Introduction
Atherosclerosis is the predominant cause of coronary
artery disease and stroke, and thus, is the leading cause of
mortality and disability, globally (1). However, the precise molecular
mechanisms involved in the initiation and progression of
atherosclerosis remain poorly defined. Vascular integrity is
critical for cardiovascular homeostasis, and endothelial barrier
dysfunction leads to the leakage and retention of low-density
lipoprotein (LDL), the extravasation of monocytes into the vassal
wall and the decrement of cholesterol efflux capacity, which
triggers the formation of atherosclerotic plaques (2–5).
Endothelial cell integrity is crucial for
maintaining lesion-free arteries. Recent evidence has indicated
that endothelial cells retain various degrees of plasticity.
Endothelial cells can achieve a mesenchymal phenotype through
endothelial-mesenchymal transition (EndMT) (6). EndMT is characterized by the loss of
specific endothelial cell marker expression, including vascular
endothelial cadherin (VE-cadherin) and platelet endothelial cell
adhesion molecule-1 (PECAM-1; also known as CD31), and the
increased expression of mesenchymal cell marker, including α-smooth
muscle actin (α-SMA) and vimentin. During EndMT, endothelial cells
lose cell polarity and cell-cell adhesion. They then detach from
the organized endothelial layer, and then migrate towards the
surrounding tissue, which damages endothelial junction stability
and increases vascular permeability (7,8).
EndMT plays an important role in various pathological conditions,
including cardiac and nephritic fibrosis (6,9),
pulmonary hypertension (10,11), vascular calcification (12), endocardial fibroelastosis
(13) and most importantly,
atherosclerosis (14–17). Recently, using an endothelial cell
fate mapping technique, EndMT was demonstrated to participate in
the process of atherosclerosis by increasing the deposition of
fibronectin and adhesion molecules (16). Additionally, EndMT-derived
fibroblast-like cells have previously been shown to contribute to
atherosclerotic plaque progression and destabilize atherosclerotic
lesions by altering the collagen-matrix metalloproteinase balance
(17).
In contrast to the consensus that EndMT promotes
atherosclerosis, the cause of EndMT during this pathological
process is largely unknown. In atherosclerotic lesions, monocytes
infiltrate into the subendothelium, differentiate into macrophages,
internalize modified lipids and then become foam cells (18–20). Macrophages and foam cells secrete
inflammatory cytokines and chemokines, which induces a
non-resolving inflammatory process and affects plaque development
(21,22). Emerging evidence has indicated
that macrophages are comprised of heterogeneous cell populations,
which is determined by the surrounding microenvironment and that
they can switch from one phenotype to another (23–25). Classically activated M1
macrophages produce tumor necrosis factor (TNF), interleukin (IL)-6
and IL-1β, exacerbating the inflammatory response and promoting the
development of atherosclerotic lesions (26). By contrast, alternatively
activated M2 macrophages are an anti-inflammatory phenotype and
have been reported to be protective in atherosclerosis (27,28). However, despite the central role
of macrophages and foam cells in the development of
atherosclerosis, their function in the process of EndMT has yet to
be determined. Thus, in the present study, we investigated the
importance of different phenotypic macrophages and foam cells in
EndMT during atherosclerosis and explored the underlying
mechanisms.
Materials and methods
Cell culture
Human aortic endothelial cells (HAECs;
ATCC® PCS-100-011™) were purchased from American Tissue
Type Culture Collection (Manassas, VA, USA) and cultured in EGM-2
BullerKit™ Medium (cat. no. 3162; Lonza, Walkersville, MD, USA).
The cells were incubated in a humidified atmosphere containing 5%
CO2 at 37°C and passaged every 4 days (split ratio, 1:3)
using 0.25% trypsin. The cells were used between passages 2 and
7.
Macrophages were induced as previously described
(29,30). Briefly, peripheral blood
mononuclear cells were isolated, as previously described (31). Peripheral blood mononuclear cells
(PBMCs) were isolated from the peripheral blood of 106 healthy
volunteers. This study was part of the project 'Macrophage derived
foam cells impair endothelial barrier function by inducing
endothelial mesenchymal transition'. All the study protocols were
approved by the Clinical Ethics Committees of Sun Yat-sen Memorial
Hospital of Sun Yat-sen University. All blood samples and
procedures were approved by the Clinical Ethics Committees of Sun
Yat-sen Memorial Hospital of Sun Yat-sen University. Briefly,
peripheral blood (20 ml) was drawn into heparinized tubes from the
106 healthy volunteers. Blood was diluted with PBS at 1:1, and then
overlaid on lymphocyte separation medium (cat. no. 0850494; MP
Biomedicals, LLC, Santa Ana, CA, USA; density at 20°C:
1.0770–1.0800 g/ml), centrifuged at 2,300 rpm for 30 min, and
progressively slowed down for the last 6 min. PBMCs were collected
and washed twice in PBS at 2,000 rpm for 5 min. Monocytes were
positively selected from PBMCs using CD14 microbeads (cat. no.
130-050-201; Miltenyi Biotec, Auburn, CA, USA), and subsequently
cultured in RPMI-1640 medium supplemented with 10% fetal bovine
serum (FBS; Gibco; Thermo Fisher Scientific, Inc., Waltham, MA,
USA) at a density of 1×106 cells/10 cm2. To
induce M1 polarization, the cells were incubated in the presence of
100 ng/ml granulocyte-macrophage colony-stimulating factor (GM-CSF;
PeproTech, Inc., Rocky Hills, NJ, USA) for 7 days, and then treated
with 100 ng/ml lipopolysac-charide (eBioscience, San Diego, CA,
USA) and 20 ng/ml interferon-γ (PeproTech, Inc.) for 24 h. To
achieve M2a and M2c polarization, the cells were incubated in the
presence of 100 ng/ml macrophage colony-stimulating factor (M-CSF;
PeproTech, Inc.) for 7 days, and then treated with 20 ng/ml IL-4 or
20 ng/ml IL-10 (PeproTech, Inc.) for 24 h. Following polarization,
the cells were stimulated with 100 μg/ml oxidized LDL
(ox-LDL; Yiyuan Biotechnology, Guangzhou, China) for 12 h to induce
differentiation into foam cells. Subsequently, the culture media
were collected and centrifuged at 2,000 × g for 30 min at 4°C to
remove the debris and frozen at −80°C.
HAECs were seeded at 60–70% confluency in 6-well
plates, and starved in EBM-2 (cat. no. 3156; Lonza) without FBS for
18 h. The HAECs were then treated with conditioned medium from
different phenotypic macrophages or foam cells at a 1:1 ratio with
EBM-2 for 6 days. The medium was changed every other day.
Following serum starvation, the HAECs were treated
with CCL-2 (0, 12.5, 25 and 50 ng/ml), CCL-3 (0, 25, 50 and 100
ng/ml), CCL-4 (0, 25, 50 and 100 ng/ml), CCL-5 (0, 10, 20 and 40
ng/ml) and CCL-7 (0, 25, 50 and 100 ng/ml) for 6 days,
respectively. HAECs were also treated with CCL-2 (50 ng/ml), CCL-3
(100 ng/ml), CCL-4 (100 ng/ml), CCL-5 (40 ng/ml) and CCL-7 (100
ng/ml) for 2, 4 and 6 days, respectively.
These recombinant human chemokines were obtained
from PeproTech, Inc. as follows: CCL-2 (cat. no. 300-04), CCL-3
(cat. no. 300-08), CCL-4 (cat. no. 300-09), CCL-5 (cat. no.
300-06), CCL-7 (cat. no. 300-17).
The conditioned medium of the M1-FCs was pre-treated
at 37°C with 10 μg/ml human CCL-4 neutralizing antibody
(cat. no. ab9675; Abcam, Cambridge, MA, USA) or isotype IgG control
(cat. no. ab37415; Abcam) for 1 h. The processed conditioned media
were then added to the HAECs at a 1:1 ratio with EBM-2. The HAECs
were treated for 6 days, and the medium was changed every other
day.
Following serum starvation, the HAECs were
pre-treated with 5 μmol/l maraviroc (cat. no. S2003; Selleck
Chemicals LLC, Houston, TX, USA) for 2 h, prior to stimulation with
100 ng/ml CCL-4. 0.1% (vol/vol) DMSO was used as a solvent control.
HAECs were treated for 6 days, and the medium was changed every
other day.
Adenovirus packaging and RNA
knockdown
To investigate the underlying mechanisms of C-C
motif chemokine ligand-4 (CCL-4)-induced EndMT in HAECs,
transforming growth factor-β (TGF-β) expression was knocked down in
the HAECs using recombinant adenoviruses with the pAdEasy packaging
system. Briefly, recombinant vectors carrying RNA interference
sequences targeting TGF-β were generated using plasmid
pRNAT-H1.1/Adeno and incision enzymes MluI and
HindIII (forward, 5′-CCAACGCGTGACTACTACGCCAAGGAttc
aagagaTCCTTGGCGTAGTAGTCTTTTTAAGCTTGCG-3′; reverse,
5′-CCGAAGCTTAAAAAGACTACTACGCCAAGGA
tctcttgaaTCCTTGGCGTAGTAGTCACGCGTCCG-3′). Cloned vectors were
linearized by PmeI and subsequently transfected into BJ5183
bacteria (containing plasmid pAD-Easy-1) using electroporation for
homologous recombination. The recombinant vector was linearized
with PacI and amplified in AD293 cells (cat. no. FH0251;
FuHeng Biology, Shanghai, China). The cells were then lysed and the
adenovirus was purified by the cesium chloride (CsCl) density
gradient ultracentrifugation method and diluted in
phosphate-buffered saline (PBS) as previously reported (32). Finally, the virals titer was
determined using the cytopathic effect (CPE) counting method. The
HAECs were infected with adenovirus at a concentration of 50
pfu/cell.
Scanning electron microscopy
Human aortas were obtained from cadaver organ
donors. We collected 1 normal and 2 atherosclerotic ascending aorta
samples, judged by medical history and Oil Red O and H&E
staining of the aortic tissues. The clinical and demographic
characteristics of the cadaver organ donors are shown in Table I. All the samples were obtained
from cadaver organ donors, who died due to car accidents. The
aortic tissues were obtained, at the same time with other donated
organs, after the clinical announcement of brain death, with
circulation stabilization, and also with signed organ donor
documents, consent and the agreement of family members, and ethics
committee approval. All procedures were approved by the Clinical
Ethics Committee of Sun Yat-Sen Memorial Hospital of Sun Yat-Sen
University (Guangzhou, China). For examination under a Hitachi
S-3400N scanning electron microscope (serial no. 341117-08;
Hitachi, Ltd., Tokyo, Japan), vessel sections were fixed in 2.5%
glutaraldehyde in PBS overnight. After washing 3 times with PBS,
the sections were dehydrated in a series of ethanol dilutions (30,
50, 70, 90 and 100%), dried by the critical-point method, sputtered
by gold-palladium and prepared for anlaysis using a Hitachi S-3400N
scanning electron microscope (Hitachi, Ltd.).
 | Table ISelected clinical and demographic
characteristics of the cadaver organ donors. |
Table I
Selected clinical and demographic
characteristics of the cadaver organ donors.
| Subject number | 1 | 2 | 3 |
|---|
| Group | Normal | AS | AS |
| Age (years) | 53 | 54 | 49 |
| Gender | Male | Male | Female |
| Smoking | No | No | No |
| History of CAD | No | Yes | Yes |
| History of DM | No | No | No |
| History of
hypertension | No | No | Yes |
| Oil Red O
staining | Negative | Positive | Positive |
| H&E
staining | Normal | AS | AS |
Immunohistofluorescence
Briefly, the samples were incubated with rabbit
anti-von Willebrand factor (vWF) polyclonal antibody (1:200; cat.
no. ab6994) and mouse anti-α-SMA monoclonal antibody (1:200; cat.
no. ab18147) (both from Abcam) at 4°C overnight. After washing 3
times with PBS-Tween-20 (PBST), they were incubated with the
appropriate Alexa Fluor 488 goat anti-rabbit or Alexa Fluor 594
goat anti-mouse antibody (Invitrogen; Thermo Fisher Scientific,
Inc.) in 5% bovine serum albumin (BSA) for 1 h at room temperature.
The nuclei were stained using 4′,6-diamidino-2-phenylindole (DAPI)
(1 μg/ml; cat. no. D9542; Sigma-Aldrich; Merck Millipore,
Darmstadt, Germany) for 3 min. EndMT was evaluated by the criteria
described in a previous study (33). All counts were performed and
compared by 2 investigators.
Western blot analysis
The cells were washed, lysed and total protein was
extracted. The total protein concentration was determined using a
BSA Protein Assay kit (Beyotime Biotechnology, Shanghai, China)
according to the manufacturer's instructions and western blot
analysis was performed. Briefly, protein extraction was separated
by sodium dodecyl sulfate-polyacrylamide gel electrophoresis
(SDS-PAGE) followed by transfer onto polyvinylidene fluoride
membranes [cat. nos. IPVH00010 (0.45 μm) and ISEQ00010 (0.2
μm); EMD Millipore Corporation, Billerica, MA, USA]. The
membrane was blocked in 5% BSA for 1 h at room temperature and
immunoblotted with the corresponding antibodies at 4°C overnight.
Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used as an
internal control. The antibodies used are listed as follows: rabbit
anti-CD31 polyclonal antibody (1:1,000; cat. no. ab28364; Abcam),
goat anti-VE-cadherin polyclonal antibody (1:1,000; cat. no.
sc-6458; Santa Cruz Biotechnology, Inc., Dallas, TX, USA), rabbit
anti-vimentin monoclonal antibody (1:1,000; cat. no. 5741; Cell
Signaling Technology, Inc., Danvers, MA, USA), rabbit anti-α-SMA
polyclonal antibody (1:1,000; cat. no. ab5694), rabbit
anti-fibroblast-specific protein-1 (FSP-1) polyclonal antibody
(1:1,000; cat. no. ab27427) (both from Abcam), rabbit anti-GAPDH
monoclonal antibody (1:1,000, cat. no. 2118), rabbit anti-TGF-β
polyclonal antibody (1:1,000; cat. no. 3711) (both from Cell
Signaling Technology, Inc.) and rabbit anti-vascular cell adhesion
molecule-1 (VCAM-1) monoclonal antibody (1:1,000; cat. no.
ab134047; Abcam). The membranes were washed 3 times with PBST and
incubated with the following appropriate secondary HRP-linked
antibodies: goat anti-rabbit IgG (1:5,000; cat. no. 7074; Cell
Signaling Technology, Inc.) or donkey anti-goat IgG (1:5,000; cat.
no. sc-2020; Santa Cruz Biotechnology, Inc.), for 1.5 h at room
temperature. After washing, detection was performed using an
advanced enhanced chemiluminescence system.
Cytokine and chemokine analysis
The TGF-β1 level was measured in the culture
supernatants of the macrophages treated with or without ox-LDL
using a commercial enzyme linked immunosorbent assay (ELISA) kit
(Neobioscience, Shenzhen, China). The levels of other cytokines and
chemokines [including IFN-γ, IL-1β, TNF-α, IL-12p70, IL-2, IL-4,
IL-5, IL-6, GM-CSF, IL-18, Eotaxin, GRO-α, IL-8, IP-10, MCP-1
(CCL-2), MIP-1α (CCL-3), MIP-1β (CCL-4), SDF-1α, IL-13 and RANTES
(CCL-5)] were measured using the ProcartaPlex™ Multiplex
Immunoassay kit (cat. no. EPX200-12173-901; Affymetrix Inc., Santa
Clara, CA, USA), according to the manufacturer's instructions.
The chemokine expression profiles of the M1
macrophages and M1-derived foam cells were assessed using a Human
Chemokine array kit (cat. no. ARY017; R&D Systems, Inc.,
Minneapolis, MN, USA), according to the manufacturer's
instructions. Briefly, the arrays were incubated in the
supernatants and detection antibody cocktail overnight at 4°C.
After washing, the arrays were incubated with
streptavidin-horseradish peroxidase and then exposed to the Chemi
Reagent Mix. The reaction intensity was analyzed using the G:BOX
XT4 imager (Syngene, Frederick, MD, USA) and the optical density
was calculated using ImageJ software.
Monocyte adhesion assay
THP-1 cells (Boster Biological Technology, Wuhan,
China) were cultured in RPMI-1640 medium supplemented with 10% FBS
and stained with 50 μM calcein-AM for 30 min at 37°C. The
labeled THP-1 cells were seeded at a density of 5.0×105
cells/ml on confluent HAECs, which were pre-treated with 100 ng/ml
CCL-4 (PeproTech, Inc.) for 6 days. Following 1 h of incubation,
non-adherent cells were removed by gentle washing with PBS and
THP-1 cell adhesion was assessed using a Nikon Ti-S inverted
fluorescence microscope (serial no. 533477; Nikon, Tokyo,
Japan).
Endothelial monolayer permeability
The HAECs were cultured on 0.2% gelatin-coated 6.5
mm-diameter Transwell inserts (0.4 μm pore size; Corning
Inc., Corning, NY, USA). Fluorescein isothiocyanate (FITC)-dextran
(1 mg/ml; molecular weight, 70 kDa; Sigma-Aldrich; Merck Millipore)
was added to the upper chamber. The medium from the lower chamber
was collected after 1 h and fluorescence measured using a
SpectraMax M5 microplate reader (Molecular Devices, LLC, Sunnyvale,
CA, USA; excitation, 485 nm; emission, 525 nm).
Statistical analysis
Data are presented as the means ± standard error of
mean. Results were analyzed by a two-tailed Student's t-test or
one-way analysis of variance with Bonferroni post-hoc test for
multiple comparisons as appropriate using GraphPad Prism software
(GraphPad Software, Inc., La Jolla, CA, USA). A value of P<0.05
was considered to indicate a statistically significant
difference.
Results
Endothelial barrier dysfunction and EndMT
occur in human atherosclerotic plaques
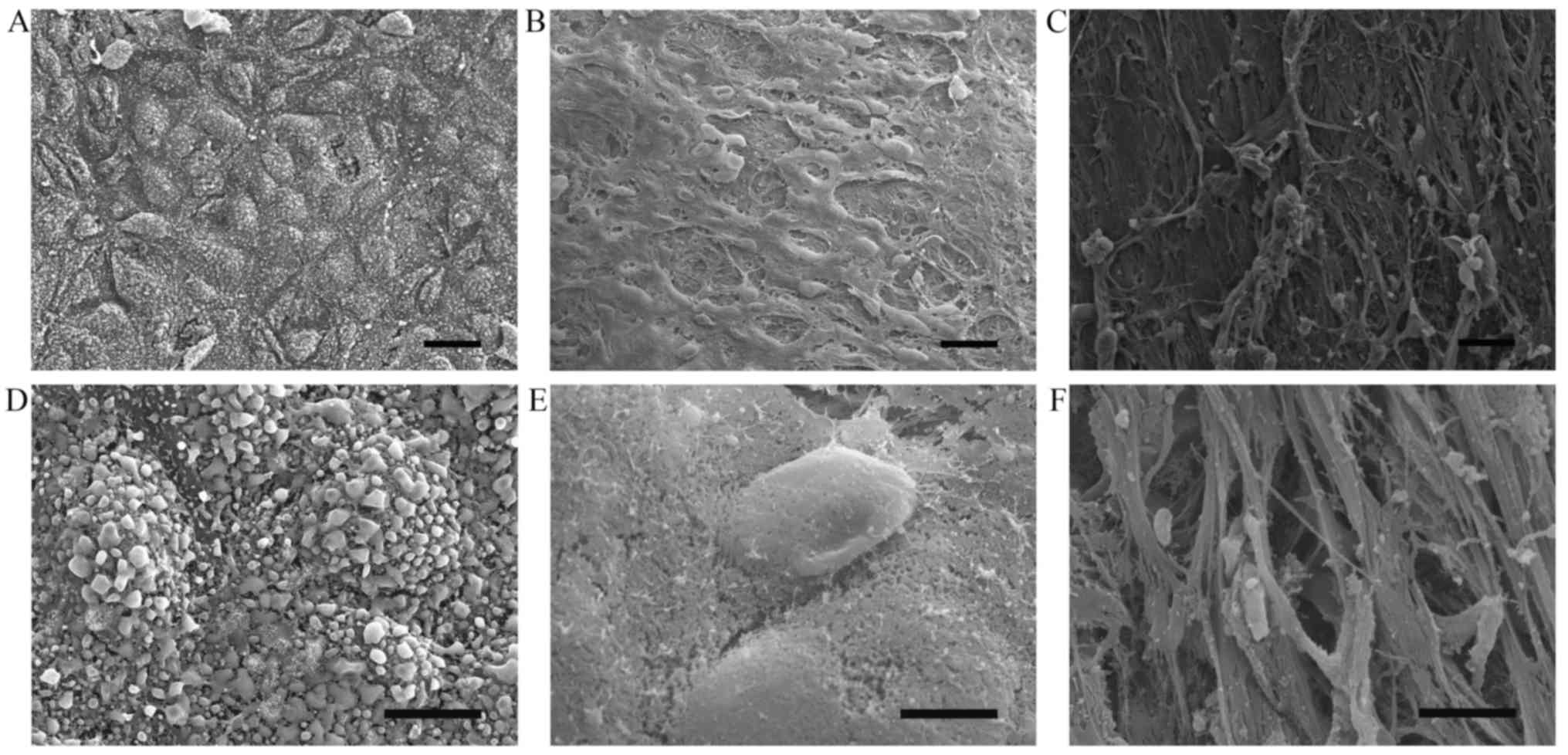
To demonstrate altered endothelial integrity in
atherosclerotic plaques, human aorta specimens were subjected to
scanning electron microscopy. The results demonstrated that
endothelial cells in the normal control aorta tissue were arranged
in an intact and regular manner (Fig.
1A and D) whereas endothelial cells were partially preserved
with altered polarity, and the subendothelial surface with a
fibrous cap was visible in the atherosclerotic lesions (Fig. 1B, C, E and F).
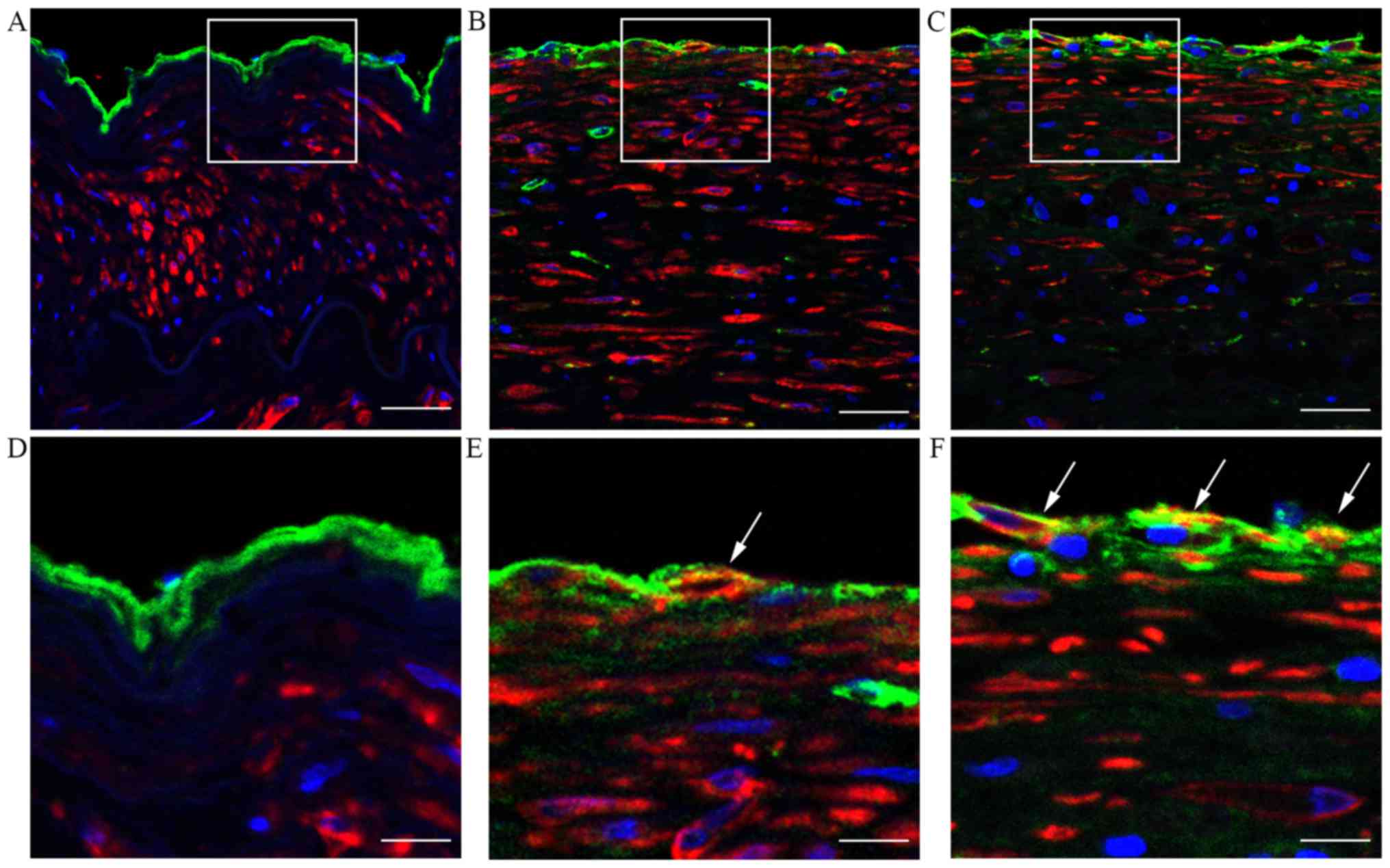
As EndMT is an important cause of the loss of
endothelial cells polarity, the endothelial changes due to EndMT in
atherosclerotic plaques were examined by immunohistofluorescence
staining. As shown in Fig. 2,
evidence of EndMT, as indicated by the co-localization of vWF and
α-SMA expression, was more pronounced in atherosclerotic lesions
compared with the normal control tissue. These data suggest that
EndMT may contribute to endothelial cell polarity changes and the
development of atherosclerosis.
Conditioned medium from M1-FCs stimulates
HAECs to undergo EndMT in vitro
The reason for EndMT and endo-thelial cell polarity
loss in atherosclerotic plaques remains unclear. Macrophages and
foam cells are the major cellular component of atherosclerotic
plaques and, thus, their functions are ciritical in the
atherosclerotic process. Therefore, the role of macrophages and
foam cells during the changes of endothelial cells in plaques was
investigated. Macrophages are classically divided into 2 groups,
namely, M1 and M2 macrophages. Different phenotypes of macrophages
have distinct properties. In this study, the effects of both
subtypes on the endothelium were examined. It was observed that
supernatants from M1 macrophage-derived foam cells (M1-FCs) induced
EndMT, with the cell morphology changing from a cobblestone-like
appearance to a spindle-shaped pattern (Fig. 3D), whereas, supernatants from
M2a-FCs or M2c-FCs did not exhibit these changes (Fig. 3E and F). Notably, the macrophages
(M1, M2a and M2c) not pre-treated with ox-LDL did not exert any
EndMT-like effects on endothelial cells (Fig. 3A–C). Western blot analysis
demonstrated that the expression of endothelial markers
(VE-cadherin and CD31) was markedly reduced, together with an
observable increase in the expression of mesenchymal markers (α-SMA
and FSP-1) in the HAECs treated with conditioned medium from M1-FCs
(Fig. 3G).
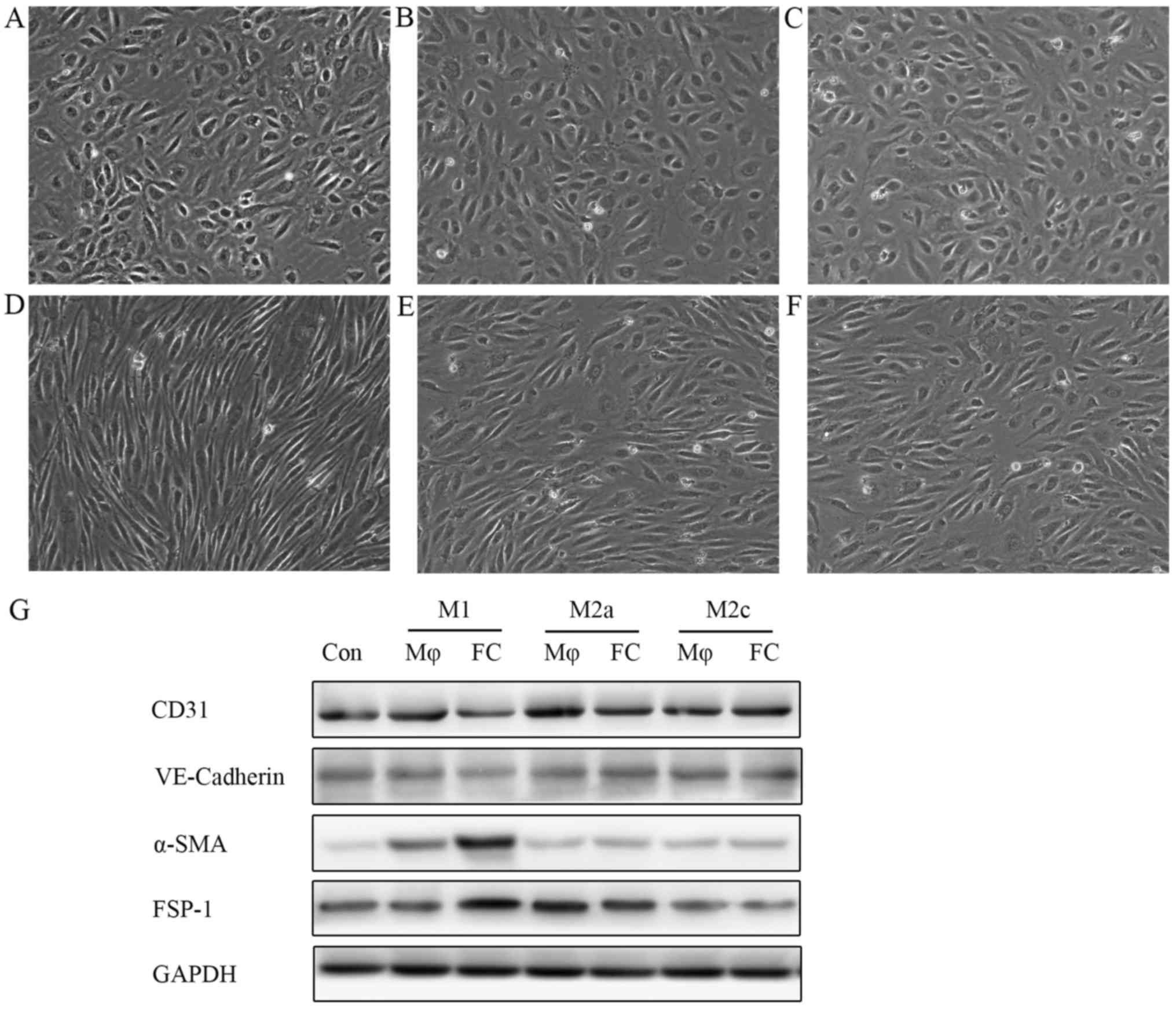 | Figure 3Conditioned medium from M1-FCs
stimulates HAECs to undergo EndMT in vitro. HAECs were
treated with conditioned medium from different phenotypic
macrophages: conditioned medium from (A) M1, (B) M2a and (C) M2c
macrophages and (D) M1, (E) M2a and (F) M2c-derived FCs at 1:1
ratio with EBM-2 for 6 days. HAECs treated with conditioned medium
from M1-FCs lost the typical cobblestone-like morphology and gained
a spindle-like appearance. (G) Western blot analysis of endothelial
and mesenchymal cell surface markers demonstrated that HAECs
incubated with conditioned medium from M1-FCs underwent EndMT.
M1-FCs, M1 macrophage-derived foam cells; Con, control; Mφ,
macrophage; FC, foam cell; VE-cadherin, vascular endothelial
cadherin; α-SMA, α-smooth muscle actin; FSP-1, fibroblast-specific
protein-1; EndMT, endothelial mesenchymal transition. |
Levels of cytokines and chemokines in the
supernatants
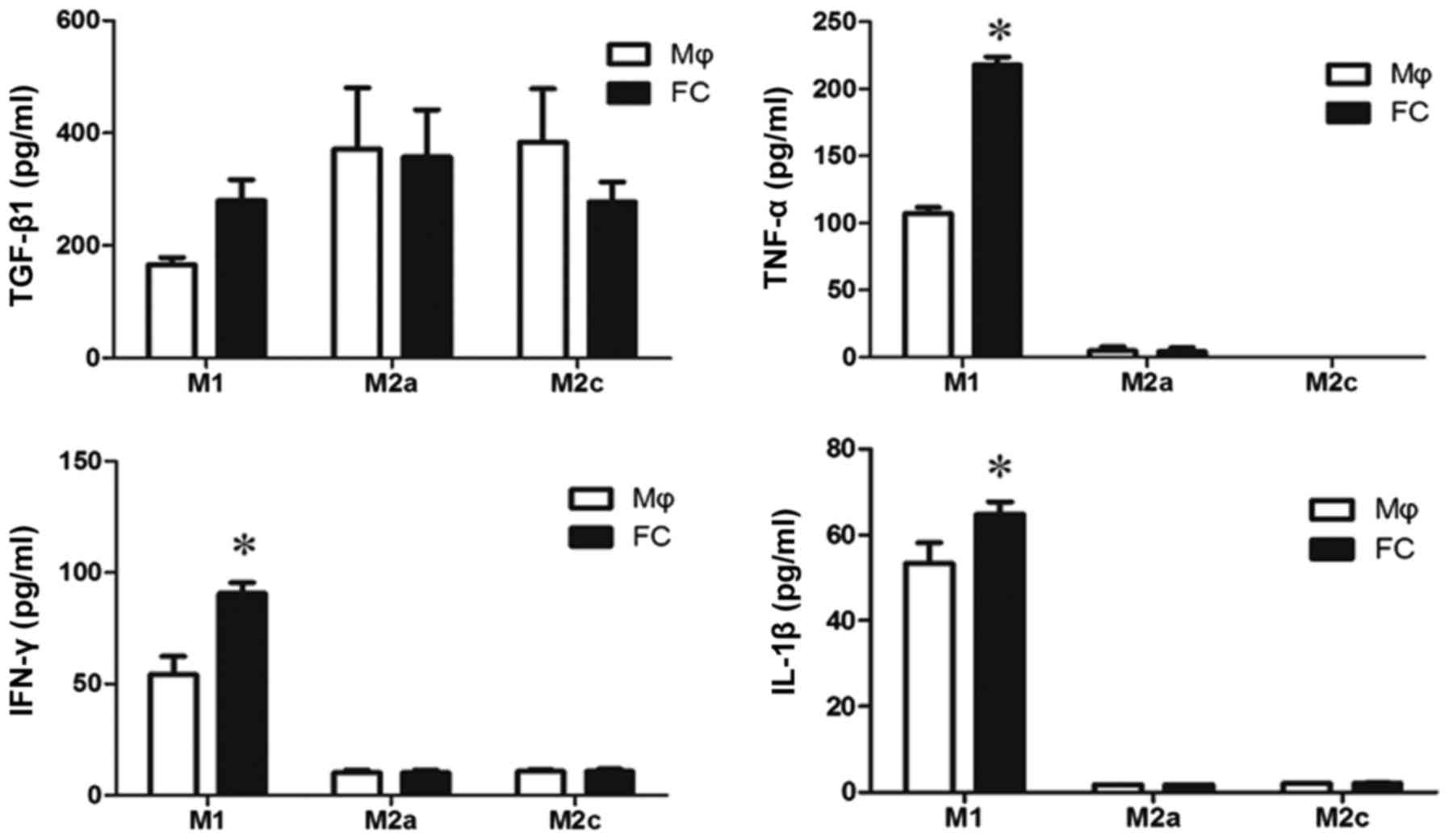
It is not clear which components in M1-FC
conditioned medium induced EndMT. TGF-β1 is one of the most
important factors for both epithelial-mesenchymal transition (EMT)
and EndMT, thus, TGF-β1 levels were examined in the supernatants of
macrophages and foam cells. The levels of TGF-β1 were not
significantly increased in the M1-FCs compared with the M1
macrophages (Fig. 4). In
addition, as inflammatory cytokines result in EndMT, the levels of
classical cytokines, including TNF-α, IL-1β and IFN-γ, were
measured. There was a significant difference in the levels of these
cytokines in the conditioned medium of M1-FCs compared with those
in M1 macrophages (Fig. 4).
However, the concentration of these cytokines was too low to induce
EndMT (16)
Secretion of CCL-4 from M1
macrophage-derived foam cells is involved in EndMT
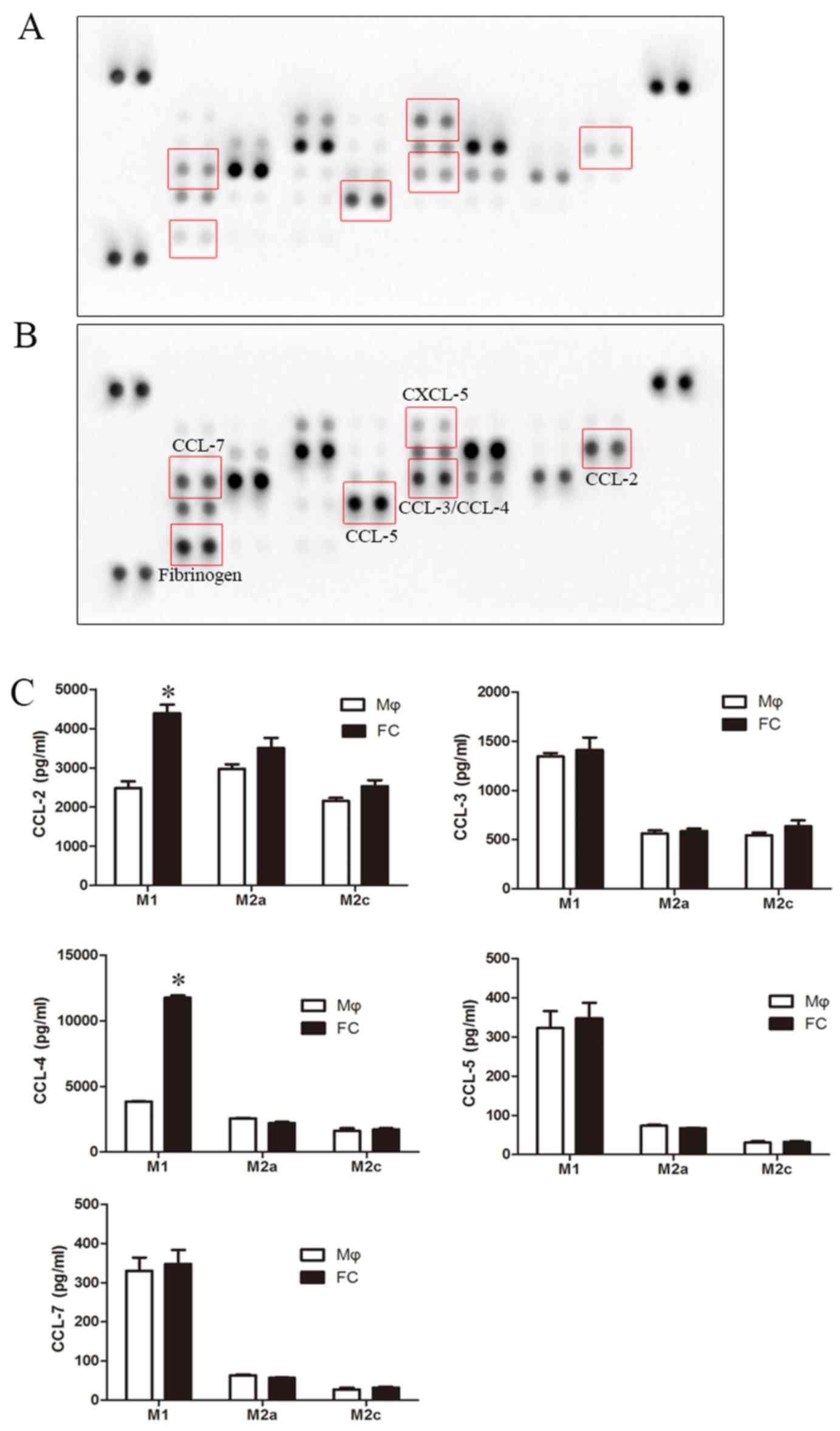
Chemokines are important inducers of EMT, and are
critical in the formation and progression of atherosclerosis
plaques (34–37); thus, a human chemokine protein
array was used to screen for alterations in the levels of
chemokines in foam cells. The protein array revealed a pronounced
increase in CCL-2, CCL-3, CCL-4, CCL-5, CCL-7 and fibrinogen
levels, and a decrease in CXCL-5 levels in the M1-FC supernatants
compared with the macrophages (Fig.
5A and B). However, ELISA confirmed that only the levels of
CCL-2 and CCL-4 exhibited a significant increase (Fig. 5C). However, the effects of these
cytokines on EndMT are unknown; thus, their effects on EndMT were
examined in the present study. CCL-2, CCL-3, CCL-4, CCL-5 and CCL-7
were used to stimulate the HAECs, and the levels of EndMT markers
were analyzed. The results presented in Fig. 6 demonstrated a marked increase in
EndMT upon CCL-4 stimulation in a time- and concentration dependent
manner (Fig. 6A and B), as
evidenced by the loss of endothelial marker expression
(VE-cadherin) and the increased expression of mesenchymal markers
(α-SMA and Vimentin). A weaker effect was observed after CCL-3 and
CCL-5 intervention (data not shown). CCL-2 and CCL-7 did not exert
any effect on EndMT (data not shown). Additionally, blocking CCL-4
with anti-CCL-4 antibody reversed the above-mentioned EndMT-related
changes induced by the M1-FC supernatant (Fig. 6C). These data suggest that CCL-4
produced by M1-FCs plays a key role in EndMT.
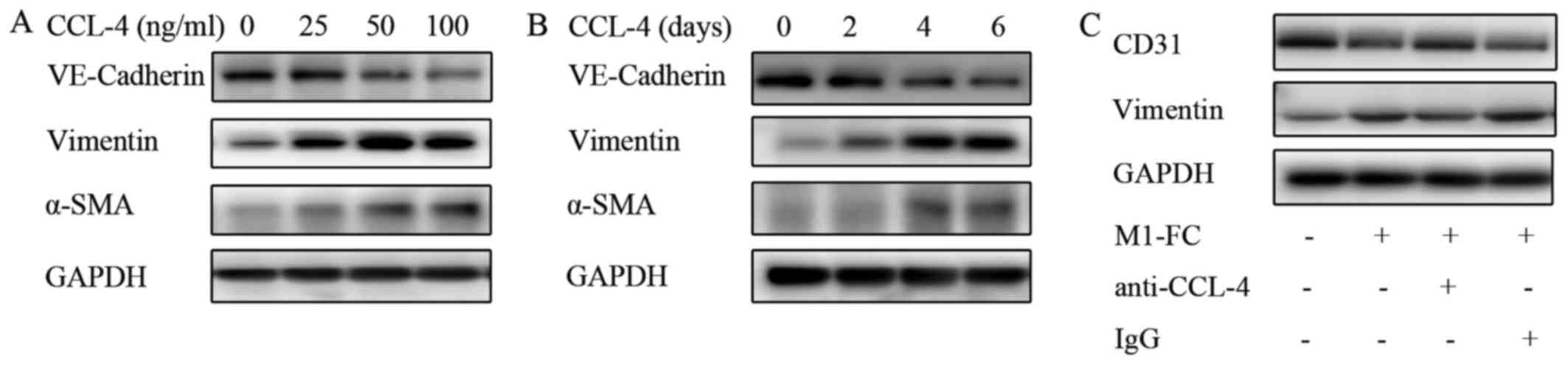 | Figure 6CCL-4 induces EndMT in a
concentration- and time-dependent manner. (A) HAECs were cultured
in medium containing 0, 25, 50 or 100 ng/ml CCL-4 for 6 days, and
(B) HAECs were cultured in medium containing 100 ng/ml CCL-4 for 2,
4 or 6 days; the levels of VE-cadherin, vimentin and α-SMA were
determined by western blot analysis (C) EndMT induced by
conditioned medium of M1-FCs was neutralized by human anti-CCL-4
antibody. Human CCL-4 neutralizing antibody (10 μg/ml) or
isotype control were incubated at 37°C with conditioned medium for
1 h before addition to HAECs. EndMT, endothelial mesenchymal
transition; HAEC, human aortic endothelial cell; CCL-4, C-C motif
chemokine ligand-4; VE-cadherin, vascular endothelial cadherin;
α-SMA, α-smooth muscle actin; M1-FC, M1 macrophage-derived foam
cell; IgG, immunoglobulin G. |
CCL-4-induced EndMT contributes to
endothelial barrier dysfunction
Monocyte adhesion, infiltration and retention in the
arteries are key features of atherosclerosis. In this study, to
investigate the effect of EndMT on monocyte adhesion, an in
vitro adhesion assay was performed to determine the adhesion of
calcein-AM-stained THP-1 cells to HAECs. We found that a small
number of THP-1 monocytes adhered to quiescent HAECs, but many more
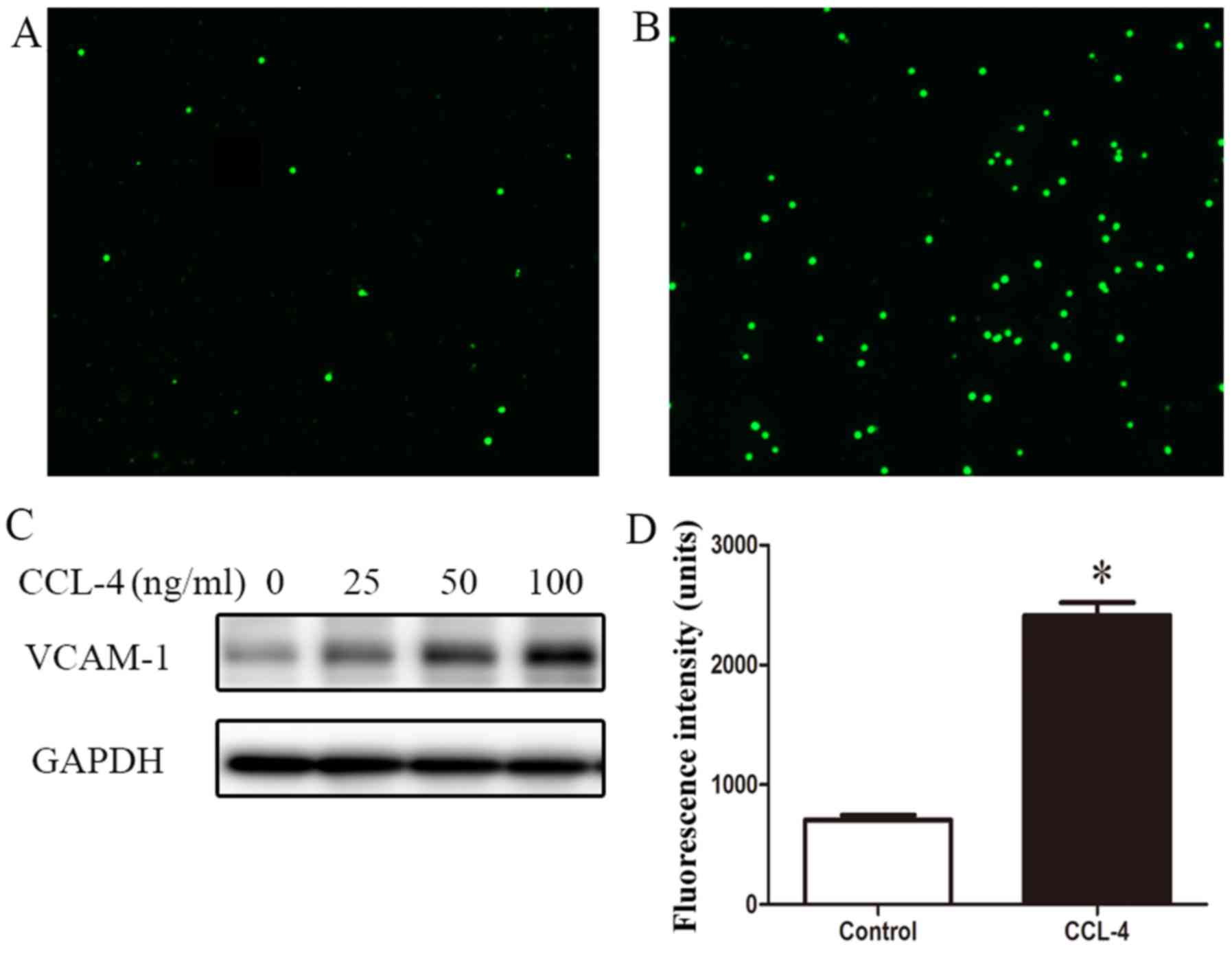
THP-1 monocytes adhered to the CCL-4-treated HAECs (Fig. 7A and B). Western blot analysis
revealed the enhanced expression of VCAM-1 in the HAECs stimulated
with CCL-4 (Fig. 7C). These
results suggest that EndMT induced by CCL-4 contributes to
inflammatory cell adhesion to the vascular endothelium.
Additionally, the loss of endothelial barrier
integrity contributes to the activation and formation of arterial
lesions. Thus, endothelial permeability was assessed using
FITC-dextran. In the cells undergoing CCL-4-induced EndMT, a
>2-fold increase in endothelial leakage compared with the
untreated cells in monolayer was observed (Fig. 7D).
C-C motif chemokine receptor-5 (CCR-5)
and TGF-β are involved in CCL-4-induced EndMT
The mechanisms through which CCL-4 induces EndMT are
not clear. Currently, CCR-5 is the unique known receptor of CCL-4.
Therefore, in this study, we investigated whether CCR-5 mediates
CCL-4-induced EndMT. Maraviroc is a widely used CCR-5 antagonist.
In this experiment, CD31 expression was restored and the vimentin
level was reduced in the HAECs treated with CCL-4 and maraviroc
compared with the cells stimulated only with CCL-4, which indicates
that CCR-5 is involved in mediating CCL-4-induced EndMT (Fig. 8A).
 | Figure 8CCL-4 increases TGF-β levels via
CCR-5. (A) Maraviroc, a C-C motif chemokine receptor-5 inhibitor,
restored CD31 expression and reduced vimentin level following
CCL-4-induced EndMT. (B) TGF-β expression was increased in HAECs by
CCL-4 in a concentration-dependent manner, and the effect was
blocked by maraviroc. (C) Knockdown of TGF-β expression in HAECs
inhibited endothelial mesenchymal transition induced by CCL-4, as
indicated by the reduced expression of vimentin and the restored
expression of CD31. CCL-4, C-C motif chemokine ligand-4; EndMT,
endothelial mesenchymal transition; DMSO, dimethyl sulfoxide; HAEC,
human aortic endothelial cell; TGF-β, transforming growth factor-β;
shRNA, short hairpin RNA; GFP, green fluorescent protein. |
The downstream mechanisms of CCR-5 in the process of
EndMT are still unclear. It was previously reported that CCR-5 may
modify the expression of TGF-β, a key EndMT protein, in endothelial
progenitor cells (38).
Therefore, the effect of the CCL-4/CCR-5 axis on TGF-β expression
was investigated in this study. We observed that TGF-β expression
was markedly increased in the HAECs stimulated with CCL-4 compared
with the untreated HAECs; however, this effect was abolished by
pre-treatment with the CCR-5 antagonist, maraviroc (Fig. 8B). Notably, the knockdown of TGF-β
expression in the HAECs inhibited the EndMT induced by CCL-4, as
indicated by the restored expression of CD31 and the decreased
expression of vimentin following TGF-β knockdown and CCL-4
stimulation (Fig. 8C). These data
indicated that CCL-4 promoted TGF-β expression via CCR-5, which
induced EndMT in HAECs. Thus, even though TGF-β expression
exhibited no significant increase inthe macrophages differentiating
into foam cells, it was upregulated by CCL-4 in the endothelium and
promoted EndMT.
Discussion
Endothelial dysfunction is a key factor that
triggers and exacerbates the formation of atherosclerotic lesions.
The present study demonstrated that the endothelial space was much
wider and EndMT occurred in the intima of human aortic
atherosclerotic plaques, however, it was absent in the normal
aorta, which indicated that the endothelial barrier was destroyed
in the atherosclerotic artery. We also observed that endothelial
barrier dysfunction associated with EndMT contributed to monocyte
adhesion to the endothelium and infiltration into the
subendothelium and a pro-inflammatory cell phenotype. Further
experiments revealed that EndMT was induced by chemokine CCL-4 in a
paracrine manner in M1-FCs in vitro by increasing TGF-β
expression.
EndMT may be a repair response or adaptation to
pathological stimulus, and contributes to the onset and progression
of vascular pathology. It has recently been confirmed that EndMT is
common during atherosclerosis and drives the progression of
atherosclerosis by increasing the deposition of fibronectin and
adhesion molecules (16) and,
altering the collagen-matrix metal-loproteinase balance (17). During the process of EndMT,
intimal endothelial cells lose their integrity followed by
increased endothelial barrier permeability and enhanced adhesion
molecule expression accompanied by increased monocyte adhesion
(7). Despite above-mentioned
effects of EndMT on atherosclerosis, the mechanisms through which
EndMT mediates the process of atherogenesis are largely unknown.
The infiltration of various inflammatory cells is an early and
crucial process during the development of atherosclerotic lesions.
Inside the vascular wall, inflammatory cells differentiate into
macrophages and ingest ox-LDL to form lipid-rich foam cells, which
is the pivotal pathological change of atherosclerosis. In the
present study, M1-FCs were demonstrated to secrete CCL-4 and
promote EndMT, which additionally increased endothelial
permeability and exacerbated monocyte infiltration, thus, forming a
positive loop to promote the development of atherosclerotic
lesions. To the best of our knowledge, the present study is the
first to report that macrophages induce EndMT, whereas other
studies have demonstrated that EMT can be induced by macrophages
(34,35).
Previous studies have demonstrated that a direct
increase in TGF-β expression is involved in EMT. Additionally, the
majority of studies have reported that EMT is induced by M2
macrophages (35,39); however, EndMT was not induced by
M2 macrophages in the present study. The most plausible explanation
for these differences between the effects of M2 macrophages on EMT
and EndMT may be that different cell types exert diverse effects.
However, M2 macrophages may promote angiogenesis in a paracrine
manner (40,41); as is already known, angiogenesis
plays an important role in the development of atherosclerotic and
plaque instability (42–44). Theoretically speaking, M2
macrophages should be pro-atherogenic due to their pro-angiogenic
effects. However, the emerging understanding of macrophage subsets
and their functions in atherosclerotic plaque has led to the
consensus that M1 macrophages are pro-atherogenic, while M2
macrophages may promote plaque stability (27,28,45), primarily though their tissue
repair and anti-inflammatory properties. Additionally, our previous
study demonstrated that EndMT damaged endothelial tube formation
capacity in an in vitro angiogenesis assay (46), which is consistent with our
present results.
The present study demonstrated that CCL-4 was
crucial for M1-FC-induced EndMT, and to the best of our knowledge,
this is the first report to investigate the association between
EndMT/EMT and CCL-4. CCL-4 has been previously reported to be
associated with the development of atherosclerosis. Population
studies have demonstrated that increased CCL-4 levels are
associated with a poor prognosis for coronary heart disease
(47–49). Mechanistic analysis has indicated
that CCL-4 is increased in atherosclerotic plaques, and reducing
its expression or inhibiting it with a CCR-5 antagonist can
attenuate the development of atherosclerosis (50). Classically, CCL-4 is regarded as a
pro-inflammatory cytokine and mediates the inflammatory cascade,
promoting atherosclerotic lesion formation. The present study
provided novel insight into EndMT and the subsequent inflammatory
cell infiltration and adhesion, explaining the adverse effects of
CCL-4 on atherosclerosis. Additionally, our study demonstrated that
the malignant effects of CCL-4 were at least partially induced
through CCR-5, the only recognized receptor of CCL-4. As expected,
CCR-5 has been reported to be involved in atherosclerosis.
Increased monocyte CCR-5 expression is associated with
atherosclerosis, and statins therapy has been demonstrated to
reduce CCR-5 concentrations (51,52). Furthermore, CCR-5 expression in
monocytes has been demonstrated to be important for migration
during the development of atherosclerosis (53) and the CCR5 antagonist, maraviroc,
has been shown to be effective in limiting plaque progression in
different atherosclerosis animal models (54). The present study also demonstrated
that monocytes were prone to adhere to the endothelial monolayer
following CCL-4 treatment, which explains the findings from the
aspect of a paracrine signaling mechanism and supports that
maraviroc, a drug used widely in clinical practice, may be a novel
potential treatment for atherosclerosis.
Certain limitations of the present study should be
noted. Firstly, the protein array was performed to measure
chemotactic cytokine family members; thus, even though classical
inflammatory cytokines promote EndMT, numerous cytokines that are
not part of this group were not examined. Additionally, the
mechanism through which CCL-4/CCR5 promotes TGF-β was not
determined, and there is currently no reported evidence regarding
this regulatory mechanism, at least to the best of our
knowledge.
Despite limitations, the present study demonstrated
that EndMT is involved in endothelial barrier dysfunction and is
associated with plaque development. This study investigated the
molecular mechanisms that mediate EndMT and elucidated a potential
paracrine mechanism involving the interaction between macrophages
and endothelial function, which may be beneficial for developing
novel treatments for atherosclerosis via targeting the CCL-4/CCR5
axis.
Acknowledgments
This study was financially supported by grants from
the National Natural Science Foundation of China (nos. 91439125,
81270212, 81570213, 81100101, 81570329 and 81500223), the Guangdong
Province Natural Science Fund (nos. S2013010014011 and
2015A030310059) and the Science and Technology Program of Guangzhou
(no. 2014Y2-00118).
References
|
1
|
Herrington W, Lacey B, Sherliker P,
Armitage J and Lewington S: Epidemiology of atherosclerosis and the
potential to reduce the global burden of atherothrombotic disease.
Circ Res. 118:535–546. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Schulte D, Küppers V, Dartsch N, Broermann
A, Li H, Zarbock A, Kamenyeva O, Kiefer F, Khandoga A, Massberg S,
et al: Stabilizing the VE-cadherin-catenin complex blocks leukocyte
extravasation and vascular permeability. EMBO J. 30:4157–4170.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Giannotta M, Trani M and Dejana E:
VE-cadherin and endothelial adherens junctions: Active guardians of
vascular integrity. Dev Cell. 26:441–454. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Steffensen LB, Mortensen MB, Kjolby M,
Hagensen MK, Oxvig C and Bentzon JF: Disturbed laminar blood flow
vastly augments lipoprotein retention in the artery wall: A key
mechanism distinguishing susceptible from resistant sites.
Arterioscler Thromb Vasc Biol. 35:1928–1935. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Monette JS, Hutchins PM, Ronsein GE,
Wimberger J, Irwin AD, Tang C, Sara JD, Shao B, Vaisar T, Lerman A,
et al: Patients with coronary endothelial dysfunction have impaired
cholesterol efflux capacity and reduced HDL particle concentration.
Circ Res. 119:83–90. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Zeisberg EM, Tarnavski O, Zeisberg M,
Dorfman AL, McMullen JR, Gustafsson E, Chandraker A, Yuan X, Pu WT,
Roberts AB, et al: Endothelial-to-mesenchymal transition
contributes to cardiac fibrosis. Nat Med. 13:952–961. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Good RB, Gilbane AJ, Trinder SL, Denton
CP, Coghlan G, Abraham DJ and Holmes AM: Endothelial to mesenchymal
transition contributes to endothelial dysfunction in pulmonary
arterial hypertension. Am J Pathol. 185:1850–1858. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Yan Z, Wang ZG, Segev N, Hu S, Minshall
RD, Dull RO, Zhang M, Malik AB and Hu G: Rab11a mediates vascular
endothelial-cadherin recycling and controls endothelial barrier
function. Arterioscler Thromb Vasc Biol. 36:339–349. 2016.
View Article : Google Scholar
|
|
9
|
Zeisberg EM, Potenta SE, Sugimoto H,
Zeisberg M and Kalluri R: Fibroblasts in kidney fibrosis emerge via
endothelial-to-mesen-chymal transition. J Am Soc Nephrol.
19:2282–2287. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ranchoux B, Antigny F, Rucker-Martin C,
Hautefort A, Péchoux C, Bogaard HJ, Dorfmüller P, Remy S, Lecerf F,
Planté S, et al: Endothelial-to-mesenchymal transition in pulmonary
hypertension. Circulation. 131:1006–1018. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Choi SH, Hong ZY, Nam JK, Lee HJ, Jang J,
Yoo RJ, Lee YJ, Lee CY, Kim KH, Park S, et al: A hypoxia-induced
vascular endothelial-to-mesenchymal transition in development of
radiation-induced pulmonary fibrosis. Clin Cancer Res.
21:3716–3726. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Yung LM, Sánchez-Duffhues G, Ten Dijke P
and Yu PB: Bone morphogenetic protein 6 and oxidized low-density
lipoprotein synergistically recruit osteogenic differentiation in
endothelial cells. Cardiovasc Res. 108:278–287. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Xu X, Friehs I, Zhong Hu T, Melnychenko I,
Tampe B, Alnour F, Iascone M, Kalluri R, Zeisberg M, Del Nido PJ,
et al: Endocardial fibroelastosis is caused by aberrant endothelial
to mesenchymal transition. Circ Res. 116:857–866. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kim M, Choi SH, Jin YB, Lee HJ, Ji YH, Kim
J, Lee YS and Lee YJ: The effect of oxidized low-density
lipoprotein (ox-LDL) on radiation-induced
endothelial-to-mesenchymal transition. Int J Radiat Biol.
89:356–363. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Moonen JR, Lee ES, Schmidt M, Maleszewska
M, Koerts JA, Brouwer LA, van Kooten TG, van Luyn MJ, Zeebregts CJ,
Krenning G, et al: Endothelial-to-mesenchymal transition
contributes to fibro-proliferative vascular disease and is
modulated by fluid shear stress. Cardiovasc Res. 108:377–386. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Chen PY, Qin L, Baeyens N, Li G, Afolabi
T, Budatha M, Tellides G, Schwartz MA and Simons M:
Endothelial-to-mesenchymal transition drives atherosclerosis
progression. J Clin Invest. 125:4514–4528. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Evrard SM, Lecce L, Michelis KC,
Nomura-Kitabayashi A, Pandey G, Purushothaman KR, d'Escamard V, Li
JR, Hadri L, Fujitani K, et al: Endothelial to mesenchymal
transition is common in atherosclerotic lesions and is associated
with plaque instability. Nat Commun. 7:118532016. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Badimon L, Storey RF and Vilahur G: Update
on lipids, inflammation and atherothrombosis. Thromb Haemost.
105(Suppl 1): S34–S42. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Libby P: Inflammation in atherosclerosis.
Arterioscler Thromb Vasc Biol. 32:2045–2051. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Webb NR and Moore KJ: Macrophage-derived
foam cells in atherosclerosis: lessons from murine models and
implications for therapy. Curr Drug Targets. 8:1249–1263. 2007.
View Article : Google Scholar
|
|
21
|
Randolph GJ: Mechanisms that regulate
macrophage burden in atherosclerosis. Circ Res. 114:1757–1771.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Hung Y, Hong M and Huang GS: Cholesterol
loading augments oxidative stress in macrophages. FEBS Lett.
580:849–861. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Mantovani A, Sica A, Sozzani S, Allavena
P, Vecchi A and Locati M: The chemokine system in diverse forms of
macrophage activation and polarization. Trends Immunol. 25:677–686.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Chinetti-Gbaguidi G, Colin S and Staels B:
Macrophage subsets in atherosclerosis. Nat Rev Cardiol. 12:10–17.
2015. View Article : Google Scholar
|
|
25
|
Leitinger N and Schulman IG: Phenotypic
polarization of macrophages in atherosclerosis. Arterioscler Thromb
Vasc Biol. 33:1120–1126. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Fang S, Xu Y, Zhang Y, Tian J, Li J, Li Z,
He Z, Chai R, Liu F, Zhang T, et al: Irgm1 promotes M1 but not M2
macrophage polarization in atherosclerosis pathogenesis and
development. Atherosclerosis. 251:282–290. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
McAlpine CS, Huang A, Emdin A, Banko NS,
Beriault DR, Shi Y and Werstuck GH: Deletion of myeloid GSK3α
attenuates atherosclerosis and promotes an M2 macrophage phenotype.
Arterioscler Thromb Vasc Biol. 35:1113–1122. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Yin K, You Y, Swier V, Tang L, Radwan MM,
Pandya AN, Agrawal DK and Vitamin D: Vitamin D protects against
atherosclerosis via regulation of cholesterol efflux and macrophage
polarization in hypercholesterolemic swine. Arterioscler Thromb
Vasc Biol. 35:2432–2442. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zhang Y, Choksi S, Chen K, Pobezinskaya Y,
Linnoila I and Liu ZG: ROS play a critical role in the
differentiation of alternatively activated macrophages and the
occurrence of tumor-associated macrophages. Cell Res. 23:898–914.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Bai J, Adriani G, Dang TM, Tu TY, Penny
HX, Wong SC, Kamm RD and Thiery JP: Contact-dependent carcinoma
aggregate dispersion by M2a macrophages via ICAM-1 and β2 integrin
interactions. Oncotarget. 6:25295–25307. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Mai J, Qiu Q, Lin YQ, Luo NS, Zhang HF,
Wen ZZ, Wang JF and YangXin C: Angiotensin II-derived reactive
oxygen species promote angiogenesis in human late endothelial
progenitor cells through heme oxygenase-1 via ERK1/2 and AKT/PI3K
pathways. Inflammation. 37:858–870. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Kim M, Neinast MD, Frank AP, Sun K, Park
J, Zehr JA, Vishvanath L, Morselli E, Amelotte M, Palmer BF, et al:
ERα upregulates Phd3 to ameliorate HIF-1 induced fibrosis and
inflammation in adipose tissue. Mol Metab. 3:642–651. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Mai J, Hu Q, Xie Y, Su S, Qiu Q, Yuan W,
Yang Y, Song E, Chen Y and Wang J: Dyssynchronous pacing triggers
endothelial-mesenchymal transition through heterogeneity of
mechanical stretch in a canine model. Circ J. 79:201–209. 2015.
View Article : Google Scholar
|
|
34
|
Su S, Liu Q, Chen J, Chen J, Chen F, He C,
Huang D, Wu W, Lin L, Huang W, et al: A positive feedback loop
between mesenchymal-like cancer cells and macrophages is essential
to breast cancer metastasis. Cancer Cell. 25:605–620. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Yeung OW, Lo CM, Ling CC, Qi X, Geng W, Li
CX, Ng KT, Forbes SJ, Guan XY, Poon RT, et al: Alternatively
activated (M2) macrophages promote tumour growth and invasiveness
in hepatocellular carcinoma. J Hepatol. 62:607–616. 2015.
View Article : Google Scholar
|
|
36
|
Zernecke A and Weber C: Chemokines in
atherosclerosis: proceedings resumed. Arterioscler Thromb Vasc
Biol. 34:742–750. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Drechsler M, Duchene J and Soehnlein O:
Chemokines control mobilization, recruitment, and fate of monocytes
in atherosclerosis. Arterioscler Thromb Vasc Biol. 35:1050–1055.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Ishida Y, Kimura A, Kuninaka Y, Inui M,
Matsushima K, Mukaida N and Kondo T: Pivotal role of the CCL5/CCR5
interaction for recruitment of endothelial progenitor cells in
mouse wound healing. J Clin Invest. 122:711–721. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Sugg KB, Lubardic J, Gumucio JP and
Mendias CL: Changes in macrophage phenotype and induction of
epithelial-to-mesenchymal transition genes following acute Achilles
tenotomy and repair. J Orthop Res. 32:944–951. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Tripathi C, Tewari BN, Kanchan RK, Baghel
KS, Nautiyal N, Shrivastava R, Kaur H, Bhatt ML and Bhadauria S:
Macrophages are recruited to hypoxic tumor areas and acquire a
pro-angiogenic M2-polarized phenotype via hypoxic cancer cell
derived cytokines Oncostatin M and Eotaxin. Oncotarget.
5:5350–5368. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Jetten N, Verbruggen S, Gijbels MJ, Post
MJ, De Winther MP and Donners MM: Anti-inflammatory M2, but not
pro-inflammatory M1 macrophages promote angiogenesis in vivo.
Angiogenesis. 17:109–118. 2014. View Article : Google Scholar
|
|
42
|
Virmani R, Kolodgie FD, Burke AP, Finn AV,
Gold HK, Tulenko TN, Wrenn SP and Narula J: Atherosclerotic plaque
progression and vulnerability to rupture: Angiogenesis as a source
of intraplaque hemorrhage. Arterioscler Thromb Vasc Biol.
25:2054–2061. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Hutter R, Speidl WS, Valdiviezo C, Sauter
B, Corti R, Fuster V and Badimon JJ: Macrophages transmit potent
proangiogenic effects of oxLDL in vitro and in vivo involving
HIF-1α activation: A novel aspect of angiogenesis in
atherosclerosis. J Cardiovasc Transl Res. 6:558–569. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Giannarelli C, Alique M, Rodriguez DT,
Yang DK, Jeong D, Calcagno C, Hutter R, Millon A, Kovacic JC, Weber
T, et al: Alternatively spliced tissue factor promotes plaque
angiogenesis through the activation of hypoxia-inducible factor-1α
and vascular endothelial growth factor signaling. Circulation.
130:1274–1286. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Ouimet M, Ediriweera HN, Gundra UM, Sheedy
FJ, Ramkhelawon B, Hutchison SB, Rinehold K, van Solingen C,
Fullerton MD, Cecchini K, et al: MicroRNA-33-dependent regulation
of macrophage metabolism directs immune cell polarization in
atherosclerosis. J Clin Invest. 125:4334–4348. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Ying R, Wang XQ, Yang Y, Gu ZJ, Mai JT,
Qiu Q, Chen YX and Wang JF: Hydrogen sulfide suppresses endoplasmic
reticulum stress-induced endothelial-to-mesenchymal transition
through Src pathway. Life Sci. 144:208–217. 2016. View Article : Google Scholar
|
|
47
|
Xu F, Lv S, Chen Y, Song X, Jin Z, Yuan F,
Zhou Y and Li H: Macrophage inflammatory protein-1β and fibrinogen
are synergistic predictive markers of prognosis of intermediate
coronary artery lesions. Cardiology. 121:12–19. 2012. View Article : Google Scholar
|
|
48
|
Edsfeldt A, Grufman H, Asciutto G,
Nitulescu M, Persson A, Nilsson M, Nilsson J and Gonçalves I:
Circulating cytokines reflect the expression of pro-inflammatory
cytokines in athero-sclerotic plaques. Atherosclerosis.
241:443–449. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Gentili A, Zaibi MS, Alomar SY, De Vuono
S, Ricci MA, Alaeddin A, Siepi D, Boni M, Vaudo G, Trayhurn P, et
al: Circulating levels of the adipokines monocyte chemotactic
protein-4 (MCP-4), macrophage inflammatory protein-1β (MIP-1β), and
eotaxin-3 in severe obesity and following bariatric surgery. Horm
Metab Res. 48:847–853. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Vistnes M: Macrophage inflammatory
protein-1β: A novel prognostic biomarker in atherosclerosis?
Cardiology. 121:149–151. 2012. View Article : Google Scholar
|
|
51
|
Salic K, Morrison MC, Verschuren L,
Wielinga PY, Wu L, Kleemann R, Gjorstrup P and Kooistra T: Resolvin
E1 attenuates atherosclerosis in absence of cholesterol-lowering
effects and on top of atorvastatin. Atherosclerosis. 250:158–165.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Hsu DC, Ma YF, Hur S, Li D, Rupert A,
Scherzer R, Kalapus SC, Deeks S, Sereti I and Hsue PY: Plasma IL-6
levels are independently associated with atherosclerosis and
mortality in HIV-infected individuals on suppressive antiretroviral
therapy. AIDS. 30:2065–2074. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Herbin O, Regelmann AG, Ramkhelawon B,
Weinstein EG, Moore KJ and Alexandropoulos K: Monocyte adhesion and
plaque recruitment during atherosclerosis development is regulated
by the adapter protein Chat-H/SHEP1. Arterioscler Thromb Vasc Biol.
36:1791–1801. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Cipriani S, Francisci D, Mencarelli A,
Renga B, Schiaroli E, D'Amore C, Baldelli F and Fiorucci S:
Efficacy of the CCR5 antagonist maraviroc in reducing early,
ritonavir-induced athero-genesis and advanced plaque progression in
mice. Circulation. 127:2114–2124. 2013. View Article : Google Scholar : PubMed/NCBI
|