Introduction
Bronchopulmonary dysplasia (BPD) is a severe
complication of preterm birth which causes considerable mortality
and morbidity in premature infants. Characterized by multifactorial
chronic lung disease, it may affect not only pulmonary function,
but also growth, cardiovascular health and neurodevelopment
(1). Although the mechanism of
BPD yet remains to be clarified, chorioamnionitis, sepsis, invasive
mechanical ventilation and hyperoxia may contribute to its
incidence (2). Modulation of
alveolarization, epithelial cell transdifferentiation, fibroblast
and myofibroblast differentiation, extracellular matrix production
and inflammation are all involved in the pathological process
(3).
Among all the pathways, Toll-like receptor 4 (TLR4)
has been demonstrated to play an important role. Hyperoxia has been
reported to upregulate TLR4 and activate the nuclear factor-κB
(NF-κB) pathway (4,5). Antagonists of TLR4 were found to
relieve apoptosis and inflammatory response in neurological
diseases (6). However, the role
of TLR4 is not univocal, as other studies have reported a
protective role of TLR4 activation (7). In this case, the exact consequence
of TLR4 activation warrants further attention.
The anti-inflammatory and anti-apoptotic effects of
vitamin D have been confirmed in many organs including the lung
(8–11). Vitamin D-knockout mice experience
a more severe inflammatory response than wild-type mice after LPS
treatment (12). The regulatory
effect of vitamin D and its receptor on TLR4 has been demonstrated
in many studies (13–15). Our previous study also found that
vitamin D could significantly relieve LPS-induced lung injury
(11). Since TLR4 is the key
receptor of LPS, we hypothesized that regulation of TLR4 may be
associated with this protective effect of vitamin D.
In this study, we established a model of
hyperoxia-induced lung injury in neonatal rats. Vitamin D treatment
was administered to investigate its effect on lung structure,
inflammatory response and apoptosis. We aimed at elucidating the
effect of vitamin D on hyperoxia-induced lung injury and the role
of TLR4 in the process.
Materials and methods
Animal preparation and hyperoxia
exposure
All animal procedures were reviewed and approved by
the Laboratory Animal Ethics Committee of China Medical University.
This study was carried out in strict compliance with the approved
protocols. Pregnant Wistar rats (200–250 g) were supplied by the
Animal Laboratory, Experimental Research Center, Shengjing
Hospital, China Medical University. All rats were maintained in
specific pathogen-free static cages with a 12-h light/dark cycle.
Chow pellets and tap water were available ad libitum.
Hyperoxia exposure was administered as previously described
(16). The oxygen concentration
was continuously monitored using a strip-chart recorder (model 572;
Servomex, Norwood, MA, USA).
Vitamin D treatment
The hyperoxia and normoxia groups were randomly
divided into two subgroups. One subgroup was treated with a vitamin
D (VD) analogue, paricalcitol (Sigma-Aldrich, St. Louis, MO, USA),
dissolved in 90:10 propylene glycol:ethanol at 0.5 µg/kg
body weight, while the vehicle subgroups received the solvent only.
Paricalcitol or vehicle was administered through i.p. injection 30
min prior to exposure to hyperoxia, and on every other day (1, 3,
5… 21) afterwards.
Histology
Lungs were collected and sectioned at a thickness of
4 µm. The slides were stained with H&E (Beyotime
Institute of Biotechnology, Haimen, China) at room temperature.
Radial alveolar counts (RAC) were measured by drawing a
perpendicular line from the center of the most peripheral
bronchiole to the pleura or the nearest interlobular septum and
counting the number of alveoli transected by this line. Every
section was evaluated by two pathologists who were blind to the
study design.
Masson-trichrome staining was performed using a
commercial kit (Beyotime Institute of Biotechnology) according to
the instructions. ECM deposition was analyzed by Image Pro Plus 6.0
(Media Cybernetics, Rockville, MD, USA).
Wet/dry ratio of the lung weight
The right upper lung lobes were excised and weighed
to determine the wet lung weight. They were dried in an oven at
80°C for 48 h until the weights stopped to change. They were then
weighed again for the dry weight. Wet/dry ratio of the lung weight
was calculated accordingly.
Bronchoalveolar lavage fluid (BALF)
collection
After anesthesia by intraperitoneal injection with a
cocktail of xylazine (Rompun 2%; Bayer AG, Leverkusen, Germany) and
ketamine (Ketavest; 100 mg/ml; Pfizer, Inc., New York, NY, USA),
the trachea was exposed and the lungs were lavaged three times with
0.2 ml sterile saline/wash. The fluids were collected and stored at
4°C. The BCA method was used to test the protein concentrations in
the fluids.
Real-time PCR
Total RNA was isolated from the lung tissues using
TRIzol reagent (Invitrogen, Carlsbad, CA, USA). First-strand cDNAs
were synthesized with a PrimeScript RT reagent kit (Takara
Biotechnology Co., Ltd., Dalian, China). Real-time PCR was
performed in a 20-µl mixture using a SYBR-Green PCR reagent
kit (Clontech Laboratories, Inc., Mountainview, CA, USA) and a
Bio-Rad IQ5 real-time system. The 2−ΔΔCq formula was
used to quantify the relative expression of mRNA. β2
microglobulin (B2M) was used as an internal control. Sequences of
the PCR primers are provided in Table
I.
 | Table IPrimer sequences used for real-time
PCR. |
Table I
Primer sequences used for real-time
PCR.
| Primer name | Forward (5′-3′) | Reverse (3′-5′) |
|---|
| Rat TNF-α |
ATGTGGAACTGGCAGAGGAG |
TGGAACTGATGAGAGGGAGC |
| Rat IL-1β |
ACTCATTGTGGCTGTGGAGA |
TAGCAGGTCGTCATCATCCC |
| Rat IL-6 |
CCACTGCCTTCCCTACTTCA |
TTCTGACAGTGCATCATCGC |
| Rat MIP-2 |
AACATCCAGAGCTTGACGGT |
ACGATCCTCTGAACCAAGGG |
| Rat IFN-γ |
GTGTCATCGAATCGCACCTG |
GGTGACAGCTGGTGAATCAC |
| Rat MCP-1 |
GCTGCTACTCATTCACTGGC |
ATTGGGGTCAGCACAGATCT |
Terminal
deoxynucleotidyltransferase-mediated dUTP nick-end labelling
(TUNEL) staining
TUNEL staining was performed using an In Situ
Cell Death Detection kit, TMR red (Roche Diagnostics, Indianapolis,
IN, USA) according to the manufacturer's instructions. The number
of TUNEL-positive cells/field was recorded and compared
statistically.
Western blot analysis
Total proteins were harvested from the lung tissue.
Equal amounts of proteins (50 µg/lane) were separated by 10%
polyacrylamide gel electrophoresis, and the proteins were
transferred electrophoretically onto polyvinylidene difluoride
membranes (EMD Millipore, Billerica, MA, USA). The membranes were
then incubated with primary antibodies. The primary antibodies and
their concentrations were as follows: anti-TLR4 (1:1,000 dilution;
ab30667), anti-caspase-3 (1:1,000 dilution; ab52293) and
anti-β-actin (1:5,000 dilution; ab8226) (all from Abcam). After
being washed with TBST 3 times, the membranes were then incubated
at room temperature for 1 h with horseradish peroxidase-conjugated
secondary antibodies (goat anti-rabbit and anti-mouse IgG-HRP; Cat.
nos. sc-2004 and sc-2005, respectively; 1:2,000 dilution; Santa
Cruz Biotechnology, Inc., Santa Cruz, CA, USA).
Statistical analyses
All continuous data are presented as mean ± standard
deviation (SD). Statistical comparison of continuous variables
between groups was performed using the Student's t-test or one-way
ANOVA (followed by Games-Howell test) with GraphPad Prism software
6.0 (GraphPad Software Inc., La Jolla, CA, USA) and SPSS 17.0 (SPSS
Inc., Chicago, IL, USA). Rank data were analyzed using Wilcoxon
rank test. P<0.05 was considered to indicate a statistically
significant result.
Results
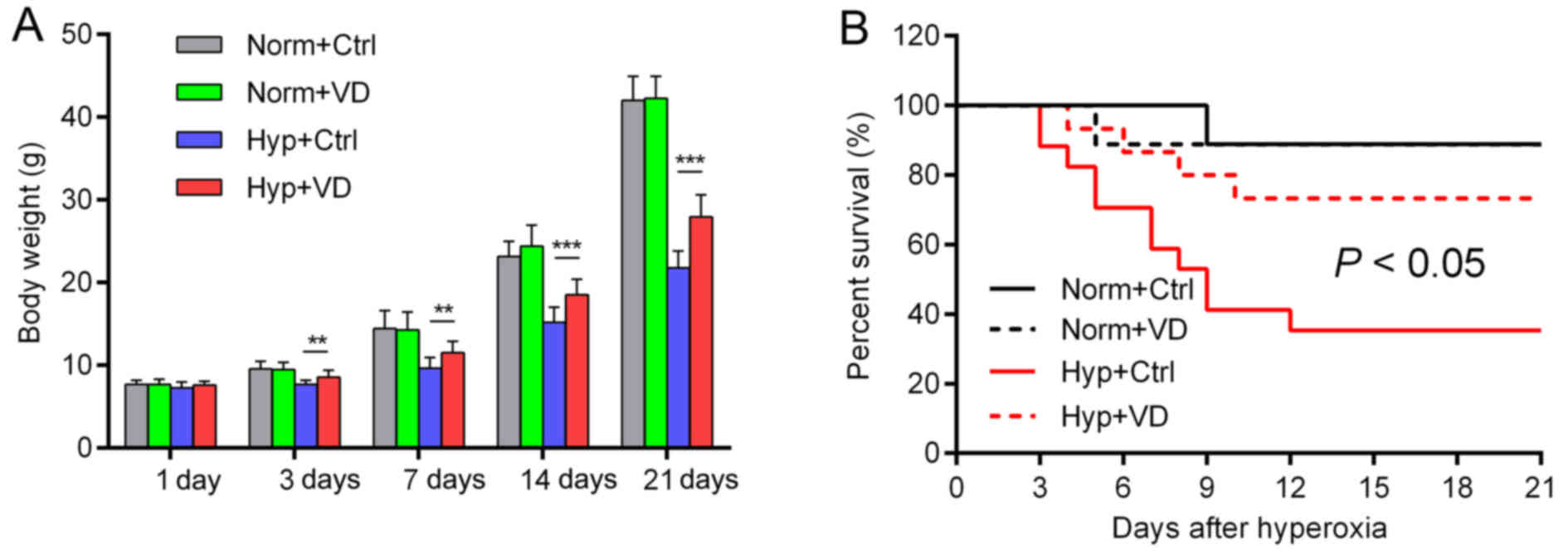
Vitamin D attenuates developmental
retardation caused by hyperoxia and promotes the survival rate
In rats treated with hyperoxia, developmental
retardation was observed from day 3, as represented by a decreased
body weight compared to the control groups. Vitamin D significantly
attenuated this retardation, but did not influence the body weights
of the normoxia groups (Fig. 1A).
Only 35.3% of the rats survived hyperoxia, while vitamin D
increased the rate to 73.3% (Fig.
1B). Vitamin D reduced the developmental retardation and
mortality caused by hyperoxia (n=9 in the normoxia group, n=15–17
in the hyperoxia group).
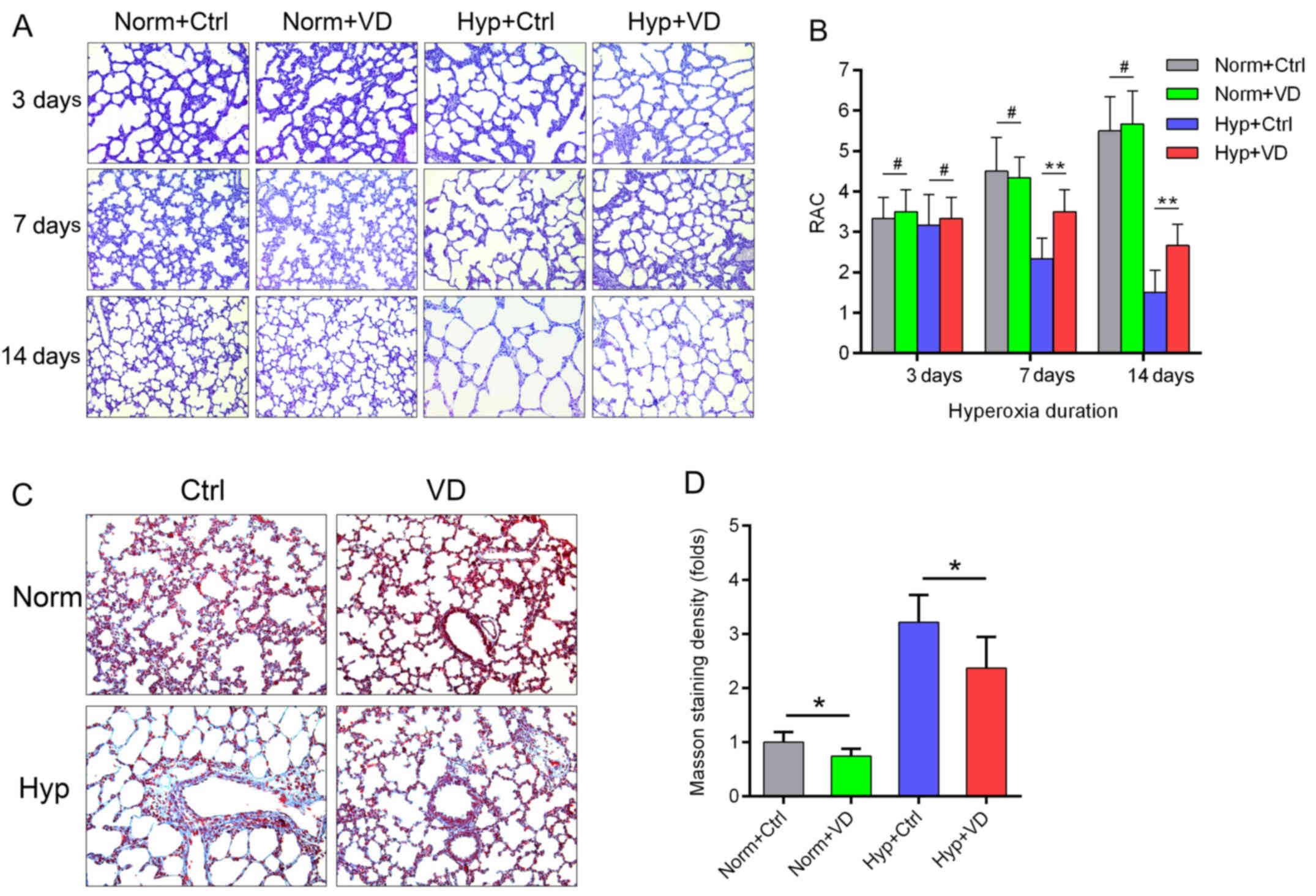
Vitamin D preserves lung structure and
decreases extracellular matrix deposition induced by hyperoxia
We investigated lung structures microscopically.
Hyperoxia led to aberrance, infiltration of neutrophils and
destruction of normal alveolar walls. Decrease in the numbers of
alveoli and secondary septa, irregular alveolar shape and
thickening of the alveolar wall were also observed. Compared to the
control groups, vitamin D-treated lungs presented with more intact
structures and less inflammation (Fig. 2A). RAC, which represent the status
of alveolarization, started to differ 7 days after hyperoxia
(Fig. 2B). Masson staining was
performed in rats after 7 days of hyperoxia. Hyperoxia induced
extracellular matrix deposition in the alveolar wall, while vitamin
D attenuated this change. This attenuating effect was also observed
in the normoxia groups (Fig. 2C and
D) (n=6 in each group).
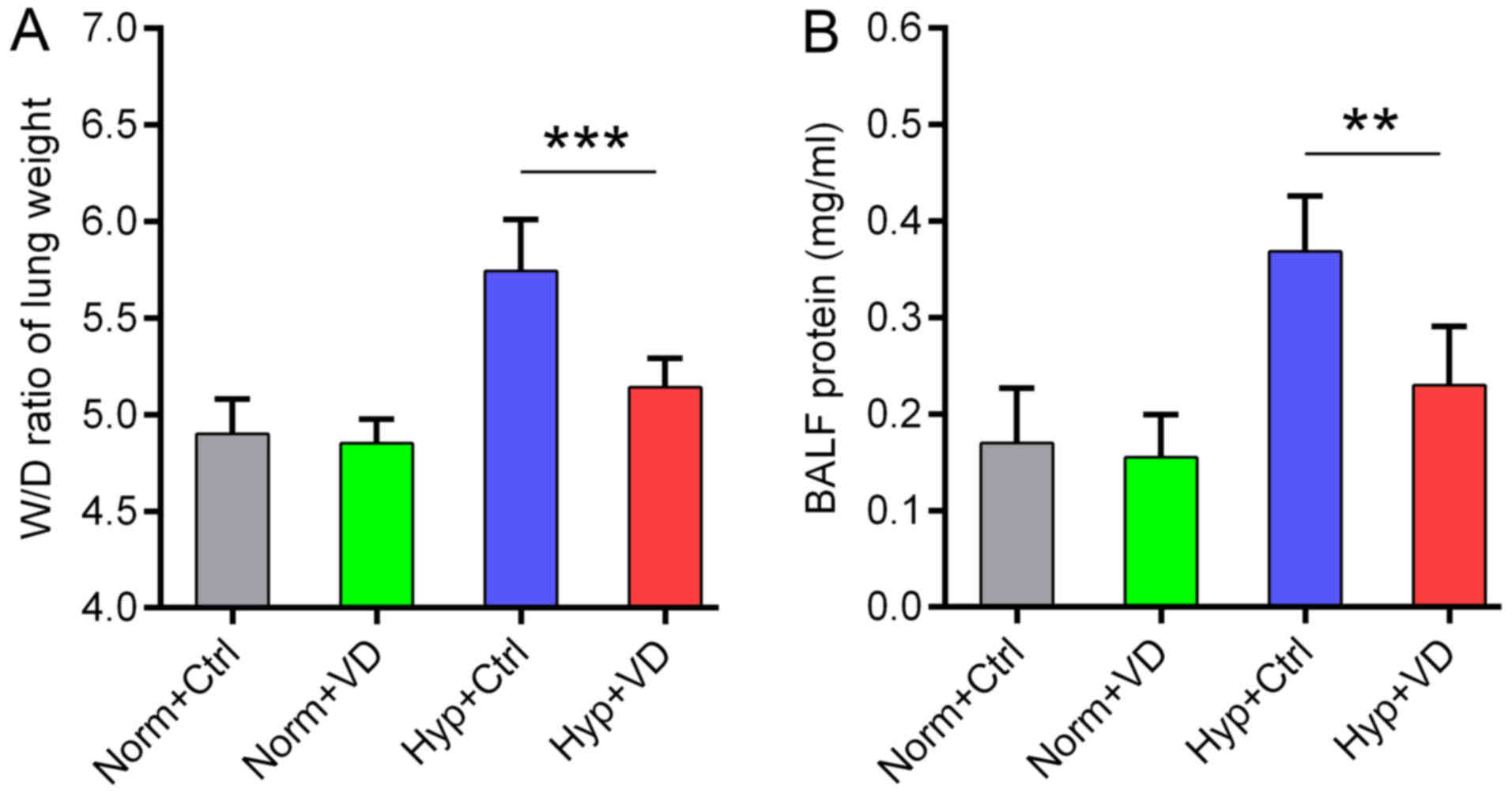
Vitamin D relieves lung edema, maintains
barrier function and inhibits inflammation in hyperoxia-treated
rats
We recorded the wet/dry ratio of the lung weight in
all the groups, which is a good indicator of lung edema. Hyperoxia
increased this ratio, indicating more water leakage into the
alveolar space. Vitamin D treatment significantly attenuated lung
edema (Fig. 3A). Similar change
was also observed in the BALF protein content, showing the
protective effect of vitamin D on the epithelial cell barrier
function (Fig. 3B) (n=6 in each
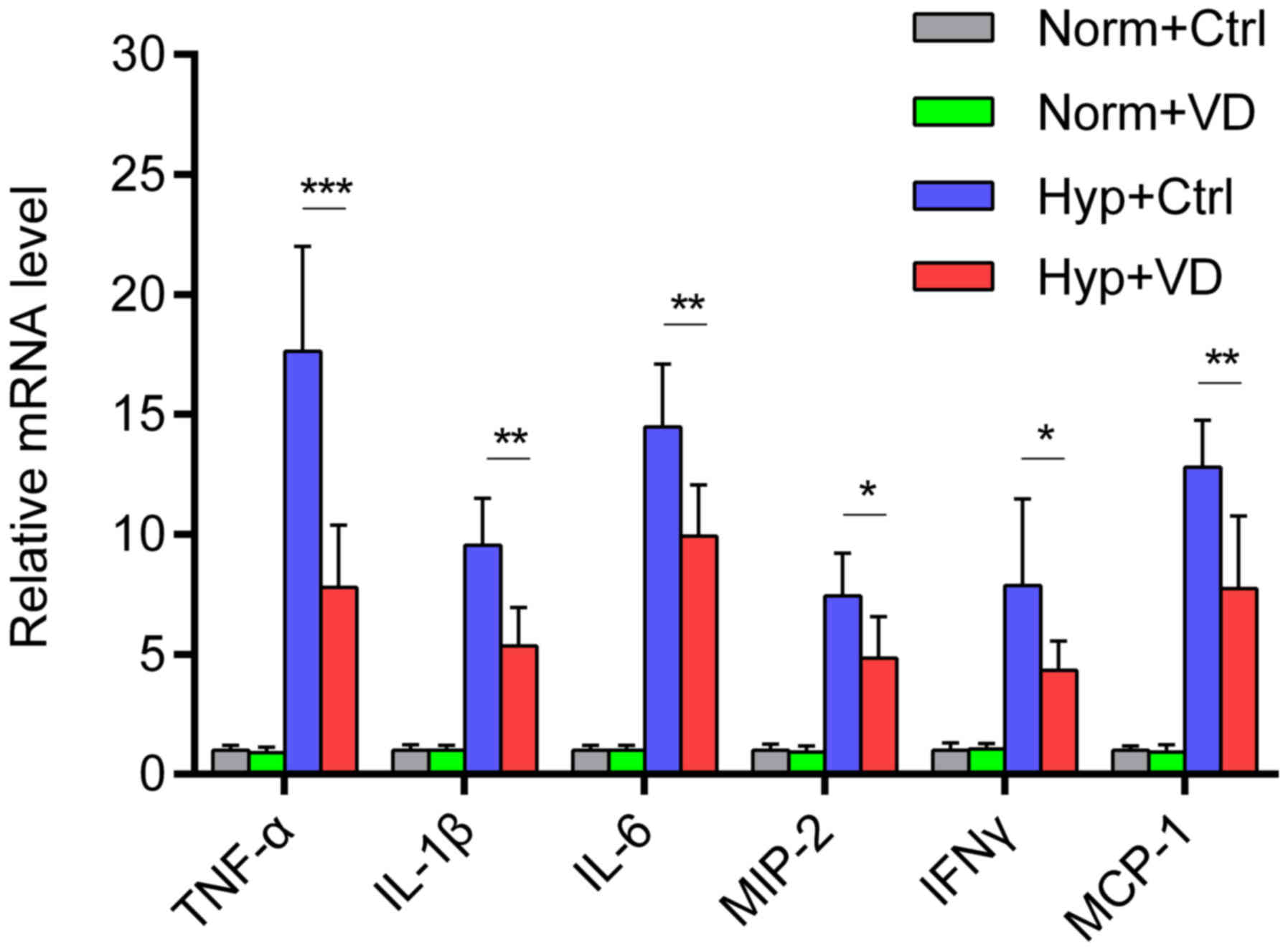
group). Then we measured the expression of inflammatory mRNAs in
the lung tissue. Hyperoxia stimulated the expression of
inflammatory cytokines and chemokines, while vitamin D
significantly inhibited the stimulation (Fig. 4) (n=6–8 in each group).
Vitamin D decreases alveolar cell
apoptosis by downregulating TLR4
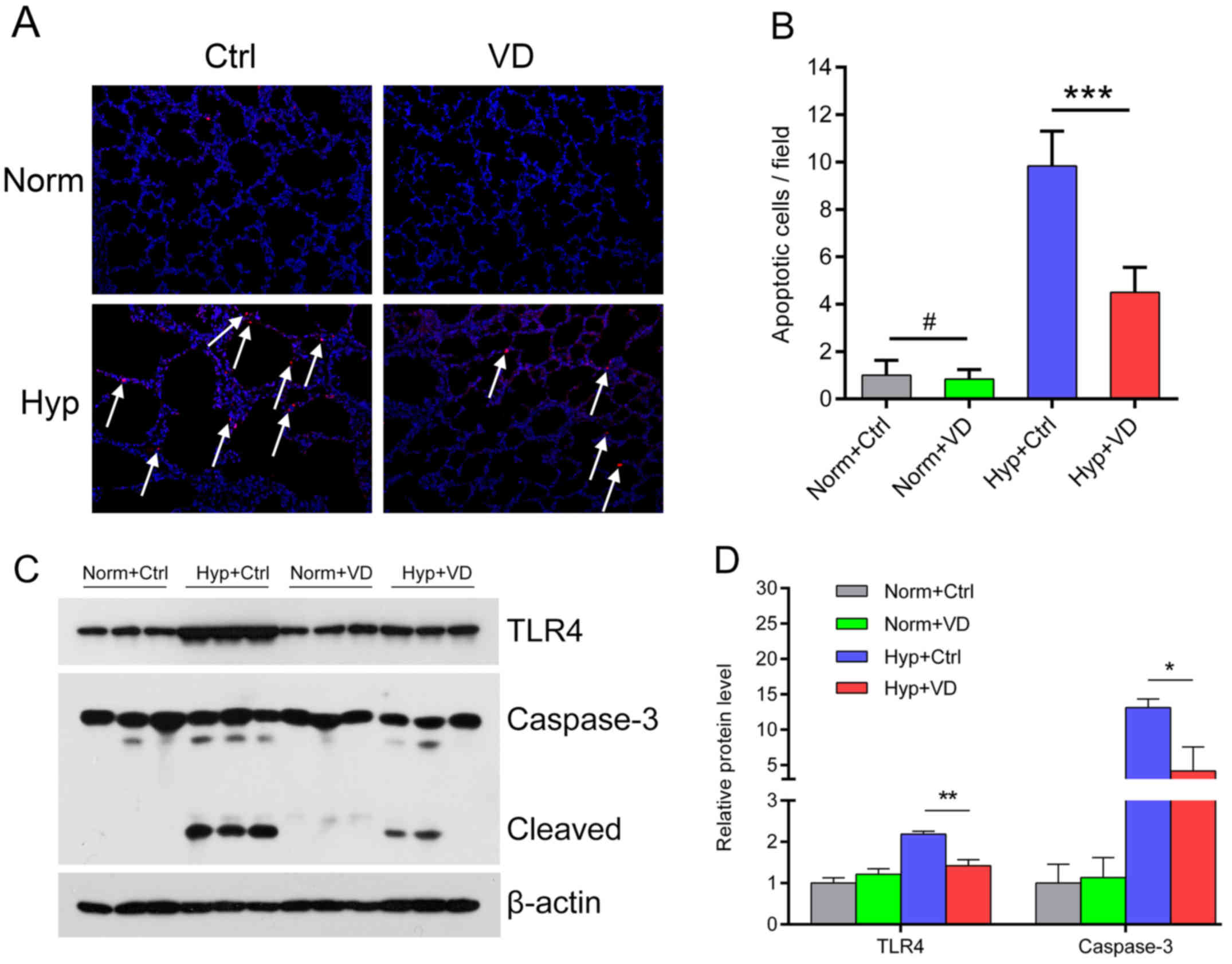
We performed TUNEL staining to investigate apoptosis
in the alveolar cells 7 days after exposure to hyperoxia. In the
normoxia groups, TUNEL-positive cells were scarce. Hyperoxia led to
a marked increase in the number of TUNEL-positive cells, while
vitamin D was protective against hyperoxia-induced apoptosis
(Fig. 5A). The apoptotic index
was calculated as the number of apoptotic cells/field, which was
significantly increased by hyperoxia and ameliorated by vitamin D
(Fig. 5B). Western blotting
showed activation of TLR4 and a subsequent increase in the
expression of caspase-3, a downstream apoptotic protein in the
hyperoxic group. Vitamin D treatment suppressed TLR4 expression and
subsequently the expression of cleaved caspase-3 (Fig. 5C and D) (n=6 in each group).
Discussion
This study demonstrated that vitamin D attenuates
hyperoxia-induced lung injury in neonatal rats, by protecting the
integrity of the lung structure, decreasing ECM deposition and
inhibiting inflammation. The mechanism may lie in the
anti-apoptotic effect of vitamin D by downregulating TLR4.
Hyperoxia-induced lung injury has been a focus of
recent research, for it best simulates BPD, a disease that severely
impairs the lung function in preterm births. The number of
macrophages, the surface volume of gas exchange and the volume of
lung parenchyma are decreased, while the area of atelectasis is
increased (17). Hyperoxia
induces the production of ROS, which subsequently activates several
downstream pathways, represented by the MAPK and NF-κB cascades
(4,18). Therefore it causes the release and
accumulation of inflammatory mediators in the alveolar space
(19). Moreover, hyperoxia may
destruct both the endothelial and the epithelial barriers by
downregulating tight junction proteins (20,21). We also observed deposition of
extracellular matrix and retarded lung development, as reported in
our preview study (22).
Through the production of ROS, hyperoxia regulates
the expression of TLRs, and thereby activates NF-κB-related
expression of inflammatory cytokines (4). TLRs are important components of the
innate immunity, which can identify all types of microbial
structures. In the lung, they protect the host from environmental
oxidants and invading microbes (23). As the receptor of LPS and a key
component in the NF-κB pathway, TLR4 is essential for survival and
lung integrity, by sensing microbial components, exogenous oxidants
and modulating inflammatory and apoptotic responses (24). Mutation or deletion of TLR4 has an
anti-inflammatory effect on endotoxin-induced inflammation and
ischemic-reperfusion injury (19). Activation of TLR4 is also
responsible for neonatal necrotizing enterocolitis-associated lung
injury, which has a similar but more severe pathological process
compared to other forms of premature-associated lung injury
(25).
Hyperoxia has been found to elevate the expression
of TLR4 in A549 cells (5), and in
this study, in neonatal rat lungs. This over-activation of TLR4 led
to increased release of inflammatory mediators including tumor
necrosis factor-α (TNF-α), interleukin-1β (IL-1β), IL-6, MIP-2,
interferon-γ (IFN-γ) and MCP-1. Over-activation of TLR4 has been
reported to promote neutrophil recruitment and microvascular and
alveolar epithelial injury in a dose-dependent manner (26). Apoptosis was induced subsequently,
as confirmed by increases in the number of TUNEL-positive cells and
the expression of caspase-3.
Vitamin D largely preserved normal lung structure in
the hyperoxia-induced lung injury. The aberrance, mal-development
of the alveoli and the deposition of extracellular matrix were
attenuated by vitamin D. We observed less infiltration of
neutrophils and reduction in the release of pro-inflammatory
cytokines, indicating an anti-inflammatory effect of vitamin D on
hyperoxia-induced lung injury. This effect has also been observed
in models of inflammatory bowel diseases and ischemic-reperfusion
injury (8,9). Our previous study found that vitamin
D receptor-knockout mice showed a more intense inflammatory
response to LPS and vitamin D treatment decreased the secretion of
pro-inflammatory cytokines in LPS-induced lung injury (11). The anti-apoptotic role of vitamin
D was also demonstrated in pulmonary tissue (10). Agonists of vitamin D receptor
relieved cardiomyocyte apoptosis in a model of myocardial
ischemic-reperfusion (27).
Vitamin D ameliorated intestinal epithelial apoptosis in
2,4,6-trinitrobenzene sulphonic acid-induced colitis by inhibiting
the expression of caspase-3 and p53 (28). Such an effect was also observed in
pulmonary epithelial cells (11).
Many studies have indicated that these
anti-inflammatory and anti-apoptotic effects of vitamin D function
through the TLR4 pathway. Vitamin D has been demonstrated to down
regulate TLR4 in liver cells, human monocytes THP-1,
antigen-presenting cells and keratinocytes, resulting in a lower
inflammatory response and reduced apoptosis (13–15,29). While hyperoxia upregulated the
expression of TLR4, vitamin D treatment significantly attenuated
this increase, and thereby exerted its anti-inflammatory and
anti-apoptotic functions.
In conclusion, hyperoxia may destruct lung
structure, retard lung development and cause deposition of
extracellular matrix. The release of pro-inflammatory cytokines and
the increase in pulmonary epithelial cell apoptosis may be caused
by activation of TLR4. Vitamin D administration can antagonize the
activation of TLR4 and therefore alleviate inflammation, reduce
apoptosis and preserve lung structure. Since hyperoxia-induced lung
injury is a typical model of BPD, vitamin D may be of curative or
preventive value to patients with BPD.
Acknowledgments
This study was supported by the National Natural
Science Foundation of China (nos. 81571479 and 81471489).
References
|
1
|
Kair LR, Leonard DT and Anderson JM:
Bronchopulmonary dysplasia. Pediatr Rev. 33:255–263. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Balany J and Bhandari V: Understanding the
impact of infection, inflammation and their persistence in the
pathogenesis of bronchopulmonary dysplasia. Front Med (Lausanne).
2:902015.
|
|
3
|
Silva DM, Nardiello C, Pozarska A and
Morty RE: Recent advances in the mechanisms of lung alveolarization
and the pathogenesis of bronchopulmonary dysplasia. Am J Physiol
Lung Cell Mol Physiol. 309:L1239–L1272. 2015.PubMed/NCBI
|
|
4
|
Chen Y, Li Q, Liu Y, Shu L, Wang N, Wu Y,
Sun X and Wang L: Attenuation of hyperoxia-induced lung injury in
neonatal rats by 1α, 25-Dihydroxyvitamin D3. Exp Lung
Res. 41:344–352. 2015. View Article : Google Scholar
|
|
5
|
Huang D, Fang F and Xu F: Hyperoxia
induces inflammation and regulates cytokine production in alveolar
epithelium through TLR2/4-NF-κB-dependent mechanism. Eur Rev Med
Pharmacol Sci. 20:1399–1410. 2016.PubMed/NCBI
|
|
6
|
Zhao Y, Zhao Y, Zhang M, Zhao J, Ma X,
Huang T, Pang H, Li J and Song J: Inhibition of TLR4
signalling-induced inflammation attenuates secondary injury after
diffuse axonal injury in rats. Mediators Inflamm. 2016:47069152016.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Perros F, Lambrecht BN and Hammad H: TLR4
signaling in pulmonary stromal cells is critical for inflammation
and immunity in the airways. Respir Res 12:. 125:2011.
|
|
8
|
Liu TJ, Shi YY, Du J, Ge X, Teng X, Liu L,
Wang EB and Zhao Q: Vitamin D treatment attenuates 2, 4,
6-trinitrobenzene sulphonic acid (TNBS)-induced colitis but not
oxazolone-induced colitis. Sci Rep. 6:328892016. View Article : Google Scholar
|
|
9
|
Lee JW, Kim SC, Ko YS, Lee HY, Cho E, Kim
MG, Jo SK, Cho WY and Kim HK: Renoprotective effect of paricalcitol
via a modulation of the TLR4-NF-κB pathway in
ischemia/reperfusion-induced acute kidney injury. Biochem Biophys
Res Commun. 444:121–127. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kose M, Bastug O, Sonmez MF, Per S,
Ozdemir A, Kaymak E, Yahşi H and Ozturk MA: Protective effect of
vitamin D against hyperoxia-induced lung injury in neonatal rats.
Pediatr Pulmonol. 52:69–76. 2017. View Article : Google Scholar
|
|
11
|
Shi YY, Liu TJ, Fu JH, Xu W, Wu LL, Hou AN
and Xue XD: Vitamin D/VDR signaling attenuates
lipopolysaccharide-induced acute lung injury by maintaining the
integrity of the pulmonary epithelial barrier. Mol Med Rep.
13:1186–1194. 2016.
|
|
12
|
Kong J, Zhu X, Shi Y, Liu T, Chen Y, Bhan
I, Zhao Q, Thadhani R and Li YC: VDR attenuates acute lung injury
by blocking Ang-2-Tie-2 pathway and renin-angiotensin system. Mol
Endocrinol. 27:2116–2125. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Verma R, Jung JH and Kim JY:
1,25-Dihydroxyvitamin D3 upregulates TLR10 while
downregulating TLR2, 4 and 5 in human monocyte THP-1. J Steroid
Biochem Mol Biol. 141:1–6. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wang H, Zhang Q, Chai Y, Liu Y, Li F, Wang
B, Zhu C, Cui J, Qu H and Zhu Ma: 1,25(OH)2D3
downregulates the Toll-like receptor 4-mediated inflammatory pahway
and ameliorates liver injury in diabetic rats. J Endocrinol Invest.
38:1083–1091. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Gambhir V, Kim J, Siddiqui S, Taylor M,
Byford V, Petrof EO, Jones G and Basta S: Influence of 1,
25-dihydroxy vitamin D3 on TLR4-induced activation of antigen
presenting cells is dependent on the order of receptor engagement.
Immunobiology. 216:988–996. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Hou AN, Fu JH, Yang HP, Zhu YT, Pan YQ, Xu
SY and Xue XD: Hyperoxia stimulates the transdifferentiation of
type II alveolar epithelial cells in newborn rats. Am J Physiol
Lung Cell Mol Physiol. 308:L861–L872. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Reis RB, Nagato AC, Nardeli CR, Matias IC,
Lima WG and Bezerra FS: Alternations in the pulmonary
histoarchitecture of neonatal mice exposed to hyperoxia. J Pediatr
(Rio J). 89:300–306. 2013. View Article : Google Scholar
|
|
18
|
Porzionato A, Sfriso MM, Mazzatenta A,
Macchi V, De Caro R and Di Giulio C: Effects of hyperoxic exposure
on signal transduction pathways in the lung. Respir Physiol
Neurobiol. 209:106–114. 2015. View Article : Google Scholar
|
|
19
|
Molteni M, Gemma S and Rossetti C: The
role of TLR4 in infectious and non-infectious inflammation.
Mediators Inflamm. 2016:69789362016. View Article : Google Scholar
|
|
20
|
You K, Xu XW, Fu JH, Xu SY, Yue XH, Yu ZL
and Xue XD: Hyperoxia disrupts pulmonary epithelial barrier in
newborn rats via the deterioration of occludin and ZO-1. Respir
Res. 13:362012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Li C, Fu JH, Liu HY, Yang HP, Yao L, You K
and Xue XD: Hyperoxia arrests pulmonary development in newborn rats
via disruption of endothelial tight junctions and downregulation of
CX40. Mol Med Rep. 10:61–67. 2014.PubMed/NCBI
|
|
22
|
Yang HP, Fu JH, Xue XD, Yao L, Qiao L, Hou
AN, Jin LL and Xing YJ: Epithelial-mesenchymal transitions in
broncho-pulmonary dysplasia of newborn rats. Pediatr Pulmonol.
49:1112–1123. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Qureshi ST, Zhang X, Aberg E, Bousette N,
Giaid A, Shan P, Medzhitov RM and Lee PJ: Inducible activation of
TLR4 confers resistance to hyperoxia-induced pulmonary apoptosis. J
Immunol. 176:4950–4958. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zhang X, Shan P, Qureshi S, Homer R,
Medzhitov R, Noble PW and Lee PJ: Cutting edge: TLR4 deficiency
confers susceptibility to lethal oxidant lung injury. J Immunol.
175:4834–4838. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Jia H, Sodhi CP, Yamaguchi Y, Lu P, Martin
LY, Good M, Zhou Q, Sung J, Fulton WB, Nino DF, et al: Pulmonary
epithelial TLR4 activation leads to lung injury in neonatal
necrotizing enterocolitis. J Immunol. 197:859–871. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Togbe D, Schnyder-Candrian S, Schnyder B,
Couillin I, Maillet I, Bihl F, Malo D, Ryffel B and Quesniaux VF:
TLR4 gene dosage contributes to endotoxin-induced acute respiratory
inflammation. J Leukoc Biol. 80:451–457. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Yao T, Ying X, Zhao Y, Yuan A, He Q, Tong
H, Ding S, Liu J, Peng X, Gao E, et al: Vitamin D receptor
activation protects against myocardial reperfusion injury through
inhibition of apoptosis and modulation of autophagy. Antioxid Redox
Signal. 22:633–650. 2015. View Article : Google Scholar :
|
|
28
|
Zhu T, Liu TJ, Shi YY and Zhao Q: Vitamin
D/VDR signaling pathway ameliorates 2, 4, 6-trinitrobenzene
sulphonic acid-induced colitis by inhibiting intestinal epithelial
apoptosis. Int J Mol Med. 35:1213–1218. 2015.PubMed/NCBI
|
|
29
|
Jeong MS, Kim JY, Lee HI and Seo SJ:
Calcitriol may down-regulate mRNA over-expression of Toll-like
receptor-2 and 4, LL-37 and proinflammatory cytokines in cultured
human keratocytes. Ann Dermatol. 26:296–302. 2014. View Article : Google Scholar : PubMed/NCBI
|