Introduction
Excessive alcohol consumption may lead to various
liver diseases, from simple steatosis to severe liver injury,
including fatty liver, steatohepatitis, liver fibrosis, cirrhosis
and even liver cancer (1).
Alcoholic liver disease (ALD) is a major medical health issue,
which not only places a heavy financial burden on individuals and
society, but also causes losses to the world economy. ALD is the
major cause of morbidity and mortality worldwide (2).
Although the pathogenesis of alcoholic liver disease
has remained to be fully elucidated, mounting evidence indicates
that oxidative stress has a key role in ALD. Ethanol-induced
oxidative stress contributes to the elevated production of reactive
oxygen species (ROS), increased lipid peroxidation and damage of
the antioxidant system, which leads to cell apoptosis and necrosis
(3). Hepatocyte cell death via
apoptosis and necrosis may be the critical process in the
exacerbation of ALD (4). Further
studies noted that oxidative stress, excessive intracellular ROS
production induced by ethanol and its metabolite have a critical
role in ethanol-induced apoptosis (5,6),
and put forward that apoptosis is mainly induced via the
Fas-mediated and mitochondria-mediated pathways (7,8).
Mounting evidence indicates that six homologues of
the transmembrane enzyme NADPH oxidase (NOX4) are highly expressed
in hepatocytes and hepatic stellate cells, and it has therefore
emerged as an important source of ROS in signal transduction,
having roles in physiological and pathological processes of ALD
(9). Based on this, it is a
reasonable assumption that ROS derived from NOX4 on the membrane
may be associated with Fas activation (10). It is well known that
mitogen-activated protein kinases (MAPKs) determine the fate of
various cells, and previous studies have demonstrated that p38 MAPK
and c-Jun N-terminal kinase (JNK) promote the mitochondrial
apoptotic pathway, leading to ethanol-induced death of SK-Hep1 or
Hepg2 cells, suggesting an interaction between apoptosis and MAPK
signalling systems (11,12).
L-02 cell is a new cell line established by the
Shanghai Biochemical Institute of the Chinese Academy of Sciences,
which originates from healthy human liver tissues. L-02 cells have
been reported to possess alcohol dehydrogenase (ADH) activity
(12). Therefore, the results may
be more exact than those from the cancerous hepatocytes such as
HepG2 or Hep3B. The L-02 cell line may be used as a model of
regular liver cells to a certain degree, and it would be reasonable
to assess ethanol-induced apoptosis in this established cell line.
In the present study, the participation of mitochondrial apoptotic
and MAPK pathways in the toxicity of ethanol to the L-02 cell line
was reported for the first time, to the best of our knowledge.
Materials and methods
Drugs and reagents
JNK inhibitor (SP600125) and p38 MAPK inhibitor
(SB202190) were obtained from Sigma-Aldrich (Merck KGaA, Darmstadt,
Germany). Fetal bovine serum (FBS), N-acetyl L-cysteine (NAC) and
apocynin were purchased from Sangon Biotech, Inc. (Shanghai,
China). Antibodies to B-cell lymphoma 2 (Bcl-2; ab59348),
Bcl-2-associated X protein (Bax; ab53154), JNK (ab124956),
phosphorylated (p)-JNK (ab207477), p38 MAPK (ab170099), p-p38 MAPK
(ab4822), NOX1 (ab55831), NOX4 (ab13303) and p22phox
(ab75941) were purchased from Abcam (Cambridge, UK). Caspase-3
antibody (9662S) was obtained from Cell Signaling Technology Inc.
(Danvers, MA, USA). β-actin antibody (C640018) was purchased from
Sangon Biotech, Inc. and oxidant-sensitive probe
2,7-dichlorodihydro fluorescein diacetate (DCFH-DA) was obtained
from Cayman Chemical Co. (Ann Arbor, MI, USA). A single-stranded
(ss) DNA Apoptosis ELISA kit was obtained from EMD Millipore
(GIR7935; Billerica, MA, USA).
Cell culture
The L-02 hepatocyte line (human normal liver cells;
no GDC079) was purchased from the China Center for Type Culture
Collection of Wuhan University (Wuhan, China). L-02 hepatocytes
were incubated with Dulbecco's modified Eagle's medium (DMEM)
containing 10% (v/v) FBS, 2 mM L-glutamine and antibiotics (100
U/ml penicillin and 100 U/ml streptomycin) maintained in a
humidified atmosphere of 5% CO2 and 95% air at 37°C.
After 24 h, the medium was replaced with fresh medium. For
subculturing purposes, cells were detached by 0.05% trypsin-EDTA
treatment.
Viability assay
L-02 cells were cultured in 96-well plates
(1.5×104 cells/well). At ~80% confluence, L-02 cells
were treated with or without (used as a control) various
concentrations of ethanol (100, 200, 300, 400, 500, 600 and 700 mM)
for 0, 3, 6, 12, 24 or 48 h. Cell viability was monitored by an MTT
assay as previously described (13).
In order to assess the effects of ethanol, L-02
cells were cultured in DMEM with 10% FBS with or without (used as a
control) ethanol for 0–48 h. Preliminary experiments had been
performed, in which caspase-3 activity and cell viability were
measured at a wide range concentrations of ethanol (100, 200, 300,
400, 500, 600 and 700 mM). These experiments indicated an obvious
enhancement of caspase-3 activity at concentrations of no less than
400 mM ethanol (data not shown). Hence, 400 mM ethanol was used as
a suitable concentration for triggering apoptosis in L-02 cells
(11). To explore the effect of
the antioxidants (NAC and apocynin) and the effect of MAPK
signalling pathway inhibitors (SP600125 and SB202190) on ROS
generation, L-02 cells were seeded in 96-well plates
(1.5×104 cells/well) and pre-treated with NAC (10 mM),
apocynin (300 µM), SP600125 (1 µM) or SB202190 (1
µM) for 1 h, followed by the addition of 400 mM ethanol and
incubation for a further 5 h. Cells individually treated with the
above agents were used as control groups.
Detection of ROS
With investigation of the role of ethanol treatment
on hydrogen peroxide generation, the oxidant-sensitive DCFH-DA,
which is a molecular probe for detection of ROS, was used. The
generation of ROS was measured by testing the fluorescence
intensity of DCF, the reduction product of DCFH-DA, as described in
a previous study (11). L-02
cells were cultured in petri dishes of 3 cm in diameter
(1×105 cells/plate) overnight and treated with or
without (as a control) transforming growth factor (TGF)-β1 (10
µg/l; Sigma Aldrich; Merck KGaA) for 3, 4, 5, 6, 7 or 8 h at
37°C, followed by incubation with 10 µM 2,7-DCFH-DA at 37°C
and away from light for 30 min. Fluorescence images were captured
under a fluorescence microscope. To assess the effect of the
antioxidants (NAC and apocynin) and the effect of MAPK signalling
pathway inhibitors (SP600125 and SB202190, respectively) on ROS
generation, L-02 cells were treated as in the cell viability assay
above. Subsequently, the fluorescence intensity was determined
using a Tecan Infinite M200 PRO Microplate reader (Tecan Group
Ltd., Männedorf, Switzerland) at an excitation wavelength of 502 nm
and an emission wavelength of 523 nm.
Detection of apoptosis
Apoptosis analysis by flow cytometry
(FCM)
To further verify that ethanol induced cell
apoptosis, an Annexin V-fluorescein isothiocyanate (FITC) apoptosis
detection kit (APT225; Sigma Aldrich; Merck KGaA) was applied with
FCM analysis. L-02 cells were cultured in petri dishes of 6 cm in
diameter (3×105 cells/plate) overnight and treated with
ethanol (400 mM) for 0, 3, 6, 9, 12 or 24 h at 37°C. The cells were
then harvested by treatment with 0.05% trypsin without EDTA and
washed with cold PBS three times. Subsequently, the cells were
suspended with 400 µl binding buffer and stained with 5
µl Annexin V-FITC for 20 min away from light, followed by
addition of 10 µl propidium iodide on the ice for 5 min and
analysis by FCM (BD Biosciences, San Jose, CA, USA).
Apoptosis analysis by ssDNA assay
ssDNA, which may be regarded as specific evidence of
the apoptotic process, was measured using a formamide-monoclonal
antibody against ssDNA using the ssDNA Apoptosis ELISA kit as
described in a previous study (11). The assay is based on the principle
that formamide denatures DNA selectively in apoptotic cells. L-02
cells were cultured in 400 mM ethanol for 5 h. The effect of NAC or
MAPK inhibitors was detected by pre-treatment with NAC, apocynin or
MAPK inhibitors followed by co-culture of cells with 400 mM ethanol
as described above in cell viability assay.
Western blot analysis
L-02 cells were cultured in petri dishes of 10 cm in
diameter (2×106 cells/plate) overnight and treated with
ethanol (400 mM) for 0, 3, 6, 9, 12 or 24 h. Cells were lysed in
radioimmunoprecipitation lysis buffer (Beyotime Institute of
Biotechnology, Shanghai, China) with protease and phosphatase
inhibitor cocktail tablets (Sangon Biotech, Inc.). The lysates were
centrifuged at 12,000 × g for 15 min at 4°C. The supernatant was
harvested and the protein concentration was measured using a
bicinchoninic acid protein assay kit (Beyotime Institute of
Biotechnology). Equivalent amounts (50 µg) of protein were
separated by 12% SDS-PAGE, transferred to a nitrocellulose membrane
and blocked for 1 h with 5% bovine serum albumin (BSA) (9048-46-8;
Sangon Biotech, Inc.) in Tris-buffered saline containing 0.05%
Tween 20 (TBST). For immune detection, the membrane was incubated
overnight at 4°C with specific rabbit antibodies to Bcl, Bax,
caspase-3, NOX1, NOX4, p22phox, JNK, p-JNK, p38 MAPK or
p-p38 (1:1,000 dilution in TBST containing 3% BSA), and then
incubated for 1 h at room temperature with horseradish
peroxidase-conjugated goat anti-rabbit antibody (D110058-0025, Cell
Signaling Technology, Inc.; 1:5,000 dilution in TBST containing 3%
BSA). Protein bands were visualized by enhanced chemiluminescence
(E002-100; Sangon Biotech, Inc.) and quantified by densitometry
using the ChemiDoc XRS imaging system (Bio-Rad Laboratories, Inc.,
Hercules, CA, USA). β-actin (1:1,000) was used as the internal
control.
Reverse transcription-polymerase chain
reaction (RT-PCR) analysis
The mRNA expression levels of NOX1, NOX4 and
p22phox were determined by RT-PCR. Total RNA was
reverse-transcribed into complementary (c)DNA using reverse
transcriptase (TIANScript cDNA; Tiangen, Beijing, China) according
to the manufacturer's instructions. Using the MJ PTC-200 PCR System
(MJ Research; Bio-Rad Laboratories, Inc.), the cDNA was amplified
with an RT-PCR kit (2X Taq PCR Master Mix; PC0902; Aidlab Co.,
Beijing China) according to the manufacturer's protocol. PCR was
performed using specific primers (Table I) provided by Sangon Biotech Inc.
The PCR products were identified using 1.5% agarose gel
electrophoresis, and the optical density of the target gene bands
in each sample was calculated using the ChemiDoc imaging system
with adjustment through β-actin correction to finally obtain the
relative expression of the target genes in each sample.
 | Table IPrimer sequences used for
determination of NOX1, NOX4, p22phox and β-actin by
polymerase chain reaction. |
Table I
Primer sequences used for
determination of NOX1, NOX4, p22phox and β-actin by
polymerase chain reaction.
| Gene | Primer
sequence | Annealing
temperature (°C) | No. cycles | Product length
(bp) |
|---|
| NOX1 | | 56 | 35 | 106 |
| F |
5′-TTGTTTGGTTAGGGCTGAATGT-3 | | | |
| R |
5′-GCCAATGTTGACCCAAGGATTTT-3 | | | |
| NOX4 | | 58 | 35 | 136 |
| F |
5′-TGTGCCGAACACTCTTGGC-3′ | | | |
| R |
5′-ATATGCACGCCTGAGAAAATA-3′ | | | |
|
p22phox | | 58 | 35 | 103 |
| F |
5′-TATTGTTGCAGGAGTGCTCA-3′ | | | |
| R |
5′-CACAGCGGTCAGGTACTTCT-3′ | | | |
| Bax | | 53 | 35 | 155 |
| F |
5′-CCCGAGAGGTCTTTTTCCGAG-3′ | | | |
| R |
5′-CCAGCCCATGATGGTTCTGAT-3′ | | | |
| Bcl-2 | | 53 | 35 | 89 |
| F |
5′-GGTGGGGTCATGTGTGTGG-3′ | | | |
| R |
5′-CGGTTCAGGTACTCAGTCATCC-3′ | | | |
| β-actin | | 56 | 35 | 199 |
| F |
5′-GGACTCCTATGTGGGTGACGA-3′ | | | |
| R |
5′-ACGGTTGGCCTTAGGGTTCA-3′ | | | |
Transfection of small interfering
(si)RNA
siRNA targeting NOX4 (NOX4-SiRNA) and the NC-siRNA
expressing plasmid were designed and synthesized by GenePharma
(Shanghai, China). L-02 cells were transfected with NOX4-siRNA or
the NC-siRNA expressing plasmid using Lipofectamine 2000
(Invitrogen; Thermo Fisher Scientific, Inc., Waltham, MA, USA)
according to the manufacturer's instructions. In brief, L-02 cells
in the logarithmic growth phase were inoculated into 6-well plates
at a density of 2×105 cells/well in 2 ml antibiotic-free
DMEM without serum. Gene transfection was performed when the cells
had grown to 70–80% confluence, and 100 pmol of siRNA and 2
µl of Lipofectamine were added to each well. Subsequently,
serum- and antibiotic-free DMEM (500 µl) was supplemented.
The cells were incubated with the transfection mixture for 6 h. In
the final phase of the incubation period, 1.5 ml of complete
culture medium without antibiotics was added and the cells were
cultured for an another 18 h. Following ethanol treatment, the
cells were collected and the expression of NOX4 was detected by
RT-quantitative PCR and immunoblotting.
NOX4 overexpression
L-02 cells were electroporated with the P5 primary
cell Nucleofector™ kit (Lonza, Basel, Switzerland). For each
transfection, NOX4-cDNA (3 µg) was dissolved in 82 µl
nucleofector solution, and 18 µl P5-supplemented primary
cell solution was then added. A total of
107–108 cells were suspended in 100 µl
cDNA plus P5 primary cell solution and immediately electroporated
in a 4D-Nucleofector X kit L cuvette (Lonza). Electroporated cells
were transferred to 6-well plates containing 2.5 ml warm complete
medium and cultured in a humidified atmosphere of 5% CO2
at 37°C. The medium was replaced with fresh medium after 24 h.
Statistical analysis
Values are expressed as the mean ± standard
deviation of three independent experiments. SPSS19.0 software (IBM
Corp., Armonk, NY, USA) was used for statistical analysis. Tukey's
test was performed following one-way analysis of variance to
evaluate significant differences between groups. P<0.05 was
considered to indicate a statistically significant difference
between groups.
Results
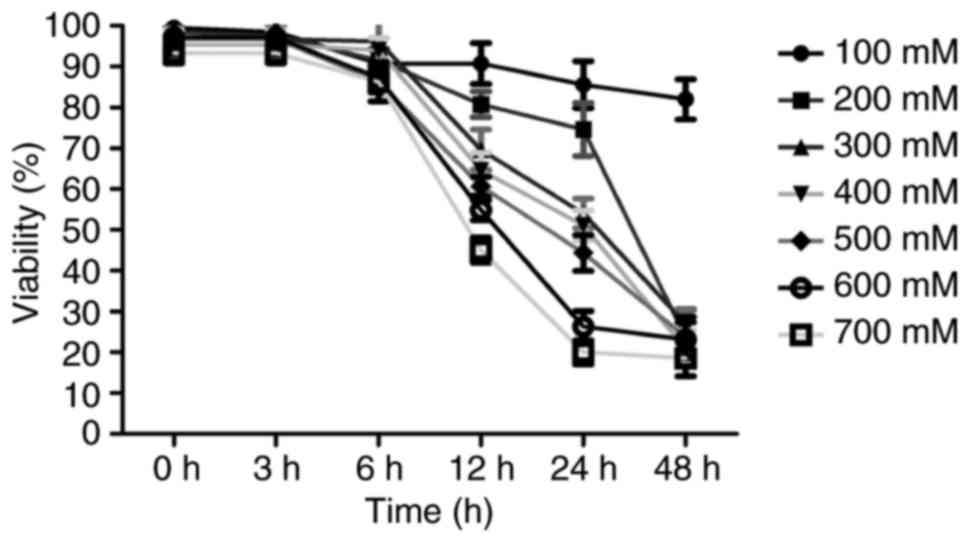
Effects of ethanol the viability of L-02
cells
To assess the effect of ethanol on the viability of
L-02 cells, an MTT assay was performed on L-02 cells in the absence
or presence of ethanol. As presented in Fig. 1, ethanol treatment reduced the
viability of L-02 cells in a dose- and time-dependent manner.
As presented in Fig.
1, addition of 400 mM ethanol led to a significant inhibition
of cell viability compared with that in the control group after 12
h. The inhibition rate was <10% at 6 h but >50% at 24 h.
These experiments indicated an obvious enhancement of caspase-3
activity at concentrations of no less than 400 mM ethanol (data not
shown). Therefore, 400 mM ethanol was indicated to be a suitable
concentration for the subsequent experiments in L-02 cells. The
ethanol concentration of 400 mM for cell treatment was verified
using phase-contrast microscopy, which indicated that no cell
necrosis occurred when L-02 cells were cultured with 400 mM ethanol
for 6 h (data not shown). However, if the L-02 cells were incubated
with 400 mM ethanol for >6 h, cell damage occurred. Therefore,
incubation for 6 h was selected to collect various experimental
data on cell apoptosis in the present study.
Effect of ethanol on apoptosis of L-02
cells
Previous studies have demonstrated that ethanol
induces apoptosis in HepG2 cells (1), SK-Hep1 cells (14) and the Chang human hepatocyte cell
line (15). In order to clarify
whether the decrease in L-02 cell viability observed in the present
study is due to ethanol-induced apoptosis, a flow cytometric assay
was used to measure cell apoptosis. The results indicated that
apoptosis induced by ethanol in L-02 cells was time-dependent
(Fig. 2A and B). It was therefore
suggested that induction of apoptosis was, at least in part, the
reason for inhibitory effect of ethanol on the viability of L-02
cells in present experiment (Fig.
1). Ethanol exposure at 400 mM for 24 h led to a marked
increase of apoptosis in treated cells as measured by flow
cytometry (Fig. 2A). The results
indicated in the presence of 400 mM ethanol for 0–9 h, L-02 cells
did not undergo apoptosis, but the apoptotic ratio of L-02 cells
was ~40% at 12 h, which was further increased to ~50% at 24 h. The
apoptotic cells were mainly in the late phase of apoptosis
(Fig. 2C). The apoptotic
population in ethanol-treated L-02 cells was increased in a
time-dependent manner.
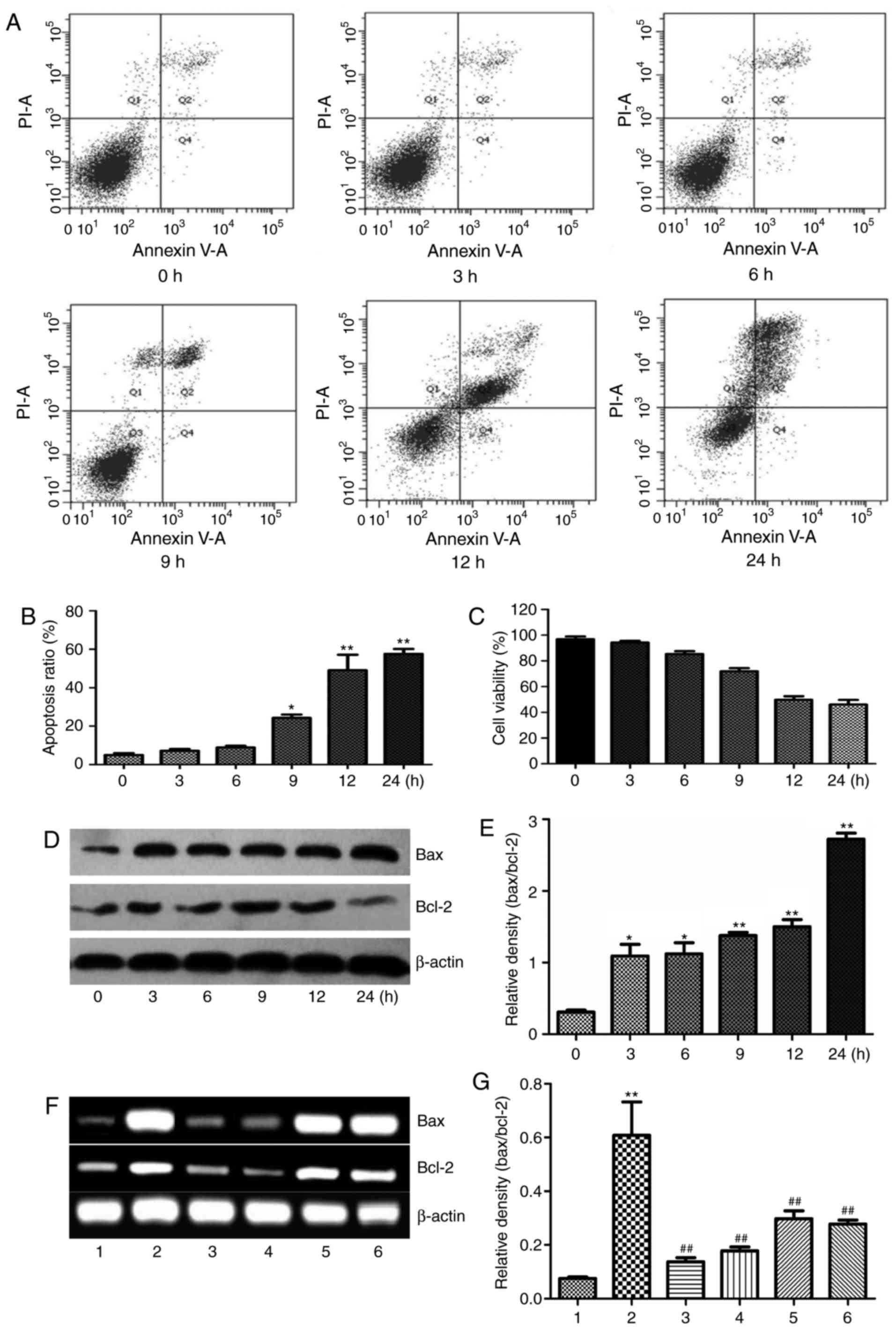 | Figure 2Effects of 400 mM ethanol on
apoptosis in L-02 cells. After treatment with 400 mM ethanol for
0–24 h, (A and B) cell apoptosis and (C) viability were measured by
a flow cytometric assay. (D) The protein expression of Bax and
Bcl-2 was measured by western blot analysis and (E) the Bax/Bcl-2
ratio was determined. L-02 cells were treated with 400 mM ethanol
with or without NAC (10 mM), Apocynin (300 µM), SB202190 (1
µM) or SP600125 (1 µM) for 6 h, and (F) the gene
expression by of Bax and Bcl-2 and (G) the Bax/Bcl-2 ratio were
determined; in addition, the protein levels of (H)
cleaved-caspase-8 and cleaved-caspase-3 were measured by western
blot analysis and (I) densitometrically quantified. (J) Apoptosis
of L-02 cells was determined using an ssDNA assay. Values are
expressed as the mean ± standard error of the mean (n=3).
*P<0.05, **P<0.01 vs. the control
group/0 h group; ##P<0.01 vs. the ethanol group.
Lanes: 1, Control; 2, ethanol; 3, ethanol + NAC (10 mM); 4, ethanol
+ apocynin (300 µM); 5, ethanol + SB202190 (1 µM); 6,
ethanol + SP600125 (1 µM). NAC, N-acetyl L-cysteine; ssDNA,
single-stranded DNA; Bcl-2, B-cell lymphoma 2; Bax,
Bcl-2-associated X protein. |
In order to verify the occurrence of apoptosis, the
effect of ethanol on apoptosis-associated proteins, Bax and Bcl-2,
were also detected. Bax and Bcl-2 are involved in the maintenance
of mitochondrial membrane stability (16). Western blot analysis demonstrated
that the expression levels of Bax and Bcl-2 in L-02 cells were
increased at 24 h after treatment with 400 mM ethanol, while the
expression of Bcl-2 was lower than that of Bax at various
time-points, which led to an increased Bax/Bcl-2 ratio with the
relative density ratio reaching 2.37 at 24 h, indicating that
apoptosis has an important role in ethanol-induced L-02 cell death
(Fig. 2D and E).
JNK and p38 MAPK were demonstrated to be involved in
ethanol-induced apoptosis, and may stimulate the activities of
pro-apoptotic proteins, such as Bax, and promote apoptosis by
inhibiting anti-apoptotic proteins, such as Bcl-2, to regulate the
release of cytochrome c and cause apoptosis in
ethanol-induced L-02 cells. The protein expression of the Bcl-2
family proteins Bax and Bcl-2, which control the release of
cytochrome c from mitochondria, were measured in L-02
treated with or without 400 mM ethanol and optional pre-treatment
with 10 mM NAC or 300 µM apocynin or 1 µM inhibitor
of p38 JNK or MAPK. The results indicated that the mRNA expression
levels of Bax were obviously increased in ethanol-treated group
(P<0.01) compared with those in the control group, and this
increase was significantly blocked when cells were pre-treated with
NAC, apocynin or inhibitors (SB202190, SP600125) of JNK and p38
MAPK. However, compared with those in the control group, after
treatment with ethanol, the mRNA levels of Bcl-2 were significantly
decreased (P<0.01), while this reduction was distinctly
inhibited by pre-treatment with NAC, apocynin or inhibitors of JNK
and p38 MAPK. These results indicated that JNK and p38 MAPK promote
apoptosis in ethanol-treated L-02 cells by regulating the
expression of Bcl-2 and Bax (Fig. 2F
and G).
The activities of caspase-8 and caspase-3 in L-02
cells treated with or without 400 mM ethanol with optional
pretreatment with 10 mM NAC or 300 µM apocynin are presented
in Fig. 2H and I. After culture
with 400 mM ethanol for 6 h, cleaved-caspase-3 and -caspase-8 in
L-02 cells were markedly increased compared with those in the
control group (P<0.01). The increases of caspase-3 and -8
activities triggered by ethanol were suppressed when L-02 cells
were pretreated with 10 mM NAC or 300 µM apopcynin as well
as MAPK inhibitors. These results indicated that the increase of
caspase-3 and -8 is triggered by oxidative stress such as ethanol
treatment in L-02 cells, as it was blocked by pre-treatment with
the antioxidant NAC or apocynin and MAPK inhibitors. The presence
of ssDNA was used to distinguish between apoptosis and necrosis in
ethanol-treated L-02 cells. The results of this assay presented in
Fig. 2J demonstrated that
compared with the control, the levels of ssDNA were markedly raised
following incubation with 400 mM ethanol (P<0.01), but this
increase was significantly repressed when the cells were
pre-treated with NAC and apocynin, as well as inhibitor of JNK or
p38 MAPK (P<0.01), indicating that JNK and p38 MAPK have at
least a partial role in the apoptotic process that was triggered by
ethanol-induced oxidative stress.
In brief, ethanol significantly reduced L-02-cell
viability in a dose- and time-dependent manner. It induced
L-02-cell apoptosis in a time-dependent manner, and this apoptosis
was significantly inhibited by NAC and apocynin, as well as the
MAPK inhibitors. Furthermore, ethanol treatment induced the
expression of Bax and the cleavage of caspase-8 and caspase-3,
while reducing the expression of Bcl-2, which was inhibited by NAC
and apocynin, as well as the MAPK inhibitors.
Effect of ethanol on ROS generation in
L-02 cells
Previous studies have indicated that ethanol
triggers ROS generation in various types of liver disease,
including cancer (17,18). To investigate whether ethanol
induces ROS generation in L-02 cells, the ROS expression levels in
ethanol-treated L-02 cells were first evaluated by testing the
oxidative conversion from non-fluorescent DCFH-DA to fluorescent
DCF. The results indicated that ethanol triggered ROS production in
L-02 cells in a time-dependent manner (Fig. 3A and B). Ethanol treatment at 400
mM for 3, 4, 5, 6 and 7 h elevated the ROS levels by 0.9, 2.1, 3.2,
3.5, 3.0-fold, respectively, of the untreated control. An increase
in ROS was also observed in SK-Hep1 cells subjected to the same
ethanol treatment (data not shown). These results indicated that
ethanol triggered ROS production in L-02 cells.
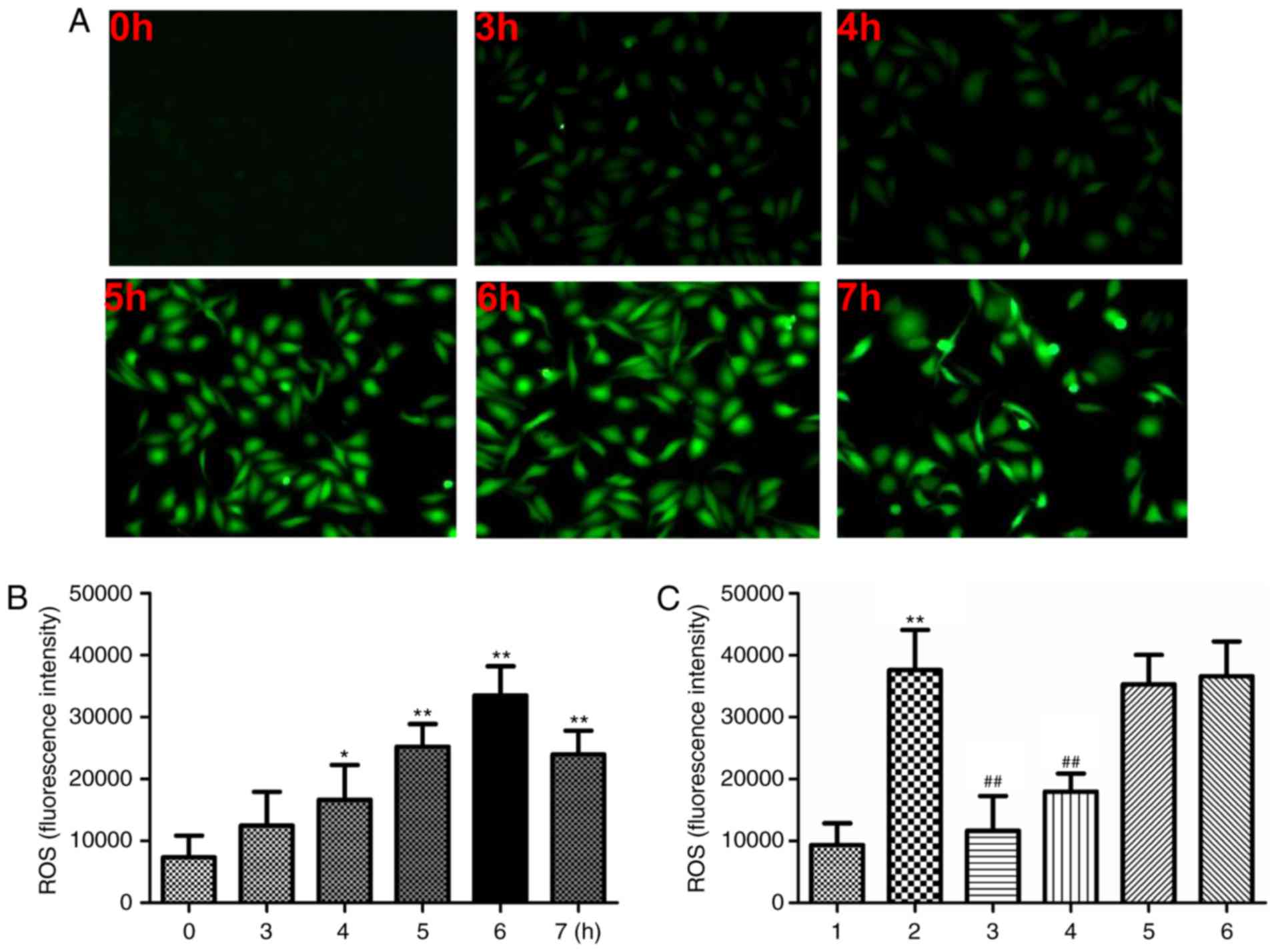 | Figure 3Effect of 400 mM ethanol on ROS
generation. (A and B) The generation of ROS in L-02 cells
(1×105 cells/plate) after treatment with 400 mM ethanol
for 0-7 h at 37°C. Magnification, ×100. (C) Generation of ROS in
L-02 cells after treatment with 400 mM ethanol with or without NAC,
Apocynin, SB202190 or SP600125 for 6 h at 37°C. Values are
expressed as the mean ± standard error of the mean (n=3).
*P<0.05, **P<0.01 vs. the control
group/0 h group; ##P<0.01 vs. the ethanol group. Groups: 1,
Control; 2, ethanol; 3, ethanol + NAC (10 mM); 4, ethanol +
apocynin (300 µM); 5, ethanol + SB202190 (1 µM); 6,
ethanol + SP600125 (1 µM). NAC, N-acetyl L-cysteine; ROS,
reactive oxygen species. |
As demonstrated in Fig. 3C, treatment with ethanol (400 mM)
for 24 h led to a marked enhancement of ROS generation in L-02
cells compared with that in the control group (P<0.01).
Pre-treatment with 10 mM NAC or 300 µM apocynin
significantly suppressed the 400 mM ethanol-induced elevation of
ROS production in L-02 cells compared with that in the group
treated with ethanol only, as indicated in Fig. 3C (P<0.01). The ethanol-induced
ROS production was also reduced by pre-treatment with the MAPK
inhibitors SB202190 and SP600125; however, no statistical
significance was reached.
In brief, ethanol induced intracellular ROS
generation compared with that in the control group, which was
dramatically suppressed by pre-treatment with antioxidant NAC or
apocynin.
Effect of ethanol on NOX in L-02
hepatocytes
NOX1 and NOX4 are highly expressed in the liver and
hepatocytes, and previous studies have indicated that NOX is a
major producer of ROS (18,19). In the present study, the
expression of NOX subunits was determined after ethanol treatment.
A time-dependent kinetic study following ethanol stimulation was
also performed. The L-02 cells were incubated with ethanol in DMEM
containing 10% FBS for 0, 3, 6, 9, 12 or 24 h. The protein
expression of NOX1, NOX4 and p22phox was examined at the
indicated time-points. The results suggested that ethanol induced
an increase in the expression of NOX1 from 9 h onwards, with a
significant increase from 12 h, increase in the expression of NOX4
and p22phox in L-02 cells after 3 h and it increased in
a time-dependent manner until 24 h (Fig. 4A and B). This demonstrated that
ethanol exposure for 3 h is sufficient for inducing an increase of
NOX1, NOX4 and p22phox expression in L-02 cells and persists for at
least 24 h.
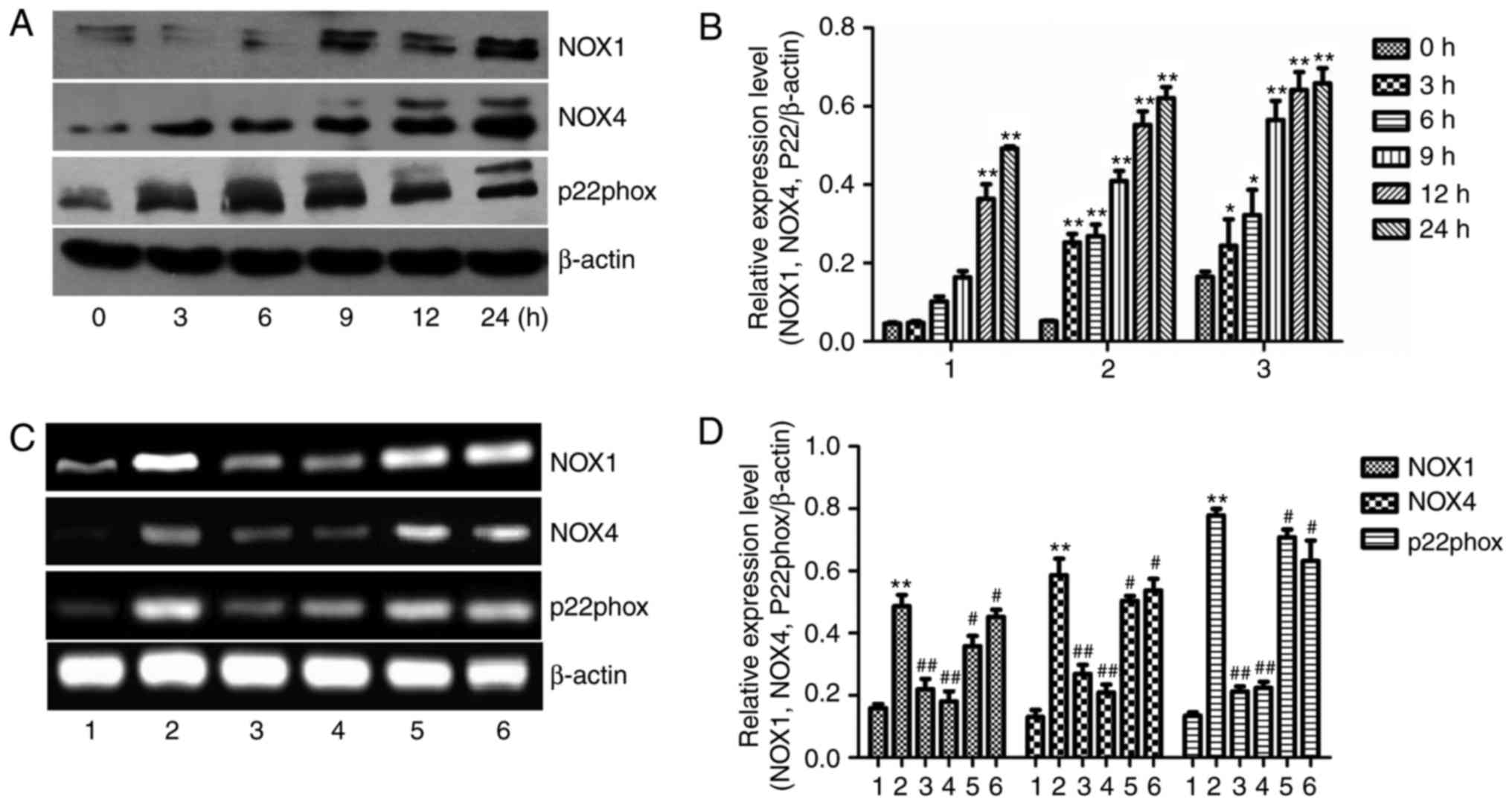 | Figure 4Effect of 400 mM ethanol on NOX1, NOX
4 and p-22phox protein expression in the L-02 cell line.
(A and B) Western blot analysis of the protein expression of NOX1,
NOX4 and p-22 in L-02 cells after treatment with 400 mM ethanol for
0–24 h. (C and D) mRNA expression of NOX1, NOX4 and
p22phox in L-02 cells after the indicated treatments for
6 h. Values are expressed as the mean ± standard error of the mean
(n=3). *P<0.05, **P<0.01 vs. the
control group/0 h group; #P<0.05,
##P<0.01 vs. the ethanol group. Groups: 1, Control;
2, ethanol (400 mM); 3, ethanol + NAC (10 mM); 4, ethanol +
apocynin (300 µM); 5, ethanol + SB202190 (1 µM); 6,
ethanol + SP600125 (1 µM). NOX, NADPH oxidase; NAC, N-acetyl
L-cysteine. |
Furthermore, RT-PCR indicated that the mRNA
expression of NOX1, NOX4 and p22phox in the
ethanol-treated group was significantly higher than that in the
control group (P<0.01; Fig. 4C and
D). By contrast, the ethanol-induced increases in the mRNA
expression of NOX1, NOX4 and p22phox in L-02 cells were
significantly suppressed by pre-treatment with NAC and apocynin
(P<0.01). NAC is an ROS scavenger and apocynin blocks the
p47phox subunit of NOX (20). As expected, SB202190 and SP600125
had no effect on the mRNA expression of the NOX subunits
(P>0.05). These results are consistent with those of previous
studies, which also indicated that increased ROS production may be
abolished by NAC and apocynin (21,22).
In short, ethanol induced intracellular ROS
generation in L-02 cells, which was markedly abolished by NAC and
apocynin but not MAPK inhibitors. Furthermore, NAC or apocynin
completely inhibited ethanol-induced activation of caspase-3 and
caspase-8 via blocking the apoptosis of L-02 cells by activating
the caspase-, MAPK- and ROS-dependent pathways. Apocynin alleviates
oxidative stress and inhibits NADPH oxidase superoxide production.
As NOX4 is one of the most important NADPH isoform in hepatocytes
that contributes to ROS production, its role was then investigated.
The protein and mRNA expression of total NOX4 was upregulated by
ethanol treatment (Fig. 4A–D).
The protein level and mRNA expression of NOX1 was also increased by
ethanol treatment (Fig. 4A–D). In
addition, the expression of p22phox, an essential
adaptor protein for functional NOX4, was also upregulated by
ethanol treatment. The ethanol-induced increase in the mRNA
expression of total NOX1, NOX4 and p22phox was reduced
by NAC or apocynin treatment.
ROS mediates P-38 and JNK activation in
ethanol-induced apoptosis of L-02 cells
Exposure of L-02 cells to ethanol induces the
production of ROS and cell apoptosis. Previous studies have
indicated that ROS production is a crucial step in the triggering
of apoptotic signalling pathways (23–25).
Further experiments were performed to assess the
association between ROS generation and apoptosis in L-02 cells
exposed to ethanol. The L-02 cells were incubated with ethanol in
DMEM containing 10% FBS for 0, 3, 6, 9, 12 or 24 h. The expression
levels of JNK, p-JNK p38 MAPK and p-p38 MAPK were examined at the
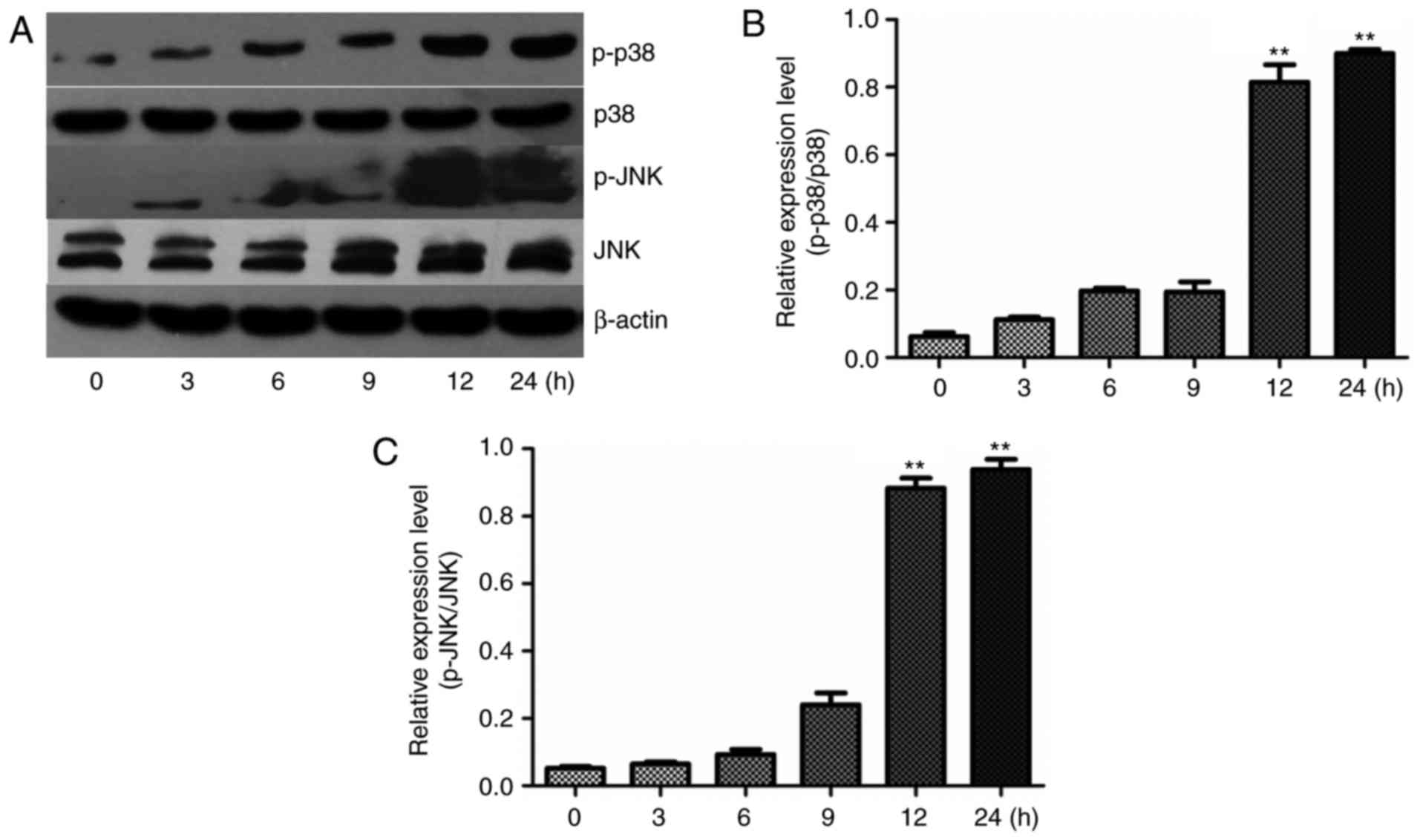
indicated time-points as presented in Fig. 5A–C. The expression levels of p-p38
MAPK and p-JNK were increased, but the levels of total p38 MAPK and
JNK remained nearly unchanged after treatment with ethanol. At 12
and 24 h, the p-p38MAPK/p38MAPK ratio and the p-JNK/JNK ratio were
significantly increased, indicating that both p38 and JNK
activation contribute to L-02 cell apoptosis.
Since activation of p38 MAPK and JNK has been
demonstrated to be associated with apoptosis, The above results
indicated that ethanol induced apoptosis in L-02 cells is
triggered, at least partially, by ROS production which acts
upstream of the apoptotic signalling pathway, leads to an increased
ratio of Bax/Bcl-2, followed by cells going into apoptosis. It
points to that at least JNK and p38 MAPK have a partial role in the
process of oxidative stress-mediated apoptosis, which was caused by
ethanol.
NOX4 has an important role in L-02
apoptosis induced by ethanol
To further confirm the role of NOX4 in
ethanol-induced L-02-cell apoptosis, NOX4 was silenced with
NOX4-siRNA or NOX4 overexpressed using plasmid transfection
technology. The results indicated that, compared with the ethanol +
NC-siRNA group, the generation of ROS was significantly decreased
in the ethanol + NOX4-siRNA-treated group, while ROS was
significantly increased in the NOX4 overexpression group (Fig. 6A). In correspondence with this,
apoptosis was significantly decreased in the ethanol + NOX4-siRNA
group, whereas apoptosis was significantly increased in the NOX4
overexpression group compared with the ethanol + NC-siRNA group
(Fig. 6B). Furthermore, NOX4
protein expression in response to ethanol was significantly
decreased in the NOX4-siRNA group but was markedly increased in the
NOX4 overexpressed group (Fig.
6C). Simultaneously, silencing of NOX4 protected L-02 cells
against ethanol-induced caspase activation (Fig. 6E and F). By contrast, NOX4
overexpression significantly increased ethanol-induced caspase
activation. To further explore the underlying mechanisms, p38 MAPK-
and JNK-dependent apoptotic pathways were investigated. Ethanol
exerted an obvious effect on p38 MAPK- and JNK-phosphorylation in
NC-siRNA cells and was further increased in NOX4-overexpressing
cells (Fig. 6F and G). In
addition, in the ethanol + NOX4-siRNA group, ethanol-induced
increases in p38 MAPK and JNK phosphorylation were markedly
inhibited. In summary, in NOX4-overexpressing L-02 cells, the
effects of ethanol were significantly enhanced, while they were
inhibited by NOX4 silencing.
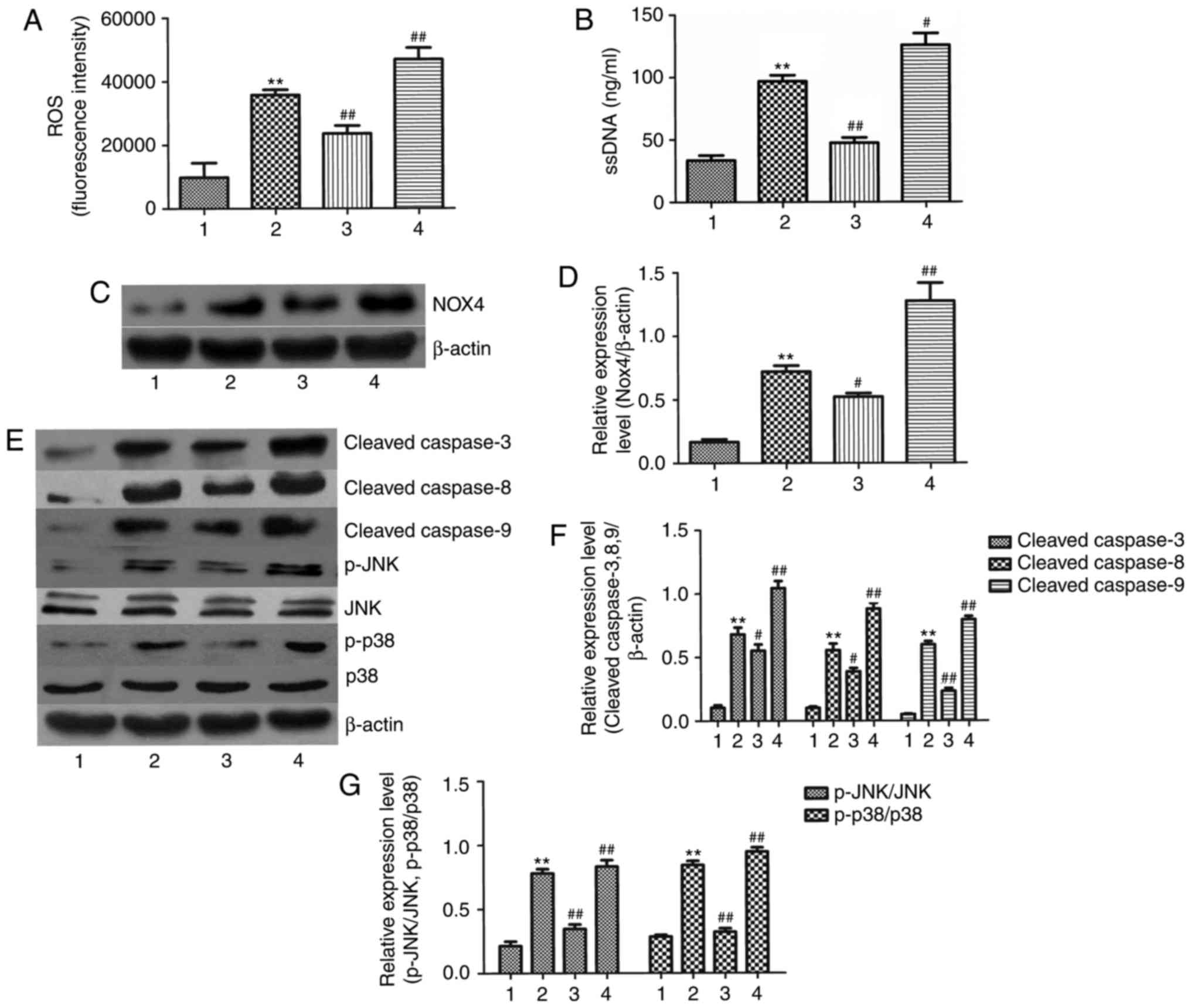 | Figure 6NOX4 has an important role in
ethanol-induced apoptosis. Effects of ethanol (400 mM) treatment of
L-02 cells with siRNA-mediated knockdown or vector-mediated
overexpression of NOX4 for 24 h on the generation of (A) ROS and
(B) ssDNA as well as the protein levels of (C and D) NOX-4, (E–G)
caspase-3, -8 and -9, p38, p-p38, JNK and p-JNK. Values are
expressed as the mean ± standard error of the mean (n=3).
**P<0.01 vs. the control group;
#P<0.05, ##P<0.01 vs. the ethanol +
NC-siRNA group. Groups: 1, NC-siRNA; 2, ethanol + NC-siRNA; 3,
ethanol + NOX4-siRNA; 4, ethanol + NOX4 overexpression. NOX, NADPH
oxidase; p-JNK, phosphorylated c-Jun N-terminal kinase; siRNA,
small interfering RNA; NC, scrambled control siRNA; ssDNA,
single-stranded DNA; ROS, reactive oxygen species. |
Discussion
Chronic ethanol consumption is a high risk factor
for hepatic disease and causes severe alcohol-associated liver
disease, including hepatitis, steatosis, fatty liver, fibrosis, and
eventually cirrhosis or even cancer, which carry a poor prognosis.
Ethanol consumption elevates the production of ROS and increases
the peroxidation of proteins, lipids and DNA (26). Prolonged alcohol abuse leads to
severe pathologies that are associated with apoptotic cell death
induced by ROS-mediated oxidative stress (17). It is important to select a cell
line whose ethanol-metabolizing enzymes are expressed as in normal
liver cells. For this purpose, the L-02 normal human hepatocyte
cell line, which has been demonstrated to express an adequate
amount of ADH, was utilized.
In the present study, the cytotoxic effects of
ethanol on L-02 cells were assessed. The major findings include
that i) ethanol induced L-02-cell apoptosis, which was dependent on
the ethanol concentration, ii) NOX4 had an important role in
ethanol-induced L-02-cell apoptosis, iii) ROS generation from NOX4
induction was involved in L-02-cell apoptosis in response to
ethanol and iv) that JNK and P38 MAPK actively participated in
ethanol-induced L-02-cell apoptosis.
The optimal ethanol concentration for treatment and
suitable experimental time-points were determined using a cell
viability assay and further preliminary experiments. The results
indicate that the 400-mM concentration of ethanol and the treatment
duration of 24 h were the optimal conditions for inducing
apoptosis. ROS generation reached a peak following ethanol
treatment at 400 mM for 6 h. Cells exposed to ethanol for 24 h
exhibited a marked increase in apoptosis, as indicated by the
Annexin V/PI assay. The apoptotic population in ethanol-treated
L-02 cells increased in a time-dependent manner.
When L-02 cells were cultured with ethanol at the
concentration of 400 mM for 0–8 h, ROS increased with the time of
incubation. NOX4, which is one of six important homologues of the
transmembrane protein NOX, is widely involved in ROS generation and
has been identified to be highly expressed in the liver and
hepatocytes (27). The results of
the present study indicated that when the L-02 cells were incubated
with ethanol, the mRNA levels of NOX4 and p22phox, which
is the protein required to activate NOX4 on the membrane, were
increased. From this, it was deduced that the generation of ROS in
L-02 cells treated with ethanol is regulated at least partially by
NOX4 on the membrane. It is well known that excessive ROS increases
oxidative stress and causes apoptosis (22). The results of the present study
indicated that in ethanol-treated L-02 cells, ROS generation
induced by NOX4 on the liver cell membrane is one of the factors
that leads to and promotes apoptosis.
Apoptosis induced via the mitochondria-dependent
pathway may be triggered by stress, chemical agents and/or drugs
(11); it is also regulated by
numerous genes (28). Bax and
Bcl-2 are important regulators; Bcl-2 has an anti-apoptotic role
and Bax has an apoptosis-promoting role (29,30). Bax and Bcl-2 serve important roles
in maintaining the stability of the mitochondrial membrane
(16). The elevated expression of
Bax leads to an increase in cell death stimulation and a shift in
the mitochondrial membrane potential to trigger the release of
cytochrome c. Bax in its cleaved form is an effector of
apoptosis, while the anti-apoptotic protein Bcl-2 restrains the
pro-apoptotic effect of Bax (30). In the present study, L-02 cells
treated with 400 mM ethanol exhibited a slight increase in the
expression of Bcl-2, while the expression of Bax was markedly
elevated, and the ratio of Bax to Bcl-2 was greatly increased in a
time-dependent manner and reached a maximum at 24 h. The increase
of the Bax/Bcl-2 ratio is likely to change the cross-membrane
potential of the mitochondria by triggering cytochrome c
release, sensitizing caspase-9 and then activating caspase-3,
inducing the occurrence of apoptosis (4,30).
These results indicated that apoptosis has a key role in L-02-cell
damage induced by ethanol and that ethanol induced mitochondrial
apoptotic pathways. However, the exact mechanisms of how ethanol
activates the mitochondrial apoptotic pathway remain to be fully
elucidated.
Cell death induced by oxidative stress is closely
associated with the increase of ROS. It has been identified that
the accumulation of ROS in the liver leads to functional disorders
in the cell membrane, oxidative DNA damage and abnormal protein
expression to finally cause liver cell injury. If the damage is
irreversible and cannot be repaired, it eventually leads to cell
apoptosis (31). Ethanol boosts
oxidative stress by increasing the formation of ROS and the
depletion of internal oxidative defences in cells (32). Oxidative stress is a vital
apoptotic stimulant for cells, particularly in those that have a
high-energy metabolism required for their rapid growth and
proliferation. As a result, ROS are overly produced by the
mitochondrial source and cause lipid peroxidation and DNA lesions,
thereby leading to cell apoptosis (32). The present study demonstrated that
ROS were gradually increased in L-02 cells from 0 to 6 h, where
they reached a maximum and were then eventually reduced. NOX4 was
also increased in line with the trends in ROS levels. Increased ROS
generation in L-02 cells triggered apoptosis. Of note,
ethanol-induced ROS formation in L-02 cells was inhibited by NAC
and apocynin. NOX and mitochondria are two major sources of ROS in
L-02 cells (4). Ethanol-induced
caspase-3 and caspase-8 activation was almost completely inhibited
by pre-treatment with NAC or apocynin, which are NOX inhibitors,
suggesting that ethanol-induced apoptosis was mediated by
NOX-derived ROS. NOX4, one of the 7 isoforms of the NADPH oxidase,
is considered to be the key enzyme of ROS production in HepG2 cell
and has a vital role in regulating the fate of HepG2 and other
cells (33,34). However, the biological roles of
NOX4 in the L-02 cells still remain elusive. In the present study,
ethanol induced NOX4 expression accompanied by the increased
expression of p22phox, an essential component for NOX4
activation. Silencing of NOX4 significantly inhibited ROS formation
and the pro-apoptotic effects of ethanol, which was consistent with
previous findings in SK-Hep1 cells and lung epithelial cells
(11,35). To investigate the underlying
mechanisms of this phenomenon, the roles of JNK and p38 MAPK were
examined (36,37). In the present study, ethanol
considerably increased the phosphorylation of JNK and p38, while
inhibition of JNK and p38 inhibited ethanol-induced apoptosis in
L-02 cells. In NOX4-overexpressing cells, JNK and p38 MAPK were
activated and ethanol-induced L-02 apoptosis and death were
promoted. Increased NOX4 expression, either by ethanol induction or
NOX4 overexpression, therefore has a role in L-02-cell
apoptosis.
The MAPK family is important for cells to regulate
proliferation and death in response to different internal stressors
(37). JNKs are known to be
involved in stimulating apoptotic signalling by activating JNK via
receptor-initiated extrinsic and mitochondrial intrinsic apoptotic
pathways (37,38). JNK and ROS stimulate the
activities of pro-apoptotic proteins (such as Bax) and promote
apoptosis by inhibiting anti-apoptotic proteins (such as Bcl-2)
(39). The present study
indicated that ethanol activates the phosphorylation of JNK and
increases the expression of Bax, suggesting that apoptosis of L-02
cells may be stimulated by the activation of JNK signalling.
p38 MAPK activation not only boosts the
mitochondrial translocation of Bim and Bax and represses the
function of Bcl-2 by increasing the phosphorylation of these
factors, but also induces the activation of caspase-3 and caspase-9
(38). The present results
demonstrated that increased activation of p38 MAPK, caspase-3 and
-9 also increased the ratio of Bax to Bcl-2 in ethanol-treated L-02
cells. p38 MAPK activation induced by ethanol was supressed by
NOX4-SiRNA, providing further evidence for the involvement of MAPK
in necrosis and apoptosis via NOX overexpression and ROS
generation. Thus, p38 MAPK may act as a pro-death effector
regardless of what type of cell death occurs in ethanol-treated
L-02 cells.
The present study assessed the effects of MAPK
inhibitors in L-02 cells exposed to ethanol and their impact on
apoptosis. The mechanisms underlying ethanol-induced apoptosis in
L-02 cells were indicated to include activation of the JNK and p38
MAPK pathways. Ethanol-induced apoptosis is likely to be mediated
via multiple pathways. For instance, ethanol elevates the
expression levels of p53, which boosts apoptosis in HepG2 cells via
the apoptotic pathway mediated by p53 (40). In addition, ethanol augments the
activities of caspase-8 and caspase-9 in HepG2 cells, thereby
mediating apoptosis by means of extrinsic and intrinsic pathways
(41). MAPK signalling is known
to be closely associated with apoptosis (42). However, the effect of the
activation of each component of the MAPK cascade is different and
depends on its cellular specificity. It was reported that
inhibition of the ERK pathway led to increased cell apoptosis and
increased activity of caspase-9 and caspase-3, while JNK inhibitor
and p38 MAPK inhibitor markedly suppressed the apoptosis induced by
ethanol and reduced caspase activity in SK-Hep1 cells (11). Thus, activation of p38 MAPK and
JNK participates in ethanol-induced L-02-cell apoptosis. Therefore,
it may be inferred that the apoptotic signal derived from ROS that
was produced by NOX, was mainly transduced through the JNK and p38
MAPK pathways. In the present study, p38 MAPK and JNK inhibitors
did not reduce the generation of ROS, rather they blocked the
transduction of the apoptotic signal induced by ROS. Inhibition of
p38 MAPK and JNK also resulted in a decrease in ethanol-induced
cell death. This was further confirmed by downregulating NOX4.
After NOX4 silencing, ROS generation was reduced followed by JNK
and p38 MAPK phosphorylation and finally apoptosis was inhibited.
By contrast, with NOX4 overexpression, ROS production was increased
and the activation of JNK and p38 MAPK was enhanced, resulting in
increased apoptosis of L-02 cells. Taken together, it may be
postulated that activation of p38 MAPK and JNK signalling pathways
to promote apoptosis were the underlying mechanisms of
ethanol-induced toxicity on L-02 cells. In summary, the present
study demonstrated that ethanol-induced apoptosis was
caspase-dependent. ROS derived from NOX were revealed to have an
important role in ethanol-induced apoptosis. L-02 cell viability
and apoptosis were affected regulated MAPK inhibitors, suggesting
that MAPK signalling pathways are involved in ethanol-induced
apoptosis.
In conclusion, apoptosis induced by ethanol in L-02
cells was accompanied with the generation of ROS and elevated
expression of NOX4. Subsequent activation of the JNK and p38
signalling pathways then promoted apoptosis in L-02 cells. MAPK
inhibitors affected the viability and apoptosis of L-02 cells,
indicating that MAPK signalling is involved in ethanol-induced cell
apoptosis. These results opened up the possibility for exploring
novel therapeutic target of organelle-based ROS to improve the
treatment of hepatocyte apoptosis-associated liver diseases.
Acknowledgments
The study was funded by The National Natural Science
Foundation of China (grant no. 81360497).
Notes
[1] Competing
interests
The authors declare that they have no competing
interests.
References
|
1
|
Duncan C: Rethinking excessive habits and
addictive behaviors. Alcohol Alcohol. 52:128–129. 2017. View Article : Google Scholar
|
|
2
|
Owens RE, Snyder HS, Twilla JD and
Satapathy SK: Pharmacologic treatment of alcoholic hepatitis:
Examining outcomes based on disease severity stratification. J Clin
Exp Hepatol. 6:275–281. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kim JW, Yang H, Kim HW, Kim HP and Sung
SH: Lignans from Opuntia ficusindica seeds protect rat primary
hepatocytes and HepG2 cells against ethanol-induced oxidative
stress. Biosci Biotechnol Biochem. 81:181–183. 2017. View Article : Google Scholar
|
|
4
|
Sun Q, Zhang W, Zhong W, Sun X and Zhou Z:
Pharmacological inhibition of NOX4 ameliorates alcohol-induced
liver injury in mice through improving oxidative stress and
mitochondrial function. Biochim Biophys Acta. 1861:2912–2921. 2017.
View Article : Google Scholar :
|
|
5
|
Zhang F, Wang X, Qiu X, Wang J, Fang H,
Wang Z, Sun Y and Xia Z: The protective effect of esculentoside a
on experimental acute liver injury in mice. PLoS One.
9:e1131072014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Sinha K, Das J, Pal PB and Sil PC:
Oxidative stress: The mitochondria-dependent and
mitochondria-independent pathways of apoptosis. Arch Toxicol.
87:1157–1180. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kim EK and Choi EJ: Compromised MAPK
signaling in human diseases: An update. Arch Toxicol. 89:867–882.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wang Y, Wang R, Wang Y, Peng R, Wu Y and
Yuan Y: Ginkgo biloba extract mitigates liver fibrosis and
apoptosis by regulating p38 MAPK, NF-κB/IκBα, and Bcl-2/Bax
signaling. Drug Des Devel Ther. 9:6303–6317. 2015.
|
|
9
|
Crosas-Molist E and Fabregat I: Role of
NADPH oxidases in the redox biology of liver fibrosis. Redox Biol.
6:106–111. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ma JQ, Ding J, Zhang L and Liu CM:
Hepatoprotective properties of sesamin against CCl4 induced
oxidative stress-mediatedapoptosisin mice via JNK pathway. Food
Chem Toxicol. 64:41–48. 2014. View Article : Google Scholar
|
|
11
|
Morio Y, Tsuji M, Inagaki M, Nakagawa M,
Asaka Y, Oyamada H, Furuya K and Oguchi K: Ethanol-induced
apoptosis in human liver adenocarcinoma cells (SK-Hep1): Fas- and
mitochondria-mediated pathways and interaction with MAPK signaling
system. Toxicol In Vitro. 27:1820–1829. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Guo X, Cui R, Zhao J, Mo R, Peng L and Yan
M: Corosolic acid protects hepatocytes against ethanol-induced
damage by modulating mitogen-activated protein kinases and
activating autophagy. Eur J Pharmacol. 791:578–588. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Liang S, Su WW, Wang YG, Peng W, Nie YC
and Li PB: Effect of quercetin 7-rhamnoside on
glycochenodeoxycholic acid-induced L-02 human normal liver cell
apoptosis. Int J Mol Med. 32:323–330. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Liu LQ, Fan ZQ, Tang YF and Ke ZJ: The
resveratrol attenuates ethanol-induced hepatocyte apoptosis via
inhibiting ER-related caspase-12 activation and PDE activity in
vitro. Alcohol Clin Exp Res. 38:683–693. 2014. View Article : Google Scholar
|
|
15
|
Andreu-Fernández V, Sancho M, Genovés A,
Lucendo E, Todt F, Lauterwasser J, Funk K, Jahreis G, Pérez-Payá E,
Mingarro I, et al: Bax transmembrane domain interacts with
prosurvival Bcl-2 proteins in biological membranes. Proc Natl Acad
Sci USA. 114:310–315. 2017. View Article : Google Scholar :
|
|
16
|
Farshori NN, Al-Sheddi ES, Al-Oqail MM,
Hassan WH, Al-Khedhairy AA, Musarrat J and Siddiqui MA:
Hepatoprotective potential of Lavandula coronopifolia extracts
against ethanol induced oxidative stress-mediated cytotoxicity in
HepG2 cells. Toxicol Ind Health. 31:727–737. 2015. View Article : Google Scholar
|
|
17
|
Liang S, Kisseleva T and Brenner DA: The
role of NADPH oxidases (NOXs) in liver fibrosis and the activation
of myofibro-blasts. Front Physiol. 7:172016. View Article : Google Scholar
|
|
18
|
Jiang JX and Török NJ: NADPH oxidases in
chronic liver diseases. Adv Hepatol. 2014:pii 7429312014.
View Article : Google Scholar
|
|
19
|
Jiao Y, Ji L, Kuang Y and Yang Q:
Cytotoxic effect of oxaloacetate on HepG2-human hepatic carcinoma
cells via apoptosis and ROS accumulation. Neoplasma. 64:192–198.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Bost ER, Frye GS, Ahn B and Ferreira LF:
Diaphragm dysfunction caused by sphingomyelinase requires the
p47(phox) subunit of NADPH oxidase. Respir Physiol Neurobiol.
205:47–52. 2015. View Article : Google Scholar
|
|
21
|
Li YY, Shi ZM, Yu XY, Feng P and Wang XJ:
Urotensin II-induced insulin resistance is mediated by NADPH
oxidase-derived reactive oxygen species in HepG2 cells. World J
Gastroenterol. 22:5769–5779. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Liu J, Wei X, Wu Y, Wang Y, Qiu Y, Shi J,
Zhou H, Lu Z, Shao M, Yu L and Tong L: Giganteaside D induces
ROS-mediated apoptosis in human hepatocellular carcinoma cells
through the MAPK pathway. Cell Oncol (Dordr). 39:333–342. 2016.
View Article : Google Scholar
|
|
23
|
He Y, Yang J, Li H, Shao H, Wei C, Wang Y,
Li M and Xu C: Exogenous spermine ameliorates high glucose-induced
cardiomyocytic apoptosis via decreasing reactive oxygen species
accumulation through inhibiting p38/JNK andJAK2 pathways. Int J
Clin Exp Pathol. 8:15537–15549. 2015.
|
|
24
|
Zhang C, Jia X, Bao J, Chen S, Wang K,
Zhang Y, Li P, Wan JB, Su H, Wang Y, et al: Polyphyllin VII induces
apoptosis in HepG2 cells through ROS-mediated mitochondrial
dysfunction and MAPK pathways. BMC Complement Altern Med.
16:582016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wang Y, Sun Z, Chen S, Jiao Y and Bai C:
ROS-mediated activation of JNK/p38 contributes partially to the
pro-apoptotic effect of ajoene on cells of lung adenocarcinoma.
Tumour Biol. 37:3727–3738. 2016. View Article : Google Scholar
|
|
26
|
Zhang Q, Cui C, Chen CQ, Hu XL, Liu YH,
Fan YH, Meng WH and Zhao QC: Anti-proliferative and pro-apoptotic
activities of Alpinia oxyphylla on HepG2 cells through ROS-mediated
signaling pathway. J Ethnopharmacol. 169:99–108. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Liang B, Liu Z, Cao Y, Zhu C, Zuo Y, Huang
L, Wen G, Shang N, Chen Y, Yue X, et al: MC37, a new mono-carbonyl
curcumin analog, induces G2/M cell cycle arrest and
mitochondria-mediated apoptosis in human colorectal cancer cells.
Eur J Pharmacol. 796:139–148. 2017. View Article : Google Scholar
|
|
28
|
Chen J, Wang Y, Hui C, Xi Y, Liu X, Qi F,
Liu H, Wang Z and Niu S: Mechanisms of Heshouwuyin in regulating
apoptosis of testicular cells in aging rats through mitochondrial
pathway. BMC Complement Altern Med. 16:3372016. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Yan X, Jiang Z, Bi L, Yang Y and Chen W:
Salvianolic acid A attenuates TNF-α- and D-GalN-induced ER
stress-mediated and mitochondrial-dependent apoptosis by modulating
Bax/Bcl-2 ratio and calcium release in hepatocyte LO2 cells. Naunyn
Schmiedebergs Arch Pharmacol. 388:817–830. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Yang Y, Zong M, Xu W, Zhang Y, Wang B,
Yang M and Tao L: Natural pyrethrins induces apoptosis in human
hepatocyte cells via Bax- and Bcl-2-mediated mitochondrial pathway.
Chem Biol Interact. 262:38–45. 2017. View Article : Google Scholar
|
|
31
|
Chen LY, Chen Q, Zhu XJ, Kong DS, Wu L,
Shao JJ and Zheng SZ: Diallyl trisulfide protects against
ethanol-induced oxidative stress and apoptosis via a hydrogen
sulfide-mediated mechanism. Int Immunopharmacol. 36:23–30. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Sumegi K, Fekete K, Antus C, Debreceni B,
Hocsak E, Gallyas F Jr, Sumegi B and Szabo A: BGP-15 protects
against oxidative stress- or lipopolysaccharide-induced
mitochondrial destabilization and reduces mitochondrial production
of reactive oxygen species. PLoS One. 12:e01693722017. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Yan F, Wang Y, Wu X, Peshavariya HM,
Dusting GJ, Zhang M and Jiang F: Nox4 and redox signaling mediate
TGF-β-induced endothelial cell apoptosis and phenotypic switch.
Cell Death Dis. 5:e10102014. View Article : Google Scholar
|
|
34
|
Amatore D, Sgarbanti R, Aquilano K,
Baldelli S, Limongi D, Civitelli L, Nencioni L, Garaci E, Ciriolo
MR and Palamara AT: Influenza virus replication in lung epithelial
cells depends on redox-sensitive pathways activated by NOX4-derived
ROS. Cell Microbiol. 17:131–145. 2015. View Article : Google Scholar :
|
|
35
|
Chuang WL, Lin PY, Lin HC and Chen YL: The
apoptotic effect of ursolic acid on SK-Hep-1 cells is regulated by
the I3K/Akt, p38 and JNK mapk signaling pathways. Molecules.
21:4602016. View Article : Google Scholar
|
|
36
|
Paik YH, Kim J, Aoyama T, De Minicis S,
Bataller R and Brenner DA: Role of NADPH oxidases in liver
fibrosis. Antioxid Redox Signal. 20:2854–2872. 2014. View Article : Google Scholar :
|
|
37
|
Nguyen KC, Willmore WG and Tayabali AF:
Cadmium telluride quantum dots cause oxidative stress leading to
extrinsic and intrinsic apoptosis in hepatocellular carcinoma HepG2
cells. Toxicology. 306:114–123. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Zhao WX, Tang SS, Jin X, Zhang CM, Zhang
T, Wang CC, Sun Y and Xiao XL: Olaquindox-induced apoptosis is
suppressed through p38 MAPK and ROS-mediated JNK pathways in HepG2
cells. Cell Biol Toxicol. 29:229–238. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Yoon J, Ham H, Sung J, Kim Y, Choi Y, Lee
JS, Jeong HS, Lee J and Kim D: Black rice extract protected HepG2
cells from oxidative stress-induced cell death via ERK1/2 and Akt
activation. Nutr Res Pract. 8:125–131. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Stiuso P, Bagarolo ML, Ilisso CP, Vanacore
D, Martino E, Caraglia M, Porcelli M and Cacciapuoti G: Protective
effect of tyrosol and S-Adenosylmethionine against ethanol-induced
oxidative stress of Hepg2 cells involves sirtuin 1, P53 and Erk1/2
Signaling. Int J Mol Sci. 17:pii: E6222016. View Article : Google Scholar
|
|
41
|
Szuster-Ciesielska A, Plewka K, Daniluk J
and Kandefer-Szerszeń M: Zinc inhibits ethanol-induced HepG2 cell
apoptosis. Toxicol Appl Pharmacol. 229:1–9. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Abid MD, Chen J, Xiang M, Zhou J, Chen X
and Gong F: Khat (Catha edulis) generates reactive oxygen species
and promotes hepatic cell apoptosis via MAPK activation. Int J Mol
Med. 32:389–395. 2013. View Article : Google Scholar : PubMed/NCBI
|