Introduction
Schmid-type metaphyseal chondrodysplasia (SMCD), a
relatively common form of metaphyseal chondrodysplasia, is an
autosomal inherited chondrodysplasia; affected patients are of
short stature and exhibit coxa vara, genu varum and a waddling gait
due to skeletal deformities, resulting from growth plate cartilage
abnormalities (1,2). Although chondrodysplasias are
considered rare [the incidence of chondrodysplasias is 1/4,000
births (3)], they severely affect
the quality of life of affected individuals. Chondrodysplasias have
diverse etiologies and there is ample evidence to indicate that
SMCD is caused by heterozygous mutations in the gene of collagen,
type X, alpha 1 (Col10a1) (4). The gene encodes a chain of type X
collagen molecule whose expression is largely restricted to zones
of calcifying or degrading growth plate cartilage, and thus the
mutations of Col10a1 may interfere with endochondral
ossification (5).
It has been demonstrated that many mutations in
genes that encode cartilage extracellular matrix (ECM) molecules
are involved in the etiology of chondrodysplasias (6). Chondrodysplasias caused by mutations
associated with ECM are due to inappropriate processing, folding,
or export of the abnormal ECM molecules, resulting in the
accumulation of these molecules within the endoplasmic reticulum
(ER) of chondrocytes (3,7). Subsequent to the cellular retention
of mutant ECM proteins, chondrocyte ER stress and the unfolded
protein response (UPR) are activated, in an attempt by the cells to
deal with the inappropriate retention of mutant proteins (8). Rajpar et al used the mouse
models phenocopying SMCD, a condition involving dwarfism and
hypertrophic zone expansion of the growth plate caused by autosomal
dominant mutations in Col10a1, and demonstrated the central
importance of ER stress in the pathology of SMCD (9). By contrast, the UPR can be activated
via ER membrane-spanning sensors, such as inositol-requiring
enzyme-1 (IRE1) and activated IRE1 can splice the coding sequence
of the X-box binding protein 1 (XBP1), a transcription
factor (TF) responsible for multiple UPR target gene expression
(10). Recently, Cameron et
al demonstrated that the IRE1/XBP1 pathway was redundant to
cartilage pathology and they proposed that the XBP1-independent
UPR-driven dysregulation of CCAAT/enhancer binding protein β
(C/EBP-β), a TF important for the transition from chondrocyte
proliferation to hypertrophy, was significant for the
pathophysiology of SMCD (11).
However, the limited studies have not yet revealed the details of
the molecule circuitry in SMCD.
In this study, we downloaded the microarray data
from GSE72261, the same gene expression profiling used in the study
by Cameron et al, from a publicly available database
(11). Mice with a SMCD collagen
X p.Asn617Lys knock-in mutation (ColXN617K) were
crossed with mice in which XBP1 activity was ablated
specifically in cartilage (Xbp1CartΔEx2),
generating the compound mutant, C/X (11). In the study by Cameron et
al (11), they mainly focused
on the analysis of role of the IRE1/XBP1 pathway in the
pathophysiology of SMCD, and thus they represented the
XBP1-dependent gene expression changes using differentially
expressed probes between ColXN617K and wild-type
(WT), but not between Xbp1CartΔEx2 and WT or C/X
and WT; the XBP1-independent gene expression changes using
differentially expressed probes between ColXN617K
and WT, and between C/X and WT, but not
Xbp1CartΔEx2 and WT. However, they did not
analyze the gene part between ColXN617K and WT,
C/X and WT, and Xbp1CartΔEx2 and WT, as well as
the gene part between ColXN617K and WT,
Xbp1CartΔEx2 and WT, but not C/X and WT.
In the present study, differentially expressed genes
(DEGs) were screened in ColXN617K,
Xbp1CartΔEx2 and C/X hypertrophic zone samples
compared with WT hypertrophic zone specimens, respectively.
Subsequently, comprehensive bioinformatics was used to analyze the
significant pathways of the DEGs, identification of TFs of the
overlapping DEGs both in the three comparison groups and
construction of the corresponding regulation networks. Furthermore,
to further analyze the more potential genes associated with SMCD,
gene co-expression modules, as well as protein-protein interaction
(PPI) networks were constructed based on the sum of DEGs. We
focused on the overlap of the results between regulatory networks
and co-expression analysis. The aim of this study was to gain a
better understanding of the molecular circuitry of SMCD, and to
identify more potential genes associated with the pathogenesis of
SMCD.
Materials and methods
Microarray data and data
preprocessing
The microarray data of GSE72261, deposited by
Cameron et al (11), were
downloaded from the National Center for Biotechnology Information
(NCBI) Gene Expression Omnibus (GEO) database (http://www.nibi.nih.gov/geo/). As described in the
original study (11),
hypertrophic zones were microdissected from one proximal tibial
growth plate from three 2-week old WT mice, three 2-week old mice
carrying a Col10a1 p.N617K mutation
(ColXN617K), three 2-week old mice lacking XBP1
activity in chondrocytes (Xbp1CartΔEx2), and
three 2-week old mice resulting from a cross between
ColXN617K and Xbp1CartΔEx2
(C/X). Thus, the GSE72261 dataset consisted of 3 WT samples, 3
Xbp1CartΔEx2 specimens (named Xbp1), 3
ColXN617K specimens (named Col) and 3 C/X specimens
(named C/X). The corresponding platform was GPL6887 Illumina
MouseWG-6 v2.0 expression beadchip. In this study, all these 12
samples were selected to carry out the follow-up analysis.
The non-normalized data were downloaded and
preprocessed using preprocessCore package (12) which is a part of Bioconductor
project (http://www.bioconductor.org/).
Besides, with the use of org.Mm.eg.db (13) and illuminaMousev2.db (14) packages of bioconductor, the probe
symbols were transformed into corresponding gene symbols. The
expression value was averaged for each gene when multiple probes
were mapped to the same gene. After data preprocessing, 20,106 gene
expression matrix were received.
Screening of DEGs
The DEGs in Xbp1 vs. WT, Col vs. WT, and CX vs. WT
were analyzed using the limma package (15) in R/Bioconductor.
|log2fold change (FC)| and p-values from an unpaired
t-test implemented in the limma package (15) were used to select the DEGs. Values
of p<0.01 and |log2FC|≥0.58 (showing >1.5-fold
differential expression) were set as the cut-off criteria.
Additionally, the DEGs identified in Xbp1 vs. WT, Col vs. WT and CX
vs. WT, respectively were clustered using the gplots package
(16) in R, evaluating whether
the DEGs identified were sample-specific. The results were
displayed as heatmaps.
By contrast, in the original study by Cameron et
al (11), DEGs were screened
in Xbp1 vs. WT, Col vs. WT and CX vs. WT, respectively with
>2-fold differential expression and with an adjusted p-value of
0.01. Moreover, Cameron et al focused on the DEGs in Col vs.
WT, but not Xbp1 vs. WT or CX vs. WT, and the DEGs in Col vs. WT
and CX vs. WT, but not Xbp1 vs. WT, in the subsequent analysis.
Pathway enrichment analysis
The database for annotation, visualization and
integrated discovery (DAVID) online software can provide a
comprehensive set of functional annotation tools (17). In order to analyze the identified
DEGs on the functional level, the Kyoto Encyclopedia of Genes and
Genomes (KEGG) pathway enrichment analysis was performed for all
DEGs using DAVID software (17).
Subsequently, p-values were calculated using the hypergeometric
test (18). Gene count ≥2 and
p-values <0.05 were set as the cut-off criterion for pathway
enrichment analysis.
Identification of TFs and construction of
the regulatory network
iRegulon (19),
available as a Cytoscape (20)
plugin, implements a genome-wide ranking-and-recovery approach to
detect enriched TF motifs and their optimal sets of direct target
genes (19). Besides, iRegulon
allows the integration of predicted regulatory binding sites
directly into a biological network (19). In the original study by Cameron
et al (11), DEGs in Col
vs. WT and CX vs. WT and Xbp1 vs. WT, as well as the DEGs between
in Col vs. WT and Xbp1 vs. WT, but not CX vs. WT, were analyzed. In
this study, we screened out the overlapping DEGs with consistent
expression changes (both upregulated or both downregulated) in the
3 groups (Xbp1 vs. WT, Col vs. WT and CX vs. WT). The lists of
overlapping DEGs with consistent expression change were subjected
to iRegulon (19) and used to
predict their transcriptional regulators using the following
parameters: minimum identity between orthologous genes, 0.05; and
maximum false discovery rate on motif similarity, 0.001. The
predicted TF-DEG pairs with normalized enrichment scores (NES)
(19) >4.5 were selected for
further analysis and the regulatory networks were constructed.
Analysis of gene co-expression
modules
To further analyze the more potential genes
associated with SMCD, we intended to perform gene co-expression
analysis of DEGs. For reducing the deviation, we used all the DEGs
as the scope of gene co-expression analysis.
Weighted correlation network analysis (WGCNA) can be
used for finding modules (clusters) of genes with high
correlations, or relating modules to external sample traits
(21). A robust correlation
coefficient emphasizes high correlations of genes and results in a
weighted network (21). In this
study, WGCNA R software package (21), which can cluster the most highly
co-expressed genes in defined modules, was applied to detect
modules of co-expressed genes of all the DEGs identified in Xbp1
vs. WT, Col vs. WT, and CX vs. WT. If the absolute value of the
correlation coefficient was high, the co-expressed genes clustered
in modules would have high gene co-expression trend consistency and
the modules would be significantly related to external sample
traits.
Functional enrichment analysis of genes
in the co-expression modules
In order to analyze gene co-expression modules on
the functional level, Gene Ontology (GO) enrichment analysis was
carried out using DAVID online tool (17) to obtain the enriched biological
process (BP) terms. A hypergeometric test (18) was applied to examine the
significance of this enrichment analysis. The count number ≥2 and
p-value <0.05 were used as the cut-off criterion.
Construction of PPI network based on
genes in the co-expression modules
The Search Tool for the Retrieval of Interacting
Genes (STRING) database can be used as it provides easy access to
known and predicted protein interactions (22). The interaction probabilities of
proteins in STRING are provided with a confidence score (22). A protein with a confidence score
>0.4 is deemed to have medium confidence of interaction with
other proteins (23). In the
present study, the STRING database was used to select the PPIs
among the DEGs identified in the co-expression modules with default
parameters (species: mus musculus). Interaction pairs of
DEGs with the confidence score ≥0.4 were selected for the PPI
network construction and the network was visualized using Cytoscape
software (24), where nodes
indicated proteins and edges indicated physical interactions. In
addition, node degrees of the DEGs were calculated. The nodes with
higher degree in the network were considered as hub proteins.
Furthermore, we integrated the results of regulatory
networks and PPI networks. The DEGs identified both in the
regulatory network and PPI network may be more potential key genes
associated with SMCD.
Results
Screening of DEGs
According to the gene expression profile, with
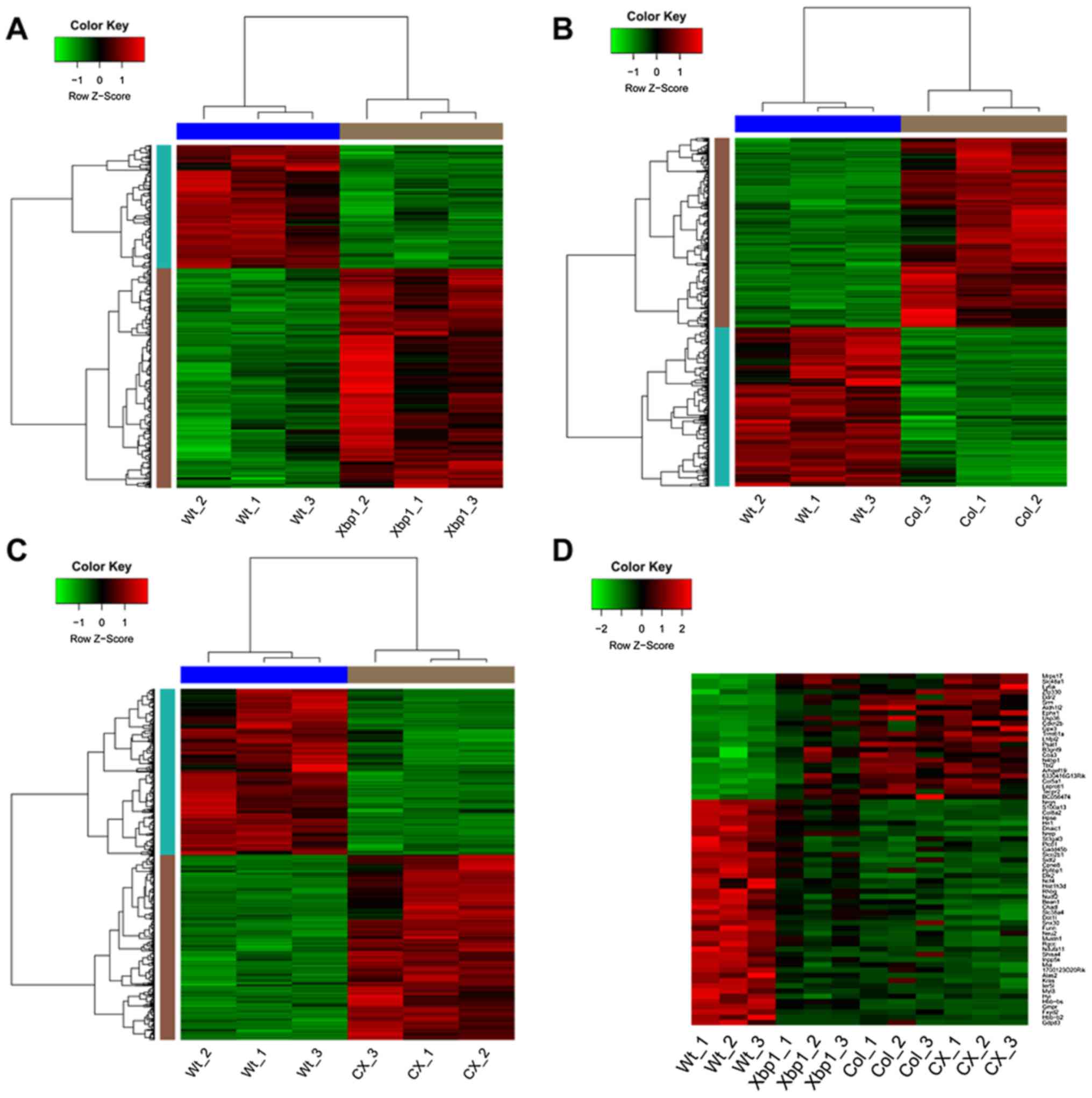
p-values <0.01 and |log2FC|≥0.58, there were 481 DEGs identified
in Xbp1 vs. WT, comprising 308 upregulated genes and 173
downregulated genes. The corresponding heatmap of the DEGs is shown
in Fig. 1A. Furthermore, 1,530
DEGs (831 up- and 699 downregulated genes) and 1,214 DEGs (640 up-
and 574 downregulated genes) were screened out in Col vs. WT and CX
vs. WT, respectively. The respective heatmaps are shown in Fig. 1B and C. From the heatmaps, we
found that the identified DEGs could distinguish the experimental
samples from the WT samples, suggesting the DEGs were eligible for
the subsequent analysis.
KEGG pathway enrichment analysis of the
DEGs identified
The KEGG pathways of the significantly upregulated
and downregulated DEGs are summarized in Table I. In total, 6 pathways were
enriched based on the DEGs identified in Xbp1 vs. WT, such as
ECM-receptor interaction and focal adhesion. Moreover, a number of
pathways were enriched by the upregulated and downregulated genes
in Col vs. WT and CX vs. WT. Table
I only displays the top 10 and 5 pathways enriched by these
DEGs in Col vs. WT and CX vs. WT, respectively. For instance, the
upregulated genes in Col vs. WT were significantly associated with
aminoacyl-tRNA biosynthesis and associated with ER stress (11), such as RNA degradation and
glutathione metabolism. The downregulated genes in Col vs. WT were
associated with focal adhesion. On the other hand, the upregulated
genes identified in CX vs. WT were significantly associated with
glycolysis/gluconeogenesis, as well as with the fructose and
mannose metabolism pathways. The downregulated genes in CX vs. WT
were found to be linked with leukocyte transendothelial
migration.
 | Table IThe pathways enriched in the Xbp1
group, and the top 10 pathways and top 5 pathways enriched in the
Col group and CX group, respectively. |
Table I
The pathways enriched in the Xbp1
group, and the top 10 pathways and top 5 pathways enriched in the
Col group and CX group, respectively.
| Gene change | KEGG term | Count | p-value |
|---|
| In Xbp1 group | | | |
| Upregulated |
mmu04512:ECM-receptor interaction | 8 | 5.23E-04 |
| mmu04510:Focal
adhesion | 11 | 0.0020 |
|
mmu04142:Lysosome | 8 | 0.0043 |
|
mmu00561:Glycerolipid metabolism | 4 | 0.0468 |
| Downregulated |
mmu03010:Ribosome | 8 | 2.69E-05 |
| mmu00860:Porphyrin
and chlorophyll metabolism | 3 | 0.0364 |
| In Col group | | | |
| Upregulated |
mmu00970:Aminoacyl-tRNA biosynthesis | 8 | 0.0011 |
| mmu00051:Fructose
and mannose metabolism | 7 | 0.0030 |
|
mmu00600:Sphingolipid metabolism | 7 | 0.0057 |
| mmu00250:Alanine,
aspartate and glutamate metabolism | 6 | 0.00594 |
|
mmu04142:Lysosome | 12 | 0.0071 |
| mmu00290:Valine,
leucine and isoleucine biosynthesis | 4 | 0.00796 |
| mmu00510:N-Glycan
biosynthesis | 7 | 0.00893 |
| mmu03018:RNA
degradation | 8 | 0.00895 |
|
mmu00480:Glutathione metabolism | 7 | 0.0160 |
|
mmu00010:Glycolysis/gluconeogenesis | 8 | 0.0172 |
| Downregulated | mmu04510:Focal
adhesion | 24 | 1.18E-05 |
|
mmu04070:Phosphatidylinositol signaling
system | 14 | 1.55E-05 |
|
mmu04142:Lysosome | 15 | 5.58E-04 |
| mmu04270:Vascular
smooth muscle contraction | 15 | 6.08E-04 |
|
mmu04512:ECM-receptor interaction | 12 | 8.03E-04 |
| mmu04912:GnRH
signaling pathway | 13 | 8.79E-04 |
| mmu05210:Colorectal
cancer | 12 | 0.00108 |
| mmu04540:Gap
junction | 12 | 0.00108 |
|
mmu05213:Endometrial cancer | 9 | 0.00157 |
| mmu04670:Leukocyte
transendothelial migration | 14 | 0.00175 |
| In CX group | | | |
| Upregulated |
mmu00010:Glycolysis/gluconeogenesis | 9 | 0.0012 |
| mmu00051:Fructose
and mannose metabolism | 6 | 0.0055 |
| mmu00030:Pentose
phosphate pathway | 5 | 0.0082 |
| mmu00520:Amino
sugar and nucleotide sugar metabolism | 6 | 0.0116 |
| mmu00750:Vitamin B6
metabolism | 3 | 0.0135 |
| Downregulated | mmu04670:Leukocyte
transendothelial migration | 16 | 1.83E-05 |
| mmu04510:Focal
adhesion | 20 | 7.17E-05 |
| mmu05210:Colorectal
cancer | 12 | 2.15E-04 |
| mmu04664:Fc epsilon
RI signaling pathway | 11 | 6.15E-04 |
| mmu04270:Vascular
smooth muscle contraction | 13 | 0.0011 |
Analysis of the regulatory network
Among all the DEGs identified in Xbp1 vs. WT, Col
vs. WT and CX vs. WT, a total of 24 common upregulated genes and 43
common downregulated genes were identified. The expression changes
of these common DEGs in all the samples are shown in the heatmap
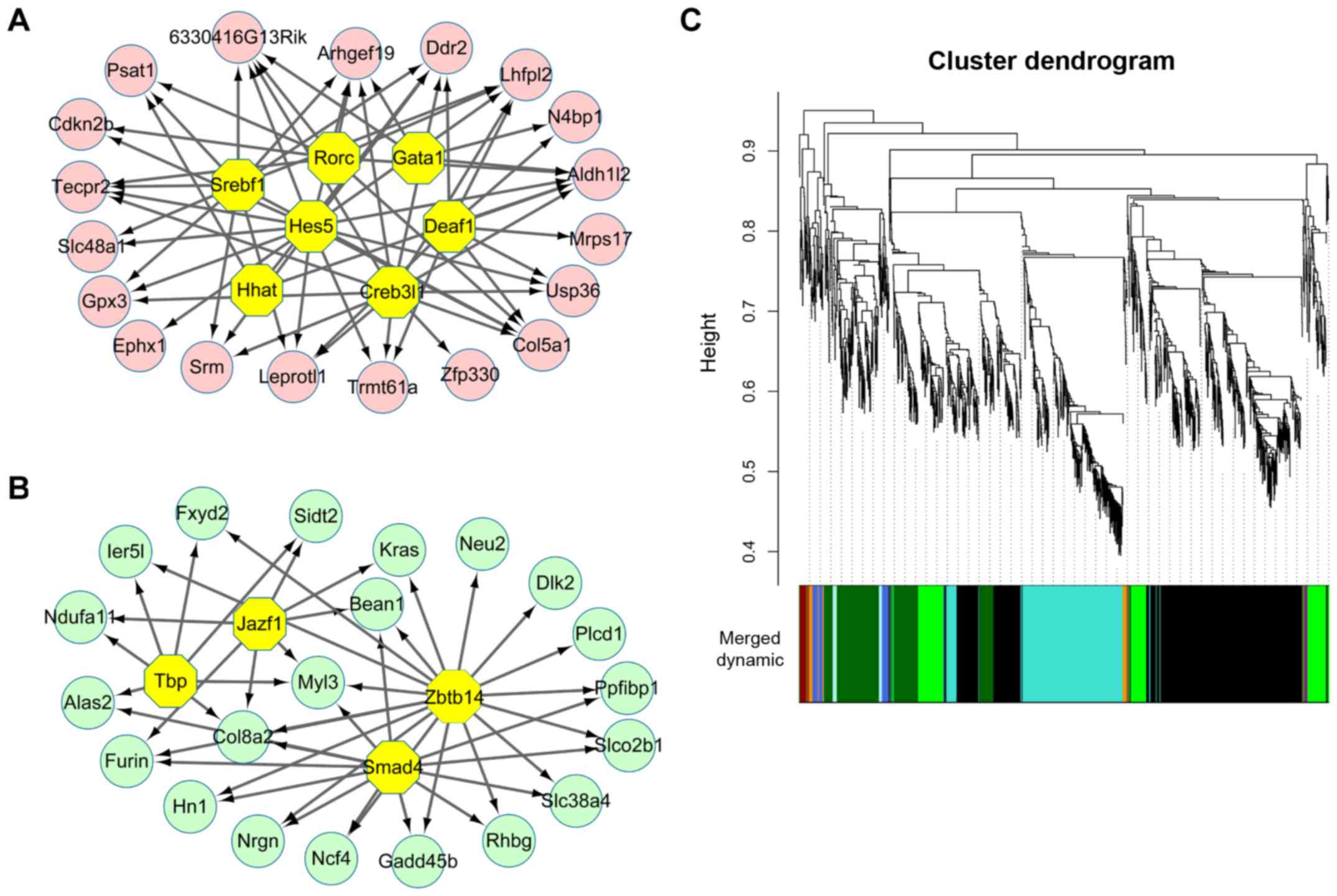
(Fig. 1D). With NES >4.5,
total 7 TFs were found to regulate 19 common upregulated genes
(Fig. 2A). These 7 TFs were DEAF1
transcription factor (Deaf1, NES=5.608), sterol regulatory element
binding transcription factor 1 (Srebf1, NES=5.579), GATA binding
protein 1 (Gata1, NES=5.315), Hes Family BHLH transcription factor
5 (Hes5, NES=5.135), RAR-related orphan receptor C (Rorc,
NES=4.74), hedgehog acyltransferase (Hhat, NES=4.593) and CAMP
responsive element binding protein 3-like 1 (Cereb3l1, NES=4.535).
By contrast, a total of 4 TFs were identified to regulate 21 common
downregulated genes (Fig. 2B).
These 4 TFs were zinc finger and BTB domain containing 14 (Zbtb14,
NES=5.722), SMAD family member 4 (Smad4, NES=5.313), TATA box
binding protein (Tbp, NES=5.137), JAZF zinc finger 1 (Jazf1,
NES=4.627). However, all the TFs that could regulate upregulated
genes or downregulate genes were not DEGs.
Gene co-expression modules and functional
enrichment analysis of genes in these modules
As already indicated, all the identified DEGs in the
3 groups (Xbp1 vs. WT, Col vs. WT and CX vs. WT) were used to
construct the co-expression network. Thus, 2,258 genes were
included (967 common genes were removed, including overlapping DEGs
in the 3 groups or only 2 groups). WGCNA analysis identified a
total of 11 modules with highly co-expressed genes (Fig. 2C). Besides, this analysis led to
the identification of dark green-colored (correlation coefficient,
0.94) and green-colored (correlation coefficient, −0.89) modules
with highest significance value (Table II). The results of several
significant GO BP terms (ranked by p-value) of DEGs in these 2
colored modules are shown in Table
III. The results demonstrated that genes enriched in the dark
green-colored module were significantly associated with the
biological processes, such as ribonucleoprotein complex biogenesis
and ribosome biogenesis. Additionally, genes enriched in the
green-colored module were significantly correlated with cell
adhesion and biological adhesion. Moreover, both the dark green-
and green-colored modules were significantly enriched for
biosynthetic processes, such as water-soluble vitamin biosynthetic
process and cholesterol biosynthetic process.
| Table IIResults of gene co-expression module
analysis. |
Table II
Results of gene co-expression module
analysis.
| Module | Correlation | Gene count | p-value |
|---|
| Turquoise | −0.6 | 503 | 0.03850043 |
| Dark orange | 0.17 | 40 | 0.590624 |
| Dark red | 0.37 | 28 | 0.2373936 |
| Dark olive
green | 0.024 | 24 | 0.9413225 |
| Dark green | 0.94 | 378 | 4.28E-06 |
| Pale turquoise | 0.69 | 31 | 0.01377187 |
| Black | 0.62 | 896 | 0.03280841 |
| Dark magenta | −0.78 | 10 | 0.002588195 |
| Green | −0.89 | 267 | 8.83E-05 |
| Royal blue | −0.73 | 66 | 0.006466701 |
| Saddle brown | −0.35 | 15 | 0.2660367 |
| Table IIIThe most significant GO terms of DEGs
identified in darkgreen and green modules. |
Table III
The most significant GO terms of DEGs
identified in darkgreen and green modules.
| Module | Term | p-value | Gene |
|---|
| Darkgreen | GO:0022613 -
ribonucleoprotein complex biogenesis | 10 | 8.91E-04 |
| GO:0042254 -
ribosome biogenesis | 9 | 9.96E-04 |
| GO:0007050 - cell
cycle arrest | 6 | 0.0037 |
| GO:0042364 -
water-soluble vitamin biosynthetic process | 4 | 0.0071 |
| GO:0034470 - ncRNA
processing | 9 | 0.0083 |
| GO:0000041 -
transition metal ion transport | 6 | 0.0088 |
| GO:0055114 -
oxidation reduction | 22 | 0.0109 |
| GO:0034660 - ncRNA
metabolic process | 10 | 0.0116 |
| GO:0016053 -
organic acid biosynthetic process | 8 | 0.0146 |
| GO:0046394 -
carboxylic acid biosynthetic process | 8 | 0.0146 |
| Green | GO:0007155 - cell
adhesion | 14 | 0.0126 |
| GO:0022610 -
biological adhesion | 14 | 0.0128 |
| GO:0006928 - cell
motion | 10 | 0.0258 |
| GO:0006695 -
cholesterol biosynthetic process | 3 | 0.0283 |
| GO:0055114 -
oxidation reduction | 14 | 0.0456 |
| GO:0016126 - sterol
biosynthetic process | 3 | 0.0462 |
| GO:0006865 - amino
acid transport | 4 | 0.0482 |
| GO:0006694 -
steroid biosynthetic process | 4 | 0.0482 |
PPI network analysis
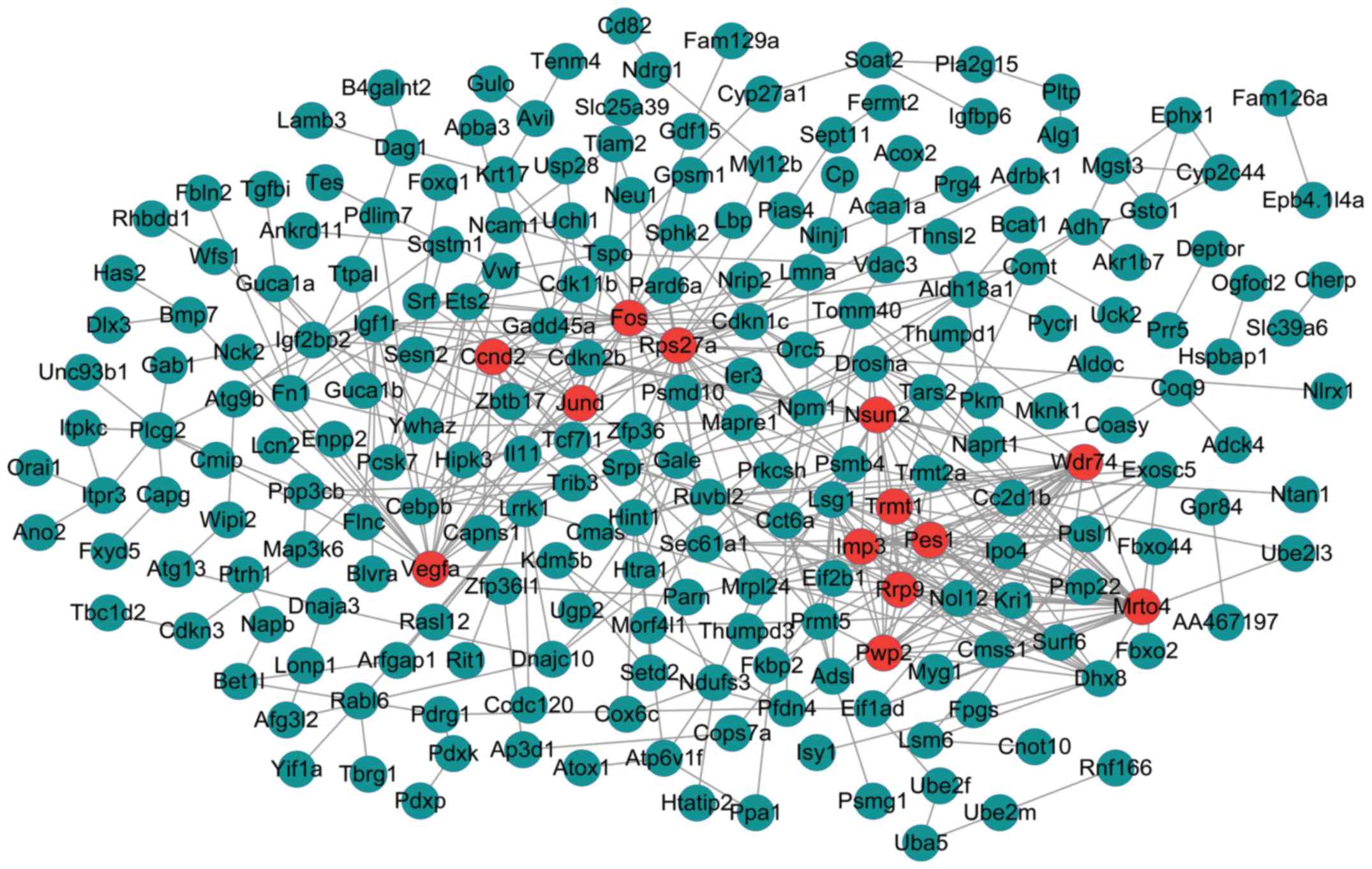
The PPI networks upon the DEGs in the dark green-
and green-colored modules are shown in Figs. 3 and 4, respectively. The PPI network of genes
in the dark green-colored module consisted of 228 nodes and 444
interactions (edges) (Fig. 3).
There were 13 DEGs with a node degree >12 in the network,
namely, MRNA turnover 4 homolog (S. cerevisiae)
(Mrto4) (degree = 27), ribosomal protein S27a
(Rps27A) (degree = 25), WD repeat domain 74 (Wdr74)
(degree = 21), FBJ murine osteosarcoma viral oncogene homolog
(Fos) (degree = 20), vascular endothelial growth factor A
(Vegfa) (degree = 19), IMP3, U3 small nucleolar
ribonucleoprotein (Imp3) (degree = 17), Jun D proto-oncogene
(Jund) (degree = 15), PWP2 periodic tryptophan protein
homolog (yeast; Pwp2) (degree = 15), pescadillo ribosomal
biogenesis factor 1 (Pes1) (degree = 15), ribosomal RNA
processing 9, small subunit (Ssu) processome component,
homolog (yeast; Rrp9) (degree = 13), NOP2/Sun RNA
methyltransferase family, member 2 (Nsun2) (degree = 13),
TRNA methyltransferase 1 homolog (S. cerevisiae)
(Trmt1) (degree = 13), cyclin D2 (Ccnd2) (degree =
13). In addition, the 13 DEGs had no overlap in the regulatory
networks.
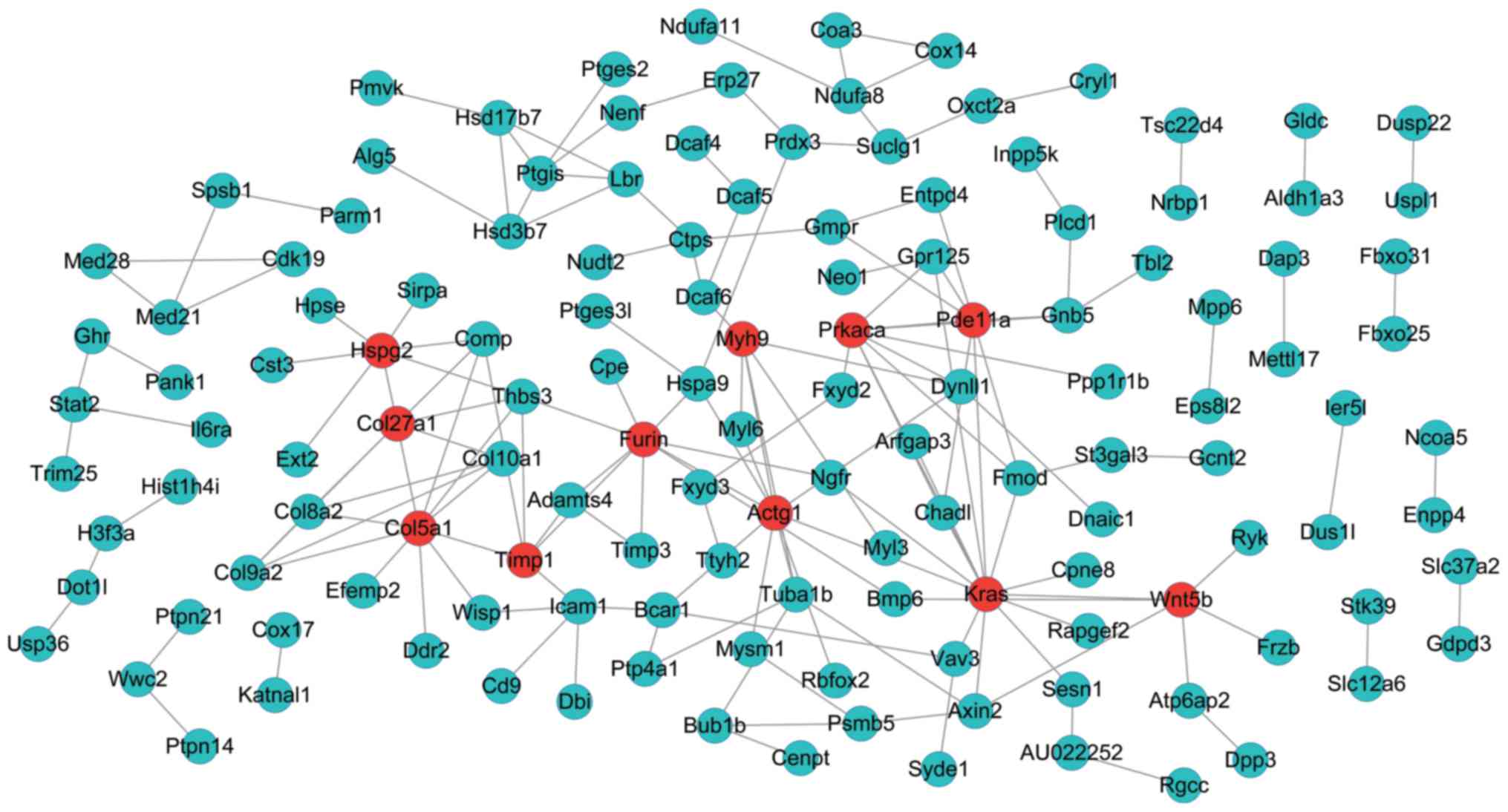
On the other hand, the PPI network of genes in the
green-colored module included 135 nodes and 166 interactions
(edges) (Fig. 4). Besides, there
were 11 DEGs with a node degree >5 in this network, namely,
Kirsten rat sarcoma viral oncogene homolog (Kras) (degree =
14), collagen, type V, alpha 1 (Col5a1) (degree = 11), actin
gamma 1 (Actg1) (degree = 10), protein kinase,
CAMP-dependent, catalytic, alpha (Prkaca) (degree = 9),
Furin (paired basic amino acid cleaving enzyme) (Furin)
(degree = 9), heparan sulfate proteoglycan 2 (Hspg2) (degree
= 8), phosphodiesterase 11A (Pde11A) (degree = 8), collagen,
type XXVII, alpha 1 (Col27a1) (degree = 7), wingless-type M
MTV integration site fam ily, member 5B (Wnt5b) (degree =
6), myosin, heavy chain 9, non-muscle (Myh9) (degree = 6),
TIMP metallopeptidase inhibitor 1 (Timp1) (degree = 6).
Fortunately, we found that 3 of these 11 DEGs, Kras
(downregulated), Col5a1 (upregulated), and Furin
(upregulated) were also included in the regulatory networks.
Discussion
In the current study, 481, 1,530 and 1,214 DEGs were
screened out in Xbp1 vs. WT, Col vs. WT and CX vs. WT,
respectively. Pathway enrichment analysis revealed that these DEGs
were enriched in different pathways, such as ECM-receptor
interaction, focal adhesion and pathways associated with
metabolism. In total, 7 TFs were found to regulate 19 common
upregulated genes that were with consistent gene change in the 3
groups, while 4 TFs were identified to regulate 21 common
downregulated genes. WGCNA demonstrated that 2 significantly
enriched gene co-expression modules (dark green- and green-colored
modules) and DEGs in the 2 modules were mainly enriched different
biological processes, such as ribosome biogenesis. Moreover,
Kras (downregulated), Col5a1 (upregulated), and
Furin (upregulated) were both in the regulatory networks and
PPI network.
With greater than 2-fold differential expression and
with an adjusted p-value of 0.01, Cameron et al (11) identified 1,337, 215 and 1,633
differentially expressed probes in CX vs. WT, Xbp1 vs. WT and Col
vs. WT, respectively. The amount of DEGs identified in this study
was slightly different from the original study of Cameron et
al (11), which may result
from different analysis methods or analytical errors. In the
present study, we first paid more attention to the DEGs which had
not been analyzed in the original study by Cameron et al
(11), including DEGs in Col vs.
WT and CX vs. WT and Xbp1 vs. WT, as well as the DEGs between in
Col vs. WT and Xbp1 vs. WT, but not CX vs. WT. Finally, our focus
was the DEGs both identified in the regulatory networks and PPI
networks.
Kras was one of the DEGs that were identified
in the regulatory networks and PPI network. Kras, a Kirsten
ras oncogene homolog from the mammalian ras gene family, encodes a
protein that is a member of the small GTPase super-family (25). Evidence has indicated that
Kras can orchestrate multiple metabolic changes, including
differential channeling of glucose intermediates, stimulation of
glucose uptake, and reprogrammed glutamine metabolism (26). Moreover, Chan et al
demonstrated that intracellular mutant collagen X could lead to
deregulated cellular metabolism (27). Taken together, we suggested that
Kras may play a critical role in the development of SMCD via
the regulation of cell metabolism.
It is well known that SMCD can be caused by
heterozygous mutations in the Col10a1 gene (4). In the present study, Col5a1
was found to be another significant DEG identified both in the
regulatory networks and PPI network. Col5a1 encodes an alpha
chain for one of the low abundance fibrillar collagens (28). It has been demonstrated that
Col5a1 is regulated by transforming growth factor-β (TGF-β)
in osteoblasts (29), and
Col5a1 is involved in the collagen biosynthesis, which is
associated with chondrocyte differentiation (11). In agreement with previous studies,
we infer that Col5a1 may play a critical role in the
pathogenesis of SMCD by participating in chondrocyte
differentiation, although further verifications are required to
confirm this result.
Furthermore, Furin was another focus in this
study, which was identified both in the regulatory networks and PPI
network. Furin is likely to represent the ubiquitous endoprotease
activity within constitutive secretory pathways (30). Evidence has indicated that Furin
is an authentic TGF-β1-converting enzyme (30). Besides, pericellular and diffuse
interterritorial distribution of TGF-β2 has been observed in
patients with SMCD and there may be a functional interaction of
TGF-β2 and type X collagen (31).
Thus, Furin may be essential in the mechanisms of SMCD,
which warrants further investigation.
However, this study has several limitations. First,
a larger sample size in the further investigations is warranted in
order to verify our findings. Second, the lack of cross-check and
experimental verification were also limitations. Validation using
other datasets in similar topic may be used to cross-check our
results. In the future, we aim to carry out experimental
verifications using different analytical approaches, such as
reverse transcription-quantitative polymerase chain reaction
(RT-qPCR) and immunohistochemistry.
In conclusion, the data of the present study
revealed several potential key genes (Kras, Col5a1
and Furin) which may play a role in the molecular mechanisms
responsible for SMCD. KRAS may play a critical role in the
development of SMCD via the regulation of cell metabolism. Besides,
Col5a1 may play a critical role in the pathogenesis of SMCD
by participating in chondrocyte differentiation. Furin may
be essential to the mechanisms of SMCD through an interaction with
type X collagen. Further characterization of the genes identified
in this study may provide deeper insight into the pathology of
SMCD.
References
|
1
|
Park H, Hong S, Cho SI, Cho TJ, Choi IH,
Jin DK, Sohn YB, Park SW, Cho HH, Cheon JE, et al: Case of mild
Schmid-type metaphyseal chondrodysplasia with novel sequence
variation involving an unusual mutational site of the COL10A1 gene.
Eur J Med Genet. 58:175–179. 2015. View Article : Google Scholar
|
|
2
|
Bateman JF, Freddi S, Nattrass G and
Savarirayan R: Tissue-specific RNA surveillance? Nonsense-mediated
mRNA decay causes collagen X haploinsufficiency in Schmid
metaphyseal chondrodysplasia cartilage. Hum Mol Genet. 12:217–225.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Patterson SE and Dealy CN: Mechanisms and
models of endoplasmic reticulum stress in chondrodysplasia. Dev
Dyn. 243:875–893. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Bogin O, Kvansakul M, Rom E, Singer J,
Yayon A and Hohenester E: Insight into Schmid metaphyseal
chondrodysplasia from the crystal structure of the collagen X NC1
domain trimer. Structure. 10:165–173. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Woelfle JV, Brenner RE, Zabel B, Reichel H
and Nelitz M: Schmid-type metaphyseal chondrodysplasia as the
result of a collagen type X defect due to a novel COL10A1 nonsense
mutation: A case report of a novel COL10A1 mutation. J Orthop Sci.
16:245–249. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Warman ML, Cormier-Daire V, Hall C, Krakow
D, Lachman R, LeMerrer M, Mortier G, Mundlos S, Nishimura G, Rimoin
DL, et al: Nosology and classification of genetic skeletal
disorders: 2010 revision. Am J Med Genet A. 155A:943–968. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Arnold WV and Fertala A: Skeletal diseases
caused by mutations that affect collagen structure and function.
Int J Biochem Cell Biol. 45:1556–1567. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Hetz C: The unfolded protein response:
Controlling cell fate decisions under ER stress and beyond. Nat Rev
Mol Cell Biol. 13:89–102. 2012.PubMed/NCBI
|
|
9
|
Rajpar MH, McDermott B, Kung L, Eardley R,
Knowles L, Heeran M, Thornton DJ, Wilson R, Bateman JF and Poulsom
R: Targeted induction of endoplasmic reticulum stress induces
cartilage pathology. PLoS Genet. 5:e10006912009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Yoshida H, Matsui T, Yamamoto A, Okada T
and Mori K: XBP1 mRNA is induced by ATF6 and spliced by IRE1 in
response to ER stress to produce a highly active transcription
factor. Cell. 107:881–891. 2001. View Article : Google Scholar
|
|
11
|
Cameron TL, Bell KM, Gresshoff IL,
Sampurno L, Mullan L, Ermann J, Glimcher LH, Boot-Handford RP and
Bateman JF: XBP1-Independent UPR Pathways Suppress C/EBP-β Mediated
Chondrocyte Differentiation in ER-Stress Related Skeletal Disease.
PLoS Genet. 11:e10055052015. View Article : Google Scholar
|
|
12
|
Bolstad BM: preprocessCore: A collection
of pre-processing functions. R package version 1. 2013, http://www.bioconductor.org/packages/release/bioc/html/preprocessCore.html.
Accessed March 27, 2013.
|
|
13
|
Gardeux V, Arslan AD, Achour I, Ho TT,
Beck WT and Lussier YA: Concordance of deregulated mechanisms
unveiled in underpowered experiments: TBP1 knockdown case study.
BMC Med Genomics. 7(Suppl 1): S12014. View Article : Google Scholar :
|
|
14
|
Hoeksema MA, Scicluna BP, Boshuizen MC,
van der Velden S, Neele AE, Van den Bossche J, Matlung HL, van den
Berg TK, Goossens P and de Winther MP: IFN-γ priming of macrophages
represses a part of the inflammatory program and attenuates
neutrophil recruitment. J Immunol. 194:3909–3916. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ritchie ME, Phipson B, Wu D, Hu Y, Law CW,
Shi W and Smyth GK: limma powers differential expression analyses
for RNA-sequencing and microarray studies. Nucleic Acids Res.
43:e472015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Warnes GR, Bolker B, Bonebakker L,
Gentleman R, Huber W, Liaw A, Lumley T, Maechler M, Magnusson A and
Moeller S: gplots: Various R programming tools for plotting data. R
package version 2.12.1. 2013, http://cran.r-project.org/web/packages/gplots/index.html.
|
|
17
|
Dennis G Jr, Sherman BT, Hosack DA, Yang
J, Gao W, Lane HC and Lempicki RA: DAVID: Database for annotation,
visualization, and integrated discovery. Genome Biol. 4:32003.
View Article : Google Scholar
|
|
18
|
Mao X, Cai T, Olyarchuk JG and Wei L:
Automated genome annotation and pathway identification using the
KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics.
21:3787–3793. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Janky R, Verfaillie A, Imrichová H, Van de
Sande B, Standaert L, Christiaens V, Hulselmans G, Herten K, Naval
Sanchez M and Potier D: iRegulon: from a gene list to a gene
regulatory network using large motif and track collections. PLoS
Comput Biol. 10:e10037312014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Smoot ME, Ono K, Ruscheinski J, Wang P-L
and Ideker T: Cytoscape 2.8: New features for data integration and
network visualization. Bioinformatics. 27:431–432. 2011. View Article : Google Scholar :
|
|
21
|
Langfelder P and Horvath S: WGCNA: An R
package for weighted correlation network analysis. BMC
Bioinformatics. 9:5592008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Franceschini A, Szklarczyk D, Frankild S,
Kuhn M, Simonovic M, Roth A, Lin J, Minguez P, Bork P, von Mering
C, et al: STRING v9.1: Protein-protein interaction networks, with
increased coverage and integration. Nucleic Acids Res.
41:D808–D815. 2013. View Article : Google Scholar :
|
|
23
|
Szklarczyk D, Franceschini A, Kuhn M,
Simonovic M, Roth A, Minguez P, Doerks T, Stark M, Muller J, Bork
P, et al: The STRING database in 2011: Functional interaction
networks of proteins, globally integrated and scored. Nucleic Acids
Res. 39:D561–D568. 2011. View Article : Google Scholar :
|
|
24
|
Spinelli L, Gambette P, Chapple CE,
Robisson B, Baudot A, Garreta H, Tichit L, Guénoche A and Brun C:
Clust&See: A Cytoscape plugin for the identification,
visualization and manipulation of network clusters. Biosystems.
113:91–95. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Vakiani E and Solit DB: KRAS and BRAF:
Drug targets and predictive biomarkers. J Pathol. 223:219–229.
2011. View Article : Google Scholar
|
|
26
|
Bryant KL, Mancias JD, Kimmelman AC and
Der CJ: KRAS: Feeding pancreatic cancer proliferation. Trends
Biochem Sci. 39:91–100. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Chan D, Ho MS and Cheah KS: Aberrant
signal peptide cleavage of collagen X in Schmid metaphyseal
chondrodysplasia. Implications for the molecular basis of the
disease. J Biol Chem. 276:7992–7997. 2001. View Article : Google Scholar
|
|
28
|
Raleigh SM, van der Merwe L, Ribbans WJ,
Smith RK, Schwellnus MP and Collins M: Variants within the MMP3
gene are associated with Achilles tendinopathy: Possible
interaction with the COL5A1 gene. Br J Sports Med. 43:514–520.
2009. View Article : Google Scholar
|
|
29
|
Kahai S, Vary CP, Gao Y and Seth A:
Collagen, type V, α1 (COL5A1) is regulated by TGF-β in osteoblasts.
Matrix Biol. 23:445–455. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Dubois CM, Blanchette F, Laprise MH, Leduc
R, Grondin F and Seidah NG: Evidence that furin is an authentic
transforming growth factor-β1-converting enzyme. Am J Pathol.
158:305–316. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Yang M, Wang X, Zhang L, Yu C, Zhang B,
Cole W, Cavey G, Davidson P and Gibson G: Demonstration of the
interaction of transforming growth factor beta 2 and type X
collagen using a modified tandem affinity purification tag. J
Chromatogr B Analyt Technol Biomed Life Sci. 875:493–501. 2008.
View Article : Google Scholar : PubMed/NCBI
|