Introduction
Cardiac hypertrophy is not only a major risk factor
in the development of heart failure, but also an independent risk
factor for cardiac morbidity and mortality (1,2).
The incidence rate of myocardial hypertrophy is >1/500 in the
general population (3–5). Due to the increased workload, the
vast majority of mammalian cardiomyocytes fail to divide soon after
birth, and this leads to hypertrophic growth (6,7).
Several factors can lead to pathological cardiac hypertrophy, such
as gene mutations, ischemic myocardial injury, diabetic
cardiomyopathy, valvular dysfunction and neurohumoral activation
(8,9). Cardiomyocyte hypertrophy may be a
compensatory response to the changes in the molecular and
biochemical microenvironment, including pressure overload. However,
clinical therapeutic strategies are still limited for the treatment
of cardiac hypertrophy (10,11). The better understanding of this
disease may contribute to the development of novel and more
effective therapeutic strategies. Thus, it is of great importance
to determine the molecular mechanisms responsible for the
development of cardiac hypertrophy in vitro.
Urotensin II (UII), an eleven amino acid cyclic
peptide, was originally isolated from the urophysis of teleost
fish, which exhibit full activity in different tissues, including
the cardiovascular system (12).
It has been demonstrated that UII is a potent vasoconstrictor. In
addition, the expression of UII and its receptor has been shown to
be increased in cardiac tissue from patients with congestive heart
failure, which positively correlates with disease severity and
inversely with cardiac function (13). It has been certified that
UII-induced cardiac hypertrophy involves multiple signaling
pathwaysis, including mitogen-activated protein kinase signaling
pathways, extracellular signal-regulated kinase (ERK), epidermal
growth factor receptor transactivation (14,15) and the Akt signaling pathways
(16). UII elicits strong
vasoconstriction via the mobilization of intracellular
Ca2+, as well as through extracellular Ca2+
entry (17). It has been shown
that UII applied in the absence of extracellular Ca2+
induces transient cytosolic Ca2+ release from the
sarcoplasmic reticulum (SR) (18). Recent studies have demonstrated
that UII-induced cardiomyocyte hypertrophy is associated with the
changes in the intracellular Ca2+ concentration
(19). However, the precise
signaling mechanisms responsible for UII-induced cardiac
hypertrophy remain to be elucidated.
The cAMP-dependent protein kinase A (PKA), a
threonine/serine kinase consists of two catalytic (C) and two
regulatory (R) subunits, each of which is capable of binding two
cAMP molecules. With the binding to cAMP, specific conformation of
the regulatory subunits occurs, causing the activation and release
of catalytic subunits (20). In
cardiomyocytes, phospholamban (PLN), a membrane protein, is the
target of PKA, which inhibits the sarco/endoplasmic reticulum
Ca2+-ATPase (SERCA). Unphosphorylated PLN binds SERCA,
reduces calcium uptake and further generates muscle contraction.
However, once PKA phosphorylates PLN at Ser16 in the cytoplasmic
helix, phosphorylated PLN relieves SERCA inhibition and initiates
muscle relaxation (21). The
inhibition of Ca2+/calcineurin-NFAT has been shown to be
involved in the attenuation of cardiac hypertrophy in vitro
and in vivo (22); thus,
the PKA signaling pathway may play a role in the development of
cardiac hypertrophy.
In cardiac muscle tissues, it has been demonstrated
that calcium (Ca2+) plays an important role in
regulating the process of excitation-contraction coupling (ECC).
When a depolarizing stimulus arrives at the sarcolemma, ECC is
activated and opens L-type Ca2+ channels in the T-tubule
region, leading to an influx of Ca2+ into the cytosol,
and further facilitating the release of Ca2+ from the
cardiac ryanodine receptor 2(RyR2), which is known as
Ca2+-induced Ca2+ release (23,24). An increase in the intracellular
free Ca2+ concentration after the intracellular
Ca2+ release stimulates the binding of Ca2+
to the troponin complex and exposes the myosin actin-binding site
in the cytosol (24). The
interaction between actin and myosin further facilitates the
generation of the power stroke and muscular contraction. To
initiate muscular relaxation, intracellular Ca2+ must be
eliminated from the cytoplasm. There are four main transporters for
Ca2+ re-uptake: sarcolemmal Ca2+-ATPase,
Na+/Ca2+ exchanger, SERCA and mitochondrial
Ca2+ uniporter (24).
In mammals, SERCA is the main transporter to transport
Ca2+ from the myoplasm into the SR (25). Moreover, 92% of Ca2+ in
rodent hearts and 70% of Ca2+ in the human heart are
removed via the SERCA pump (25,26). Thus, there may be a tight
association between intracellular Ca2+ and cardiomyocyte
hypertrophy.
To date, at least to the best of our knowledge, the
effect of UII on the PKA signaling pathway in cardiomyocytes has
not been documented. In this study, we therefore aimed to determine
whether UII-induced cardiomyocyte hypertrophy is associated with
the activation of the PKA signaling pathway in an in vitro
model, and whether PLN/SERCA is involved downstream. The treatment
of cardiomyocytes with a PKA inhibitor (KT-5720), allowed us to
identify the key role for the activation of PKA in UII-induced
hypertrophy.
Materials and methods
Reagents and antibodies
Dulbecco's modified Eagle's medium (DMEM), tissue
culture reagents and fetal calf serum (FCS) were purchased from
Invitrogen Life Technologies (Carlsbad, CA, USA). Antidodies
[anti-PKA (SAB1300370), anti-p-PKA (SAB4301240), anti-PLN
(HPA026900), anti-p-PLN (SAB1305590) and anti-SERCA (S7199)] were
purchased from Sigma-Aldrich (St. Louis, MO, USA). UII was obtained
from Sigma-Aldrich. KT-5720 was obtained from Tocris Bioscience
(Bristol, UK).
Neonatal cardiac myocyte culture and
treatment protocol
We obtained ventricles from 1-day-old Sprague-Dawley
(SD) rats and isolated cardiac myocytes through digestion. All of
the experiments were performed in accordance with the Guide for the
Care and Use of Laboratory Animals and approved by the ethics
committee of Shanxi Medical University. Briefly, SD rats were
anesthetized and decapitated. The ventricles were rapidly extracted
and minced into sections in Hanks' balanced salt solution and
digested with 1% collagenase (Sigma-Aldrich) in an incubator with
5% CO2 at 37°C for 15 min. The cell suspension was
centrifuged at 90 rpm for 8 min and the supernatant was resuspended
in PBS containing 15% fetal bovine serum (FBS) (both from Wisent,
Inc., St-Jean-Baptiste, QC, Canada). The cells were cultured for 2
h to allow the fibroblasts to attach to the dish. Cardiomyocytes
were collected from the supernatants and cultured with DMEM
containing 1 g/l glucose plus 10% FBS and 1%
penicillin/streptomycin (Gibco, Carlsbad, CA, USA). The
cardiomyocytes were divided into 4 groups as follows: i) the
control group, in which the cells were treated with PBS in 2 ml
DMEM complete medium as a placebo for 2 h, and then with 2 ml DMEM
complete medium for 48 h; ii) the UII group, in which the cells
were treated with PBS in 2 ml DMEM complete medium as a placebo for
2 h, and then exposed to 100 nM UII in 2 ml DMEM complete medium
for 48 h; iii) the UII + KT-5720 group, in which the cells were
treated with 5 μM KT-5720 in 2 ml DMEM complete medium for 2
h, and then exposed to 100 nM UII also in 2 ml DMEM complete medium
for 48 h; and iv) the KT-5720 group, in which the cells were
treated with 5 μM KT-5720 in 2 ml DMEM complete medium for 2
h, and then with 2 ml DMEM complete medium for 48 h.
Measurement of cell planimetric area
To determine the involvement of the PKA signaling
pathway in UII-induced cardiac hypertrophy, the cells were plated
in 6-well plates at about 30/cm2 density. The cell
grouping and culture were performed as mentioned above. The
pre-treated cells were washed with PBS twice prior to fixation in
4% paraformaldehyde solution. After 10 min, the residual
paraformaldehyde solutions were removed with three 5 min washes of
0.1% Triton X-100 (v/v, in PBS). The cells were double-labeled with
phalloidin (for F-actin labeling) conjugated with Alexa Fluor 488
(The Jackson Laboratory, Bar Harbor, ME, USA) and DAPI
(Sigma-Aldrich) (for DNA nuclear labeling). Measurements were
calibrated by the measurement of a known standard and results were
determined from 3 independent experiments.
Measurement of protein/DNA content
The cells were collected and then split into 2 equal
aliquots for total protein and DNA measurements. One aliquot was
used to extract protein and the protein concentration was detected
using a BCA protein assay (Pierce, Rockford, IL, USA) (27). The other aliquot was used to
extract DNA and the DNA concentration was investigated using
Hoechst 33258 with calf thymus DNA as a standard (both
Sigma-Aldrich) (28). The ratio
of protein to DNA was then calculated to estimate potential protein
synthesis.
Detection of the intracellular
Ca2+ concentration
For the measurement of the Ca2+
concentration, the cells in the 4 groups were incubated with 10
μM of Fura-3AM (Molecular Probes, Eugene, OR, USA) in
recording solution (in mM: 135 NaCl, 5.6 KCl, 1.8 CaCl2,
1.2 MgCl2, 10 HEPES, and 10 glucose, pH 7.4) for 30 min
at 37°C and then washed in recording solution. The staining cells
were then co-labeled with DAPI for DNA nuclear staining. The
coverslips were mounted in a 1 ml plastic chamber and observed
under a confocal microscope (LSM510; Carl Zeiss, Thornwood, NY,
USA) to measure fluorescence. Fluo-3AM was excited at 488 nm, and
fluorescence emission was measured using a 505- to 530-nm band-pass
filter. Fluorescence images with Fura-3 AM and DAPI were collected
and fluorescence quantification was carried out using the MetaFluor
Imaging system (Molecular Devices).
Western blot analysis of protein
expression
The cells were washed with cold PBS and the protein
concentration was assessed using a BCA protein assay (Pierce).
Proteins were separated on polyacrylamide gels and then
electrotransferd onto nitrocellulose membranes (Amersham,
Buckinghamshire, UK). After being blocked for 3 h in Tris-buffered
saline with 0.1% Tween-20 (TBST) and 3% bovine serum albumin (BSA),
the membranes were incubated overnight at 4°C with an optimized
dilution of 1:1,000 of rabbit polyclonal antibodies against
phospho-PKA [p-PKA (Thr197)], PKA, phospho-PLN [p-PLN (serine-16)],
PLN and SERCA2a. The membranes were then washed and incubated with
alkaline phosphatase-conjugated secondary antibodies (SAB3700854;
Sigma-Aldrich) in TBST for 2 h and developed with NBT/BCIP color
substrate (Promega, Madison, WI, USA). The densities of the bands
on the membranes were scanned and analyzed using an image analyzer
(LabWorks Software; UVP, Upland, CA, USA).
Statistical analysis
Data are presented as the means ± standard error of
mean (SEM). Statistical analyses were performed using one-way
analysis of variance (ANOVA), followed by adjusted t-tests with
P-values corrected by the Newman-Keuls test. A p-value <0.05 was
considered to indicate a statistically significant difference
(two-tailed statistics). Data were analyzed using statistical
package for social science (SPSS) (SPSS, Inc., Chicago, IL, USA)
software version 14.0 for Microsoft Windows.
Results
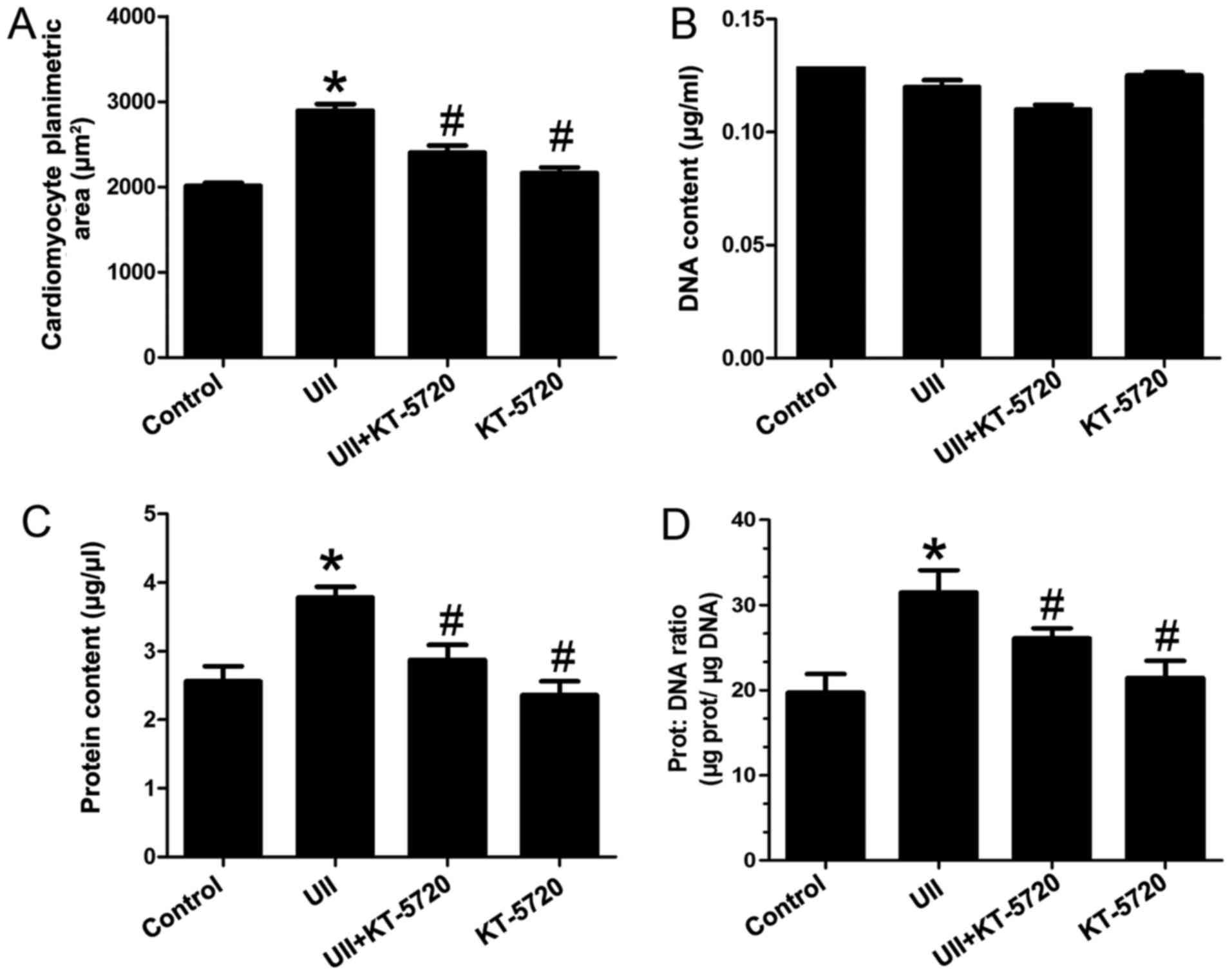
KT-5720 inhibits UII-induced hypertrophy
in neonatal rat cardiomyocytes
In order to gain further insight into the role
played by UII and the effect of pre-treatment with KT-5720, the
candomyocyte planimetric area, DNA content, protein content and
protein/DNA ratio were measured. As shown in Fig. 1A and C, UII induced a significant
increase in cell area and in the protein content, confirming the
occurrence of cardiomyocyte hypertrophy compared with the untreated
cells (p<0.05). On the contrary, the DNA content of the neonatal
cardiomyocytes only changed slightly under the different treatment
conditions (Fig. 1B; p>0.05).
This led to a significant increase in the protein to DNA ratio in
the UII-exposed cells, in comparison to the untreated cells
(Fig. 1D; p<0.05). At the same
time, pre-treatment of the UII-exposed cells with KT-5720 (a
selective inhibitor of PKA) prevented the hypertrophic response.
KT-5720 abolished the UII-induced increase in the cell area,
protein content and protein to DNA ratio, suggesting that the PKA
signaling pathway is involved in the effects of UII. Of note,
neither UII nor the PKA inhibitor used exerted an effect on the DNA
content. KT-5720 alone exerted similar effects as those observed
with the co-incubation of UII and KT-5720.
KT-5720 decreases the high level of
intracellular Ca2+ induced by UII in cardiomyocytes
To determine the mechanisms responsible for
UII-induced cardiomyocyte hypertrophy, the double-labeling with
DAPI and Fura-3AM immunofluorescence staining was applied to
measure the intracellular Ca2+ intensity in
cardiomyocytes. As is shown in Fig.
2A and B, UII induced a significant increase in the
Ca2+ content in the cardiomyocytes compared with the
control group. However, the effect of UII was markedly inhibited by
pre-treatment with KT-5720. Incubation with KT-5720 alone exerted
greater inhibitory effects than with co-incubation (UII and
KT-5720) (Fig. 2C and D).
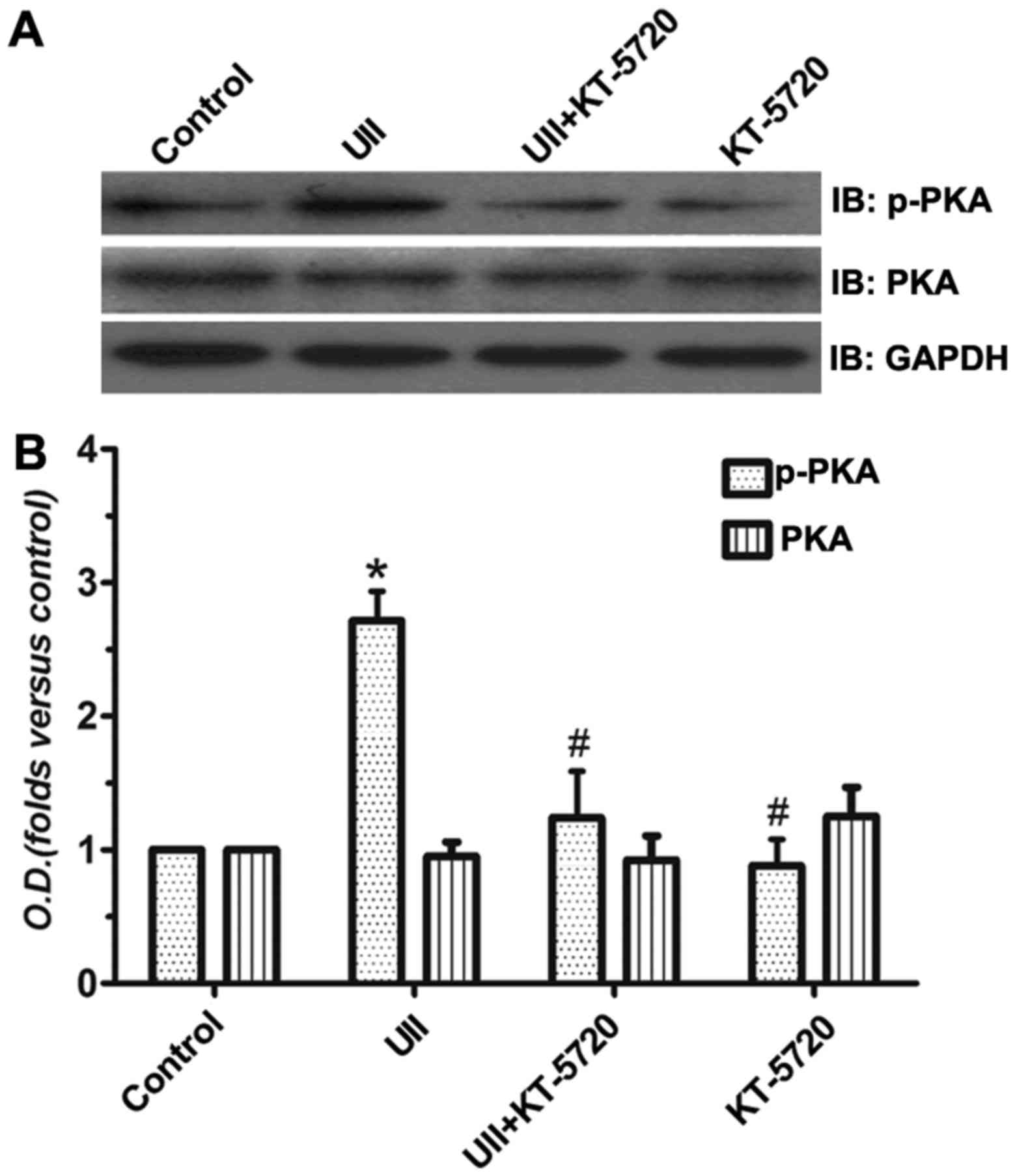
UII activates the PKA signaling
pathway
We examined the involvement of the PKA signaling
pathway in hypertrophy by western blot analysis. As is shown in
Fig. 3, the protein expression
level of p-PKA in the UII group was significantly increased
compared with that in the control group. Pre-treatment with KT-5720
reversed this increasing phenomenon. KT-5720 incubation alone
exerted similar effects as those observed with with co-incubation
(UII and KT-5720). The total PKA protein expression remained
unaltered. These data directly suggest that the PKA signaling
pathway plays an important role in UII-induced cardiomyocyte
hypertrophy.
Downstream mechanisms of PKA activation
induced by UII
In cardiomyocytes, PLN phosphorylation at Ser16 by
PKA is sufficient to reverse SERCA inhibition and increase the
enzyme affinity for calcium that is pumped into the SR, initiating
muscle relaxation (29). SERCA is
a 110 kDa membrane-embedded ATP-driven calcium pump and is
regulated by intramembrane interaction with PLN.
In this study, in order to further investigate the
downstream changes of p-PKA, the expression of PLN/SERCA was
detected by western blot analysis. Compared with the control group,
the results revealed that the p-PLN and SERCA2a bands were both
dramatically increased in the cells incubated with UII (Figs. 4 and 5). The results also revealed that UII
had did not affect the total PLN levels compared with the other
groups. Pre-treatment with KT-5720 exerted an opposite effect to
UII. Incubation with KT-5720 alone exerted similar effects as those
observed with co-incubation (UII and KT-5720). These findings
suggest that the PLN/SERCA2a signaling pathway is involved in the
UII-induced neonatal cardiomyocyte hypertrophy by the regulation of
the intracellular Ca2+ content.
Discussion
In this study, we demonstrated that UII-induced
neonatal cardiomyocyte hypertrophy involves the phosphorylation of
PKA. Calcium is the primary determinant of cardiac force production
(30). Alterations in the
phosphorylation levels of Ca2+-handing proteins
downstream of PKA not only control myocardial contraction and
relaxation, but also represent molecular signatures of failing
hearts. UII is associated with cardiovascular function and may act
as autocrine/paracrine neurohormonal factor within the heart. UII
exerts inotropic effects via the G-protein-coupled receptor (GPCR)
UTR in cardiac and peripheral vascular tissue. Stimulation of Gβ
receptor prompts the activation of adenylyl cyclase, leading to an
increase in the intracellular cAMP levels and finally, the
activation of PKA. PKA substrate phosphorylation is involved in
many cellular processes, such as gene expression, metabolism and
contraction (31).
In cardiomyocytes, PKA phosphorylates many of the
components associated with the excitation-coupling mechanism,
including PLN. The phosphorylation of PLN promotes the relaxation
of cardiomyocytes by accelerating Ca2+ re-uptake into
the SR by SERCA2a. The rise of cytosolic Ca2+ initiates
contraction by the binding of Ca2+ to cardiac troponin
C, which in turn binds and displaces cardiac troponin I from its
actin-binding site, thereby allowing myosin heads to interact with
actin to form cross bridges (30). The sustained increase of
intracellular Ca2+ levels induces left ventricular
hypertrophy and dysfunction (32), which ultimately leads to the
development of heart failure.
Even though cardiac hypertrophy is a compensatory
response to external stimulation for maintaining the function of
the heart, it is finally involved in disorders of contraction and
relaxation (33). Many signaling
pathways which respond to extracellular stimuli are involved in the
molecular mechanisms of cardiomyocyte hypertrophy. It has been
indicated that PKC, as one of the major signal transduction
molecules, plays an significant role in regulating the
differentiation and proliferation of different types of cells
(34). It has been demonstrated
that Ca2+ influx induced by PKA may activate the
Raf-1/MAP kinase cascade (33).
Our previous study has confirmed that CaMKII signaling pathway also
plays an important role in UII-induced cardiomyocyte hypertrophy
(35). In the present study, we
demonstrate that PKA signaling also plays a crucial role in
modulating intracellar Ca2+ influx through the
phospho-PLN protein to maintain cardiac contractility. These data
reinforce two possible roles of CaMKII and PKA signaling pathways
as potential treatment agents in pathological cardiac hypertrophy.
The activity of SERCA pump regulated by PLN is essential to
preserve intracellar Ca2+ homeostasis and meet the
physiological function requirements of the heart. In the future, we
aim to focus on the effects of UII, its receptor, and the related
signaling pathways in cardiac health and disease.
The effect of UII on the cardiovascular system has
been investigated in vivo (36). Human UII (hUII) has also been used
in the cynomolgus monkey to detect its cardiovascular effects
(37). A complex dose-dependent
hemodynamic response is excited after hUII systemic applications.
Following exposure to 30–300 pmol/kg hUII, a marked systemic
vasoconstriction and a severe myocardial contractile dysfunction
occur, which differs from the effects of other vasoconstrictors,
such as endothelin-1 (ET-1) and angiotensin (Ang)II (38). In addition, the effects of hUII
have been investigated in rats (39). These results suggest that the
increase in the levels of UII in myocardial cells leads to
myocardial dysfunction under disease conditions, such as aortic
stenosis (40). Taken together,
our in vitro results, as well as other in vivo data
demonstrate that UII induces significant cardiac hypertrophy.
In conclusion, the present study demonstrates that
UII induces neonatal cardiomyocyte hypertrophy and this involves
the activation of the PKA/PLN/SERCA2a signaling pathway. Our study
established that the blockade of the PKA signaling pathway inhibits
hypertrophy. These data provide a new experimental foundation for
pathological cardiac hypertrophy, and may aid in the development of
novel treatment strategies for cardiac hypertrophy.
Acknowledgments
This study was supported by grants from the Natural
Science Foundation of Shanxi Province (no. 2012011036-1), the
Shanxi Provincial Scientific Research Projects Foundation of
Abroad-Studying Personnel (no. 2012-7), the Shanxi Provincial
University Scientific Research Projects Foundation of
Abroad-Studying and Returning Personnel (no. 2011-63), the Selected
Scientific Research Projects Foundation of Abroad-Studying
Personnel, Office of Human Resources, Shanxi Province (no.
2013-68), the Selected Scientific Research Projects Foundation of
Abroad-Studying and Returning Personnel, the Shanxi Province (no.
2010-97), Technology Innovation Foundation of Shanxi Medical
University (no. 2010-7) and the Shanxi Provincial Scientific
Research Projects Foundation of Abroad-Studying and Returning
Personnel (no. 2009-9), the Technology Innovation Foundation of
Shanxi Medical University (no. 01200912), the Shanxi Provincial
University Scientific Research Projects Foundation of
Abroad-Studying Personnel (no. 201128), and the Shanxi Provincial
Technology Projects Foundation of Abroad-Studying Personnel
(2012-084).
References
|
1
|
Levy D, Garrison RJ, Savage DD, Kannel WB
and Castelli WP: Prognostic implications of echocardiographically
determined left ventricular mass in the Framingham Heart Study. N
Engl J Med. 322:1561–1566. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Tang S, Chen H, Cheng Y, Nasir MA, Kemper
N and Bao E: The interactive association between heat shock factor
1 and heat shock proteins in primary myocardial cells subjected to
heat stress. Int J Mol Med. 37:56–62. 2016.PubMed/NCBI
|
|
3
|
Wu C, Dong S and Li Y: Effects of
miRNA-455 on cardiac hypertrophy induced by pressure overload. Int
J Mol Med. 35:893–900. 2015.PubMed/NCBI
|
|
4
|
Liu P, Yan S, Chen M, Chen A, Yao D, Xu X,
Cai X, Wang L and Huang X: Effects of baicalin on collagen I and
collagen III expression in pulmonary arteries of rats with hypoxic
pulmonary hypertension. Int J Mol Med. 35:901–908. 2015.PubMed/NCBI
|
|
5
|
Chen P, Li F, Xu Z, Li Z and Yi XP:
Expression and distribution of Src in the nucleus of myocytes in
cardiac hypertrophy. Int J Mol Med. 32:165–173. 2013.PubMed/NCBI
|
|
6
|
Heineke J and Molkentin JD: Regulation of
cardiac hypertrophy by intracellular signalling pathways. Nat Rev
Mol Cell Biol. 7:589–600. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Guo H, Liu B, Hou L, The E, Li G, Wang D,
Jie Q, Che W and Wei Y: The role of mAKAPβ in the process of
cardiomyocyte hypertrophy induced by angiotensin II. Int J Mol Med.
35:1159–1168. 2015.PubMed/NCBI
|
|
8
|
Hill JA and Olson EN: Cardiac plasticity.
N Engl J Med. 358:1370–1380. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Chuang CT, Guh JY, Lu CY, Chen HC and
Chuang LY: S100B is required for high glucose-induced pro-fibrotic
gene expression and hypertrophy in mesangial cells. Int J Mol Med.
35:546–552. 2015.
|
|
10
|
Zhang Y, Si Y and Ma N: Meis1 promotes
poly (rC)-binding protein 2 expression and inhibits angiotensin
II-induced cardiomyocyte hypertrophy. IUBMB Life. 68:13–22. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zhou HM, Zhong ML, Zhang YF, Cui WY, Long
CL and Wang H: Natakalim improves post-infarction left ventricular
remodeling by restoring the coordinated balance between endothelial
function and cardiac hypertrophy. Int J Mol Med. 34:1209–1218.
2014.PubMed/NCBI
|
|
12
|
Gong H, Ma H, Liu M, Zhou B, Zhang G, Chen
Z, Jiang G, Yan Y, Yang C, Kanda M, et al: Urotensin II inhibits
the proliferation but not the differentiation of cardiac side
population cells. Peptides. 32:1035–1041. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Douglas SA, Tayara L, Ohlstein EH, Halawa
N and Giaid A: Congestive heart failure and expression of
myocardial urotensin II. Lancet. 359:1990–1997. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Liu JC, Chen CH, Chen JJ and Cheng TH:
Urotensin II induces rat cardiomyocyte hypertrophy via the
transient oxidization of Src homology 2-containing tyrosine
phosphatase and transactivation of epidermal growth factor
receptor. Mol Pharmacol. 76:1186–1195. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Onan D, Pipolo L, Yang E, Hannan RD and
Thomas WG: Urotensin II promotes hypertrophy of cardiac myocytes
via mitogen-activated protein kinases. Mol Endocrinol.
18:2344–2354. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Gruson D, Ginion A, Decroly N, Lause P,
Vanoverschelde JL, Ketelslegers JM, Bertrand L and Thissen JP:
Urotensin II induction of adult cardiomyocytes hypertrophy involves
the Akt/GSK-3beta signaling pathway. Peptides. 31:1326–1333. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Maguire JJ and Davenport AP: Is
urotensin-II the new endothelin? Br J Pharmacol. 137:579–588. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Domínguez-Rodríguez A, Díaz I,
Rodríguez-Moyano M, Calderón-Sánchez E, Rosado JA, Ordóñez A and
Smani T: Urotensin-II signaling mechanism in rat coronary artery:
role of STIM1 and Orai1-dependent store operated calcium influx in
vasoconstriction. Arterioscler Thromb Vasc Biol. 32:1325–1332.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhang Y, Ying J, Jiang D, Chang Z, Li H,
Zhang G, Gong S, Jiang X and Tao J: Urotensin-II receptor
stimulation of cardiac L-type Ca2+ channels requires the
βγ subunits of Gi/o-protein and phosphatidylinositol
3-kinase-dependent protein kinase C β1 isoform. J Biol Chem.
290:8644–8655. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Rababa'h A, Singh S, Suryavanshi SV,
Altarabsheh SE, Deo SV and McConnell BK: Compartmentalization role
of A-kinase anchoring proteins (AKAPs) in mediating protein kinase
A (PKA) signaling and cardiomyocyte hypertrophy. Int J Mol Sci.
16:218–229. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Masterson LR, Yu T, Shi L, Wang Y,
Gustavsson M, Mueller MM and Veglia G: cAMP-dependent protein
kinase A selects the excited state of the membrane substrate
phospholamban. J Mol Biol. 412:155–164. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Yin Z, Wang X, Zhang L, Zhou H, Wei L and
Dong X: Aspirin attenuates angiotensin II-induced cardiomyocyte
hypertrophy by inhibiting the Ca(2+)/Calcineurin-NFAT signaling
pathway. Cardiovasc Ther. 34:21–29. 2016. View Article : Google Scholar
|
|
23
|
Fabiato A: Calcium-induced release of
calcium from the cardiac sarcoplasmic reticulum. Am J Physiol.
245:C1–C14. 1983.PubMed/NCBI
|
|
24
|
Bers DM: Cardiac excitation-contraction
coupling. Nature. 415:198–205. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
25
|
Bers DM: Ca transport during contraction
and relaxation in mammalian ventricular muscle. Basic Res Cardiol.
92(Suppl 1): 1–10. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
MacLennan DH and Kranias EG:
Phospholamban: a crucial regulator of cardiac contractility. Nat
Rev Mol Cell Biol. 4:566–577. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zhang C, Shan XL, Liao YL, Zhao P, Guo W,
Wei HC and Lu R: Effects of stachydrine on norepinephrine-induced
neonatal rat cardiac myocytes hypertrophy and intracellular calcium
transients. BMC Complement Altern Med. 14:4742014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Giordano A, Romano S, Mallardo M,
D'Angelillo A, Calì G, Corcione N, Ferraro P and Romano MF: FK506
can activate transforming growth factor-beta signalling in vascular
smooth muscle cells and promote proliferation. Cardiovasc Res.
79:519–526. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Maclennan DH: Interactions of the calcium
ATPase with phospholamban and sarcolipin: structure, physiology and
pathophysiology. J Muscle Res Cell Motil. 25:600–601. 2004.
|
|
30
|
Irie T, Sips PY, Kai S, Kida K, Ikeda K,
Hirai S, Moazzami K, Jiramongkolchai P, Bloch DB, Doulias PT, et
al: S-nitrosylation of calcium-handling proteins in cardiac
adrenergic signaling and hypertrophy. Circ Res. 117:793–803. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Walsh DA and Van Patten SM: Multiple
pathway signal transduction by the cAMP-dependent protein kinase.
FASEB J. 8:1227–1236. 1994.PubMed/NCBI
|
|
32
|
Shioya T: A simple technique for isolating
healthy heart cells from mouse models. J Physiol Sci. 57:327–335.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Yamazaki T, Komuro I, Zou Y, Kudoh S,
Mizuno T, Hiroi Y, Shiojima I, Takano H, Kinugawa K, Kohmoto O, et
al: Protein kinase A and protein kinase C synergistically activate
the Raf-1 kinase/mitogen-activated protein kinase cascade in
neonatal rat cardiomyocytes. J Mol Cell Cardiol. 29:2491–2501.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Komuro I and Yazaki Y: Control of cardiac
gene expression by mechanical stress. Annu Rev Physiol. 55:55–75.
1993. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Shi H, Han Q, Xu J, Liu W, Chu T and Zhao
L: Urotensin II induction of neonatal cardiomyocyte hypertrophy
involves the CaMKII/PLN/SERCA 2a signaling pathway. Gene. 583:8–14.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Tölle M and van der Giet M:
Cardiorenovascular effects of urotensin II and the relevance of the
UT receptor. Peptides. 29:743–763. 2008. View Article : Google Scholar
|
|
37
|
Douglas SA, Sulpizio AC, Piercy V, Sarau
HM, Ames RS, Aiyar NV, Ohlstein EH and Willette RN: Differential
vasoconstrictor activity of human urotensin-II in vascular tissue
isolated from the rat, mouse, dog, pig, marmoset and cynomolgus
monkey. Br J Pharmacol. 131:1262–1274. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Bai XY, Liu XC, Yang Q, Tang XD and He GW:
The interaction between human urotensin II and vasodilator agents
in human internal mammary artery with possible clinical
implications. Ann Thorac Surg. 92:610–616. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Shyu KG, Wang BW, Chen WJ, Kuan P and Lin
CM: Angiotensin II mediates urotensin II expression by hypoxia in
cultured cardiac fibroblast. Eur J Clin Invest. 42:17–26. 2012.
View Article : Google Scholar
|
|
40
|
Hassan GS, Chouiali F, Saito T, Hu F,
Douglas SA, Ao Z, Willette RN, Ohlstein EH and Giaid A: Effect of
human urotensin-II infusion on hemodynamics and cardiac function.
Can J Physiol Pharmacol. 81:125–128. 2003. View Article : Google Scholar : PubMed/NCBI
|