rat hepatic stellate
cell line, which has a stable phenotype and biochemical
characteristics, was kindly provided by Dr S.L. Friedman (Liver
Center Laboratory, San Francisco General Hospital, San Francisco,
CA, USA). Cells were cultured at 37°C in a humidified incubator
under a 5% CO2 atmosphere. HSC-T6 cells were seeded in
complete medium for 24 h. The cells were changed to fresh
serum-free media containing the indicated concentrations of apamin
(0.5, 1 and 2 µg/ml). After 24 h, the cells were replaced
with fresh serum-free media containing 2 ng/ml of TGF-β1 for 24
h.
Histopathology and
immunohistochemistry
Hematoxylin and eosin (H&E), Masson's trichrome
and immunohistochemical staining were performed according to a
previously described procedure (15). Sections were stained with H&E
and Masson's trichrome. For immunohistochemical analysis, sections
were incubated with anti-fibroblast specific protein-1 (FSP-1)
(ab41532; Abcam, Cambridge, UK) for 1 h at 37°C, processed by an
indirect immunoperoxidase technique using a commercial kit (Dako,
Carpinteria, CA, USA). The slides were examined with an Eclipse 80i
microscope (Nikon, Tokyo, Japan) and analyzed with iSolution DT
software (IMT i-Solution, Vancouver, BC, Canada).
Immunofluorescence staining
Paraffin-embedded mouse liver sections (3-µm
thickness) were prepared by a routine procedure. After blocking
with 10% donkey serum for 30 min, the slides were immunostained
with primary antibodies against cytokeratin (CK)7 (ab9021), CK19
(ab52625) (Abcam), and proliferating cell nuclear antigen (PCNA)
(sc-56; Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA). After
washing, they were incubated with the secondary antibodies (Alexa
Flour 488 and/or Alexa Fluor 594) for 30 min at 37°C. Sections were
then counterstained with Hoechst 33342. Stained slides were imaged
using a Nikon A1+ confocal microscope (Nikon).
Immunocytochemistry
HSC-T6 cells were grown on chamber slides and were
fixed with 3.7% paraformaldehyde for 10 min. Cells were permeated
with 0.5% Triton X-100 for 10 min and then incubated with primary
antibodies against SMA (A5228; Sigma) for 1 h at 37°C. After
washing, the cells were incubated with the secondary antibodies
(Alexa Fluor 594) for 30 min at 37°C. Cells were counterstained
with Hoechst 33342. The cells were imaged using a Nikon A1+
confocal microscope.
Enzyme-linked immunosorbent assay
(ELISA)
Concentrations of interleukin-6 (IL-6) and
interferon-γ (IFN-γ) in serum were measured with ELISA kit (R&D
Systems). The OD was measured at 450 nm in an ELISA reader (BMG
Labtech, Mornington, Germany).
Western blot analysis
Western blotting was performed as previously
described (16). Primary
antibodies used in this study were the following; anti-p-Smad2/3
(#8828), anti-Smad2/3 (#8685) (Cell Signaling Technology, Danvers,
MA, USA), anti-IL-1β (sc-7884), anti-Smad4 (sc-7966),
anti-glyceraldehyde 3-phosphate dehydrogenase (GAPDH; sc-25778)
(Santa Cruz Biotechnology, Inc.), anti-SMA (A5228; Sigma),
anti-tumor necrosis factor-α (TNF-α; ab6671), anti-collagen I
(ab34710) (Abcam), TGF-β1 (MAB240-100; R&D Systems) and
anti-fibronectin (610077; BD Biosciences, San Diego, CA, USA).
Statistical analysis
The experimental results are expressed as mean ± SE.
ANOVA and paired or unpaired t-test were performed for statistical
analysis as appropriate. p-value <0.05 was considered to
indicate a statistically significant result. All experiments were
performed at least three times.
Results
Apamin ameliorates liver damage and
inflammatory hepatic injury
To investigate the effects of apamin treatment on
liver fibrosis, a mouse model induced by DDC diet feeding was used.
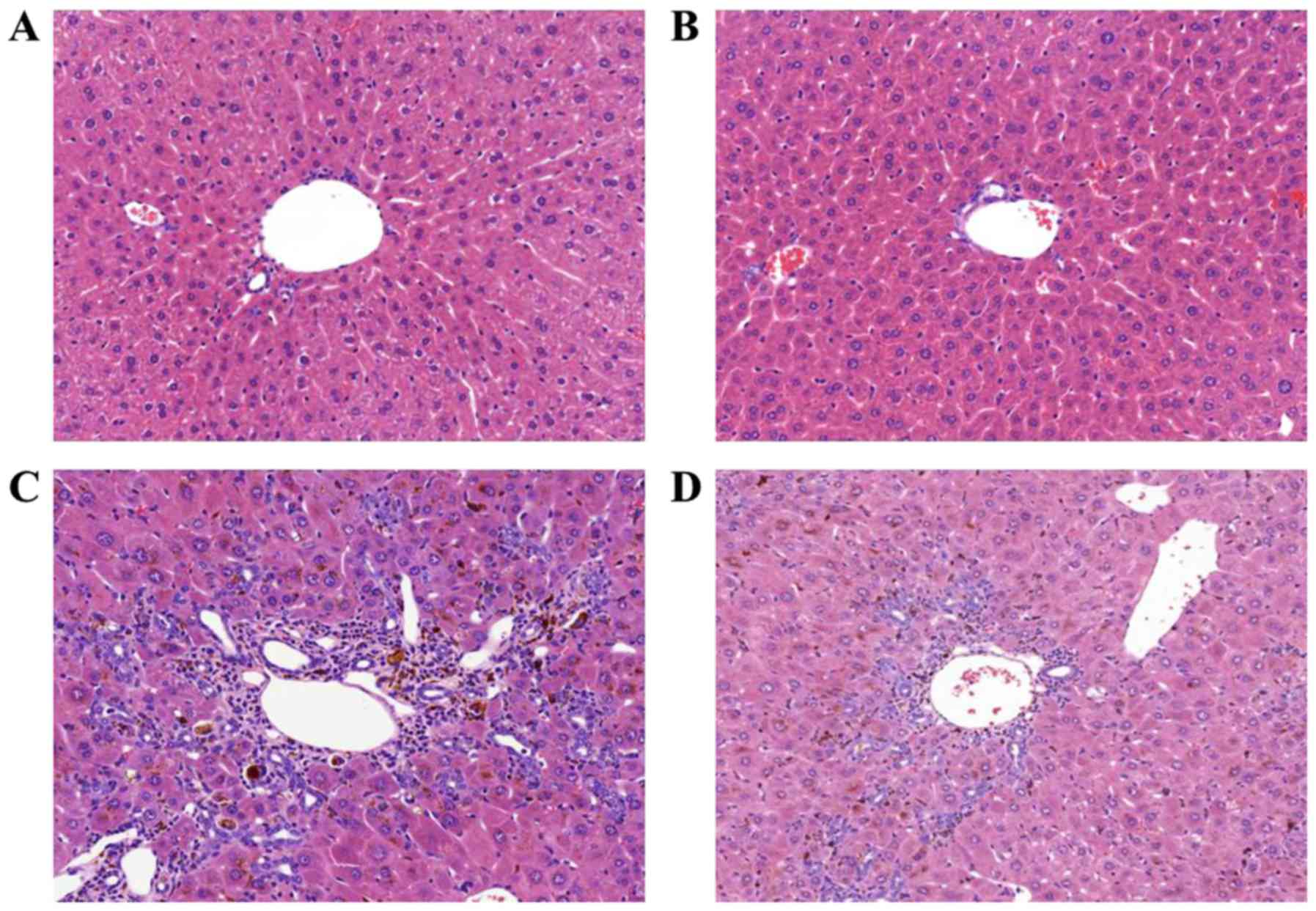
When challenged with the DDC diet for 4 weeks, the structure of the
hepatic lobule was clear in the NC group (Fig. 1A) and there was a large amount of
bile duct proliferation, accompanied by inflammatory cell
infiltration in the DDC-fed group as shown by H&E staining
(Fig. 1C). In addition, the above
pathological changes were reduced in the apamin-treated group
(Fig. 1D) compared with these
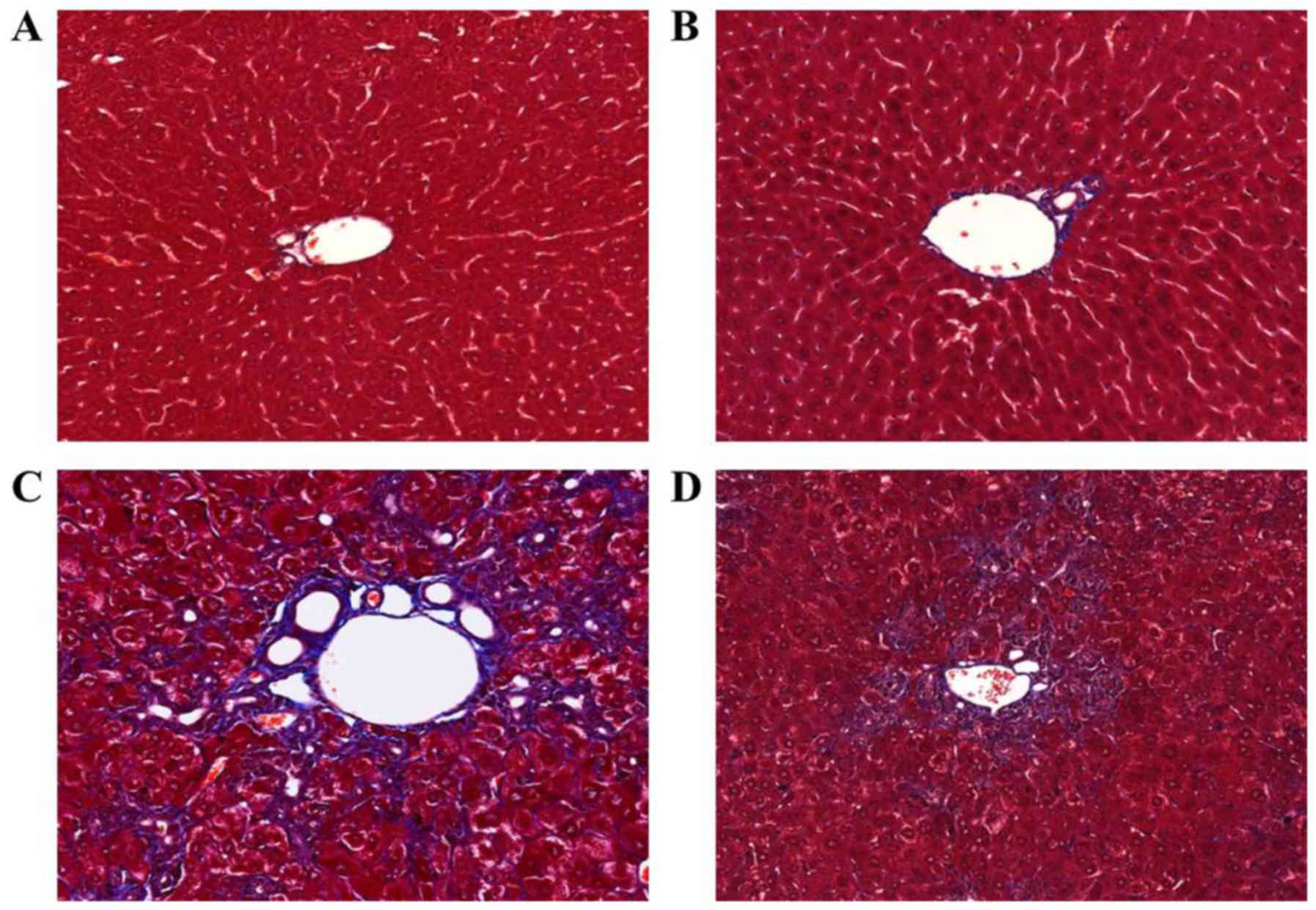
changes noted in the DDC-fed group. Masson's trichrome staining
indicated collagen deposition surrounding the proliferated bile
duct in the DDC-fed group (Fig.
2C). In contrast, apamin treatment resulted in diminished
fibrosis and collagen deposition (Fig. 2D).
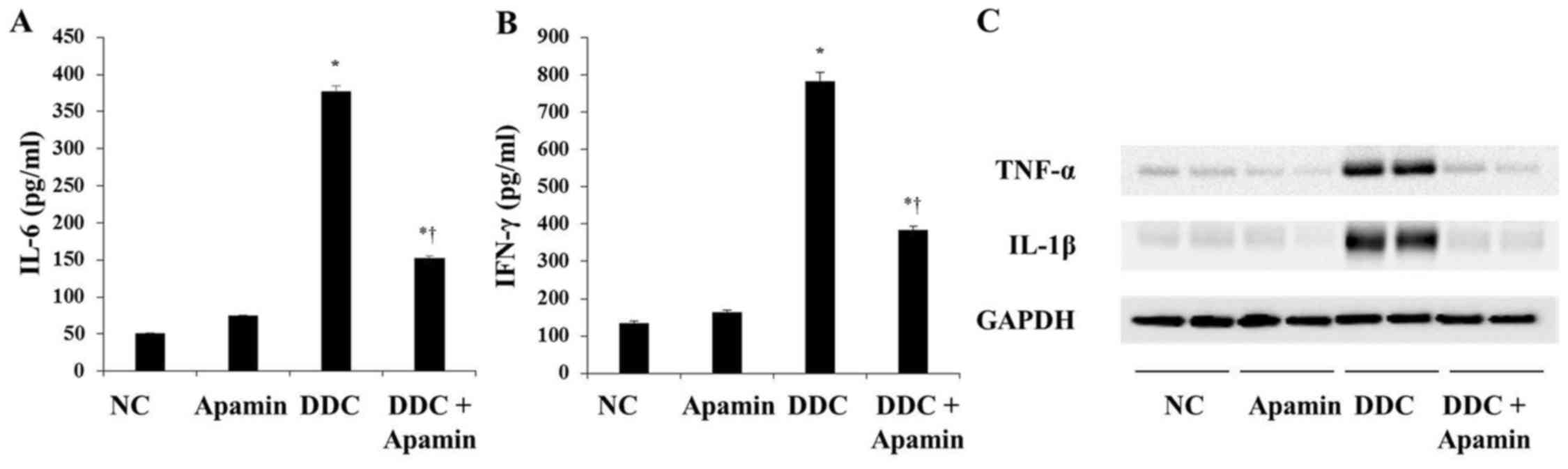
In cholangiopathies, inflammation and reactive
proliferation of bile ducts are closely related with the
development of biliary fibrosis (17). ELISA and western blot analyses
indicated that expression levels of IL-6, IFN-γ, TNF-α and IL-1β
were significantly higher in the DDC-fed group compared with these
levels in the NC group (Fig. 3).
However, apamin treatment attenuated inflammatory cytokine
expression, including IL-6, IFN-γ, TNF-α and IL-1β compared with
expression levels in the DDC-fed group. Taken together, these data
confirm the anti-inflammatory and moderate anti-fibrotic effects of
apamin on the DDC-fed mice.
Effect of apamin on BEC proliferation in
DDC-fed mice
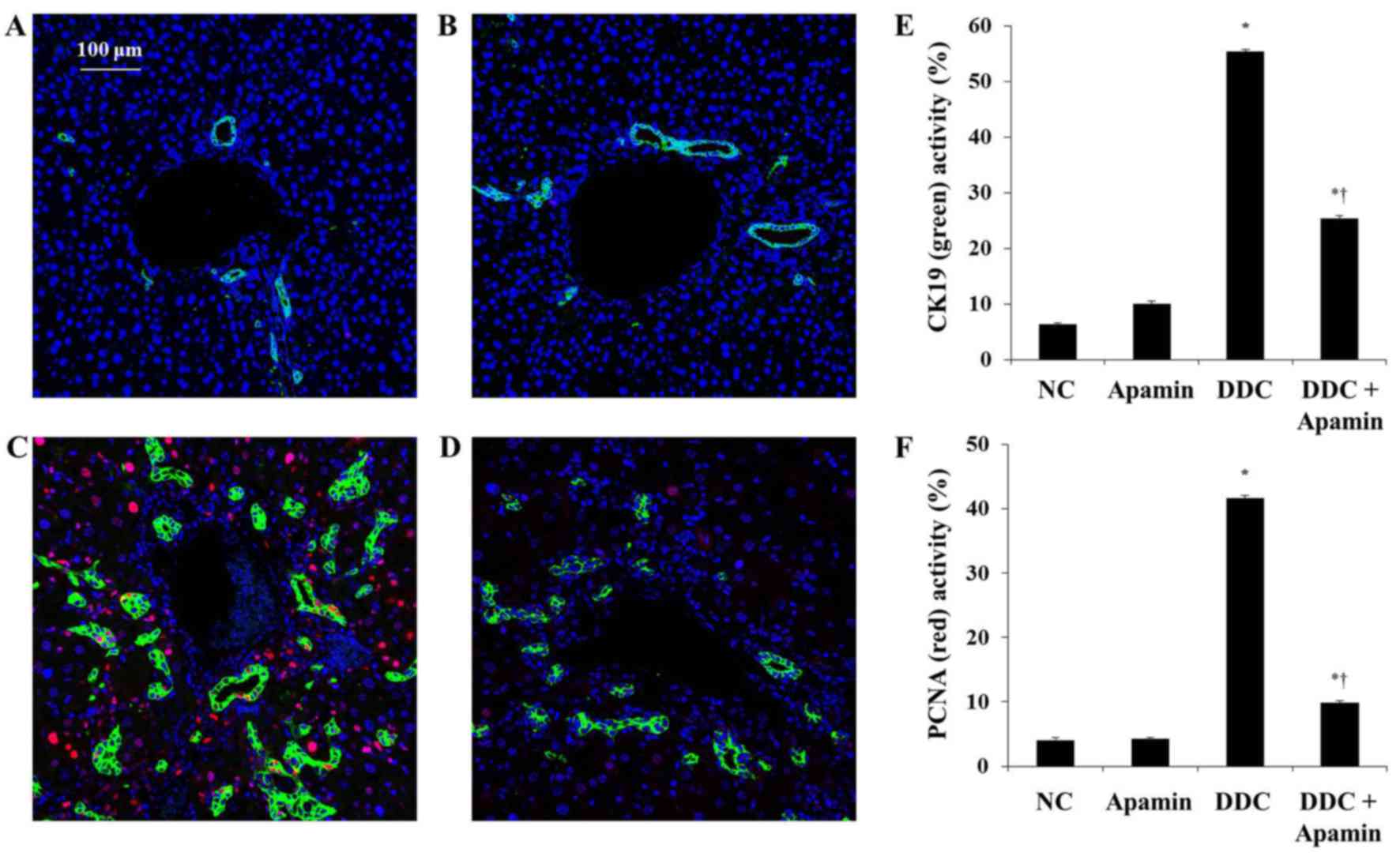
We next determined the effect of apamin on ductular
reaction in the DDC-fed mice by immunofluorescence of CK19
expression. Chronic DDC feeding in mice was previously demonstrated
to result in cholangitis and immune responses against BECs with the
destruction of bile ducts and ductules (18). CK19 is regarded as a hallmark of
bile epithelial cells (19).
Immunofluorescence staining showed that CK19 was highly expressed
in the BECs in bile ductules in enlarged portal tracts (Fig. 4). The DDC-fed group had
significantly increased expression of CK19 compared with the NC
group. In contrast, apamin treatment significantly reduced biliary
activation and proliferation as evidenced by CK19 staining,
indicating a defect in the ductular reaction. In addition,
immunofluorescence staining of PCNA showed that apamin treatment
suppressed the proliferation of BECs compared with the DDC-fed
group. These results indicate that apamin may inhibit cholestatic
liver fibrosis by suppressing BEC proliferation and ductular
reaction induced by the DDC diet.
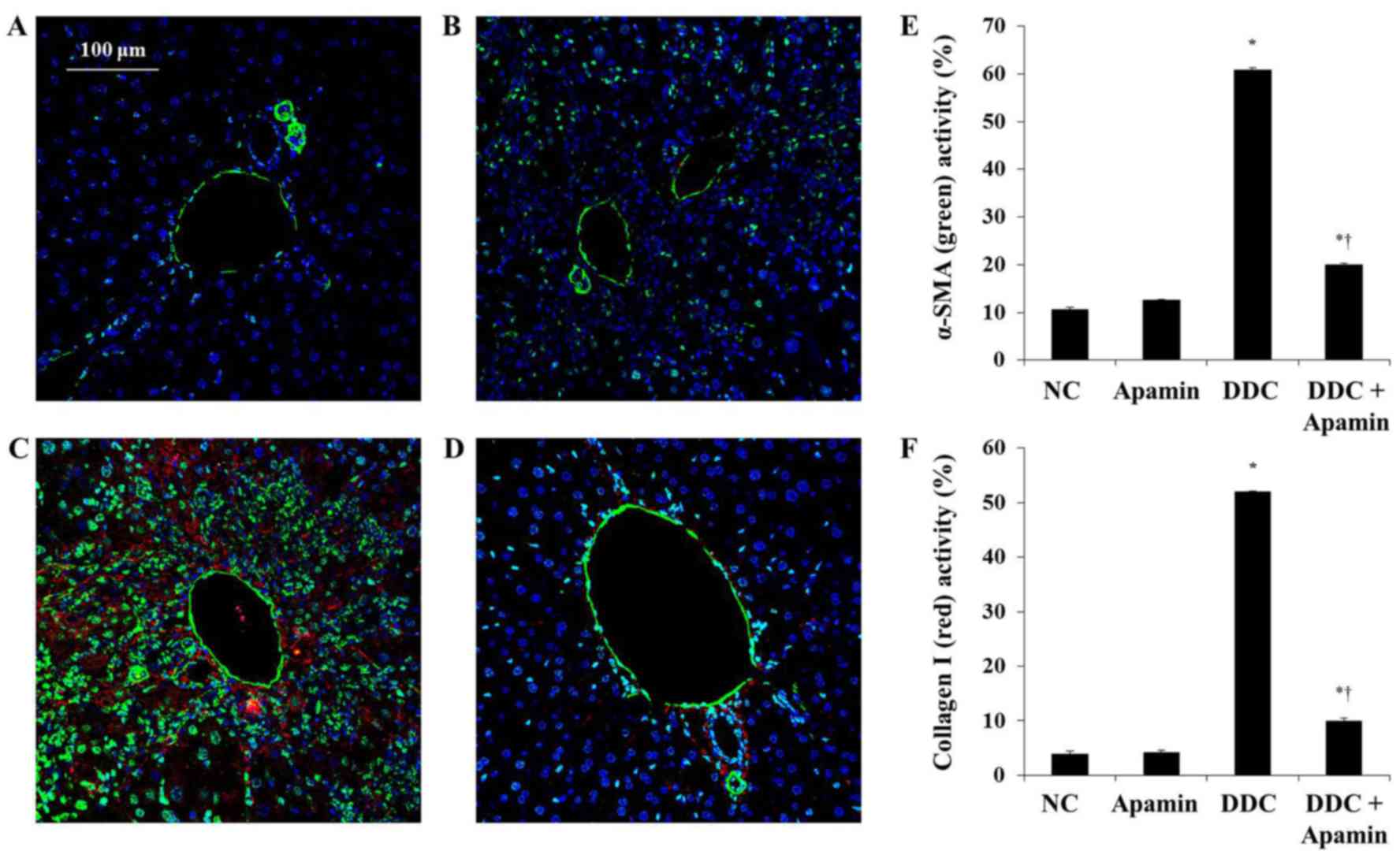
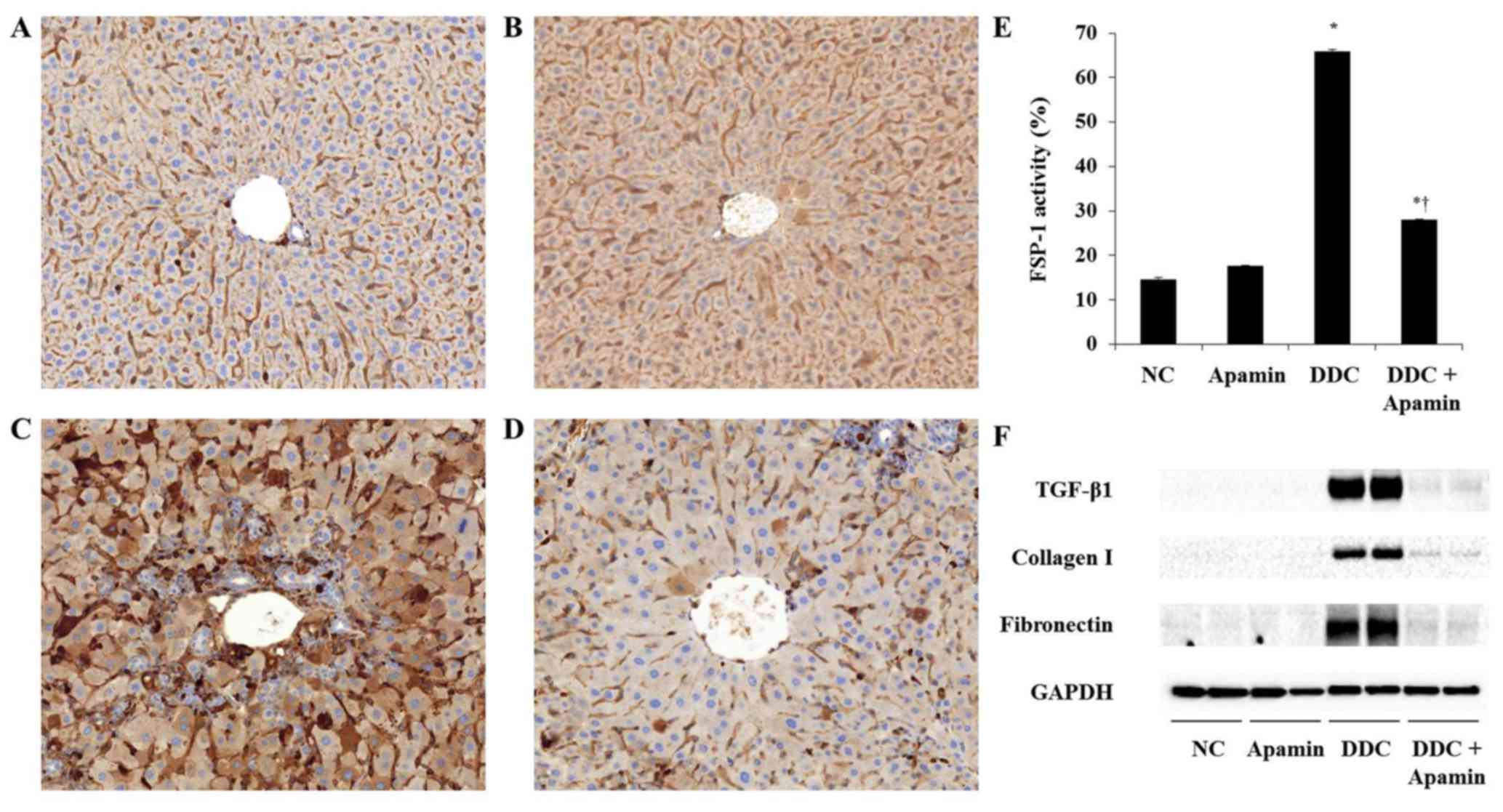
Apamin inhibits ECM deposition in the
livers of DDC-fed mice
To investigate the anti-fibrotic effect of apamin on
ECM deposition in the DDC-fed mice, we used western blot analysis,
immunohistochemistry and immunofluorescence assay to determine the
effects of this compound on ECM molecules. Liver fibrosis induced
by DDC was confirmed by induction of fibrogenic genes, FSP-1,
α-smooth muscle actin (α-SMA) and collagen I expression. Expression
of α-SMA was strongly expressed in the myofibroblasts and HSCs
around the proliferated bile duct in the DDC-fed group and clearly
with the apamin treatment (Fig.
5). Moreover, expression of collagen I in the DDC-fed group was
significantly increased, especially in the portal tracts. Compared
to the DDC group, apamin treatment inhibited collagen I expression.
During tissue remodeling in liver fibrosis, FSP-1 is considered as
a marker of fibroblasts in the fibrotic liver. DDC feeding
increased the number of cells positive for FSP-1 expression
(Fig. 6C). In contrast, apamin
treatment resulted in a reduction in FSP-1-positive cells (Fig. 6D). Furthermore, western blot
results showed that the expression levels of TGF-β1, collagen I,
and fibronectin were significantly higher in the DDC-fed group,
whereas apamin treatment markedly decreased the protein level of
TGF-β1, collagen I, and fibronectin compared with the DDC-fed group
(Fig. 6F). Taken together, the
data suggest that apamin may protect liver fibrosis during DDC
feeding by suppressing fibrotic gene expression.
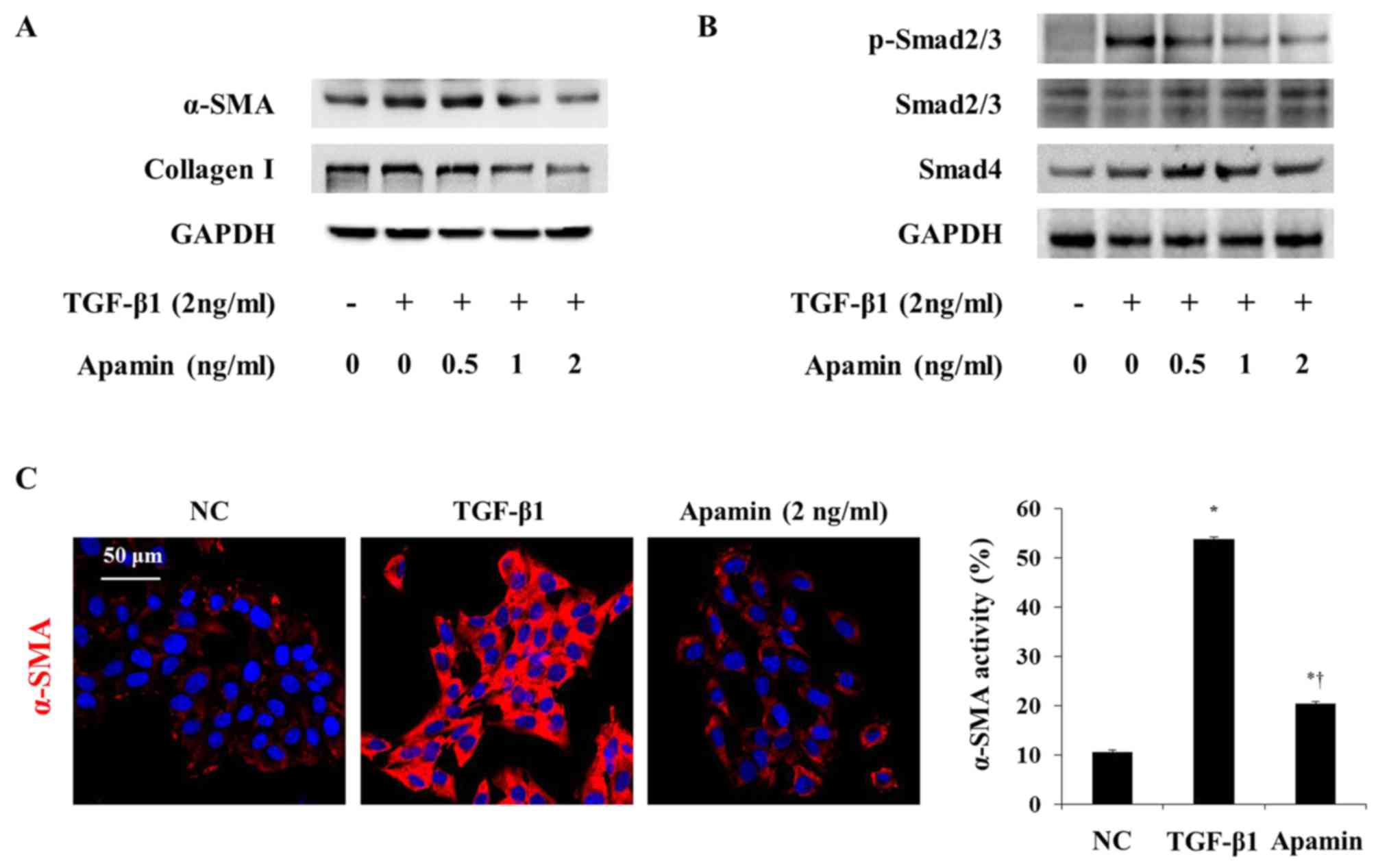
Apamin inhibits activation of HSCs
through the Smad signaling pathway
TGF-β1 is a Smad family member, and it is known to
stimulate the activation of HSCs (20). We next investigated whether the
inhibitory effect of apamin on the activation of HSCs by TGF-β1 is
through the Smad signaling pathway. α-SMA and collagen I expression
was increased by TGF-β1 and was decreased by apamin treatment in
the HSC-T6 cells (Fig. 7A). Also,
immunofluorescence showed that TGF-β1 induced activation of HSC-T6
cells through increased α-SMA expression (Fig. 7C). However, apamin treatment
markedly reduced the expression of α-SMA in the TGF-β1-induced
HSC-T6 cells. Western blot analysis showed that phosphorylation of
Smad2/3 and Smad4 were stimulated by 2 ng/ml TGF-β1 (Fig. 7B). Apamin treatment abrogated the
activation of p-Smad2/3 and Smad4 induced by TGF-β1. These results
indicated that apamin may attenuate TGF-β1-activated HSCs by
inhibiting the Smad signaling pathway.
Discussion
Liver fibrosis is a wound healing process that
results in increased levels of ECM protein. Bee venom and its
components are known to have anti-inflammatory and anti-fibrotic
effects on liver fibrosis (21–24). Recent studies have shown that
melittin inhibited cholangitis and biliary fibrosis in a
xenobiotic-induced mouse model (15). Apamin comprises 2–3% of the dry
weigh of bee venom (25).
Moreover, apamin is a specific blocker of SK channels. It has been
reported that apamin-sensitive K+ channel is present in
biliary cells and contributes to secretion in response to increased
intracellular Ca2+ (11,26). Hence, we investigated the effects
of apamin on the pathogenesis of DDC-induced biliary fibrosis and
the suppression of activated HSCs.
The biliary system is lined by BECs, which make up
~5% of all liver cells and function as important bile modifiers
(4,27). Hepatic fibrosis induced by DDC
feeding is related to increased biliary pyophyrin secretion and the
activation of BECs with development of bile duct injury, leading to
pericholangitis and ductular reaction resulting in portal-portal
fibrosis (28). Proliferating
BECs can secrete a variety of profibrogenic cytokines, which
promote the activation and proliferation of HSCs and also promote
the synthesis of ECM, leading to the initiation and development of
cholestatic hepatic fibrosis (6).
Moreover, CK19 is particularly important for the proliferation of
BECs during the ductular reaction, which is generally considered an
attempt by the liver to restore bile flow between the lobules and
the terminal bile ducts (29). In
the present study, apamin significantly inhibited bile duct
proliferation through a decrease in CK7, CK19 and PCNA expression
in the DDC-fed mice.
TGF-β1 is a major fibrogenic mediator involved in
the activation and transdifferentiation of HSCs (30). TGF-β1 binds to the TGF-β1 type II
receptor (TβRII) and phosphorylates Smad2/3, and then p-Smad2/3
interacts with Smad4 to trans-activate target genes in the nucleus
(31). Smad activation is
critical for the induction of many TGF-β-responsive genes including
collagen I, fibronectin and α-SMA (32). In the present study, TGF-β1,
collagen I, fibronectin and α-SMA levels were significantly
increased in the DDC-fed mouse group as determined by western blot
analysis and immunofluorescence (Figs. 5 and 6F). Treatment with apamin resulted in a
significant reduction in TGF-β1, collagen I, fibronectin and α-SMA
levels. Moreover, apamin treatment also inhibited α-SMA expression
through the Smad signaling pathway in the TGF-β1-induced HSCs
(Fig. 7). These findings suggest
that the HSCs are transformed into myofibroblasts and secrete
TGF-β1, thus stimulating the production of ECM in the DDC-fed mouse
model (15).
The principal finding of this study is the
anti-fibrotic effects of apamin. Apamin suppressed the
proliferation of BECs and activation of HSCs. In the present study,
apamin significantly inhibited bile duct proliferation and reduced
ECM deposition in the DDC-fed mice. Furthermore, apamin suppressed
the protein expression of p-Smad2/3 and Smad4 induced by TGF-β1 in
the HSCs. These results suggest that apamin inhibits the
proliferation of BECs and activation of HSCs by suppressing the
TGF-β1 signaling pathway in hepatic fibrosis.
Acknowledgments
This study was supported by the National Research
Foundation of Korea grant funded by the Korean Government (no.
NRF-2015R1D1A1A01061026).
References
|
1
|
Friedman SL: Mechanisms of hepatic
fibrogenesis. Gastroenterology. 134:1655–1669. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Gonzalez-Sanchez E, Firrincieli D, Housset
C and Chignard N: Nuclear receptors in acute and chronic
cholestasis. Dig Dis. 33:357–366. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Zollner G, Marschall HU, Wagner M and
Trauner M: Role of nuclear receptors in the adaptive response to
bile acids and cholestasis: Pathogenetic and therapeutic
considerations. Mol Pharm. 3:231–251. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Glaser SS, Gaudio E, Miller T, Alvaro D
and Alpini G: Cholangiocyte proliferation and liver fibrosis.
Expert Rev Mol Med. 11:e72009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Lindor KD, Gershwin ME, Poupon R, Kaplan
M, Bergasa V and Heathcote EJ; American Association for Study of
Liver Diseases: Primary biliary cirrhosis. Hepatology. 50:291–308.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Glaser SS, Onori P, Wise C, Yang F,
Marzioni M, Alvaro D, Franchitto A, Mancinelli R, Alpini G, Munshi
MK, et al: Recent advances in the regulation of cholangiocyte
proliferation and function during extrahepatic cholestasis. Dig
Liver Dis. 42:245–252. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Matsumoto K, Fujii H, Michalopoulos G,
Fung JJ and Demetris AJ: Human biliary epithelial cells secrete and
respond to cytokines and hepatocyte growth factors in vitro:
Interleukin-6, hepatocyte growth factor and epidermal growth factor
promote DNA synthesis in vitro. Hepatology. 20:376–382. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Popov Y and Schuppan D: Targeting liver
fibrosis: Strategies for development and validation of antifibrotic
therapies. Hepatology. 50:1294–1306. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Friedman SL: Molecular regulation of
hepatic fibrosis, an integrated cellular response to tissue injury.
J Biol Chem. 275:2247–2250. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Moreno M and Giralt E: Three valuable
peptides from bee and wasp venoms for therapeutic and
biotechnological use: Melittin, apamin and mastoparan. Toxins
(Basel). 7:1126–1150. 2015. View Article : Google Scholar
|
|
11
|
Mourre C, Fournier C and Soumireu-Mourat
B: Apamin, a blocker of the calcium-activated potassium channel,
induces neurodegeneration of Purkinje cells exclusively. Brain Res.
778:405–408. 1997. View Article : Google Scholar
|
|
12
|
Feranchak AP, Doctor RB, Troetsch M,
Brookman K, Johnson SM and Fitz JG: Calcium-dependent regulation of
secretion in biliary epithelial cells: The role of apamin-sensitive
SK channels. Gastroenterology. 127:903–913. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Lee WR, Kim KH, An HJ, Kim JY, Lee SJ, Han
SM, Pak SC and Park KK: Apamin inhibits hepatic fibrosis through
suppression of transforming growth factor β1-induced hepatocyte
epithelial-mesenchymal transition. Biochem Biophys Res Commun.
450:195–201. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kim SJ, Park JH, Kim KH, Lee WR, An HJ,
Min BK, Han SM, Kim KS and Park KK: Apamin inhibits THP-1-derived
macrophage apoptosis via mitochondria-related apoptotic pathway.
Exp Mol Pathol. 93:129–134. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kim KH, Sung HJ, Lee WR, An HJ, Kim JY,
Pak SC, Han SM and Park KK: Effects of melittin treatment in
cholangitis and biliary fibrosis in a model of xenobiotic-induced
cholestasis in mice. Toxins (Basel). 7:3372–3387. 2015. View Article : Google Scholar
|
|
16
|
Kim JY, Kim KH, Lee WR, An HJ, Lee SJ, Han
SM, Lee KG, Park YY, Kim KS, Lee YS, et al: Apamin inhibits
PDGF-BB-induced vascular smooth muscle cell proliferation and
migration through suppressions of activated Akt and Erk signaling
pathway. Vascul Pharmacol. 70:8–14. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Baghdasaryan A, Fuchs CD, Österreicher CH,
Lemberger UJ, Halilbasic E, Påhlman I, Graffner H, Krones E,
Fickert P, Wahlström A, et al: Inhibition of intestinal bile acid
absorption improves cholestatic liver and bile duct injury in a
mouse model of sclerosing cholangitis. J Hepatol. 64:674–681. 2016.
View Article : Google Scholar
|
|
18
|
Pollheimer MJ, Fickert P and Stieger B:
Chronic cholestatic liver diseases: Clues from histopathology for
pathogenesis. Mol Aspects Med. 37:35–56. 2014. View Article : Google Scholar
|
|
19
|
Yongping M, Zhang X, Xuewei L, Fan W, Chen
J, Zhang H, Chen G, Liu C and Liu P: Astragaloside prevents
BDL-induced liver fibrosis through inhibition of notch signaling
activation. J Ethnopharmacol. 169:200–209. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Yang JW, Hien TT, Lim SC, Jun DW, Choi HS,
Yoon JH, Cho IJ and Kang KW: Pin1 induction in the fibrotic liver
and its roles in TGF-β1 expression and Smad2/3 phosphorylation. J
Hepatol. 60:1235–1241. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kim SJ, Park JH, Kim KH, Lee WR, Chang YC,
Park KK, Lee KG, Han SM, Yeo JH and Pak SC: Bee venom inhibits
hepatic fibrosis through suppression of pro-fibrogenic cytokine
expression. Am J Chin Med. 38:921–935. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Lee WR, Pak SC and Park KK: The protective
effect of bee venom on fibrosis causing inflammatory diseases.
Toxins (Basel). 7:4758–4772. 2015. View Article : Google Scholar
|
|
23
|
Lee WR, Park JH, Kim KH, Park YY, Han SM
and Park KK: Protective effects of melittin on transforming growth
factor-β1 injury to hepatocytes via anti-apoptotic mechanism.
Toxicol Appl Pharmacol. 256:209–215. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Park JH, Kum YS, Lee TI, Kim SJ, Lee WR,
Kim BI, Kim HS, Kim KH and Park KK: Melittin attenuates liver
injury in thioacetamide-treated mice through modulating
inflammation and fibrogenesis. Exp Biol Med (Maywood).
236:1306–1313. 2011. View Article : Google Scholar
|
|
25
|
Son DJ, Lee JW, Lee YH, Song HS, Lee CK
and Hong JT: Therapeutic application of anti-arthritis,
pain-releasing, and anti-cancer effects of bee venom and its
constituent compounds. Pharmacol Ther. 115:246–270. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Dutta AK, Khimji AK, Sathe M, Kresge C,
Parameswara V, Esser V, Rockey DC and Feranchak AP: Identification
and functional characterization of the intermediate-conductance
Ca(2+)-activated K(+) channel (IK-1) in biliary epithelium. Am J
Physiol Gastrointest Liver Physiol. 297:G1009–G1018. 2009.
View Article : Google Scholar
|
|
27
|
Tabibian JH, Masyuk AI, Masyuk TV, O'Hara
SP and LaRusso NF: Physiology of cholangiocytes. Compr Physiol.
3:541–565. 2013.PubMed/NCBI
|
|
28
|
Fickert P, Stöger U, Fuchsbichler A,
Moustafa T, Marschall HU, Weiglein AH, Tsybrovskyy O, Jaeschke H,
Zatloukal K, Denk H, et al: A new xenobiotic-induced mouse model of
sclerosing cholangitis and biliary fibrosis. Am J Pathol.
171:525–536. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Chen Y, Guldiken N, Spurny M, Mohammed HH,
Haybaeck J, Pollheimer MJ, Fickert P, Gassler N, Jeon MK, Trautwein
C, et al: Loss of keratin 19 favours the development of cholestatic
liver disease through decreased ductular reaction. J Pathol.
237:343–354. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Leask A and Abraham DJ: TGF-beta signaling
and the fibrotic response. FASEB J. 18:816–827. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Flanders KC: Smad3 as a mediator of the
fibrotic response. Int J Exp Pathol. 85:47–64. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Inagaki Y and Okazaki I: Emerging insights
into Transforming growth factor beta Smad signal in hepatic
fibrogenesis. Gut. 56:284–292. 2007. View Article : Google Scholar : PubMed/NCBI
|