Introduction
A number of empirical studies have demonstrated that
hypoxic and ischemic stress play a role in multiple human central
nervous system (CNS) diseases including ischemic stroke, which is
becoming one of the leading causes of mortality worldwide (1). Although various pathogenic
mechanisms associated with ischemic stroke, such as oxidative
stress, apoptosis, inflammation, calcium overload, endoplasmic
reticulum (ER) stress and the disruption of the blood-brain barrier
(BBB) have been put forward (2,3),
the underlying mechanisms of hypoxic/ischemic-induced cerebral
injury are unclear and remain to be fully explored. According to
recent studies, ER stress is an essential step in neuronal injury
resulting from cerebral ischemia and the modulation of ER stress
provides a remarkable protective function in the ischemic brain
(4,5). Therefore, it is of realistic
significance to further investigate the potential mechanisms
associated with ER stress under conditions of cerebral ischemia. It
is generally known that oxygen-glucose deprivation (OGD) induces
neuronal cell injury, and this model is commonly used in research
to examine cerebral ischemic injury (6). Thus, in this study, we used PC12
cells, which were induced to differentiate by nerve growth factor,
and were then exposed to OGD to establish a cerebral
hypoxia-ischemia model, in order to investigate the underlying
mechanisms of OGD-induced ER stress.
It is well established that nitric oxide (NO) is a
physiological intercellular messenger in the CNS, synthesized by
the nitric oxide synthase (NOS)-catalyzed reaction (7,8).
There are three isoforms of NOS characterized in brain cells,
namely neuronal NOS (nNOS), inducible NOS (iNOS) and endothelial
NOS (eNOS) (9). Increasing
evidence has confirmed that particularly excessive NO production by
nNOS mediates excitotoxicity by promoting a cascade reaction under
energy depletion-induced neuronal brain injury (10,11). However, the mechanisms through
which elevated levels of NO result in neuronal cell death in
ischemic brain injury are still unclear. AMP-activated protein
kinase (AMPK) is a serine threonine kinase, considered as a key
metabolic and stress sensor/effector (12). AMPK is activated under
pathological conditions, incuding nutrient deprivation, vigorous
exercise, or heat shock. It has been demonstrated that AMPK plays
an essential role in cerebral ischemia; however, its role remains
controversial (13,14). It has been shown that AMPK
activation is detrimental and the inhibition of AMPK activation is
protective under conditions of cerebral ischemia (15). By contrast, others have suggested
that the activation of AMPK leads to neuroprotection (16). Notably, increasing evidence
indicates that NO is an important activator of AMPK, and the
activation of AMPK is muted in mice lacking nNOS under conditions
of cerebral ischemia (17,18).
However, the association between AMPK and NOS/NO in OGD-induced
PC12 cell injury remains unclear.
Hydrogen sulfide (H2S) is a third
signaling gaseous mediator followed by CO and NO, predominantly
produced from L-cysteine in the CNS by cystathionine β-synthase
(CBS), and has a variety of physiological and pathophysiological
functions in the CNS (19).
Emerging evidence indicates that H2S is considered as
not only a neuromodulator, but also a neuroprotectant (20). It has also been shown that in
vivo and in vitro models of cerebral ischemia injury,
H2S treatment significantly reduces the infarct size and
ameliorates neurological function via its antioxidant,
anti-apoptotic and anti-inflammatory effects, implying the
therapeutic role of H2S in cerebral ischemic stroke
(21,22). In addition, H2S reduces
ER stress induced by multiple neurotoxins, such as
6-hydroxy-dopamine (6-OHDA) (23)
and homocysteine (24), resulting
in neuroprotective effects. Thus, we wished to investigate whether
the disruption of endogenous H2S generation is involved
in OGD-induced ER stress. Furthermore, it has been shown that the
complex interaction between CBS/H2S and NO signaling
plays an important role in ischemia/reperfusion (I/R)-related brain
damage (25). The activity of CBS
can be suppressed by NO (26,27), while H2S increases eNOS
activation and NO generation (28). Hence, the associatoin between
these in cerebral ischemic injury is worthy of research.
In the present study, we observed that OGD induced
ER stress accompanied by the upregulation of nNOS/NO/AMPK signaling
and the downregulation of the CBS/H2S system in PC12
cells. Furthermore, NaHS or inhibitor of the nNOS pathway
attenuated the ER stress induced by OGD. Simultaneously, the
promotion of the CBS/H2S system attenuated OGD-induced
nNOS/AMPK activation. These results demonstrated that OGD induced
ER stress through the activation of nNOS/NO/AMPK signaling as a
result of CBS/H2S system blockage.
Materials and methods
Reagents
3-Bromo-7-nitroindazole (3-Br-7-NI) was purchased
from Tocris Cookson, Ltd. (Avonmouth, UK). 5-Aminoimid
azole-4-carboxamide-1-β-d-ribofuranoside (AICAR) was
obtained from Toronto Research Chemicals, Inc. (North York, ON,
Canada). NaHS and S-adenosyl-L-methionine (SAM) were purchased from
Sigma-Aldrich (St. Louis, MO, USA). Dulbecco's modified Eagle's
medium (DMEM) and fetal bovine serum (FBS) were purchased from
Gibco (Grand Island, NY, USA). Hoechst 33258,
penicillin/streptomycin, RIPA lysis buffer and enhanced
chemiluminescence (ECL) reagents were supplied by Beyotime
Biotechnology (Shanghai, China). Cell counting kit-8 (CCK-8) was
purchased from Dojindo Molecular Technologies (Rockville, MD, USA).
The Nitric Oxide Assay kit and Total Nitric Oxide Synthase Assay
kit were purchased from Nanjing Jiancheng Bioengineering Institute
(Nanjing, China). The primary antibodies against glucose-regulated
protein 78 (GRP78), C/EBP homologous protein (CHOP), cleaved
caspase-12 and phosphorylated AMP-activated protein kinase (p-AMPK)
were supplied by Cell Signaling Technology, Inc. (Beverly, MA,
USA). Specific antibody to AMPK was obtained from Santa Cruz
Biotechnology, Inc. (San Diego, CA, USA). All reagents were of the
purest commercial grade.
Cell culture and model of OGD-induced
cell injury and treatment
Highly differentiated rat adrenal pheochromocytoma
(PC12) cells, obtained from the American Type Culture Collection
(ATCC; Manassas, VA, USA), were maintained in DMEM supplemented
with 10% FBS and 1% penicillin/streptomycin at 37°C in a humidified
atmosphere with 5% CO2/95% air. To establish the model
of OGD-induced cell injury, PC12 cells were exposed to OGD for 12 h
by incubation in deoxygenated glucose-free and serum-free DMEM at
37°C in a humidified atmosphere with 5% CO2, 94%
N2, and 1% O2 for 12 h. To investigate the
effect of nNOS/NO signaling on OGD-induced injury, the PC12 cells
were pre-treated with 3-Br-7-NI (a relatively selective nNOS
inhibitor, 10 µM) for 30 min and then co-incubated with OGD
for 12 h. To investigate the effect of the AMPK pathway on
3-Br-7-NI-caused the inhibition of ODG-induced injury in PC12
cells, PC12 cells ware pretreated with AICAR (20 µM) for 30
min and then incubated with 3-Br-7-NI (10 µM) for 30 min
followed by exposure to OGD for 12 h.
Measurement of cell viability
The viability of the PC12 cells was evaluated by
CCK-8 assay according to the manufacturer's instructions. In brief,
the PC12 cells in the logarithmic phase were seeded into 96-well
plate at a density of approximately 1×104 cells/well
overnight. At the end of drug treatment, 10 µl of CCK-8
reagent were added to each well, followed by incubation for 4 h at
37°C. The absorbance at 450 nm was measured using a microplate
reader (Epoch; BioTek, Winooski, VT, USA). The viability of the
cells was expressed as a percentage of that of the control cells.
The assays were performed in duplicate for 3 times.
Determination of intracellular NO levels
and NOS activity
PC12 cells at the logarithmic growth phase were
seeded in 6-well plates a density of approximately 1×106
cells/well overnight. Following the different interventions, total
protein was extracted in PBS with Ultrasonic Cell Disruption System
(5 sec, 15 times, 4°C) and quantified using the BCA Protein Assay
kit. The NO level and total NOS activity were assessed using the
Nitric Oxide Assay kit and Total Nitric Oxide Synthase Assay kit
following the instructions provided by the respective
manufacturers. Data related to the NO level are expressed as
pmol/mg protein. Data related to total NOS activity are expressed
as U/mg protein. Each independent experiment was repeated at least
in triplicate.
Measurement of H2S
concentration
The level of H2S in the cell culture
supernatant was measured using the methylene blue
spectrophotometric method in that H2S and zinc acetate
were co-incubated to form zinc sulfide which then dissolved in
hydrochloric acid solution supplemented with
N,N-dimethyl-p-phenylenediamine sulphate
yielding and ferric chloride (FeCl3), resulting in the
formation of methylene blue, which was quantified
spectrophotometrically. Briefly, the culture supernatant of PC12
cells with different interventions was collected following
centrifugation for 5 min at 1,000 rpm. A total of 500 µl of
supernatant was combined with 250 µl of zinc acetate (1%),
250 µl of
N,N-dimethyl-p-phenylenediamine sulfate (20
mmol/l) and 200 µl of FeCl3 (30 mmol/l) in 500
µl of hydrochloric acid solution (10%). Following incubation
for 15 min at room temperature, the absorbance of at 670 nm was
measured by spectrophotometry (LAS-3000; Fujifilm, Tokyo, Japan).
The H2S concentration was calculated based on the
standard curve which was generated by serial dilution of NaHS and
was expressed in µmol/l.
Measurement of CBS activity
The PC12 cells subjected to the different treatments
were collected and homogenized in potassium phosphate buffer (50
mmol/l, pH 6.8). Following centrifugation at 14,000 × g for 60 min
at 4°C, the supernatant was collected for enzyme assays. A mixing
system comprising L-cysteine (0.5 mol/l), enzyme protein (0–100
µg), 5-pyridoxal phosphate/potassium phosphate buffer
solution (100 mmol/l) and potassium phosphate buffer (100 mM, pH
7.4) was used, transferring the Eppendorf tubes from ice to a
shaking water bath at 37°C. Following incubation for 60 min, the
reaction was terminated by the addition of w/v zinc acetate (1%) to
trap H2S followed by v/v trichloroacetic acid (10%) to
precipitate proteins. Subsequently,
N,N-dimethyl-p-phenylenediamine-sulfate in HCl
(7.2 M) was immediately added to the reaction system followed by
addition of FeCl3 in HCl (1.2 M). The absorbance of at
670 nm was measured by spectrophotometry (LAS-3000; Fujifilm) and
the H2S content was calculated against a calibration
curve of standard NaHS solutions. The CBS activity was expressed as
the amount of H2S generated per mg of reaction samples,
with a unit of nmol/mg protein.
Measurement of caspase-3 activity
The activity of capsase-3 was detected using a
commercial kit (Nanjing Jiancheng Bioengineering Institute)
according to the manufacturer's instructions. The absorbance at 405
nm was detected using a microplate reader (Epoch; BioTek). Data
were expressed as the fold of the control cells.
Western blot analysis
The PC12 cells were homogenized in RIPA lysis buffer
at 4°C for 30 min and the supernatant was collected following
centrifugation at 12,000 rpm for 10 min at 4°C. The protein
concentration was measured using the BCA Protein Assay kit. Equal
amounts of proteins were separated by 10–15% sodium dodecyl
sulfate-polyacrylamide gel (SDS-PAGE), and then transferred onto
polyvinylidene fluoride (PVDF) membranes (Millipore Corp., Bedford,
MA, USA). After blocking for 2 h in TBS with 0.01% Tween-20 (TBST)
containing 5% skim milk at room temperature, the membranes were
incubated with primary antibodies against CBS (1:2,000; ab96252;
Abcam, Cambridge, UK), AMPK (sc-19128; Santa Cruz Biotechnology,
Inc., Santa Cruz, CA, USA), p-AMPK (1:2,000; cat. no. #2537), GRP78
(1:2000; cat. no. #3183) and CHOP (1:2,000; cat. no. #2895) (all
from Cell Signaling Technology, Inc), cleaved caspase-12 (1:1,000;
ab62484; Abcam), and β-actin (1:1,000; cat. no. #4970; Cell
Signaling Technology, Inc.) overnight at 4°C, respectively. After
washing with TBST for 3 times, the membranes were incubated with
the appropriate diluted horseradish peroxidase (HRP)-conjugated
secondary antibody for 2 h at room temperature. Subsequently, the
membranes were washed again and the protein bands were detected
using the ECL system (ZsBio, Beijing, China). The integrated
optical density was calculated using ImageJ 1.4 6i software. The
amount of protein was represented as a percentage of that of the
control cells.
Statistical analysis
Data are expressed as the means ± SEM. The analysis
of significant differences groups was performed by one-way analysis
of variance (ANOVA) followed by the least significant difference
(LSD) test. Differences were considered statistically significant
at P<0.05.
Results
OGD induces cytotoxicity and ER stress by
promoting nNOS/NO signaling in PC12 cells
As NOS/NO signaling is known to play a role in
cerebral ischemic injury (10,29), in this study, we first
investigated the alternations of intracellular NO generation in
OGD-exposed PC12 cells. We found that the exposure of PC12 cells to
OGD for 12 h caused an obvious increase in the level of NO in PC12
cells (Fig. 1A). To investigate
the possible role of nNOS activity in mediating OGD-induced NO
generation in PC12 cells, we detected the activity of nNOS and
found an increase in the activity of nNOS following the exposure of
the PC12 cells to OGD for 12 h (Fig.
1B). In addition, we examined the effect of the OGD-induced
increase in nNOS/NO signaling on ER stress in PC12 cells. We found
that incubation of the PC12 cells with 3-Br-7-NI (10 µM), a
relatively selective nNOS inhibitor, for 30 min significantly
abrogated the OGD-induced decrease in the viability of the PC12
cells (Fig. 1C). Simultaneously,
pre-treatment with 3-Br-7-NI attenuated OGD-induced ER stress, as
evidenced by the decreased expression of ER-related proteins,
including GRP78 (Fig. 1D), CHOP
(Fig. 1E) and cleaved caspase-12
(Fig. 1F) proteins in PC12 cells.
Of note, 3-Br-7-NI treatment alone had no effect on cell viability
and ER-related protein expression. Taken together, these results
suggest that the OGD-mediated NO overproduction via the increased
activity of nNOS, is in part, responsible for nerve cytotoxicity
and ER stress during ischemia-induced cerebral injury.
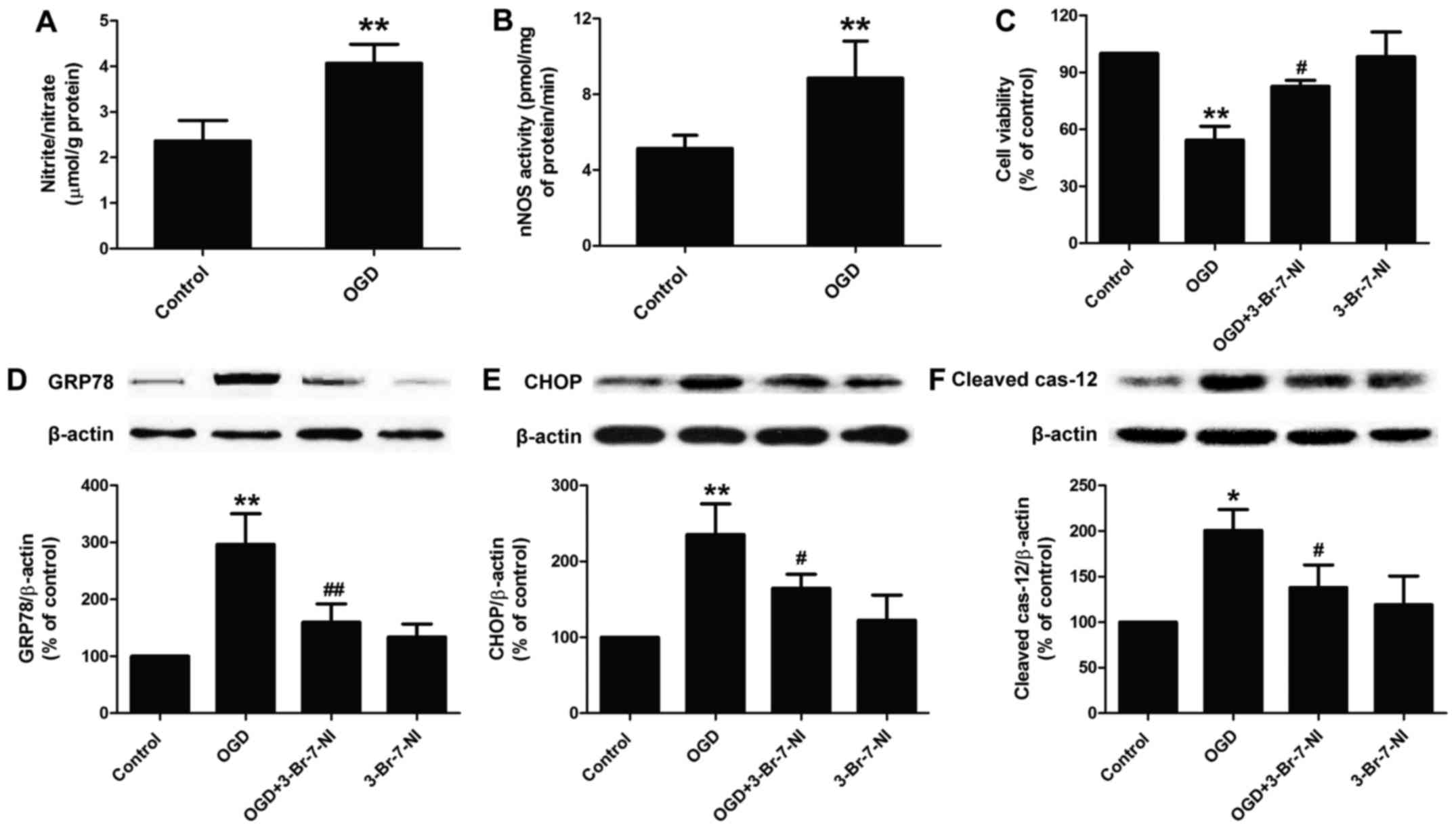 | Figure 1Effect of nNOS/NO signaling on
OGD-induced cytotoxicity and ER stress in PC12 cells. PC12 cells
were incubated with OGD for 12 h in the presence or absence of
3-Br-7-NI (a relatively selective nNOS inhibitor, 10 µM).
(A) The NO levels were measured by Nitric Oxide Assay kit as
described in the Materials and methods. (B) NOS activity was
detected by Total Nitric Oxide Synthase Assay kit as described in
the Materials and methods. (C) Cell viability was measured by CCK-8
assay. The expression levels of (D) GRP78, (E) CHOP, and (F)
cleaved caspase-12 proteins were detected by western blot analysis.
Data are presented as the means ± SEM from independent experiments
performed in triplicate. *P<0.05,
**P<0.01 as compared to untreated control group;
#P<0.05, ##P<0.01 as compared to the
group exposed to OGD alone. nNOS, neuronal nitric oxide synthase;
NO, nitric oxide; OGD, oxygen-glucose deprivation; ER, endoplasmic
reticulum; NOS, nitric oxide synthase; GRP78, glucose-regulated
protein 78; CHOP, C/EBP homologous protein. |
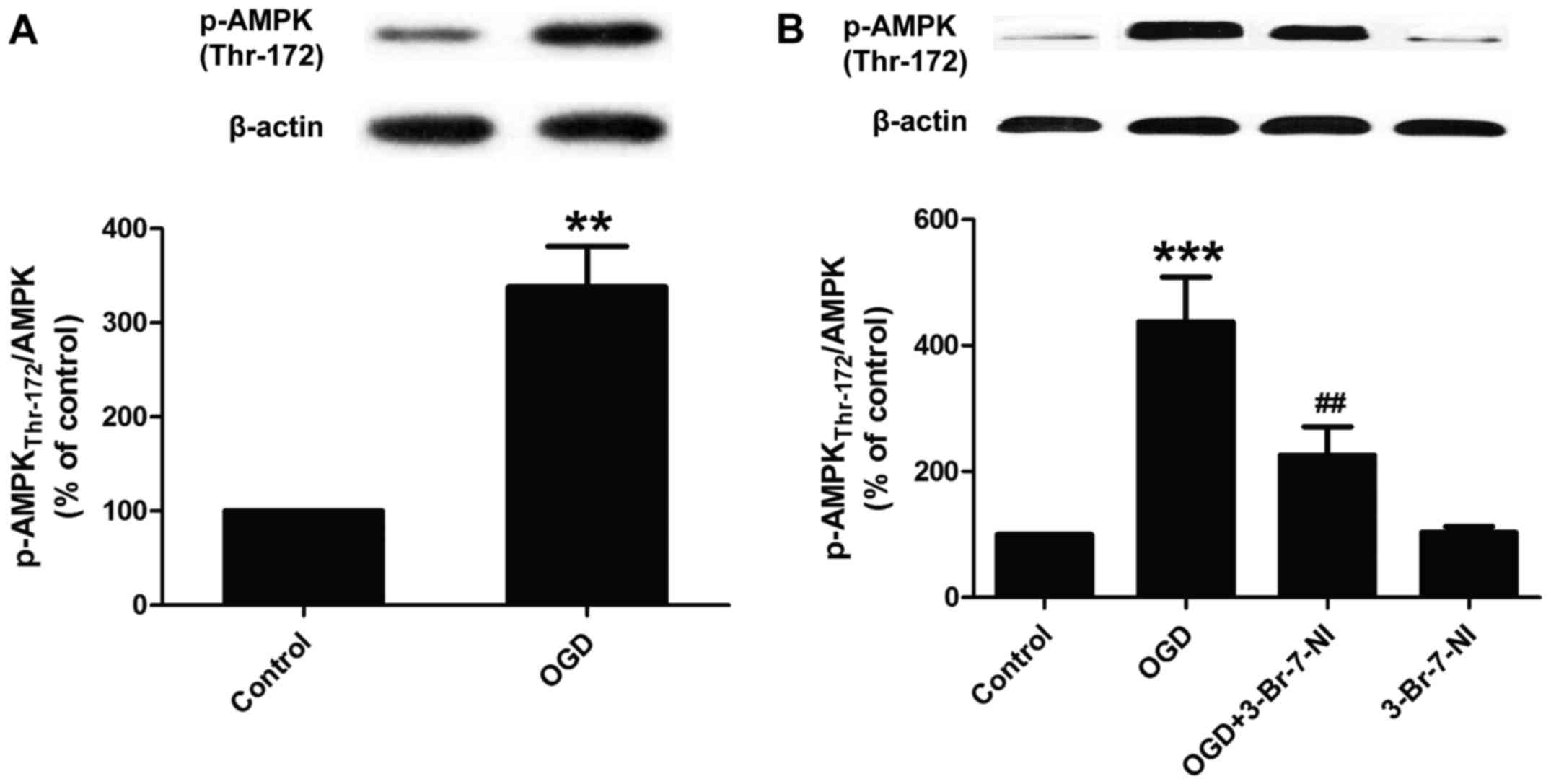
nNOS/NO signaling is involved in
OGD-induced AMPK activation in PC12 cells
A previous demonstrated the interaction between NO
and AMPK activation in stroke (17). Thus, we wished to determine
whether this interaction exists in OGD-exposed PC12 cells. We found
that exposure to OGD for 12 h led to a marked increase in the
expression of phosphorylated AMPK (p-AMPK Thr-172) and in the ratio
of p-AMPK/AMPK in the PC12 cells (Fig. 2A), indicating that OGD induced
AMPK activation. Subsequently, in order to determine whether the
OGD-induced AMPK activation is dependent on the enhancement of
nNOS/NO signaling, the PC12 cells were pre-treated with 3-Br-7-NI
(a nNOS inhibitor, 10 µM) for 30 min and then co-exposed to
OGD for 12 h. We found that 3-Br-7-NI markedly abolished the
OGD-induced increase in the phosphorylation levels of AMPK
(Fig. 2B) in the PC12 cells.
These results indicated that the OGD-induced activation of AMPK was
dependent on nNOS/NO signaling; this in turn, may potentially
promote neuronal injury during cerebral ischemia and anoxia.
Activation of AMPK signaling attenuates
the inhibitory effects of 3-Br-7-NI on OGD-induced excessive ER
stress in PC12 cells
To further demonstrate whether AMPK activation is
involved in OGD-induced ER stress mediated by the nNOS/NO system,
the effect of AICAR, an AMPK activator, on ER stress was
investigated. As shown in Fig.
3A, pre-treatment with AICAR (20 µM) attenuated the
promoting effects of 3-Br-7-NI on the viability of the OGD-exposed
PC12 cells, indicating that OGD induced cytotoxicity via
NOS-activated AMPK. In addition, we found that pre-treatment with
AICAR markedly abolished the inhibitory effects of 3-Br-7-NI on
OGD-induced ER stress, as evidenced by the upregulated expression
of GRP-78 (Fig. 3B), CHOP
(Fig. 3C) and cleaved caspase-12
(Fig. 3D). These results
indicated that the OGD-induced increase in nNOS/NO/AMPK signaling
contributed to OGD-induced ER stress.
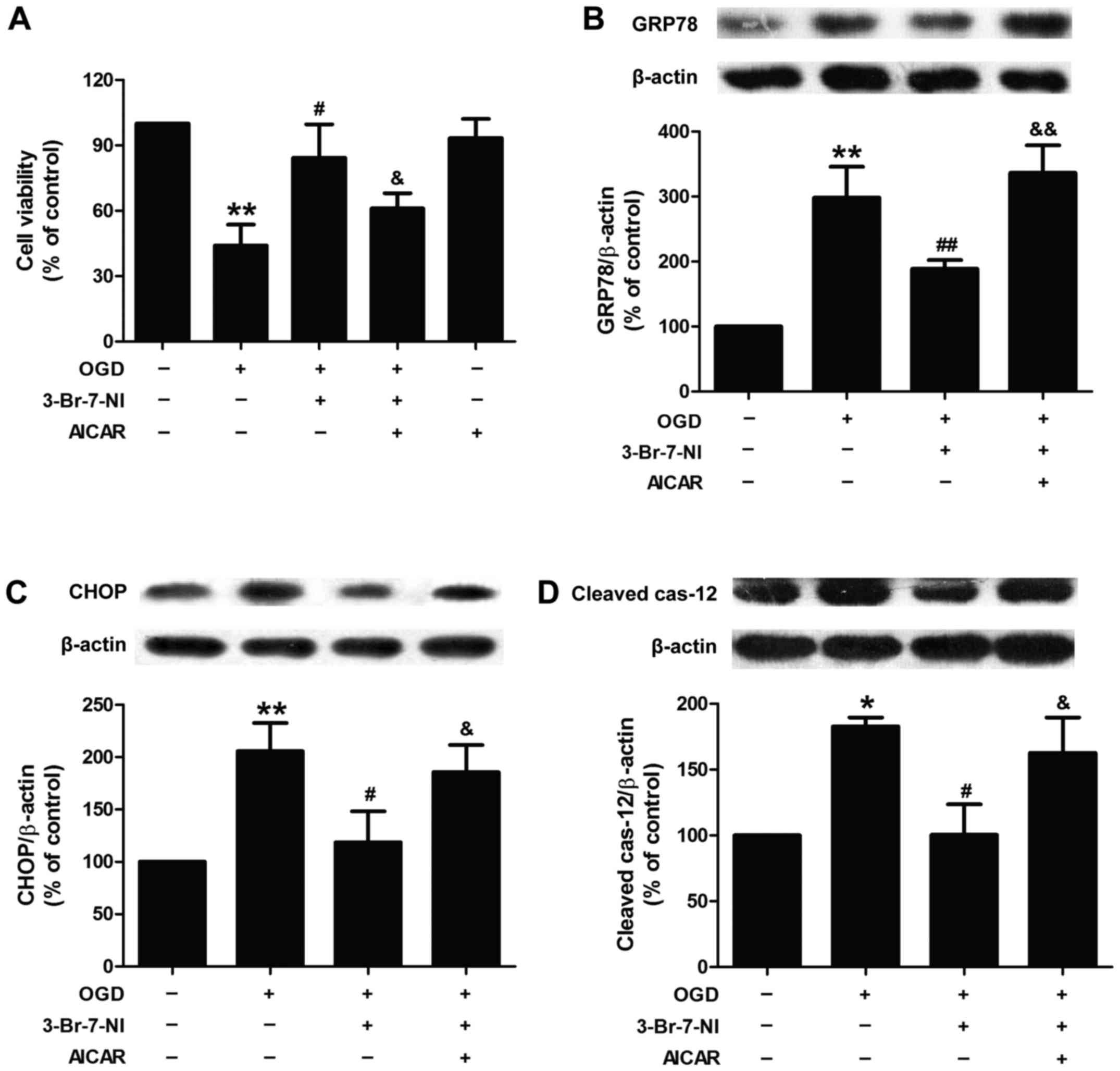 | Figure 3Effect of AMPK activator (AICAR) on
the inhibition of ODG-induced ER stress in PC12 cells by a
relatively selective nNOS inhibitor (3-Br-7-NI). PC12 cells were
pre-treated with AICAR (20 µM) for 30 min and then incubated
with 3-Br-7-NI (10 µM) for 30 min followed by exposure to
OGD for 12 h. (A) Cell viability was measured by CCK-8 assay. The
expression levels of (B) CRP78, (C) CHOP, and (D) cleaved
caspase-12 proteins were measured by western blot analysis and
β-actin was used as a loading control. Data are presented as the
means ± SEM from independent experiments performed in triplicate.
*P<0.05, **P<0.01 as compared to the
untreated control group; #P<0.05,
##P<0.01 as compared to the group exposed to OGD
alone; &P<0.05, &&P<0.01 as
compared to the group exposed to OGD and treated with 3-Br-7-NI.
nNOS, neuronal nitric oxide synthase; OGD, oxygen-glucose
deprivation; ER, endoplasmic reticulum; GRP78, glucose-regulated
protein 78; CHOP, C/EBP homologous protein. |
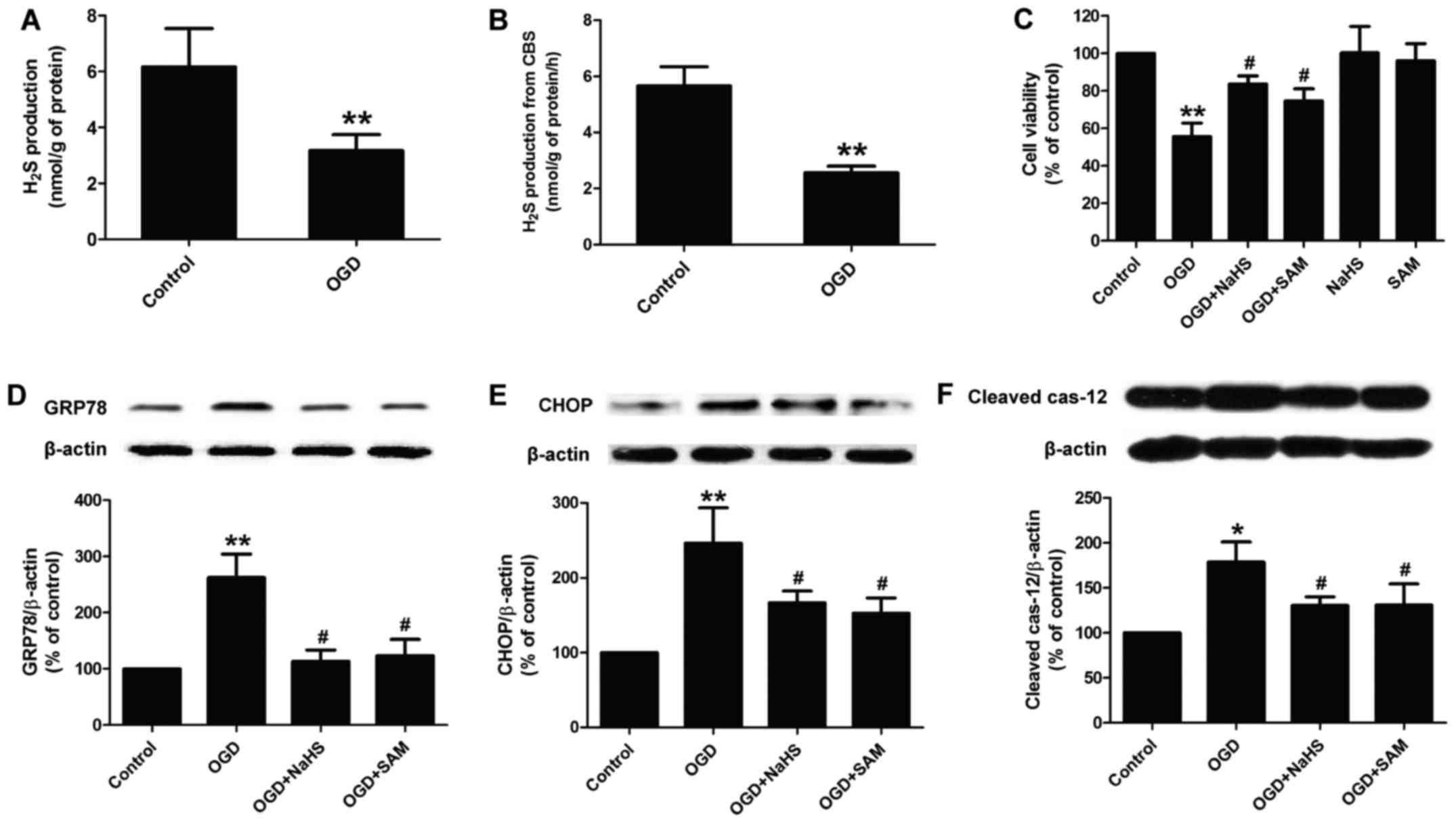
Blockage of the CBS/H2S system
contributes to OGD-induced ER stress in PC12 cells
We then investigated the role of the
CBS/H2S system in OGD-treated PC12 cells. As shown in
Fig. 4, we found that exposure of
the PC12 cells to OGD for 12 h markedly reduced the level of
H2S in the culture supernatant (Fig. 4A). In addition, the activity of
CBS was also attenuated by exposure to OGD (Fig. 4B). These results suggest that
exposure to OGD results in the downregulation of the
CBS/H2S system. In order to further confirm the role of
the CBS/H2S system in OGD-induced neuronal injury, NaHS
(a donor of H2S) and SAM (a CBS agonist) were used. We
found that pre-treatment with NaHS (200 µM) for 30 min and
SAM (100 µM) for 1 h markedly attenuated the OGD-induced
decrease in cell viability, while NaHS or SAM treatment alone had
no effect on the viability of PC12 cells (Fig. 4C). At the same time, both NaHS and
SAM abolished the OGD-induced increase in the expression of ER
stress-related marker proteins, including GRP78 (Fig. 4D), CHOP (Fig. 4E) and cleaved caspase-12 (Fig. 4F) in PC12 cells. These results
indicate that OGD causes cytotoxicity and ER stress via the
inhibition of the CBS/H2S system.
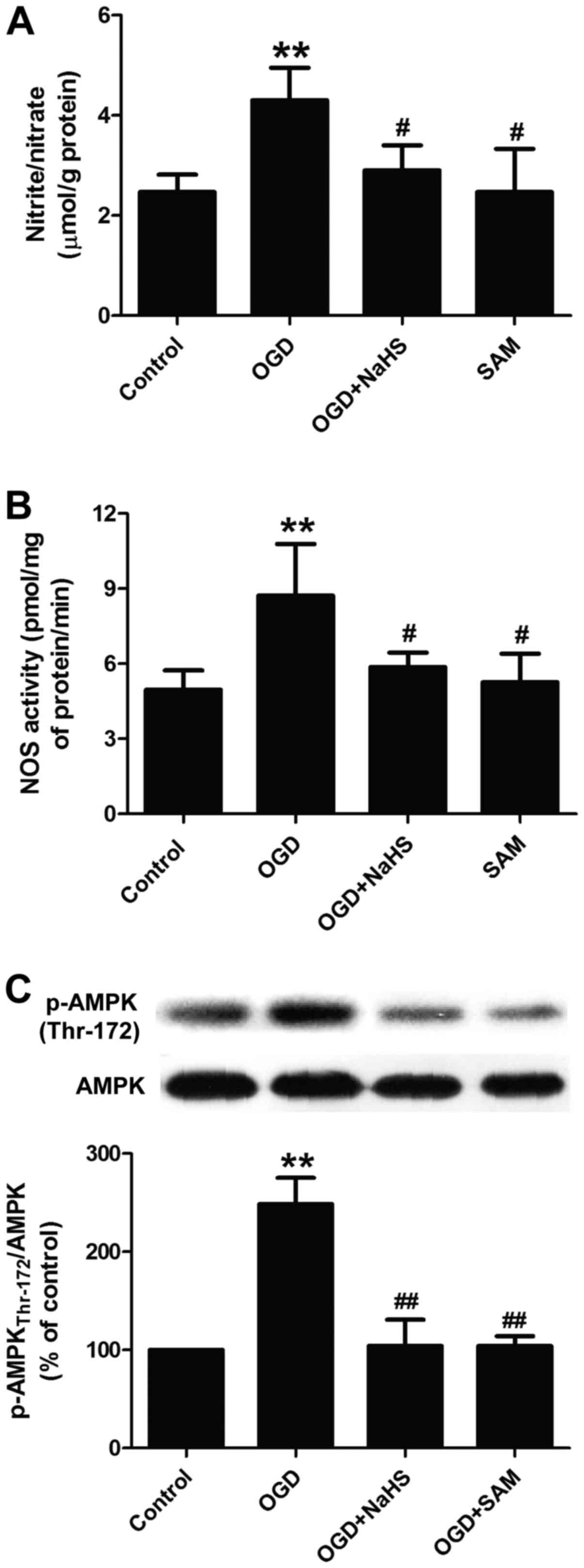
Enhancement of CBS/H2S by NaHS
and SAM mitigates the OGD-induced activation of nNOS/NO and AMPK in
PC12 cells
Finally, we further investigated the association of
CBS/H2S, nNOS/NO and AMPK in the PC12 cells exposed to
OGD. We found that the increased levels of H2S induced
by NaHS (200 µM) and the enhanced activity of CBS induced by
SAM (100 µM) distinctly reversed the OGD-induced NO
overproduction, as evidenced by a decrease in the levels of NO
(Fig. 5A) and in the activity of
nNOS (Fig. 5B), indicating that
the inhibition of the CBS/H2S system mediates the
OGD-induced upregulation of the nNOS/NO system. Simultaneously,
NaHS or SAM treatment also reduced the ratio of p-AMPK
(Thr-172)/AMPK in the PC12 cells exposed to OGD (Fig. 5C), suggesting that OGD induced
AMPK activation by suppressing the CBS/H2S system.
Discussion
In the present study, we explored the roles of
nNOS/NO/AMPK signaling and the CBS/H2S system in
OGD-induced ER stress in PC12 cells. In this study, to the best of
our knowledge, we demonstrate for the first time that OGD causes ER
stress through the activation of the nNOS/NO/AMPK pathway resulting
from the inhibition of the CBS/H2S system.
According to the World Health Organization,
approximately 15 million individuals each year suffer from cerebral
ischemic injury, such as stroke (30). Despite this, the understanding of
the mechanisms underlying cerebral ischemic injury remains
extremely limited. Accumulating evidence has indicated that ER
stress plays an important role in the progression of brain neuronal
injury resulting from I/R (31–33). Certain stimuli, such as ischemia
and hypoxia may trigger the accumulation of unfolded proteins in
the ER, leading to excessive or aberrant ER stress and accelerated
nerve damage (34). These
findings suggest that the elucidation of mechamisms responsible for
ER stress signaling may provide a novel target for effective
therapeutic approaches for cerebral ischemia.
NO is a physiological mediator generated from
L-arginine and oxygen by various forms of NOS, including eNOS, nNOS
and iNOS in the brain (35).
Increasing evidence reveals a wide range of roles for NOS/NO
signaling involved in the occurrence and development of ischemic
brain or brain ischemic injury (36). However, NO acts in a protective or
deleterious manner depended upon the NOS isoform (37). It has been reported that
hypoxic-ischemic injury is attenuated in mice deficient in nNOS,
but is exacerbated in eNOS-deficient mice (38,39), indicating that NO overproduction
released from nNOS contributes to brain damage. Other studies have
further demonstrated that NO is a mediator of neuronal damage
following cerebral ischemia (40,41) and the inhibition of NOS decreases
BBB disruption, leading to neuroprotective effects under conditions
of I/R injury during acute hypertension in rats (42). Furthermore, Nadjafi et al
found that NO production was increased during OGD/reperfusion in
OLN-93 oligodendrocytes (43).
Consistent with these studies, our study demonstrated that exposure
to OGD markedly increased the level of NO and the activity of nNOS
in PC12 cells. Notably, an experimental study detected that
3-Br-7-NI, a potent and selective nNOS inhibitor, attenuated brain
ischemic injury in diabetic stroke via the inhibition of the ER
stress pathway (44), implying
the mediation of NOS/NO signaling in ER stress under conditions of
cerebral ischemia. Similar to this, the current study demonstrated
that 3-Br-7-NI pre-treatment also mitigated OGD-induced ER stress,
as evidenced by the down-regulated expression of ER-related
proteins, such as GRP78, CHOP and cleaved caspase-12 in PC12 cells.
Thus, these results suggest that OGD induces ER stress through the
enhancement of nNOS/NO signaling.
AMPK has been reported to be present in most
mammalian tissues, including the brain (45). It is increasingly becoming
recognized that alternations in AMPK activation are not only
related to metabolic needs, but are also related to sensing and
responding to ʻvarious cell stressʼ, such as ischemia, hypoxia and
energy depletion (46). However,
little is known regarding the physiological and pathological
functions of AMPK and the mechanisms through which AMPK activation
occurs in the brain during ischemia. Of note, some studies have
shown that cerebral ischemia increases the activation of AMPK in
the brain in an NO-dependent manner, which is muted in mice lacking
nNOS (17,18), indicating the important role of
NOS/NO signaling in AMPK activation. Nevertheless, the interactions
between AMPK, NO and NOS warrant further investigation,
particularly under the condition of cerebral ischemic injury. Our
finds indicated that exposure to OGD increased the phosphorylation
of AMPK in PC12 cells, while this effect was abolished by
pre-treatment with 3-Br-7-NI, indicating that the OGD-induced AMPK
activation is dependent on nNOS/NO signaling. In addition, we found
that the AMPK activator, AICAR, attenuated the protective effects
of 3-Br-7-NI against OGD-induced cytotoxicity and ER stress in PC12
cells. These results suggest that OGD causes ER stress by
increasing the activity of AMPK, as a result of nNOS/NO signaling
promotion.
Emerging evidence indicates that H2S
plays a broad range of roles in cerebral ischemic injury (20,47). In in vivo model of cerebral
I/R injury, H2S pre-conditioning improved neurological
function and decreased the infarct size, implying that
H2S plays a therapeutic role in cerebral ischemic stroke
(21). In addition, a number of
studies have confirmed that H2S attenuates ER stress
induced by multiple stresses and neurotoxins, including chronic
unpredictable mild stress, homocysteine and 6-OHDA (23,24,48), exerting neuroprotective effects.
However, the association of H2S and ER stress in
cerebral ischemic injury is not yet understood. Thus, we
hypothesized that the disruption of H2S may be also
involved in ER stress in PC12 cells exposed to OGD. In the current
study, we found that exposure to OGD markedly reduced the level of
H2S in the culture supernatant, as well as the activity
and expression of CBS, indicating that the CBS/H2S
system was inhibited by OGD. These findings are consistent with
those of the study by Shen et al, who demonstrated that in
acute ischemic conditions, CBS is upregulated and activated
followed by causing an increased production of H2S
(49). In addition, we found that
NaHS abolished the OGD-induced decrease in the viability of the
PC12 cells and the increase in ER stress, as evidenced by the
increased expression of GRP78, CHOP and cleaved caspase-12 in the
PC12 cells following treatment with NaHS. These results indicated
that blocking the CBS/H2S system contributes to
OGD-induced ER stress.
An increasing number of studies have revealed the
complex association between H2S and NO, which has an
indispensable effect in multiple diseases (50,51). In another study, Grossi proved
that H2S is regarded as a co-factor responsible for the
generation of NO (52). In
addition, another finding is that H2S directly inhibits
eNOS, as well as nNOS, contributing to the dual modulation of
vascular tension (53). In the
present study, we further investigated the association between the
CBS/H2S system and nNOS/NO signaling in OGD-exposed PC12
cells. We found that pre-treatment with NaHS markedly attenuated
OGD-induced NO overproduction by the excessive activation of nNOS
and AMPK activation in PC12 cells. These results indicated that OGD
induced the activation of nNOS/NO/AMPK signaling activation through
the inhibition of CBS/H2S, leading to ER stress.
In conclusion, our findings demonstrate that
exposure to OGD induces ER stress, and the activation of the
nNOS/NO/AMPK pathway results from the attenuation of the
CBS/H2S system. Our findings provide a better
understanding of the signal transduction mechanisms involved in the
pathophysiological process of cerebral ischemia-induced ER stress,
which provides novel targets and guidance for the development of
neuroprotective agents.
References
|
1
|
Adeoye O, Hornung R, Khatri P and
Kleindorfer D: Recombinant tissue-type plasminogen activator use
for ischemic stroke in the United States: A doubling of treatment
rates over the course of 5 years. Stroke. 42:1952–1955. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Manzanero S, Santro T and Arumugam TV:
Neuronal oxidative stress in acute ischemic stroke: Sources and
contribution to cell injury. Neurochem Int. 62:712–718. 2013.
View Article : Google Scholar
|
|
3
|
Sierra C, Coca A and Schiffrin EL:
Vascular mechanisms in the pathogenesis of stroke. Curr Hypertens
Rep. 13:200–207. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Xin Q, Ji B, Cheng B, Wang C, Liu H, Chen
X, Chen J and Bai B: Endoplasmic reticulum stress in cerebral
ischemia. Neurochem Int. 68:18–27. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
DeGracia DJ and Montie HL: Cerebral
ischemia and the unfolded protein response. J Neurochem. 91:1–8.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wang CH, Lee WJ, Ghanta VK, Wang WT, Cheng
SY and Hsueh CM: Molecules involve in the self-protection of
neurons against glucose-oxygen-serum deprivation (GOSD)-induced
cell damage. Brain Res Bull. 79:169–176. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Vincent SR: Nitric oxide: A radical
neurotransmitter in the central nervous system. Prog Neurobiol.
42:129–160. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Garthwaite J, Charles SL and
Chess-Williams R: Endothelium-derived relaxing factor release on
activation of NMDA receptors suggests role as intercellular
messenger in the brain. Nature. 336:385–388. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Qi SH, Hao LY, Yue J, Zong YY and Zhang
GY: Exogenous nitric oxide negatively regulates the S-nitrosylation
p38 mitogen-activated protein kinase activation during cerebral
ischaemia and reperfusion. Neuropathol Appl Neurobiol. 39:284–297.
2013. View Article : Google Scholar
|
|
10
|
Liu H, Li J, Zhao F, Wang H, Qu Y and Mu
D: Nitric oxide synthase in hypoxic or ischemic brain injury. Rev
Neurosci. 26:105–117. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Brown GC: Nitric oxide and neuronal death.
Nitric Oxide. 23:153–165. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Tzatsos A and Tsichlis PN: Energy
depletion inhibits phosphatidylinositol 3-kinase/Akt signaling and
induces apoptosis via AMP-activated protein kinase-dependent
phosphorylation of IRS-1 at Ser-794. J Biol Chem. 282:18069–18082.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Li J and McCullough LD: Effects of
AMP-activated protein kinase in cerebral ischemia. J Cereb Blood
Flow Metab. 30:480–492. 2010. View Article : Google Scholar :
|
|
14
|
Hardie DG and Frenguelli BG: A neural
protection racket: AMPK and the GABA(B) receptor. Neuron.
53:159–162. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Culmsee C, Monnig J, Kemp BE and Mattson
MP: AMP-activated protein kinase is highly expressed in neurons in
the developing rat brain and promotes neuronal survival following
glucose deprivation. J Mol Neurosci. 17:45–58. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kuramoto N, Wilkins ME, Fairfax BP,
Revilla-Sanchez R, Terunuma M, Tamaki K, Iemata M, Warren N, Couve
A, Calver A, et al: Phospho-dependent functional modulation of
GABA(B) receptors by the metabolic sensor AMP-dependent protein
kinase. Neuron. 53:233–247. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
McCullough LD, Zeng Z, Li H, Landree LE,
McFadden J and Ronnett GV: Pharmacological inhibition of
AMP-activated protein kinase provides neuroprotection in stroke. J
Biol Chem. 280:20493–20502. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Heales SJ, Bolaños JP, Stewart VC, Brookes
PS, Land JM and Clark JB: Nitric oxide, mitochondria and
neurological disease. Biochim Biophys Acta. 1410:215–228. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Stein A and Bailey SM: Redox biology of
hydrogen sulfide: Implications for physiology, pathophysiology, and
pharmacology. Redox Biol. 1:32–39. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhang X and Bian JS: Hydrogen sulfide: A
neuromodulator and neuroprotectant in the central nervous system.
ACS Chem Neurosci. 5:876–883. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Gheibi S, Aboutaleb N, Khaksari M,
Kalalian-Moghaddam H, Vakili A, Asadi Y, Mehrjerdi FZ and Gheibi A:
Hydrogen sulfide protects the brain against ischemic reperfusion
injury in a transient model of focal cerebral ischemia. J Mol
Neurosci. 54:264–270. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Yin J, Tu C, Zhao J, Ou D, Chen G, Liu Y
and Xiao X: Exogenous hydrogen sulfide protects against global
cerebral ischemia/reperfusion injury via its anti-oxidative,
anti-inflammatory and anti-apoptotic effects in rats. Brain Res.
1491:188–196. 2013. View Article : Google Scholar
|
|
23
|
Xie L, Tiong CX and Bian JS: Hydrogen
sulfide protects SH-SY5Y cells against 6-hydroxydopamine-induced
endoplasmic reticulum stress. Am J Physiol Cell Physiol.
303:C81–C91. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wei HJ, Xu JH, Li MH, Tang JP, Zou W,
Zhang P, Wang L, Wang CY and Tang XQ: Hydrogen sulfide inhibits
homocysteine-induced endoplasmic reticulum stress and neuronal
apoptosis in rat hippocampus via upregulation of the BDNF-TrkB
pathway. Acta Pharmacol Sin. 35:707–715. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Yin W, Lan L, Huang Z, Ji J, Fang J, Wang
X, Ji H, Peng S, Xu J and Zhang Y: Discovery of a ring-opened
derivative of 3-n-butylphthalide bearing NO/H2S-donating
moieties as a potential anti-ischemic stroke agent. Eur J Med Chem.
115:369–380. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Morikawa T, Kajimura M, Nakamura T,
Hishiki T, Nakanishi T, Yukutake Y, Nagahata Y, Ishikawa M, Hattori
K, Takenouchi T, et al: Hypoxic regulation of the cerebral
microcirculation is mediated by a carbon monoxide-sensitive
hydrogen sulfide pathway. Proc Natl Acad Sci USA. 109:1293–1298.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Taoka S and Banerjee R: Characterization
of NO binding to human cystathionine beta-synthase: Possible
implications of the effects of CO and NO binding to the human
enzyme. J Inorg Biochem. 87:245–251. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zhao W, Zhang J, Lu Y and Wang R: The
vasorelaxant effect of H(2)S as a novel endogenous gaseous K(ATP)
channel opener. EMBO J. 20:6008–6016. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Liu HT and Mu DZ: Inducible nitric oxide
synthase and brain hypoxic-ischemic brain damage. Zhongguo Dang dai
er ke za zhi. 16:962–967. 2014.In Chinese. PubMed/NCBI
|
|
30
|
Moskowitz MA, Lo EH and Iadecola C: The
science of stroke: Mechanisms in search of treatments. Neuron.
67:181–198. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Bai X, Liu S, Yuan L, Xie Y, Li T, Wang L,
Wang X, Zhang T, Qin S, Song G, et al: Hydrogen-rich saline
mediates neuroprotection through the regulation of endoplasmic
reticulum stress and autophagy under hypoxia-ischemia neonatal
brain injury in mice. Brain Res. 1646:410–417. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Cao G, Zhou H, Jiang N, Han Y, Hu Y, Zhang
Y, Qi J, Kou J and Yu B: YiQiFuMai powder injection ameliorates
cerebral ischemia by inhibiting endoplasmic reticulum
stress-mediated neuronal apoptosis. Oxid Med Cell Longev.
2016:54932792016. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Osada N, Kosuge Y, Ishige K and Ito Y:
Characterization of neuronal and astroglial responses to ER stress
in the hippocampal CA1 area in mice following transient forebrain
ischemia. Neurochem Int. 57:1–7. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Herrmann AG, Deighton RF, Le Bihan T,
McCulloch MC, Searcy JL, Kerr LE and McCulloch J: Adaptive changes
in the neuronal proteome: Mitochondrial energy production,
endoplasmic reticulum stress, and ribosomal dysfunction in the
cellular response to metabolic stress. J Cereb Blood Flow Metab.
33:673–683. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Edelman GM and Gally JA: Nitric oxide:
Linking space and time in the brain. Proc Natl Acad Sci USA.
89:11651–11652. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Lu Q, Harris VA, Rafikov R, Sun X, Kumar S
and Black SM: Nitric oxide induces hypoxia ischemic injury in the
neonatal brain via the disruption of neuronal iron metabolism.
Redox Biol. 6:112–121. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Pei DS, Song YJ, Yu HM, Hu WW, Du Y and
Zhang GY: Exogenous nitric oxide negatively regulates c-Jun
N-terminal kinase activation via inhibiting endogenous NO-induced
S-nitrosylation during cerebral ischemia and reperfusion in rat
hippocampus. J Neurochem. 106:1952–1963. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Huang Z, Huang PL, Ma J, Meng W, Ayata C,
Fishman MC and Moskowitz MA: Enlarged infarcts in endothelial
nitric oxide synthase knockout mice are attenuated by
nitro-L-arginine. J Cereb Blood Flow Metab. 16:981–987. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Huang Z, Huang PL, Panahian N, Dalkara T,
Fishman MC and Moskowitz MA: Effects of cerebral ischemia in mice
deficient in neuronal nitric oxide synthase. Science.
265:1883–1885. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Ferriero DM, Holtzman DM, Black SM and
Sheldon RA: Neonatal mice lacking neuronal nitric oxide synthase
are less vulnerable to hypoxic-ischemic injury. Neurobiol Dis.
3:64–71. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Nowicki JP, Duval D, Poignet H and Scatton
B: Nitric oxide mediates neuronal death after focal cerebral
ischemia in the mouse. Eur J Pharmacol. 204:339–340. 1991.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Mohammadi MT, Shid Moosavi SM and Dehghani
GA: Contribution of nitric oxide synthase (NOS) activity in
blood-brain barrier disruption and edema after acute
ischemia/reperfusion in aortic coarctation-induced hypertensive
rats. Iran Biomed J. 15:22–30. 2011.PubMed/NCBI
|
|
43
|
Nadjafi S, Ebrahimi SA and
Rahbar-Roshandel N: Effect of berberine on nitric oxide production
during oxygen-glucose deprivation/reperfusion in OLN-93
oligodendrocytes. Pak J Biol Sci. 17:1185–1189. 2014. View Article : Google Scholar
|
|
44
|
Srinivasan K and Sharma SS:
3-Bromo-7-nitroindazole attenuates brain ischemic injury in
diabetic stroke via inhibition of endoplasmic reticulum stress
pathway involving CHOP. Life Sci. 90:154–160. 2012. View Article : Google Scholar
|
|
45
|
Gao G, Widmer J, Stapleton D, Teh T, Cox
T, Kemp BE and Witters LA: Catalytic subunits of the porcine and
rat 5′-AMP-activated protein kinase are members of the SNF1 protein
kinase family. Biochim Biophys Acta. 1266:73–82. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Ramamurthy S and Ronnett GV: Developing a
head for energy sensing: AMP-activated protein kinase as a
multifunctional metabolic sensor in the brain. J Physiol.
574:85–93. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Ji K, Xue L, Cheng J and Bai Y:
Preconditioning of H2S inha-lation protects against
cerebral ischemia/reperfusion injury by induction of HSP70 through
PI3K/Akt/Nrf2 pathway. Brain Res Bull. 121:68–74. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Tan H, Zou W, Jiang J, Tian Y, Xiao Z, Bi
L, Zeng H and Tang X: Disturbance of hippocampal H2S
generation contributes to CUMS-induced depression-like behavior:
Involvement in endoplasmic reticulum stress of hippocampus. Acta
Biochim Biophys Sin (Shanghai). 47:285–291. 2015. View Article : Google Scholar
|
|
49
|
Shen Y, Shen Z, Miao L, Xin X, Lin S, Zhu
Y, Guo W and Zhu YZ: miRNA-30 family inhibition protects against
cardiac ischemic injury by regulating cystathionine-gamma-lyase
expression. Antioxid Redox Signal. 22:224–240. 2015. View Article : Google Scholar :
|
|
50
|
Chatzianastasiou A, Bibli SI, Andreadou I,
Efentakis P, Kaludercic N, Wood ME, Whiteman M, Di Lisa F, Daiber
A, Manolopoulos VG, et al: Cardioprotection by H2S
donors: Nitric oxide-dependent and -independent mechanisms. J
Pharmacol Exp Ther. 358:431–440. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Duan XC, Liu SY, Guo R, Xiao L, Xue HM,
Guo Q, Jin S and Wu YM: Cystathionine-β-synthase gene transfer into
rostral ventrolateral medulla exacerbates hypertension via nitric
oxide in spontaneously hypertensive rats. Am J Hypertens.
28:1106–1113. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Grossi L: Hydrogen sulfide induces nitric
oxide release from nitrite. Bioorg Med Chem Lett. 19:6092–6094.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Kubo S, Doe I, Kurokawa Y, Nishikawa H and
Kawabata A: Direct inhibition of endothelial nitric oxide synthase
by hydrogen sulfide: Contribution to dual modulation of vascular
tension. Toxicology. 232:138–146. 2007. View Article : Google Scholar : PubMed/NCBI
|