Introduction
Chronic wounds or impaired acute wounds severely
affect the physical and mental health of patients. This non-healing
wound is generally caused by bacterial infection or diabetes, as
well as subsequent sustained inflammatory responses, defective
matrix remodelling and impaired re-epithelialization (1). Lipopolysaccharide (LPS), a main
ingredient of the Gram-negative bacterial cell wall, can trigger
immune responses by stimulating macrophages to secrete inflammatory
cytokines, such as interleukin (IL)-1β and tumor necrosis factor α
(TNFα). TNFα is considered to be a central pathogenic factor in
chronic wounds, and anti-TNFα therapy has shown promising effects
in improving these injuries (2,3).
Skin-resident cells, particularly keratinocytes play
an active role in cutaneous inflammation by producing various
cytokines, chemokines and adhesion molecules (4). Chemokines, such as monocyte
chemotactic protein-1 (MCP-1), regulated upon activation normal T
cell expressed and secreted factor (RANTES), IL-8,
interferon-γ-inducible protein-10 (IP-10) and intercellular
adhesion molecule intercellular cell adhesion molecule-1 (ICAM-1)
promote the recruitment of excessive immunocytes into skin lesions,
which is a key mechanism in the deterioration of chronic wounds
(3,5,6).
In addition, the persistent deficiency in cell-cell and cell-matrix
adhesion initiated by hyperinflammation can result in defects in
tissue remodeling. Matrix metalloproteinases (MMPs) secreted by
keratinocytes, fibroblasts and immune cells contribute to the
degradation of extracellular matrix (ECM), and are responsible for
this defect (7). Excessive MMP9
production has been strongly linked to various inflammatory skin
diseases (8). Keratinocyte
migration and proliferation play fundamental roles in
re-epithelialization. Previous studies have reported that the
capacities of migration and proliferation of keratinocytes are
impaired in chronic wounds (9–12);
however, the roles of TNFα are not yet completely clear.
Atypical protein kinase C (aPKC), a subfamily of
atypical serine/threonine protein kinase C, requires neither
Ca2+ nor diacylglycerol for optimal activity compared to
conventional PKC and novel PKC subfamilies (13). PKCζ, a member of the aPKC family,
is an emerging regulator of various biological processes, such as
cell polarity, tumorigenesis and chemotaxis (14–19). In several cell types, PKCζ has
been implicated in nuclear factor-κB (NF-κB) activation (20–23). The functions of PKCζ in
LPS-stimulated macrophages and TNFα-stimulated keratinocytes have
not yet been clearly examined.
The present study confirmed that PKCζ is crucial in
LPS-induced TNFα expression in macrophages. Moreover, importantly,
to the best of our knowledge, we demonstrate for the first time
that in human HaCaT keratinocytes PKCζ regulates the TNFα-induced
expression of the inflammatory related factors, IL-8, MCP-1, ICAM-1
and MMP9, which to a large degree is mediated via NF-κB activation.
In addition, this study also demonstrates that TNFα at pathological
concentrations (10 or 100 ng/ml) inhibits HaCaT cell migration and
proliferation in vitro; however, PKCζ was not found to be
involved in these processes. In vivo experiments revealed
that TNFα administration to the surrounding wound tissue at the
late phase of wound healing resulted in the delay of wound closure
and inopportune upregulations in the levels of IL-8, MCP-1, ICAM-1
and MMP9; however, PKCζ inhibitor attenauted TNFα-initiated wound
closure impairment and inflammatory disorders. Therefore, our
findings provide novel insight into the prevention of the aberrant
activity of skin keratinocytes induced by TNFα in chronic
wounds.
Materials and methods
Cell culture
The immortalized mouse macrophage cell line,
RAW264.7, and the human keratinocyte cell line, HaCaT, were
obtained from the American Type Culture Collection (Manassas, Va,
USA) and the China Center for Type Culture Collection (Wuhan,
Hubei, China), respectively. The cells were cultured in Dulbecco's
modified Eagle's medium (DMEM; Gibco, Gaithersburg, MD, USA)
supplemented with 10% fetal bovine serum (FBS; Gibco, Grand Island,
NY, USA). The cells were incubated in an incubator at 37°C, 5%
CO2.
Cell cytotoxicity assay
The cell counting kit-8 (CCK-8) (7Sea Biotech,
Shanghai, China) was used to examine the cytotoxicity according to
the instructions provide by the manufacturer. The RAW264.7 cells
were seeded in a 96-well plate at a density of 5.0×103
cells/well and incubated at 37°C for 12 h. The cells were then
treated with fresh medium alone or with LPS (100 ng/ml; Sigma,
Santa Clara, CA, USA) with or without PKCζ-specific pseudosubstrate
inhibitor (1, 5 and 10 µM; Invitrogen, Carlsbad, CA, USA)
for 48 h. Subseqently, 10 µl CCK-8 reagent were added to
each well and the cells were cultured for a further 2 h at 37°C, 5%
CO2. The absorbance was measured at 450 nm using a Tecan
Infinite M200 Pro Nano Quant microplate reader (Tecan, Männedorf,
Switzerland).
RNA interference
The PKCζ-specific siRNA duplexes (PKCζ-siRNA) and
scramble control siRNA duplexes (scramble-siRNA) were purchased
from GenePharma Co. (Shanghai, China). The sequences of these
siRNAs are listed in Table I. The
HaCaT cells were transfected with either PKCζ-siRNA (100 nM) or
scramble-siRNA (100 nM) using Lipofectamine 2000 transfection
reagent (Invitrogen) in accordance with the instructions of the
manufacturer. After 6 h, the transfection mixture was removed and
supplemented with fresh complete medium. The transfected cells were
incubated in an incubator at 37°C, 5% CO2 for a further
20 or 40 h, and the efficiency of siRNA interference was examined
by reverse transcription-quantitative PCR (RT-qPCR) or western blot
analysis.
 | Table ISequences of siRNA used in this
study. |
Table I
Sequences of siRNA used in this
study.
| siRNA | Sense (5′→3′) | Antisense
(5′→3′) |
|---|
| PKCζ |
GACACAACGAGCACUUUCUTT |
AGAAAGUGCUCGUUGUGUCTT |
| Scramble |
UUCUCCGAACGUGUCACGUTT |
ACGUGACACGUUCGGAGAATT |
RT-qPCR
The RAW264.7 cells or HaCaT cells were lysed in
TRIzol reagent (Takara, Shiga, Japan) and total RNA was extracted
in accordance with the instructions of the manufacturer. Total RNA
(500 ng) was reverse transcribed into cDNA on the basis of the
instructions of Prime Script™ RT reagent kit (Takara). The obtained
cDNA was subjected to amplification using the SYBR®
Premix Ex Taq™ kit (Takara) and Bio-Rad IQ5 Real-Time system
(Bio-Rad, Hercules, CA, USA). The primers sequences are listed in
Table II. The PCR amplification
condition 4 stages: i) initial denaturation, 95°C for 30 sec; ii)
denaturation, 95°C for 30 sec; iii) annealing, 58°C or 60°C for 10
sec; and iv) elongation, 72°C for 15 sec for total 38 cycles. The
results were normalized against the mean Cq-values of
glyceraldehyde 3-phosphate dehydrogenase (GAPDH), namely ΔCq, and
the fold increases were calculated as 2−ΔΔCq.
| Table IISequences of primers used in this
study. |
Table II
Sequences of primers used in this
study.
| Primers | Forward primer
(5′→3′) | Reverse primer
(5′→3′) |
|---|
| Mouse-TNFα |
CTATCTCCAGGTTCTCTTCAA |
GCAGAGAGGAGGTTGACTTTC |
| Mouse-IL-1β |
GCTCATCTGGGATCCTCTCC |
CCTGCCTGAAGCTCTTGTTG |
| Mouse-GAPDH |
ATTGTCAGCAATGCATCCTG |
ATGGACTGTGGtcATGAGCC |
| Human-PKCζ |
TGCTTACATTTCCTCATCCC |
TCATTCTTCTTCAACCGCAC |
| Human-MMP2 |
TGAAGTATGGGAACGCCGAT |
CGTACTTGCCATCCTTCTCA |
| Human-MMP9 |
TCTTCCCCTTCACTTTCCTG |
GCCACGAGGAACAAACTGTA |
| Human-IL-8 |
TACTCCAAACCTTTCCACCC |
AACTTCTCCACAACCCTCTG |
| Human-MCP-1 |
GCAATCAATGCCCCAGTCA |
TGCTGCTGGTGATTCTTCTATAGCT |
| Human-ICAM-1 |
CACAGTCACCTATGGCAACGA |
TGGCTTCGTCAGAATCACGTT |
| Human-GAPDH |
CTCCTCCACCTTTGACGCTG |
TCCTCTTGTGCTCTTGCTGG |
| Mouse-IL-8 |
TTGCCTTGACCCTGAAGCCCCC |
GGCACATCAGGTACGATCCAGGC |
| Mouse-MCP-1 |
CATCCACGTGTTGGCTCA |
GATCATCTTGCTGGTGAATGAGT |
| Mouse-ICAM-1 |
GGCACCCAGCAGAAGTTGTT |
CCTCAGTCACCTCTACCAAG |
| Mouse-MMP9 |
TCTGTATGGTCGTGGCTCTA |
CCTGTAATGGGCTTCCTCTATG |
Western blot analysis
The cells were lysed in RIPA buffer (Heart
Biological Technology Co., Ltd., Xi'an, China) containing 1%
aprotinin, 1% activated Na3VO4 and 1% PMSF on
ice. The protein concentration was detected according to the
instructions of the Pierce™ BCA protein assay kit (Thermo Fisher
Scientific, Rockford, IL, USA). Total proteins (50 µg) were
loaded onto a 10% sodium dodecyl sulfate (SDS)-polyacrylamide gel,
separated by electrophoresis and transfered onto PVDF membranes
(Millipore, Boston, MA, USA). The membranes were blocked with 5%
skim milk at room temperature for 2 h and then incubated with
rabbit anti-human PKCζ (1:1,000, #sc-216; Santa Cruz Biotechnology,
Inc., CA, USA), rabbit anti-human MMP2 (1:1,000, #10373-2-AP), MMP9
(1:500, #10375-2-AP), mouse anti-human β-actin (1:1,000,
#60008-1-lg) (both from China Branch of Proteintech Co., Hubei,
China) and rabbit anti-human phospho-PKCζ antibodies (1:500, #9378;
Cell Signaling Technology, Boston, MA, USA) at 4°C overnight. The
following day, the membranes were sequentially incubated with
HRP-conjugated goat anti-rabbit IgG (1:3,000, #A0208) or
HRP-conjugated goat anti-mouse IgG (1:3,000, #A0216) (Beyotime,
Shanghai, China) at 37°C for 1 h. Immunoreactive proteins were
detected using ECL reagent (Millipore) and the FluorChem FC system
(Alpha Innotech, San Leandro, CA, USA). Bands were quantitated
using ImageJ software.
Nuclear and cytoplasmic protein
extraction
The cells were seeded in a 6-well plate and
incubated at 37°C for 24 h. The cells were pre-treated with or
without the NF-κB inhibitor, BAY11-7082 (5 µM; Selleck,
Houston, TX, USA), for 2 h or PKCζ I (5 µM; Invitrogen) for
3 h and then stimulated with TNFα (rhTNFα, 10 ng/ml; Shanghai
Bioleaf Biotech Co., Ltd., Shanghai, China) for 2 h in the presence
of BAY11-7082 or PKCζ I. The nuclear proteins were isolated on the
basis of the intructions of a commercial nuclear and cytoplasmic
protein extraction kit (Beyotime). Separated nuclear proteins were
analyzed by immunoblotting. The internal reference protein of
nuclear protein was histone H3. The primary antibodies included
rabbit anti-human NF-κB-p65 antibody (1:500, #sc-372) and goat
anti-human histone H3 antibody (1:500, #sc-8654) (Santa Cruz
Biotechnology, Inc.). The secondary antibodies included
HRP-conjugated goat anti-rabbit IgG (1:1,000, #A0208) and donkey
anti-goat IgG (1:1,000, #A0108) (Beyotime).
NF-κB immunofluorescence analysis
All operations were executed in accordance with the
instructions of the NF-κB activation, Nuclear Translocation assay
kit (Beyotime). Cells growing on coverslip were fixed with 4%
paraformaldehyde for 20 min, and then incubated with 5% BSA for 30
min at room temperature to suppress non-specific binding of IgG.
The cells were then incubated with rabbit anti-human NF-κB-p65
antibody overnight at 4°C. The following day, the NF-κB-p65
antibody was removed and the cells were sequentially incubated with
anti-rabbit Cy3 IgG at room temperature for 1 h. The nucleus was
stained with DAPI. Images were captured using a FSX100 microscope
(Olympus, Tokyo, Japan).
Cell proliferation assay
Cell proliferation was evaluated by CCK-8 assay
(7Sea Biotech) according to the instructions provided by the
manufacturer. Briefly, the cells were seeded in a 96-well plate at
a density of 4.0×103 cells/well and incubated at 37°C
for 24 h. The cells were then treated with or without PKCζ-specific
pseudosubstrate inhibitor (10 µM; Invitrogen), After 3 h,
the inhibitor mixture was removed and replaced with fresh medium
alone or with TNFα (rhTNFα, 10 or 100 ng/ml; Shanghai Bioleaf
Biotech Co., Ltd.) for 24, 48 and 72 h. At the indicated time
point, the medium was removed and supplemented with the medium
containing CCK-8 reagent (10 µl/well) and the cells were
cultured for a further 1–1.5 h at 37°C, 5% CO2.
Subsequently, the absorbance was measured at 450 nm using a Tecan
Infinite M200 Pro Nano Quant micro-plate reader (Tecan).
Scratch wound healing assay
The HaCaT cells were seeded in 35-mm culture dishes
in DMEM supplemented with 10% FBS (both from Gibco) at 37°C, 5%
CO2 for 24 h, until full confluence. Subsequently, 10
µg/ml mitomycin C (Invitrogen) was added for 1 h to
completely inhibit cell proliferation. A scratch of 500 µm
in width was created on the confluent cells with a 10 µl
pipette tip. The cells were washed 3 times with phosphate-buffered
saline (PBS) and pre-treated with or without PKCζ inhibitor (10
µM; Invitrogen) for 3 h, then incubated in serum-free DMEM
alone (control group), or serum-free DMEM containing TNFα (10 or
100 ng/ml; Bioleaf) at 37°C, 5% CO2 for 24 h. The cells
were photographed under a microscope and the gap width of scratch
was measured using Image Pro Plus software. The cell migration
distance was the difference value between the gap width at 0 and 24
h. The migration distance at control group was set as 1.
Detection of the activity of PKCζ in
vivo
Male 6-week old BALB/c mice (n=40) were obtained
from The Center of Experimental Animal, the Fourth Military Medical
University. The experiment was repeated 3 times. All animal
experiments were performed in accordance with the guidelines from
the Administration of Animal Experiments for Medical Research
Purposes issued by the Ministry of Health of China, which is in
accordance with the principle of ARRIVE. The protocol was approved
by the Animal Experiment Administration Committee of the Fourth
Military Medical University. The mice were randomly divided into 2
groups (n=8) as follows: the PBS control group and TNFα group. To
examine the differences in the phosphorylation level of PKCζ
between the PBS- and TNFα-treated wounds, a 1.0×1.0 cm
full-thickness wound was created on the back of each BALB/c mouse
following anesthetization by an intraperitoneal injection of 1%
pentobarbital sodium (40 mg/kg). Subsequently, 100 µl PBS or
100 µl TNFα (200 ng, dissolved in PBS) was injected into the
tissue around the wound, and after 2 h, the samples of the
wound-edge skin (WES) and normal skin (NS) in each mouse were
collected for the examination of the levels of PKCζ and
phosphorylated PKCζ by western blot analysis.
In vivo wound closure assay
The BALB/c mice were randomly divided into 3 groups
(n=8) as follows: the PBS control group, TNFα group and TNFα + PKCζ
inhibitor group. Following anesthetization, a 1.0×1.0 cm
full-thickness wound was created on the back of each BALB/c mouse.
Subsequenlty, 100 µl PBS, 100 µl TNFα solution
(rmTNFα, 50 ng, dissolved in PBS; Bioleaf) or 100 µl TNFα
solution + PKCζ inhibitor (20 µM) was injected into the skin
tissue around the wound each day from day 6 post-incision (days 6,
7, 8, 9, 10, 11 and 12 post-incision). The wound area of each mouse
was digitally photographed and measured using Photoshop software or
the skin tissue samples of the wound edges in each mouse were
collected for mRNA extraction and RT-qPCR analysis on days 7, 9 and
13 post-wounding.
Statistical analysis
All experiments were repeated at least 3 times, and
the results are the means ± SD. The analysis of statistically
significant differences between groups was performed by one-way
analysis of variance (ANOVA) with the Student's t-test or the
Mann-Whitney U test using SPSS 17.0 software. A value of p<0.05
was considered to indicate a statistically significant
difference.
Results
PKCζ mediates LPS-induced inflammatory
responses in RAW264.7 cells
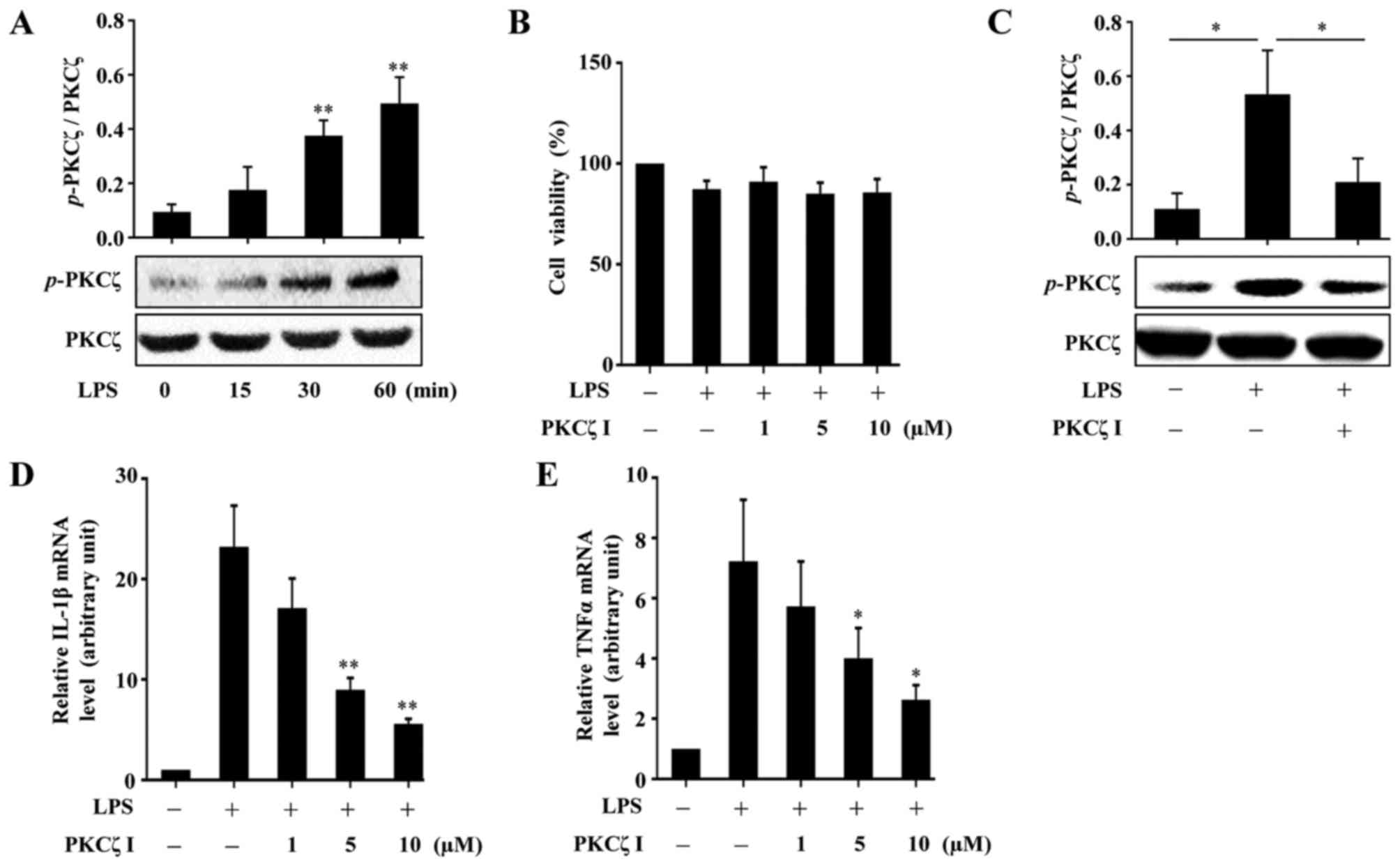
A previous study reported that PKCζ activity is
required for LPS-induced IL-1β production in RAW264.7 cells
(22); however, whether PKCζ
mediates the expression of another important inflammatory factor,
namely TNFα, has not yet been examined, at least to the best of our
knowledge. PKCζ can be activated through phosphorylating its
threonine 410 site. In the present study, we validated that
stimulation with LPS (100 ng/ml) resulted in an increase in the
phosphorylation level of PKCζ in the RAW264.7 cells (Fig. 1A). To measure the cytotoxicity of
LPS or PKCζ-specific pseudosubstrate inhibitor (PKCζ I) in the
RAW264.7 cells, CCK-8 assay was performed. The results revealed
that both LPS at a concentration of 100 ng/ml and PKCζ I up to 10
µM did not damage the viability of the RAW264.7 cells
(Fig. 1B). Pre-treatment of the
cells with PKCζ I inhibited the LPS-induced phosphorylation of PKCζ
(Fig. 1C) and suppressed the
LPS-induced increase in the mRNA expression levels of IL-1β and
TNFα (Fig. 1D and E).
TNFα promotes the activation of PKCζ in
HaCaT keratinocytes
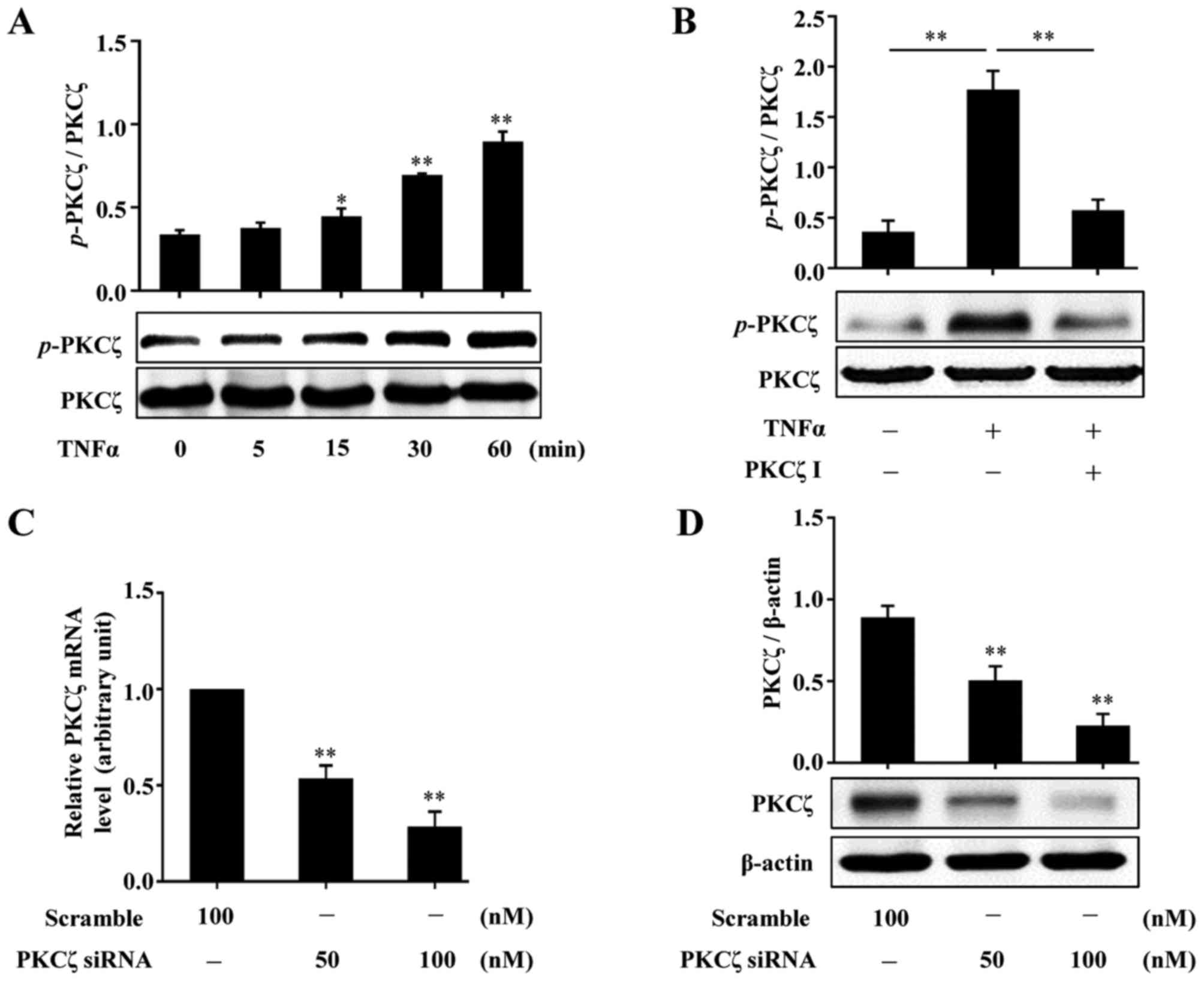
To examine whether PKCζ is involed in the effects of
TNFα on HaCaT keratinocytes, we firstly examined whether TNFα
stimulates the activation of PKCζ. Following treatment with TNFα
(10 ng/ml) and at the indicated time points (0, 5, 15, 30 and 60
min), the HaCaT cells were lysed and the lysates were assessed by
western blot analysis. The results revealed that exposure to TNFα
resulted in increased phosphorylation levels of PKCζ, which was
firstly observed at 15 min and reached peak levels at 60 min
(Fig. 2A). We then found that
pre-treatment of the cells with PKCζ I (10 µM) for 3 h
distinctly suppressed the TNFα-induced PKCζ phosphorylation
(Fig. 2B). In addition, the
effects of PKCζ-specific small interfering RNA (PKCζ-siRNA) were
examined by RT-qPCR and western blot analysis. The results revealed
that the mRNA and protein expression levels of PKCζ were all
effectively suppressed (Fig. 2C and
D).
PKCζ is involved in TNFα-induced
inflammatory responses in HaCaT cells
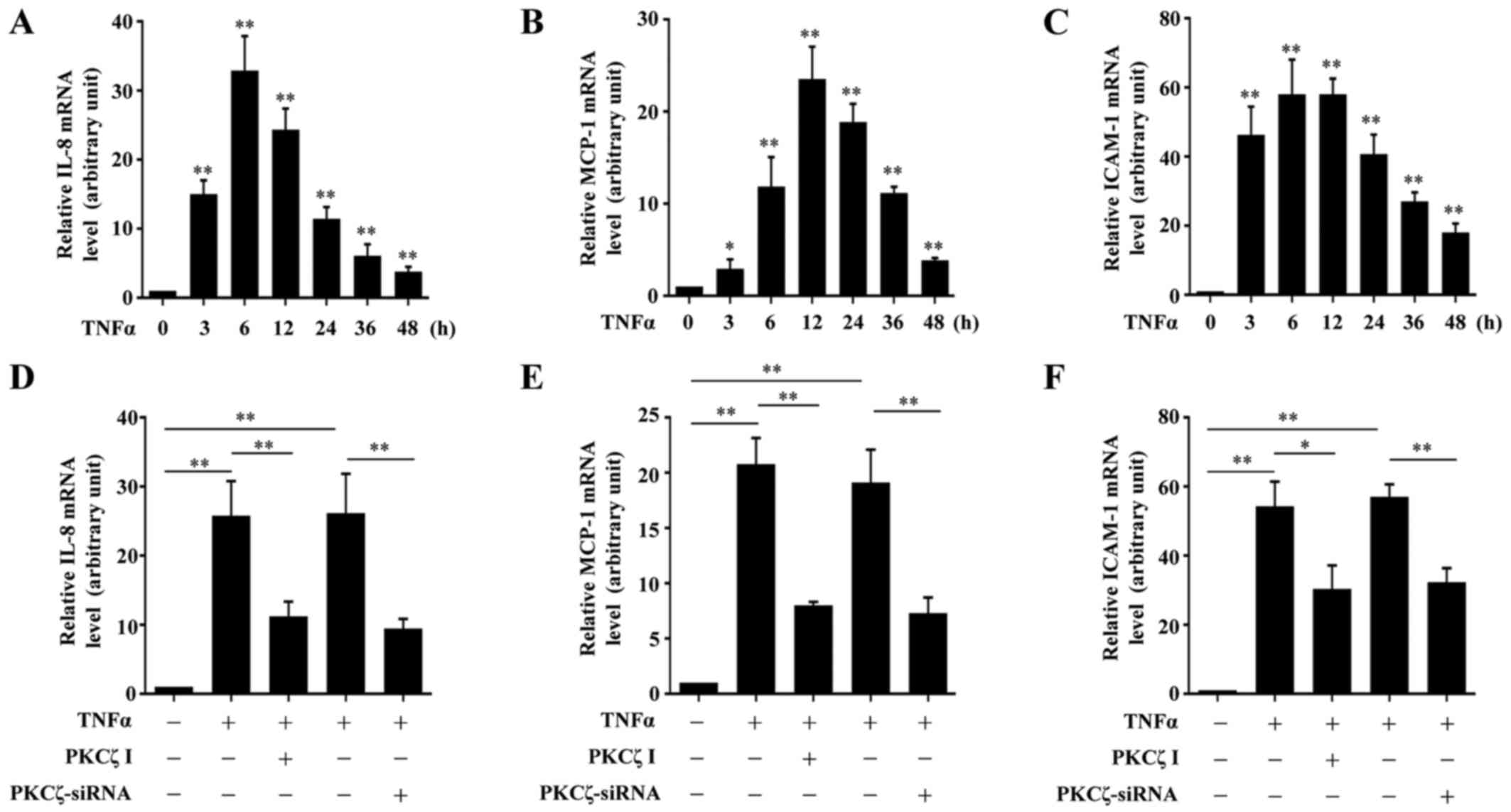
It is known that the chemokines, MCP-1 and IL-8, and
the adhesion molecule, ICAM-1, are crucial in the pathogenesis of
inflammatory disorders in chronic wounds. Thus, to determine
whether PKCζ is involved in the TNFα-induced inflammatory responses
in HaCaT cells, we examined the effects of PKCζ I on the mRNA
expression levels of IL-8, MCP-1 and ICAM-1. Exposure to TNFα led
to an increase in the mRNA expression levels of IL-8, MCP-1 and
ICAM-1 over time which respectively peaked at 6, 12 and 6 h
post-TNFα treatment, and then gradually declined, but remained at
significantly high levels at 48 h (Fig. 3A–C). The cells were pre-treated
with PKCζ I for 3 h in advance, and then stimulated with TNFα for
12 h, which largely inhibited the TNFα-induced upregulation of
IL-8, MCP-1 and ICAM-1. The silencing of PKCζ by transfection with
siRNA for 6 h also led to similar results (Fig. 3D–F).
PKCζ mediates the TNFα-induced
upregulation of MMP9 in HaCaT cells
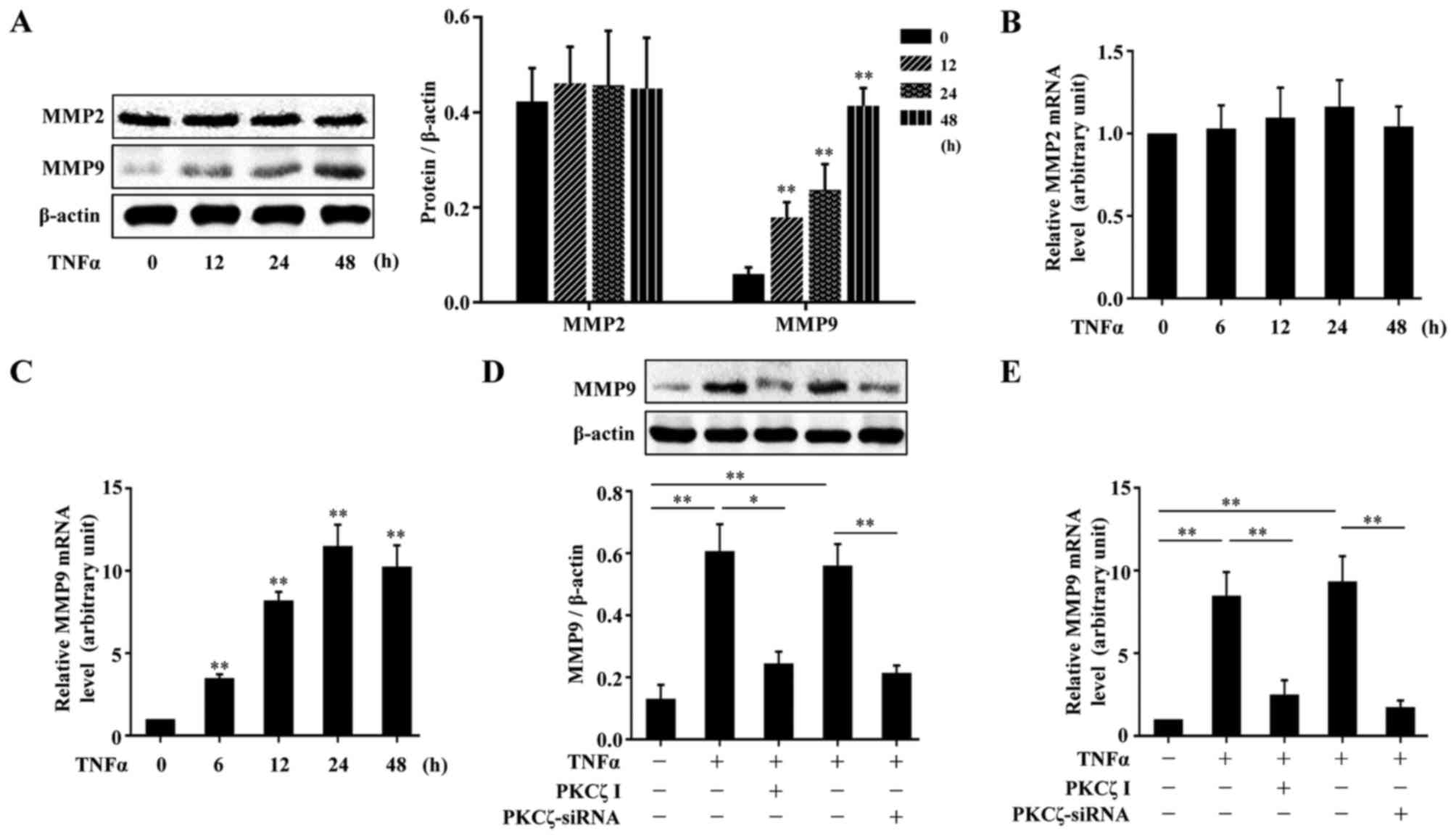
MMPs, are a crucial class of protease, and have been
reported to be abnormally upregulated in impaired wound healing
(7,8). We firstly examined the effects of
TNFα on the expressions of MMP2 and MMP9 in HaCaT cells. The cells
were treated with TNFα for various periods of time (0, 12, 24 and
48 h), and the cells were lysed and the lysates were assessed by
western blot analysis. The results revealed that TNFα had no
significant effect on the protein expression of MMP2; however, MMP9
protein expression was indeed elevated by exposure to TNFα
(Fig. 4A). The analysis of MMP2
and MMP9 mRNA expression levels by RT-qPCR also revealed similar
results (Fig. 4B and C). To
explore the role of PKCζ in the TNFα-induced changes in the levels
of MMP9 in the HaCaT cells, we used PKCζ I or PKCζ siRNA to disrupt
the normal function of PKCζ. The results revealed that the
TNFα-induced upregulation of MMP9 at both the protein and mRNA
level tightly depended on the activity of PKCζ, as treatment with
PKCζ I or PKCζ siRNA decreased MMP9 expression (Fig. 4D–E).
Inhibition of the activity of PKCζ
impairs the TNFα-induced nuclear translocation of NF-κB-p65 that is
required for the upregulation of MCP-1, IL-8, ICAM-1 and MMP9
following stimulation with TNFα
From the above-mentioned results, we found that PKCζ
regulated the expression of MCP-1, IL-8, ICAM-1 and MMP9 beginning
from the transcriptional level. Moreover, several studies have
suggested that PKCζ is likely to be an upstream regulator of NF-κB,
which is an important transcription factor involved in the
regulation of many biological processes (20–23). Thus, we hypothesized that the
PKCζ-mediated inflammatory responses and MMP9 expression in HaCaT
cells may depend on the activation of NF-κB. The cells were
pre-treated with the NF-κB inhibitor, BAY11-7082, for 2 h or PKCζ I
for 3 h, and then stimulated with TNFα for 2 h in the presence of
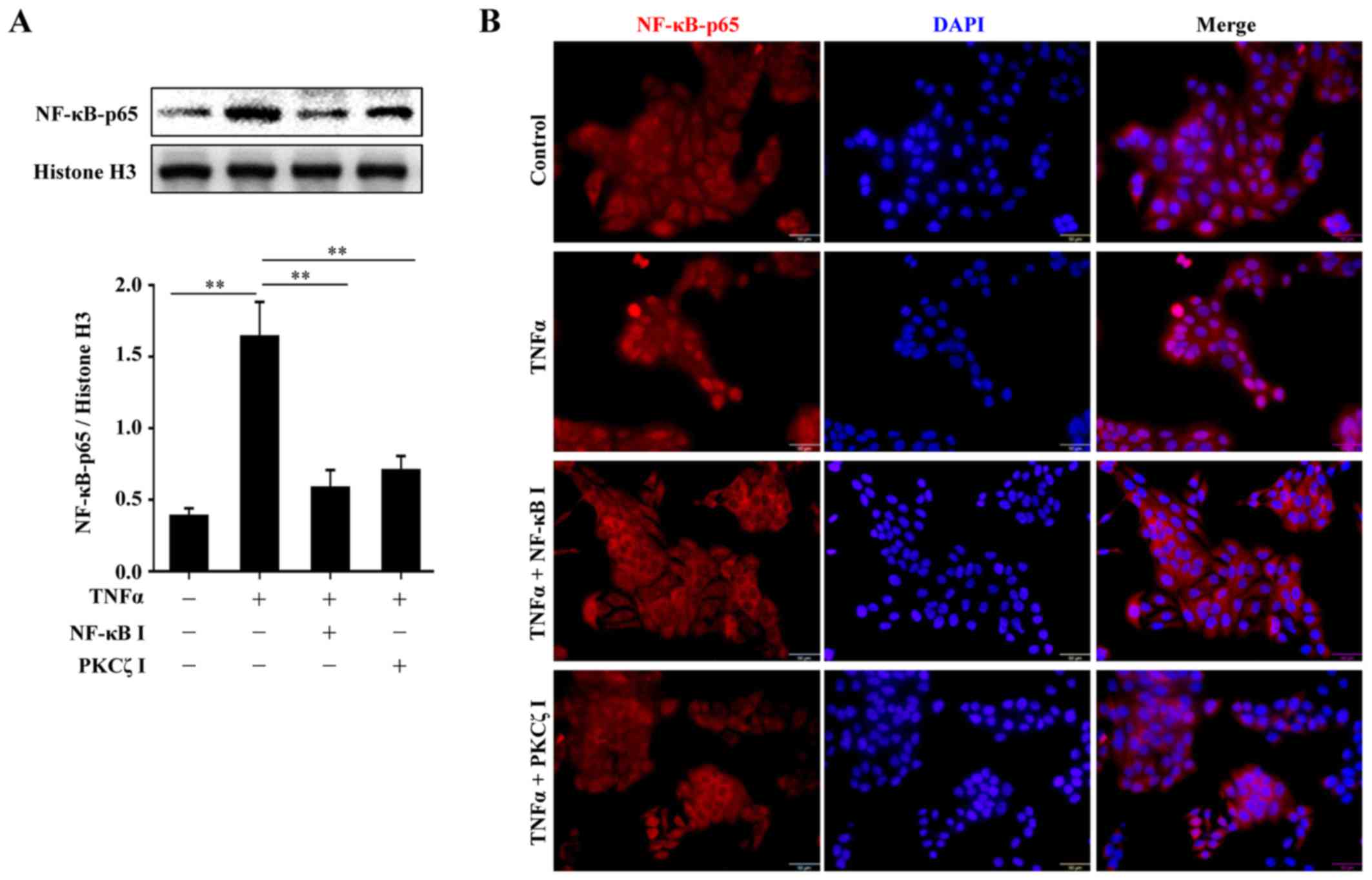
BAY11-7082 or PKCζ I. The nuclear transport of p65, a key subunit
of NF-κB, was assessed by western blot analysis or
immunofluorescence. The results revealed that following TNFα
stimulation, there was a distinct nuclear translocation of p65;
however, this nuclear import was prevented by PKCζ I, which was in
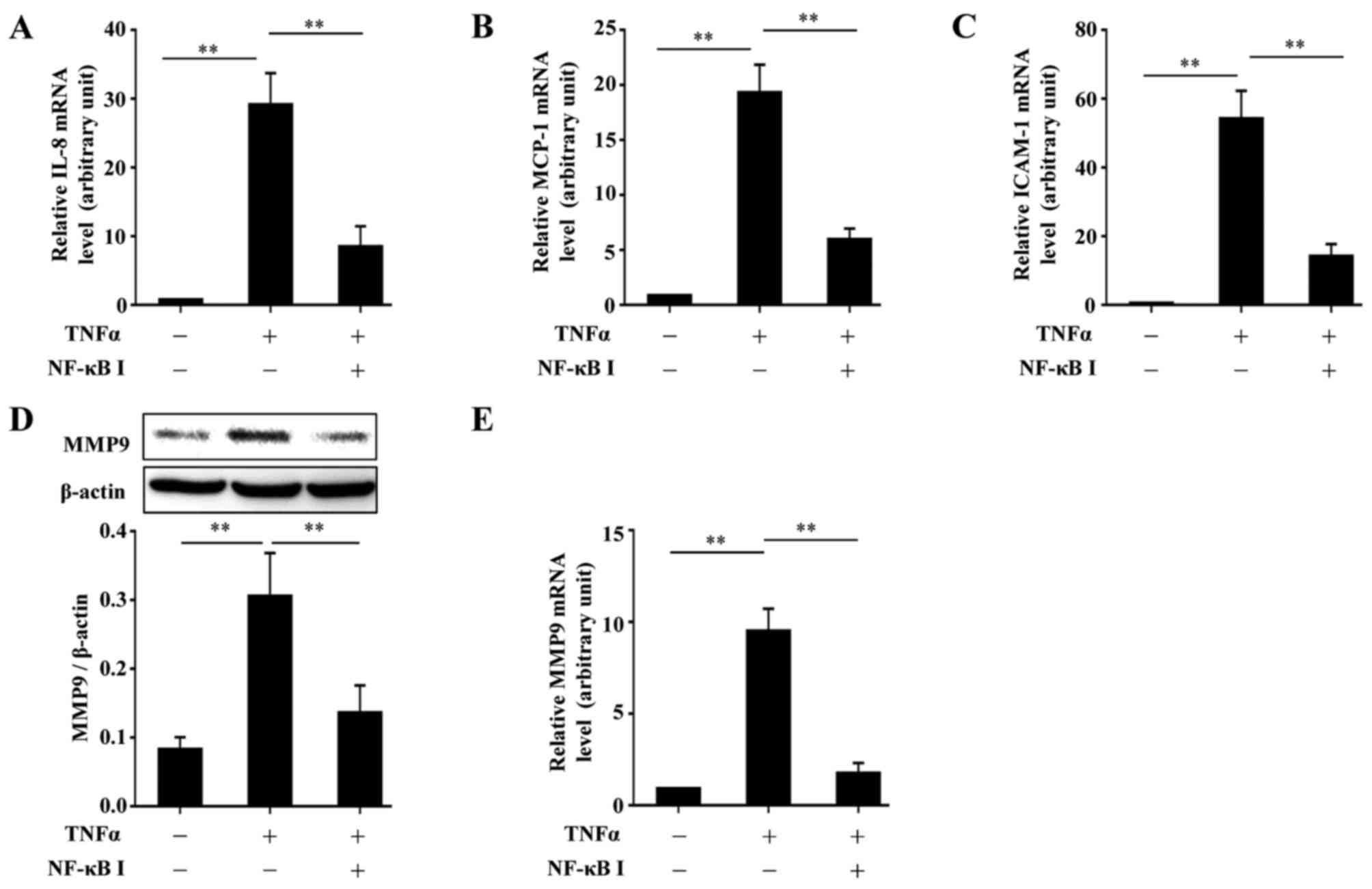
accordance with the function of BAY11-7082 (Fig. 5). Further experiments confirmed
the requirement of NF-κB in the TNFα-induced upregulation of MCP-1,
IL-8, ICAM-1 and MMP9 by inhibiting the function of NF-κB with
BAY11-7082. The use of BAY11-7082 decreased the levels of MCP-1,
IL-8, ICAM-1 and MMP9 induced by TNFα (Fig. 6).
PKCζ does not significantly affect
TNFα-weakened HaCaT keratinocyte migration and proliferation in
vitro
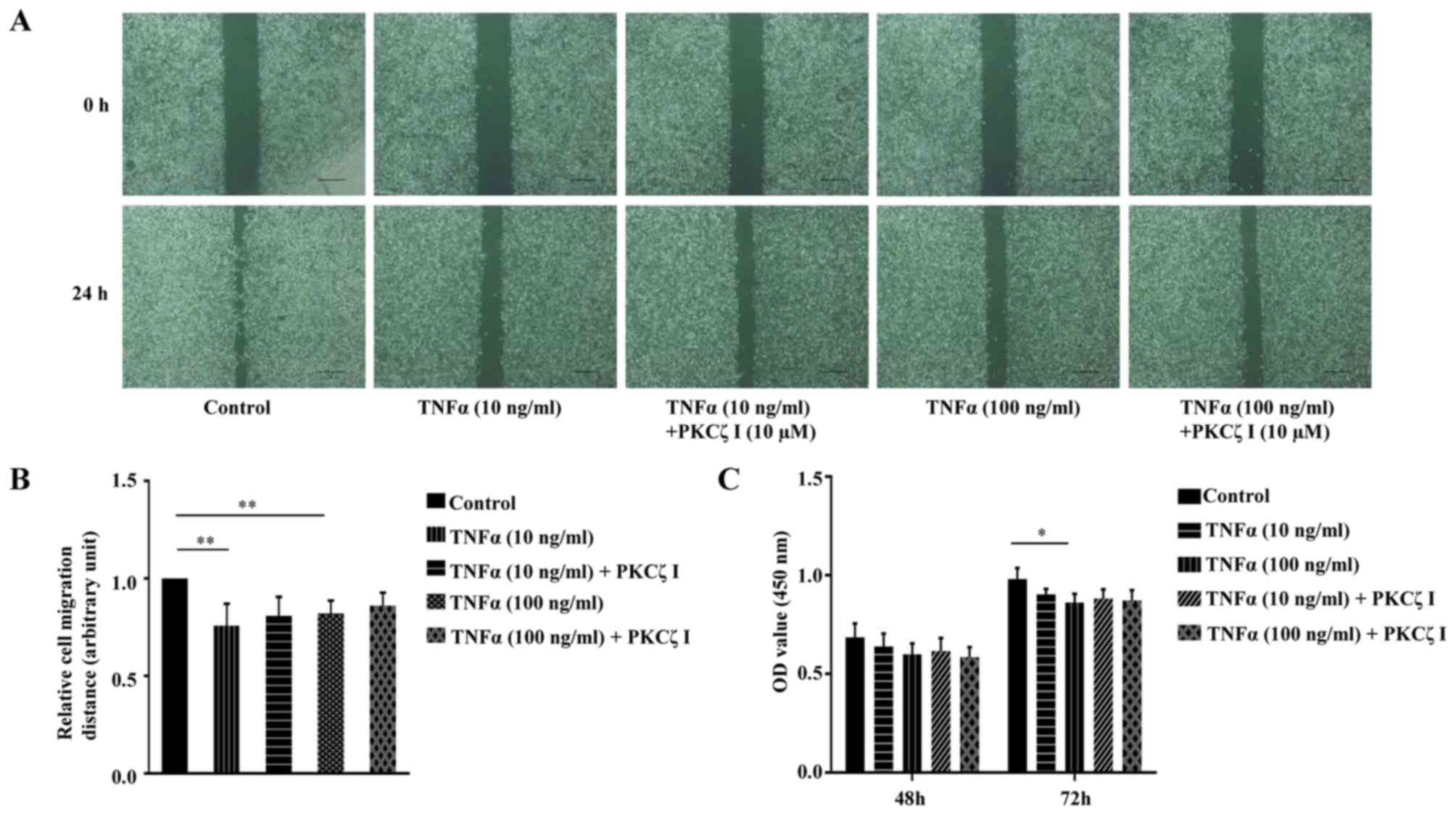
Keratinocyte migration and proliferation are
important to the rate of wound closure. We thus examined the effect
of TNFα and PKCζ I on HaCaT cell migration using a scratch wound
healing model. The cells were treated with 10 µg/ml
mitomycin C for 1 h to inhibit cell proliferation. A scratch was
made on fully confluent cells. Subsequently, following
pre-treatment with or without PKCζ I for 3 h, the cells were
stimulated with TNFα at pathological concentrations (10 or 100
ng/ml) for 24 h. The results revealed that exposure to TNFα
markedly decreased the rate of wound healing and the inhibition of
the function of PKCζ by PKCζ I did not significantly attenuate the
TNFα-induced decrease in keratinocyte migration (Fig. 7A and B). CCK-8 assay was then used
to examine the effect of TNFα and PKCζ on HaCaT cell proliferation.
The absorbance at 450 nm was directly proportional to the cell
proliferation capacity. The results revealed that pathological
concentrations of TNFα caused a small degree of inhibition on cell
proliferation at 48 h; however, this difference was not
statistically significant. Until 72 h, exposure to 100 ng/ml TNFα
inhibited cell proliferation by approximately 11.6%, which was
statistically significant (Fig.
7C). We also found that PKCζ I pre-treatment did not
signifincantly affect the TNFα-induced impairment of keratinocyte
proliferation (Fig. 7C).
PKCζ inhibition attenuates the
TNFα-induced wound closure delay and inflammatory disorders in
vivo
Previous studies have reported that from the 5th day
after normal acute wounding, the mRNA levels of 2 pro-inflammatory
cytokines, IL-1β and TNFα, and the chemokine, MCP-1, in wound
tissues are declined and cannot be detected on day 13
post-wounding; however, in the wounds of diabetic mice, the mRNA
levels of IL-1β, TNFα and MCP-1 are still strongly elevated and
sustained during the late phase (days 7–13 after wounding) of the
wound repair process (5,24). It has become a consensus that
sustained inflammation triggers the prolonged infiltration of
immune cells at the late stage of cutaneous wound repair and can
cause defects in wound healing. Furthermore, a number of studies
have confirmed that the local injection of potent pro-inflammatory
cytokines, IL-1α or TNFα or IFN-γ, in the skin of various animals,
such as mice, dogs, rats, rabbits and human volunteers can cause
significant inflammatory responses and the rapid infiltration of
immunocytes (25–30). In the present study, we locally
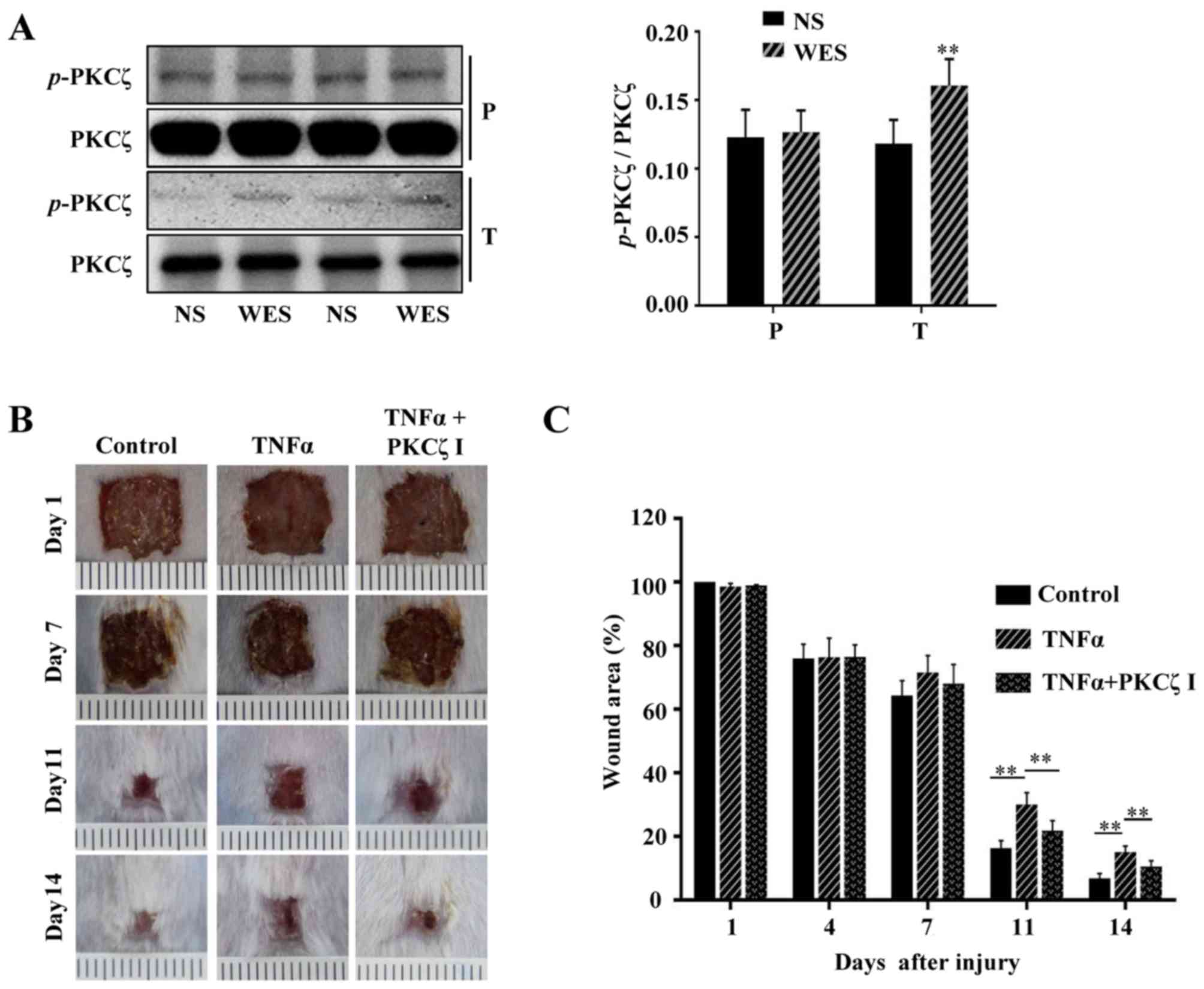
injected PBS or TNFα into the wound edges of BALB/c mice, and after
2 h we found that compared with the normal cutaneous tissue, the
phosphorylation of PKCζ was increased in the wound tissue treated
with TNFα (Fig. 8A). To explore
whether prolonged TNFα exposure can result in wound closure delays
in mice and whether PKCζ is involved in this process, 100 µl
PBS, 100 µl TNFα solution (50 ng, dissolved in PBS) or 100
µl TNFα solution + PKCζ inhibitor (20 µM) were
injected into the skin tissue around the wound each day on days
6–12 post-wounding. The results revealed that the TNFα injection at
the late phase of wound closure caused a delay in wound repair;
however, the administration of PKCζ inhibitor led to a favourable
improvement, attenuating this impairment (Fig. 8B and C). Our in vitro
experiments revealed that PKCζ played an indispensable role in
TNFα-induced inflammatory responses in keratinocytes. Total RNA was
extracted from the wound tissues of the PBS, TNFα and TNFα + PKCζ
I-treated mice at different intervals (days 7, 9 and 13 after
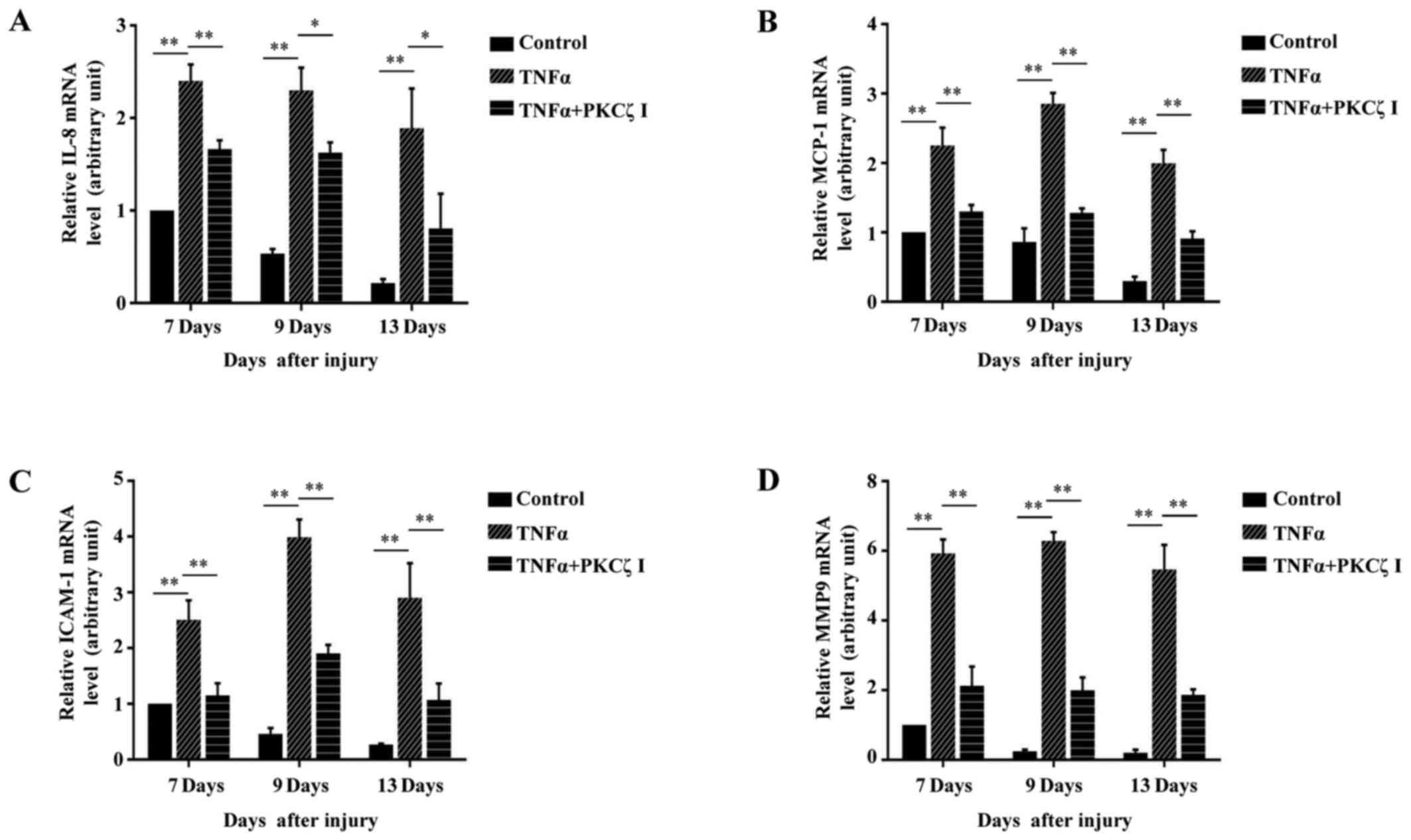
wounding) for RT-qPCR analysis. We observed a gradual derease in
the levels of IL-8, MCP-1, ICAM-1 and MMP9 to very low levels on
day 13 post-wounding in the PBS control mice. However, in the
TNFα-treated mice, until day 13 post-wounding, the expression
levels of these molecules remained at markedly high levels, which
may be one of the causes that resulted in defecs in wound closure.
However, as expected, PKCζ inhibition markedly inhibited the
TNFα-induced upregulation in the levels of IL-8, MCP-1, ICAM-1 and
MMP9 (Fig. 9).
Discussion
A severely enhanced and prolonged TNFα environment
in chronic wounds, such as diabetic foot ulcers can trigger
abnormally excessive inflammatory responses, which leads to severe
defects in tissue remodeling and re-epithelialization (1). This notoriously non-healing wound is
a threat to human health and the improvement of this injury is
challenging.
Previous studies have suggested that the roles of
PKCζ in different biological processes depend on cell types, milieu
and specific binding partners (31,32). Bacterial LPS can powerfully
activate the innate immune system of the body (33). It has been shown that PKCζ
activity is required for LPS-stimulated IL-1β gene expression in
RAW264.7 cells (22). This study
demonstrated that not only IL-1β, but also the expression of TNFα
was regulated by PKCζ activity in LPS-stimulated RAW264.7 cells
(Fig. 1).
TNFα is considered to be one of the main culprits in
non-healing wounds. Keratinocytes exposed to TNFα show inflammatory
related functions other than maintaining epithelization. In the
inflammatory epidermis, the chemokines, IL-8 and MCP-1, and the
intercellular adhesion molecule-1 (ICAM-1), play important roles in
mediating the interaction between immune cells and keratinocytes
(34–36). These infiltrating immune cells
synthesize more and more inflammatory cytokines and chemokines to
further lead to the amplification of inflammation, which
contributes to the pathogenesis of chronic wounds. Therefore, the
in-depth mechanisms of the effects of TNFα on keratinocytes are
worthy of exploration. Previous studies have indicated that PKCζ is
an important regulator of TNFα signaling via the NF-κB and MAPK
signaling pathways in retinal endothelial cells and human umbilical
vein endothelial cells (23,37,38); however, the function of PKCζ in
human keratinocytes has not been fully investigated. Moreover, a
previous study indicated that PKCζ expression was relatively
increased and that it was involved in the upregulation of CD1d in
the lesions of psoriasis, which is another chronic inflammatory
skin disorder characterized by abnormal TNFα stimulation (39). This study demonstrated that TNFα
enhanced the phosphorylation level (Thr410) of PKCζ in a
time-dependent manner in HaCaT keratinocytes (Fig. 2A), which led us to explore whether
PKCζ plays a role in TNFα-induced inflammatory responses.
PKCζ-specific pseudosubstrate peptide inhibitor (PKCζ I) or siRNA
was used to block the activaty of PKCζ (Fig. 2B) or to silence its expression
(Fig. 2C and D). The present
study demonstrated that the mRNA expression levels of IL-8, MCP-1
and ICAM-1 in the HaCaT keratinocytes were all elevated by TNFα
stimulation, and peaked respectively at 6, 12 and 6 h post-TNFα
treatment and gradually reduced with time (Fig. 3A–C). Pre-treatment with PKCζ I or
PKCζ-siRNA significantly prevented the TNFα-induced upregulation in
the levels of IL-8, MCP-1 and ICAM-1 (Fig. 3D–F). These results provide new
insight, indicating that PKCζ I is a positive regulator of the
pro-inflammatory effects of TNFα on keratinocytes.
Compared with MMP2, MMP9 is poorly expressed in
normal skin (40). In acute
wounds, MMP9 is rapidly increased as an early response to injury,
and then is rapidly decreased to the basic levels (29). The tight regulation and control of
MMP9 are important since the persistent and high expression of MMP9
initiated by excessive inflammation can lead to a sustained
deficiency in cell-cell and cell-matrix adhesion, which is the
subsequent cause of non-healing wounds (8,41,42). Moreover, active MMP9 has been
reported that to cleave and activate IL-8 (43,44), which may result in the further
dysregulation of inflammation. Our results revealed that the
exposure of HaCaT cells to TNFα incured a time-dependent
upregulation in the levels of MMP9; however, MMP2 was was not
affected (Fig. 4A–C). PKCζ has
been reported to be involved in the regulation of MMP9 in rabbit
smooth muscle cells (45).
Therefore, we speculated that PKCζ may be an important mediater in
the TNFα-induced expression of MMP9 in keratinocytes. As expected,
inhibiting the function of PKCζ using an inhibitor or siRNA
significantly suppressed the increased expression of MMP9
stimulated by TNFα at both the mRNA and protein level (Fig. 4D and E).
In previous studies PKCζ was reported to be involved
in NF-κB activation by affecting the Ser311 site phosphorylation or
nuclear translocation of p65, a key subunit of the NF-κB complex
(20–23). However, the association between
PKCζ and NF-κB has not been explored in human keratinocytes, at
least to the best of our knowledge. Our results of western blot
analysis of nucleoproteins demonstrated that in HaCaT cells, PKCζ
inhibitor exerted similar effects to the NF-κB inhibitor,
BAY11-7082, by partly preventing the TNFα-induced nuclear import of
p65 (Fig. 5A), and the results
from immunofluorescence assay also confirmed the above conclusion
(Fig. 5B). Further experiments
indicated that BAY11-7082 inhibited the TNFα-induced increase in
the expression of IL-8, MCP-1, ICAM-1 and MMP9 (Fig. 6A–D). Therefore, our results
indicated that in HaCaT keratinocytes, PKCζ regulates the
expression of IL-8, MCP-1, ICAM-1 and MMP9 by influencing NF-κB
activation.
Keratinocytes require the transition from a
quiescent phenotype to an actively migratory and proliferative
phenotype during wound healing. Accumulating evidence has indicated
that keratinocytes at the diabetic wound edges have an impaired
migration and proliferative activity (9,10).
Our scratch wound assay demonstrated that TNFα at pathological
concentrations significantly inhibited HaCaT cell migration
(Fig. 7A and B), which was
consistent with the results of a previous study by Zhang et
al (10). PKCζ inhibitor
faintly improved this inhibition of cell migration; however, its
effect was not significant (Fig. 7A
and B). TNFα has been reported to actively participate in the
promotion of apoptosis and inhibition of proliferation in many
other epithelial cell types (46,47). Our CCK-8 proliferation assay
revealed that exposure to 100 ng/ml TNFα for 72 h inhibited the
proliferation of HaCaT cells; however, this inhibitory effect was
not strong, and thus HaCaT cells were not very sensitive to the
TNFα-induced inhibition of proliferation as other epithelial cell
types (Fig. 7C). Of note, we
found that the presence of PKCζ I appeared to faintly increase this
sensitivity (Fig. 7C). This may
be due to the fact that TNFα/PKCζ/NF-κB can also induce the
expression of some anti-apoptotic proteins (48).
In the present study, PBS or TNFα was locally
administered to the wound edges of BALB/c mice, and we found that
after 2 h, TNFα treatment effectively upregulated the
phosphorylation level of PKCζ in wound tissue (Fig. 8A). Previous studies have shown
that in chronic wounds, the expression of TNFα does not return to
the basic level, but maintains a persistent high level at the late
stage (5,24). Local injections of inflammatory
cytokines in the skin can induce rapid inflammatory reaction
(25–30), thus mimicking the environment of
various inflammatory skin disease. To further examine whether the
prolonged presence of TNFα at the late phase of wound closure
influences the rate of wound healing, exogenous TNFα was injected
into the tissue around the wound on day 6–12 post-wounding. We
demonstrated that the administration of TNFα markedly inhibited
wound closure (Fig. 8B and C) and
stimulated the sustained high expression of IL-8, MCP-1, ICAM-1 and
MMP9 in wounds (Fig. 9). The
concomitant administration of PKCζ I significantly attenuated this
non-healing condition (Fig. 8B and
C) and resulted in the attenuation of the TNFα-induced
upregulation of the above-mentioned molecules (Fig. 9). These findings suggest that PKCζ
is involved in TNFα-triggered inflammatory disorders in unhealed
wounds. In the future, in order to elucidate the mechanisms through
which PKCζ regulates chronic wound healing, further investigations
are clearly warranted.
In conclusion, to the best of our knowledge, this
study, for the first time indicates that PKCζ effectively regulates
TNFα-induced inflammatory responses and MMP9 expression in
vitro and in vivo, which may provide a promising target
for researching the interventional therapy of chronic inflammatory
skin defects.
Abbreviations:
|
PKCζ
|
protein kinase Cζ
|
|
IL-1β
|
interleukin-1β
|
|
TNFα
|
tumor necrosis factor α
|
|
LPS
|
lipopolysaccharide
|
|
IL-8
|
interleukin-8
|
|
MCP-1
|
monocyte chemotactic protein-1
|
|
RANTES
|
regulated upon activation normal T
cell expressed and secreted factor
|
|
IP-10
|
interferon-γ-inducible protein-10
|
|
ICAM-1
|
intercellular cell adhesion
molecule-1
|
|
MMP2
|
matrix metalloproteinase 2
|
|
MMP9
|
matrix metalloproteinase 9
|
|
NF-κB
|
nuclear factor-κB
|
References
|
1
|
Martin P and Nunan R: Cellular and
molecular mechanisms of repair in acute and chronic wound healing.
Br J Dermatol. 173:370–378. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Streit M, Beleznay Z and Braathen LR:
Topical application of the tumour necrosis factor-alpha antibody
infliximab improves healing of chronic wounds. Int Wound J.
3:171–179. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Xu F, Zhang C and Graves DT: Abnormal cell
responses and role of TNF-α in impaired diabetic wound healing.
BioMed Res Int. 2013:7548022013. View Article : Google Scholar
|
|
4
|
Barker JN, Mitra RS, Griffiths CE, Dixit
VM and Nickoloff BJ: Keratinocytes as initiators of inflammation.
Lancet. 337:211–214. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Wetzler C, Kämpfer H, Stallmeyer B,
Pfeilschifter J and Frank S: Large and sustained induction of
chemokines during impaired wound healing in the genetically
diabetic mouse: Prolonged persistence of neutrophils and
macrophages during the late phase of repair. J Invest Dermatol.
115:245–253. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Lan CC, Wu CS, Huang SM, Wu IH and Chen
GS: High-glucose environment enhanced oxidative stress and
increased interleukin-8 secretion from keratinocytes: New insights
into impaired diabetic wound healing. Diabetes. 62:2530–2538. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Trengove NJ, Stacey MC, MacAuley S,
Bennett N, Gibson J, Burslem F, Murphy G and Schultz G: Analysis of
the acute and chronic wound environments: The role of proteases and
their inhibitors. Wound Repair Regen. 7:442–452. 1999. View Article : Google Scholar
|
|
8
|
Rayment EA, Upton Z and Shooter GK:
Increased matrix metal-loproteinase-9 (MMP-9) activity observed in
chronic wound fluid is related to the clinical severity of the
ulcer. Br J Dermatol. 158:951–961. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lan CC, Liu IH, Fang AH, Wen CH and Wu CS:
Hyperglycaemic conditions decrease cultured keratinocyte mobility:
Implications for impaired wound healing in patients with diabetes.
Br J Dermatol. 159:1103–1115. 2008.PubMed/NCBI
|
|
10
|
Zhang C, Ponugoti B, Tian C, Xu F,
Tarapore R, Batres A, Alsadun S, Lim J, Dong G and Graves DT: FOXO1
differentially regulates both normal and diabetic wound healing. J
Cell Biol. 209:289–303. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Galkowska H, Olszewsk WL, Wojewodzka U,
Mijal J and Filipiuk E: Expression of apoptosis- and cell
cycle-related proteins in epidermis of venous leg and diabetic foot
ulcers. Surgery. 134:213–220. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Spravchikov N, Sizyakov G, Gartsbein M,
Accili D, Tennenbaum T and Wertheimer E: Glucose effects on skin
keratinocytes: Implications for diabetes skin complications.
Diabetes. 50:1627–1635. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Rosse C, Linch M, Kermorgant S, Cameron
AJ, Boeckeler K and Parker PJ: PKC and the control of localized
signal dynamics. Nat Rev Mol Cell Biol. 11:103–112. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ohno S: Intercellular junctions and
cellular polarity: The PAR-aPKC complex, a conserved core cassette
playing fundamental roles in cell polarity. Curr Opin Cell Biol.
13:641–648. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Yin J, Liu Z, Li H, Sun J, Chang X, Liu J,
He S and Li B: Association of PKCζ expression with
clinicopathological characteristics of breast cancer. PLoS One.
9:e908112014. View Article : Google Scholar
|
|
16
|
Butler AM, Scotti Buzhardt ML, Li S, Smith
KE, Fields AP and Murray NR: Protein kinase C zeta regulates human
pancreatic cancer cell transformed growth and invasion through a
STAT3-dependent mechanism. PLoS One. 8:e720612013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Sun R, Gao P, Chen L, Ma D, Wang J,
Oppenheim JJ and Zhang N: Protein kinase C zeta is required for
epidermal growth factor-induced chemotaxis of human breast cancer
cells. Cancer Res. 65:1433–1441. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Huang S, Ouyang N, Lin L, Chen L, Wu W, Su
F, Yao Y and Yao H: HGF-induced PKCζ activation increases
functional CXCR4 expression in human breast cancer cells. PLoS One.
7:e291242012. View Article : Google Scholar
|
|
19
|
Chen R, Wang Y, Liu Y, Zhang Q, Zhang X,
Zhang F, Shieh CH, Yang D and Zhang N: Quantitative study of the
interactome of PKCζ involved in the EGF-induced tumor cell
chemotaxis. J Proteome Res. 12:1478–1486. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Yao H, Hwang JW, Moscat J, Diaz-Meco MT,
Leitges M, Kishore N, Li X and Rahman I: Protein kinase C zeta
mediates cigarette smoke/aldehyde- and lipopolysaccharide-induced
lung inflammation and histone modifications. J Biol Chem.
285:5405–5416. 2010. View Article : Google Scholar
|
|
21
|
Leverence JT, Medhora M, Konduri GG and
Sampath V: Lipopolysaccharide-induced cytokine expression in
alveolar epithelial cells: Role of PKCζ-mediated p47 phox
phosphorylation. Chem Biol Interact. 189:72–81. 2011. View Article : Google Scholar
|
|
22
|
Huang X, Chen LY, Doerner AM, Pan WW,
Smith L, Huang S, Papadimos TJ and Pan ZK: An atypical protein
kinase C (PKC zeta) plays a critical role in
lipopolysaccharide-activated NF-kappa B in human peripheral blood
monocytes and macrophages. J Immunol. 182:5810–5815. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Aveleira CA, Lin CM, Abcouwer SF, Ambrósio
AF and Antonetti DA: TNF-α signals through PKCζ/NF-κB to alter the
tight junction complex and increase retinal endothelial cell
permeability. Diabetes. 59:2872–2882. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Goren I, Müller E, Schiefelbein D,
Christen U, Pfeilschifter J, Mühl H and Frank S: Systemic
anti-TNFalpha treatment restores diabetes-impaired skin repair in
ob/ob mice by inactivation of macrophages. J Invest Dermatol.
127:2259–2267. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Nakayama T, Fujisawa R, Yamada H, Horikawa
T, Kawasaki H, Hieshima K, Izawa D, Fujiie S, Tezuka T and Yoshie
O: Inducible expression of a CC chemokine liver- and
activation-regulated chemokine (LARC)/macrophage inflammatory
protein (MIP)-3 alpha/CCL20 by epidermal keratinocytes and its role
in atopic dermatitis. Int Immunol. 13:95–103. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Sanz MJ, Hartnell A, Chisholm P, Williams
C, Davies D, Weg VB, Feldmann M, Bolanowski MA, Lobb RR and
Nourshargh S: Tumor necrosis factor alpha-induced eosinophil
accumulation in rat skin is dependent on alpha4 integrin/vascular
cell adhesion molecule-1 adhesion pathways. Blood. 90:4144–4152.
1997.PubMed/NCBI
|
|
27
|
Tremblay C, Paradis M and Doré M:
Expression of E- and P-selectin in tumor necrosis factor-induced
dermatitis in dogs. Vet Pathol. 38:261–268. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Speiser W, Kapiotis S, Kopp CW, Simonitsch
I, Jilma B, Jansen B, Exner M and Chott A: Effect of intradermal
tumor necrosis factor-alpha-induced inflammation on coagulation
factors in dermal vessel endothelium. An in vivo study of human
skin biopsies. Thromb Haemost. 85:362–367. 2001.PubMed/NCBI
|
|
29
|
Movat HZ, Burrowes CE, Cybulsky MI and
Dinarello CA: Acute inflammation and a Shwartzman-like reaction
induced by interleukin-1 and tumor necrosis factor. Synergistic
action of the cytokines in the induction of inflammation and
microvascular injury. Am J Pathol. 129:463–476. 1987.PubMed/NCBI
|
|
30
|
Sharpe RJ, Margolis RJ, Askari M, Amento
EP and Granstein RD: Induction of dermal and subcutaneous
inflammation by recombinant cachectin/tumor necrosis factor (TNF
alpha) in the mouse. J Invest Dermatol. 91:353–357. 1988.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Moscat J, Diaz-Meco MT and Wooten MW: Of
the atypical PKCs, Par-4 and p62: Recent understandings of the
biology and pathology of a PB1-dominated complex. Cell Death
Differ. 16:1426–1437. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Diaz-Meco MT and Moscat J: The atypical
PKCs in inflammation: NF-κB and beyond. Immunol Rev. 246:154–167.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Maldonado RF, Sá-Correia I and Valvano MA:
Lipopolysaccharide modification in Gram-negative bacteria during
chronic infection. FEMS Microbiol Rev. 40:480–493. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Harada Y, Edamatsu H and Kataoka T: PLCε
cooperates with the NF-κB pathway to augment TNFα-stimulated
CCL2/MCP1 expression in human keratinocyte. Biochem Biophys Res
Commun. 414:106–111. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Raingeaud J and Pierre J: Interleukin-4
downregulates TNFalpha-induced IL-8 production in keratinocytes.
FEBS Lett. 579:3953–3959. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Youn GS, Kwon DJ, Ju SM, Choi SY and Park
J: Curcumin ameliorates TNF-α-induced ICAM-1 expression and
subsequent THP-1 adhesiveness via the induction of heme oxygenase-1
in the HaCaT cells. BMB Rep. 46:410–415. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Nigro P, Abe J, Woo CH, Satoh K, McClain
C, O' Dell MR, Lee H, Lim JH, Li JD, Heo KS, et al: PKCzeta
decreases eNOS protein stability via inhibitory phosphorylation of
ERK5. Blood. 116:1971–1979. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Liang H, Baudouin C, Behar-Cohen F,
Crisanti P and Omri B: Protein kinase C-zeta mediates retinal
degeneration in response to TNF. J Neuroimmunol. 183:104–110. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Zhao Y, Fishelevich R, Petrali JP, Zheng
L, Anatolievna MA, Deng A, Eckert RL and Gaspari AA: Activation of
keratinocyte protein kinase C zeta in psoriasis plaques. J Invest
Dermatol. 128:2190–2197. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Vaalamo M, Weckroth M, Puolakkainen P,
Kere J, Saarinen P, Lauharanta J and Saarialho-Kere UK: Patterns of
matrix metal-loproteinase and TIMP-1 expression in chronic and
normally healing human cutaneous wounds. Br J Dermatol. 135:52–59.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Wong VW, Garg RK, Sorkin M, Rustad KC,
Akaishi S, Levi K, Nelson ER, Tran M, Rennert R, Liu W, et al: Loss
of keratinocyte focal adhesion kinase stimulates dermal proteolysis
through upregulation of MMP9 in wound healing. Ann Surg.
260:1138–1146. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Reiss MJ, Han YP, Garcia E, Goldberg M, Yu
H and Garner WL: Matrix metalloproteinase-9 delays wound healing in
a murine wound model. Surgery. 147:295–302. 2010. View Article : Google Scholar :
|
|
43
|
Van den Steen PE, Proost P, Wuyts A, Van
Damme J and Opdenakker G: Neutrophil gelatinase B potentiates
interleukin-8 tenfold by aminoterminal processing, whereas it
degrades CTAP-III, PF-4, and GRO-alpha and leaves RANTES and MCP-2
intact. Blood. 96:2673–2681. 2000.PubMed/NCBI
|
|
44
|
Opdenakker G, Van den Steen PE, Dubois B,
Nelissen I, Van Coillie E, Masure S, Proost P and Van Damme J:
Gelatinase B functions as regulator and effector in leukocyte
biology. J Leukoc Biol. 69:851–859. 2001.PubMed/NCBI
|
|
45
|
Hussain S, Assender JW, Bond M, Wong LF,
Murphy D and Newby AC: Activation of protein kinase Czeta is
essential for cytokine-induced metalloproteinase-1, -3, and -9
secretion from rabbit smooth muscle cells and inhibits
proliferation. J Biol Chem. 277:27345–27352. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Basso FG, Pansani TN, Turrioni AP, Soares
DG, de Souza Costa CA and Hebling J: Tumor necrosis factor-α and
interleukin (IL)-1β, IL-6, and IL-8 impair in vitro migration and
induce apoptosis of gingival fibroblasts and epithelial cells,
delaying wound healing. J Periodontol. 87:990–996. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Lei M, Bai X, Yang T, Lai X, Qiu W, Yang L
and Lian X: Gsdma3 is a new factor needed for TNF-α-mediated
apoptosis signal pathway in mouse skin keratinocytes. Histochem
Cell Biol. 138:385–396. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Sun J, Han J, Zhao Y, Zhu Q and Hu J:
Curcumin induces apoptosis in tumor necrosis factor-alpha-treated
HaCaT cells. Int Immunopharmacol. 13:170–174. 2012. View Article : Google Scholar : PubMed/NCBI
|