Introduction
Adenosine kinase (ADK) is the key metabolic
regulator of the purine ribonucleoside adenosine and is essential
for intracellular adenosine metabolic clearance via phosphorylating
adenosine to AMP based on its low Km(1). ADK is expressed in most vital organs
that require metabolic adenosine clearance, in particular the liver
and placenta (2). ADK controls
specific organ functions via a combination of adenosine
receptor-dependent and -independent mechanisms (1). Changes in adenosine homeostasis may
activate the four adenosine-associated G-protein-coupled receptors,
alter energy metabolism and regulate DNA methylation (3). A number of studies have demonstrated
that ADK serves important roles in several diseases, including
epilepsy (4), traumatic brain
injury (5), stroke (6), diabetes (7) and cancer (8).
Bone is a highly dynamic organ that is constantly
undergoing remodeling by means of bone formation and resorption in
the adult skeleton (9). Previous
studies have investigated the role of adenosine and its four G
protein-coupled receptors in the maintenance of bone homeostasis
in vivo and in vitro, particularly adenosines
association with osteoclastogenesis and bone resorption in rodents
and humans (10). Previous
reports suggest that activating adenosine A1 receptor
(A1R) and adenosine A2B receptor (A2BR) promotes
osteoclastogenesis; however, adenosine A2A receptor
(A2AR) activation has the opposite effect (11–14) and adenosine A3 receptor
(A3R) activation indirectly inhibits osteoclastogenesis due to its
anti-inflammatory effect (15).
ADK deficiency results in the accumulation of
intracellular adenosine and a decrease in AMP, ADP and ATP, which
in turn leads to disordered purinergic signaling (1). A recent clinical investigation based
on next generation sequencing revealed that patients deficient in
ADK with intellectual disabilities also presented with short
stature, frontal bossing and slender hands and fingers (16). In a mouse model, genetic deletion
of ADK resulted in mice succumbing within 14 days of birth whereas
mice with ADK deficiency exhibited reduced body weight and length
(17). However, the function of
ADK in bone metabolism is still unknown.
Osteoclasts are the primary bone-resorbing cells and
are thought to be derived from the monocyte-macrophage lineage
(18). In the present study, a
mouse model with ADK knockout in myeloid monocyte cells
(ADKMMC-KO) was established via Cre/LoxP site-specific
recombination. The effect of ADK on bone metabolism was assessed by
gross observation and bone histomorphometric analysis in
vivo to ascertain whether there were changes in bone
homeostasis. In addition, osteoclast differentiation and bone
resorption were examined in vitro by genetic deletion and
pharmacologic inhibition of ADK in osteoclasts.
Materials and methods
Animals
All animal experiments in this study were approved
and supervised by the Institutional Animal Care and Use Committee
(IACUC No. 2014027) of Guangdong Laboratory Animals Monitoring
Institute (Guangzhou, China). ADKflox/flox (C57BL/6
background) mice (hereafter referred to as ADKWT)
provided by Professor Yuqing Huo (Vascular Biology Center,
Department of Cellular Biology and Anatomy, Medical College of
Georgia, Augusta University, Augusta, GA, USA) (19) were mated to lysozyme 2-Cre mice
(lysMcre; stock no. 004781; Jackson Laboratory,
Farmington, CT, USA), in which the Cre recombinase gene is knocked
into the lysozyme 2 locus and is specifically expressed in myeloid
monocyte cells (20). Mice with
ADK-deficient myeloid monocyte cells (ADKMMC-KO) were
generated by mating ADKflox/floxxlysMcre mice
with ADKflox/flox mice. All mice were provided with
water and a standard chow diet ad libitum. Mice were
maintained in a specific pathogen-free facility with a 12 h
light/dark cycle at 23±2°C and 50±10% relative humidity.
For the in vivo experiment, 4-month-old male
ADKMMC-KO (23–25 g; n=10) and ADKWT (27–29 g;
n=10) mice were used for gross apperance and bone histomorphometric
analysis, with ADKWT as the control. For the in
vitro experiment, 2-month-old male ADKMMC-KO (21–23
g; n=10) and ADKWT mice (25–27 g; n=10) were used for
osteoclastogenesis and bone resorption analysis. In addition,
2-month-old male ADKWT mice (25–27 g; n=20) were
assigned to either an ABT-702-treated group (n=10) or a vehicle
control group (n=10) for in vitro osteoclastogenesis and
bone resorption analysis.
Cell culture
Murine osteoclasts were generated from bone marrow
cells as previously described (21). Briefly, following CO2
euthanasia, primary ADKMMC-KO or ADKWT mouse
bone marrow cells were isolated by flushing the tibia and femur of
2-month-old male mice with α-modified Eagle's medium (α-MEM; Thermo
Fisher Scientific, Inc., Waltham, MA, USA). Cells were subsequently
centrifuged (1,000 × g) at 4°C for 5 min and incubated in α-MEM
containing 10% (v/v) fetal bovine serum (Thermo Fisher Scientific,
Inc.), penicillin-streptomycin (100 U/ml) and 1% (w/v) glutamine
(Sigma Aldrich; Merck KGaA, Darmstadt, Germany) overnight at 37°C
in a humidified atmosphere containing 5% CO2. To
generate bone marrow-derived macrophages, non-adherent cells were
collected, plated onto 24-well plates (1×106 cells/well)
and cultured at 37°C in a humidified atmosphere containing 5%
CO2 with 25 ng/ml macrophage colony-stimulating factor
(M-CSF; R&D Systems, Inc., Minneapolis, MN, USA) for 3 days in
α-MEM. Cells were then used for osteoclast differentiation or ADK
western blot analysis. To generate osteoclasts, bone marrow-derived
macrophages were incubated at 37°C in a humidified atmosphere
containing 5% CO2 with α-MEM containing 25 ng/ml M-CSF
and 30 ng/ml receptor activator of nuclear factor kappa-B ligand
(RANKL; R&D Systems, Inc.) for 5 days. To evaluate the effect
of ADK inhibition on osteoclast differentiation, ABT-702
dihydrochloride (Tocris; Bio-Techne, Minneapolis, MN, USA), a
potent non-nucleoside ADK inhibitor, was pre-dissolved in 5% EtOH
and the solution was diluted with osteoclast differentiation medium
as described for cell culture. All assays were performed in
triplicate.
Western blotting
Total protein was extracted from bone marrow-derived
macrophages using radioimmunoprecipitation assay buffer (Sigma
Aldrich; Merck KGaA) according to the manufacturer's protocol.
Protein was quantified using a bicinchoninic acid assay and 20 µg
samples were separated by 10% SDS-PAGE and transferred onto
nitrocellulose membranes. The membranes were blocked with 5% (w/v)
non-fat dry milk in TBST buffer for 1 h at 4°C. Rabbit monoclonal
antibodies against ADK (Sigma Aldrich; Merck KGaA; cat no.
SAB2701969; 1:1,000) and GAPDH (Sigma Aldrich; Merck KGaA; cat. no.
G9545, 1:2,500) were used to assess ADK protein levels in cultured
cell lysates. The membranes were incubated with the primary
antibodies in 1% bovine serum albumin (Sigma Aldrich; Merck KGaA)
overnight at 4°C and washed three times with TBST. Subsequently,
membranes were incubated with horseradish peroxidase-conjugated
secondary goat anti rabbit antibody (Invitrogen; Thermo Fisher
Scientific, Inc.; cat. no. 31460; 1:2,000) in non-fat dry milk for
1 h at room temperature and washed three times with TBST. Membranes
were visualized using West Pico Chemiluminescent Substrate (Thermo
Fisher Scientific, Inc.). Bands were quantified using Image Lab 5.0
(Bio-Rad Laboratories, Inc., Hercules, CA, USA).
Tartrate-resistant acid phosphatase
(TRAP) activity assay
TRAP staining was performed using an Acid
Phosphatase Leukocyte (TRAP) kit (Sigma Aldrich; Merck KGaA)
according to the manufacturer's protocol. Briefly, osteoclasts
differentiated from bone marrow cells were fixed in 37%
formaldehyde for 30 sec at 4°C and rinsed thoroughly in deionized
water. TRAP staining solution was mixed and preheated to 37°C and
the fixed cells were added and incubated for 1 h at 37°C. Cells
were counterstained with hematoxylin for 30 sec at room temperature
and the number of osteoclasts (defined as multinuclear
TRAP-positive cells) per well was counted under a bright field
inverted microscope (magnification, ×200; DMI3000; Leica
Microsystems GmbH, Wetzlar, Germany).
Bone resorption assay and
assessments
A bone resorption assay was performed as previously
described (22). Briefly, bone
marrow-derived macrophages (1×106 cells/well) were
seeded in Osteo-Assay Surface 24-well plates (Sigma Aldrich; Merck
KGaA); osteoclastic differentiation and culture were performed as
above. Plates were examined under a bright field inverted
microscope (magnification, ×10) and images of 10 random fields/well
were captured. The percentage and number of resorption areas were
calculated using with ImageJ software 1.50i (National Institutes of
Health, Bethesda, MD, USA).
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was extracted from cultured cells using
TRIzol (Invitrogen; Thermo Fisher Scientific, Inc.) and RT was
performed using the PrimeScript™ RT reagent kit (Takara Bio, Inc.,
Otsu, Japan) at 37°C for 30 min and 85°C for 1 min according to the
manufacturer's protocol. All primers were provided by Invitrogen
(Thermo Fisher Scientific, Inc.) and were as follows: TRAP, forward
5′-ACCATTGTTAGCCACATACG-3′ and reverse 5′-GTGAAACCGCAAGTAGCC-3;
matrix metalloproteinase-9 (MMP-9), forward
5′-AGTTTGGTGTCGCGGAGCAC-3′ and reverse 5′-TACATGAGCGCTTCCGGCAC-3′;
nuclear factor of activated T-cells, cytoplasmic 1 (NFATc1),
forward 5′-GGGTCAGTGTGACCGAAGAT-3′ and reverse
5′-GGAAGTCAGAAGTGGGTGGA-3′; proto-oncogene tyrosine-protein kinase
Src (c-Src), forward 5′-CCAGGCTGAGGAGTGGTACT-3′ and reverse
5′-CAGCTTGCGGATCTTGTAGT-3′; FBJ osteosarcoma oncogene (c-FOS),
forward 5′-CCAGTCAAGAGCATCAGCAA-3′ and reverse
5′-AAGTAGTGCAGCCCGGAGTA-3′; Cathepsin K (CtsK), forward
5′-GAAGAAGACTCACCAGAAGCAG-3′ and reverse
5′-TCCAGGTTATGGGCAGAGATT-3′; calcitonin receptor (CTR), forward
5′-CCTGACAGCAACCGAACC-3′ and reverse 5′-GCAACCAAAGCAGCAATC-3′;
GAPDH, forward 5′-TGTAGACCATGTAGTTGAGGTCA-3′ and reverse
5′-AGGTCGGTGTGAACGGATTTG-3′. qPCR reactions were performed as
follows: 95°C for 30 sec followed by 40 cycles of 95°C for 5 sec
and 60°C for 34 sec, using an ABI-7500 Fast Real-Time PCR System
(Applied Biosystems; Thermo Fisher Scientific, Inc.) in a total
volume of 25 µl containing 2 µM primers and 12.5 µl of the
SYBR® Premix Ex Taq™ (Takara Bio, Inc.). Results were
quantified using the 2−ΔΔCq method (23).
Bone histomorphometric analysis
The proximal tibia metaphysis and tibia shaft were
fixed at 4°C for 48 h in 4% paraformaldehyde and decalcified in 10%
EDTA for 3–4 weeks. Tibias were subsequently embedded in paraffin
and cut into 4-µm sections. Following deparaffinization and
rehydration, sections were stained with hematoxylin and eosin
(H&E) for 1 min at room temperature. Terminology recommended by
the Histomorphometry Nomenclature Committee of the American Society
for Bone and Mineral Research (24) and the BIOQUANT OSTEO 2009
morphometric measuring system (Bioquant Image Analysis Corporation,
Nashville, TN, USA) were used in the present study. The proximal
tibia metaphysis histomorphometric perimeters were determined by
measuring the areas under 1–3 µm from the growth plate excluding
the primary spongiosa and trabeculae connected to the cortical
bone. The lower end of the tibia was separated from the fibula and
prepared for tibia shaft transection and histomorphometric
perimeters were determined.
Statistical analysis
Two-tailed unpaired t-tests with Welch's correction
were used for comparisons between the two groups, using GraphPad
Prism 6.01 (GraphPad Software, Inc., La Jolla, CA, USA). Data are
presented as the mean + standard error of the mean of three
independent experiments. P<0.05 was considered to indicate a
statistically significant difference.
Results
Spontaneous dwarfism in
ADKMMC-KO mice
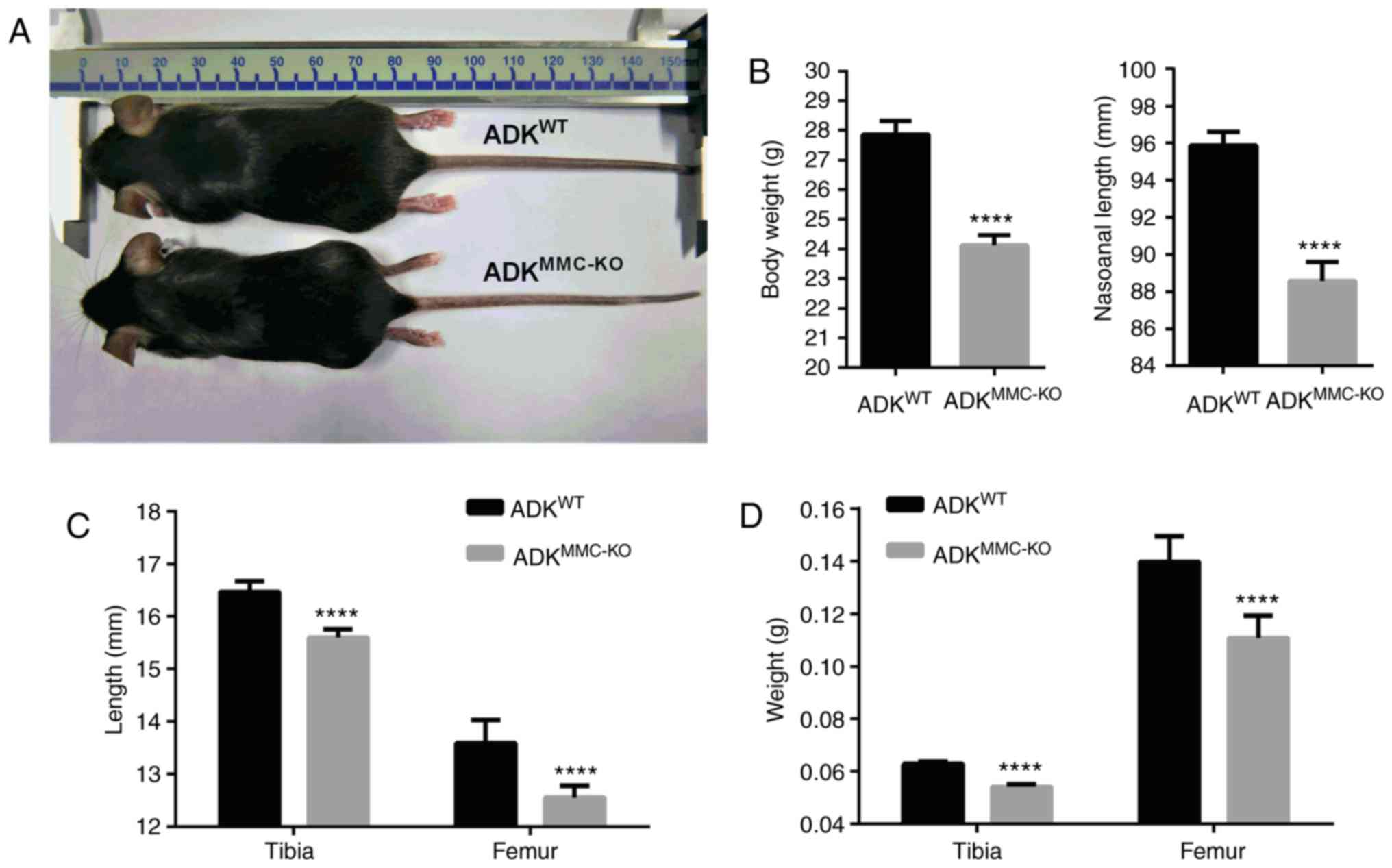
Primordial dwarfism was observed in
ADKMMC-KO mice. Compared with male ADKWT
mice, the appearance of ADKMMC-KO mice was not markedly
different, however the mice appeared markedly smaller at 4 months
old (Fig. 1A). Measurements
revealed that the body weight and nasoanal length of
ADKMMC-KO mice was significantly decreased compared with
ADKWT mice (P<0.0001; Fig. 1B).
To investigate whether the dwarfism observed in
ADKMMC-KO mice was associated with skeletal growth, the
tibia and femur were assessed. The results demonstrated that the
tibia and femur lengths were significantly shorter in
ADKMMC-KO mice compared with ADKWT mice
(P<0.0001; Fig. 1C).
Furthermore, it was demonstrated that the tibia and femur weigh
significantly less in ADKMMC-KO mice compared with
ADKWT mice (P<0.0001; Fig. 1D). These results suggest that
conditional ADK knockout in myeloid monocyte cells causes skeletal
dysostosis in mice.
Abnormal bone formation in
ADKMMC-KO mice
To further investigate the causes of skeletal
dysostosis in ADKMMC-KO mice, bone samples underwent
histomorphometric analysis. In the proximal tibia metaphysis, fewer
bone trabeculae and a smaller volume of trabecular bones was
observed in ADKMMC-KO mice compared with
ADKWT mice (Fig. 2A).
Quantitative analysis of bone trabecular parameters revealed that
the bone area (%Tb.Ar; P<0.01), trabecular thickness (Tb.Th;
P<0.05) and trabecular number (Tb.N; P<0.05) were
significantly decreased in ADKMMC-KO mice compared with
ADKWT mice (Fig. 2B).
Furthermore, trabecular separation (Tb.Sp; P<0.05) was
significantly increased in ADKMMC-KO mice compared with
ADKWT mice (Fig. 2B).
Analysis of osteoclastic parameters revealed that the percentage of
trabecular osteoclast surface to bone surface (%Oc.S/BS; P<0.01)
and ratio of osteoclast number to bone surface (N.Oc/BS; P<0.01)
were significantly increased in ADKMMC-KO mice compared
with ADKWT mice (Fig.
2B). However, the adherent perimeter of osteoblasts (%Ob.S/BS;
P<0.01) and number of osteoblasts per unit bone surface
(N.Ob/BS; P<0.01), were significantly lower in in
ADKMMC-KO mice compared with ADKWT mice
(Fig. 2B).
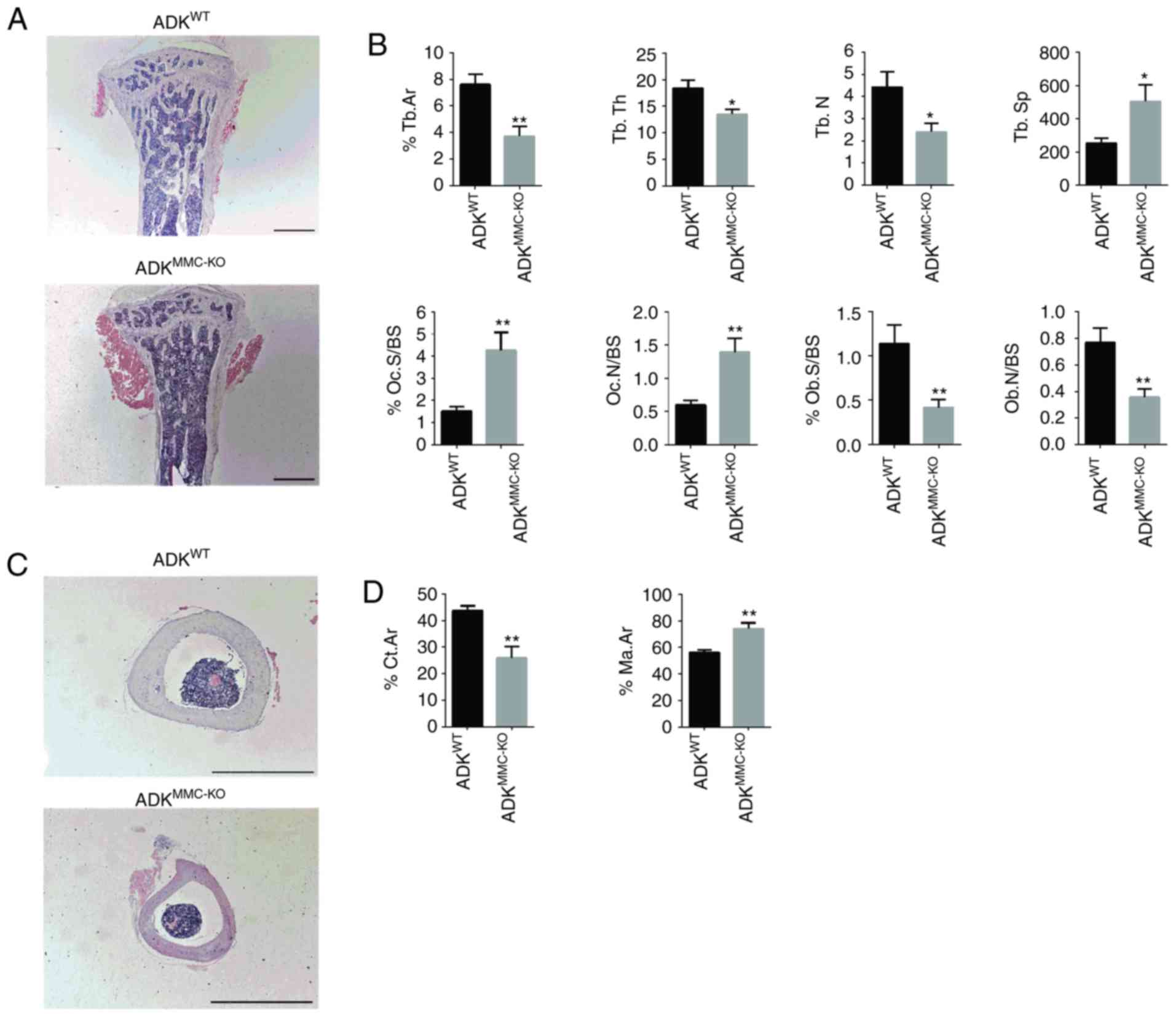 | Figure 2Histological analysis of the tibia
from ADKMMC-KO mice. (A) H&E staining of the
proximal tibia metaphysis of ADKMMC-KO and
ADKWT mice. (B) Bone histomorphometric analysis of
%Tb.Ar, Tb.Th, Tb.N, Tb.Sp, %Oc.S/BS, N.Oc/BS, %Ob.S/BS and N.Ob/BS
for ADKWT and ADKMMC-KO mice. (C) H&E
staining of the tibia shaft from ADKMMC-KO and
ADKWT mice. (D) Bone histomorphometric analysis of
%Ct.Ar and %Ma.Ar in the middle part of tibia shaft from
ADKWT and ADKMMC-KO mice. n=10 per group.
Scale bar=200 µm. *P<0.005 and **P<0.01
vs. ADKWT. ADK, adenosine kinase; ADKWT, ADK
wild type; ADKMMC-KO, ADK knockout in myeloid monocyte
cells; H&E, hematoxylin and eosin; %Tb.Ar, percentage of bone
area; Tb.Th, trabecular thickness; Tb.N, trabecular number; Tb.Sp,
trabecular separation; %Oc.S/BS, percentage of adherent perimeter
cells; N.Oc/BS, number of osteoclasts per unit bone surface;
%Ob.S/BS, percentage of adherent perimeter osteoblasts; N.Ob/OS,
number of osteoblasts per unit bone surface; %Ct.Ar, percentage of
cortical bone area; %Ma.Ar, bone marrow cavity area. |
The tibial shaft of in ADKMMC-KO mice was
also examined and, compared with ADKWT mice, the
cortical bone was markedly reduced and an expansive bone marrow
cavity was observed (Fig. 2C).
Quantitative analysis revealed that the percentage of cortical bone
area was significantly decreased (%Ct.Ar, P<0.01) and the bone
marrow cavity area was significantly increased (%Ma.Ar; P<0.01)
in ADKMMC-KO mice (Fig.
2D). These results suggest that conditional ADK knockout in
myeloid monocyte cells causes defective bone formation in
ADKMMC-KO mice.
Inhibition of ADK promotes
osteoclastogenesis
To further understand whether spontaneous
osteoporosis in ADKMMC-KO mice is due to ADK inhibition,
bone marrow macrophages (BMMs) were harvested from 2-month-old male
ADKWT and ADKMMC-KO mice. Western blotting
revealed that the expression of ADK protein was significantly
decreased in ADKMMC-KO compared with ADKWT
mice (Fig. 3A, P<0.001). BMMs
were subsequently exposed to M-CSF and RANKL for 8 days to induce
osteoclastic differentiation. A TRAP assay was performed to reveal
TRAP-positive cells and the typical multinucleated characteristics
of in vitro differentiated osteoclasts were observed
(Fig. 3B). Notably, larger
TRAP-positive osteoclasts were observed in ADKMMC-KO
cultures compared with those from ADKWT mice (Fig. 3B). It was also demonstrated that
BMMs derived from ADKMMC-KO mice formed significantly
more osteoclasts compared with those derived from ADKWT
mice (P<0.01; Fig. 3B).
Factors associated with osteoclast formation were evaluated using
RT-qPCR and the results indicated that relative levels of TRAP
(P<0.01) and NFATc1 (P<0.05) mRNA were significantly
increased in ADKMMC-KO osteoclasts compared with
ADKWT osteoclasts (Fig.
3C); however, no significant difference was observed in c-FOS
expression (Fig. 3C).
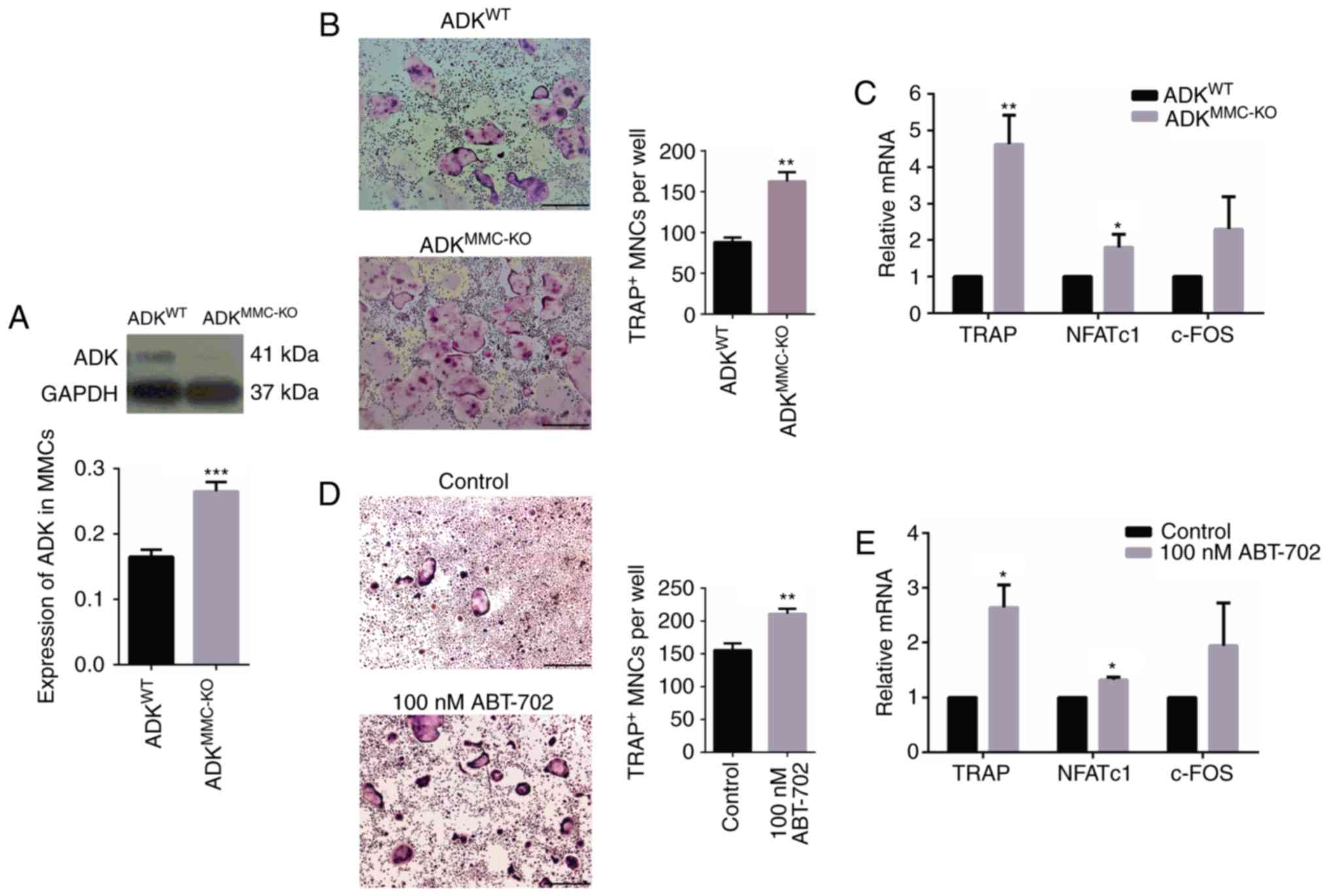 | Figure 3ADK knockdown enhances
osteoclastogenesis and the expression of associated genes. (A)
Expression of ADK protein in BMMs from 2-month male
ADKWT and ADKMMC-KO mice. (B) TRAP staining
of osteoclasts generated from BMMs of ADKWT and
ADKMMC-KO mice. (C) Relative TRAP, NFATc1 and c-FOS mRNA
in osteoclasts generated from BMMs of ADKWT and
ADKMMC-KO mice. (D) TRAP staining analysis of
osteoclasts generated from BMMs of ADKWT mice treated
with 100 nM ABT-702. (E) Relative TRAP, NFATc1 and c-FOS mRNA
levels in osteoclasts generated from BMMs of ADKWT mice treated
with 100 nM ABT-702 Scale bar=500 µm. *P<0.05,
**P<0.01 and ***P<0.01 vs. control.
ADK, adenosine kinase; BMMs, bone marrow macrophages;
ADKWT, ADK wild type; ADKMMC-KO, ADK knockout
in myeloid monocyte cells; TRAP, tartrate-resistant acid
phosphatase; NFATc1, nuclear factor of activated T-cells,
cytoplasmic 1; Control, untreated ADKWT mice. |
To further verify the role of ADK in osteoclast
formation, the ADK inhibitor ABT-702 was applied to BMMs from
ADKWT mice to induce osteoclastic differentiation
(Fig. 3D). Treatment with 100 nM
ABT-702 significantly induced TRAP-positive osteoclast formation
compared with untreated ADKWT BMMs (P<0.01; Fig. 3D). The expression of TRAP
(P<0.05) and NFATc1 (P<0.05) mRNA was significantly increased
by treatment with 100 nM ABT0702 compared with untreated
ADKWT BMMs, similar to ADKMMC-KO cultures
(Fig. 3E). These results suggest
that ADK inhibition induces osteoclast formation.
ADK inhibition increases osteoclastic
resorption activity
To investigate whether ADK inhibition affects
osteoclastic resorption function, BMMs derived from 2-month-old
male ADKWT and ADKMMC-KO mice were incubated
with M-CSF and RANKL. Following 7 days of culture, the resorption
area of osteoclasts induced from ADKMMC-KO BMMs was
significantly greater than that of osteoclasts induced from
ADKWT BMMs (P<0.05; Fig. 4A). Similarly, the number of
resorption areas per well was significantly greater in the
ADKMMC-KO group compared with the ADKWT GROUP
(P<0.05; Fig. 4A). Factors
associated with osteoclast formation were further evaluated in
these cultures using RT-qPCR the results demonstrated that relative
levels of MMP9 (P<0.01), CtsK (P<0.001) and CTR (P<0.01)
mRNA were significantly increased in ADKMMC-KO
osteoclasts compared with ADKWT osteoclasts (Fig. 4B). However, no significant
difference was observed in c-Src expression (Fig. 4B).
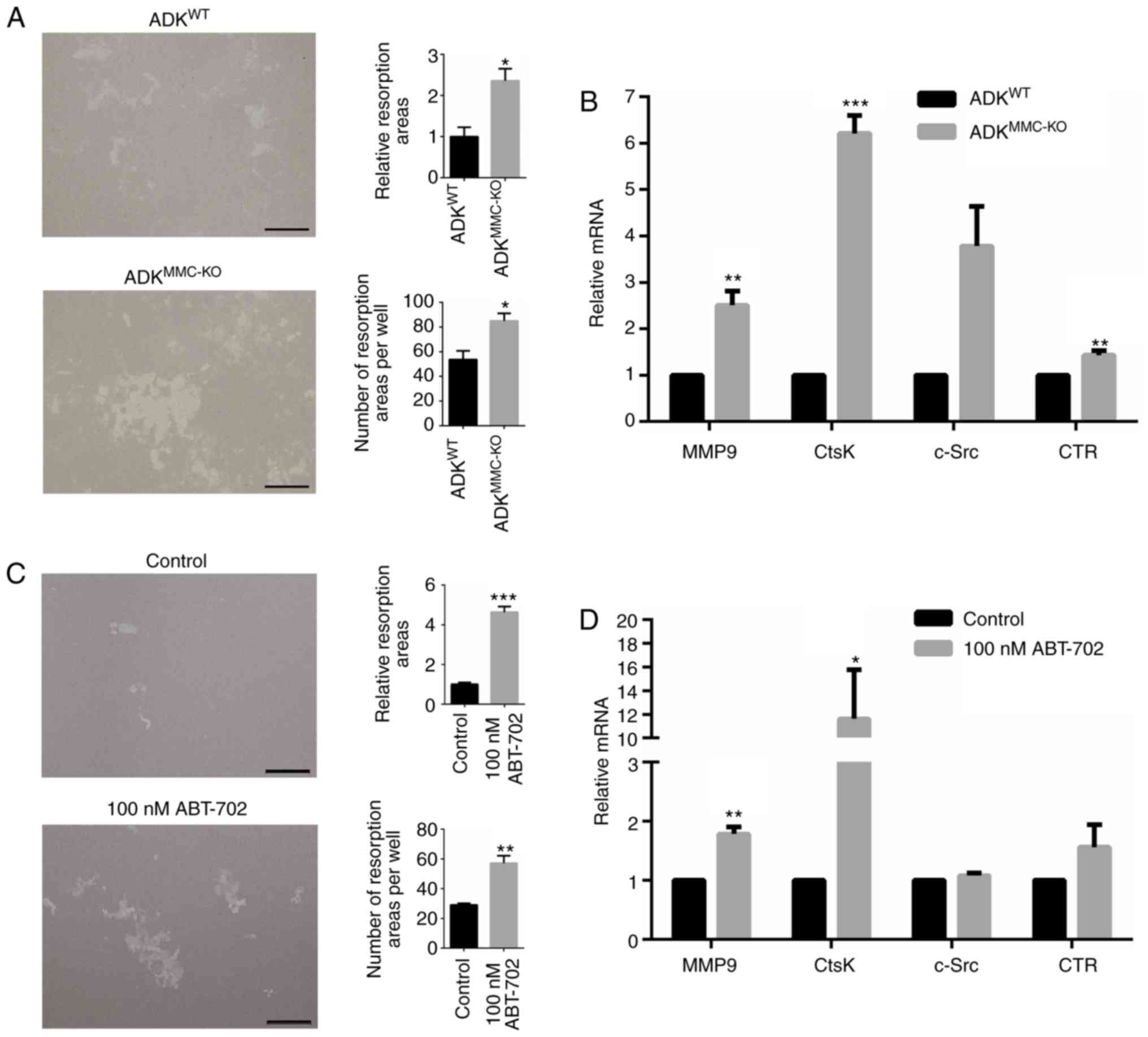 | Figure 4ADK knockdown enhances osteoclastic
resorption activity and the expression of associated genes. (A) The
resorption activity of osteoclasts generated from BMMs of
ADKWT and ADKMMC-KO mice. (B) Relative MMP9,
CtsK, c-Src and CTR mRNA expression in osteoclasts generated from
BMMs of ADKWT and ADKMMC-KO mice. (C) The
resorption activity of osteoclasts generated from BMMs of
ADKWT treated with 100 nM ABT-702. (D) Relative MMP9,
CtsK, c-Src and CTR mRNA expression in osteoclasts generated from
BMMs of ADKWT mice treated with 100 nM ABT-702. Scale
bar=200 µm. *P<0.005, **P<0.01 and
***P<0.001 vs. control. ADK, adenosine kinase; BMMs,
bone marrow macrophages; ADKWT, ADK wild type;
ADKMMC-KO, ADK knockout in myeloid monocyte cells; MMP9,
matrix metalloproteinase 9; c-FOS, FBJ osteosarcoma oncogene; CtsK,
Cathepsin K; c-Src, proto-oncogene tyrosine-protein kinase Src;
CTR, calcitonin receptor; Control, untreated ADKWT mice. |
BMMs derived from ADKWT mice were also
treated with 100 nM ABT-702 to further evaluate the effect of ADK
inhibition on osteoclastic resorption. The relative resorption area
(P<0.001) and number of resorption areas per well (P<0.01)
was significantly increased in cells treated with ABT-702 compared
with untreated BMMs derived from ADKWT mice (Fig. 4C). The relative expression of MMP9
(P<0.01) and CtsK (P<0.05) mRNA was significantly increased
in ABT-702 cultures compared with untreated BMMs derived from
ADKWT mice (Fig. 4D).
However, no significant difference was observed in c-Src and CTR
levels (Fig. 4D). These results
suggest that ADK inhibition may promote osteoclastic resorption
activity
Discussion
In the present study, a mouse model with ADK
deficient myeloid monocyte cells (ADKMMC-KO) was
established by Cre/LoxP site-specific recombination. As a result of
this conditional ADK knockout, spontaneous dwarfism associated with
decreased bone mass was observed in adult mice. Further in
vitro investigations based on genetic and pharmaceutical ADK
disruption in bone marrow macrophages revealed that ADK inhibition
significantly increases osteoclastogenesis and bone resorption in
mouse BMMs. Collectively, these results suggest that ADK may be a
potential target for the treatment of metabolic bone diseases
associated with osteoclast.
Previous clinical studies have reported that human
patients with global ADK deficiency due to homozygous point
mutations exhibit developmental delay, including short stature,
frontal bossing and slender hands and fingers (16,25). A study involving homozygous
disruption of the ADK gene in mice also reported reduced body size
and weight within a few days of birth (1). In the present study,
ADKMMC-KO mice were characterized by spontaneous
dwarfism, including reduced body weight, nasoanal length, and
weight and length of tibia and femur. Histomorphometric analysis
revealed significantly decreased bone mass in proximal tibia
metaphysis and the middle of the tibial shaft in
ADKMMC-KO mice. Combined with previous reports, these
results indicate that ADK may serve an important role in bone
metabolism homeostasis.
Osteoclasts are highly specialized cells that are
derived from the monocyte/macrophage lineage of the bone marrow
(26). lysMcre mice
have been used extensively to investigate osteoclast function in
previous studies (27–29). In the present study, osteoclastic
changes in ADKMMC-KO mice were assessed in vivo
using histomorphometric analysis and in vitro using a BMM
cell culture. The results revealed increased osteoclastic
parameters, including osteoclastogenesis and bone resorption,
associated with ADK deficiency. Histomorphometric analysis also
demonstrated that the function of osteoblasts derived from
pluripotent mesenchymal stem cells was inhibited in
ADKMMC-KO mice, suggesting that purinergic signaling
from ADK-deficient activated osteoclasts may affect osteoblasts by
interacting with adenosine receptors (10). Furthermore, the exosomes derived
from activated osteoclasts may also inhibit osteoblast activity
(30).
ADK converts intracellular adenosine to AMP; ADK
deficiency or dysfunction results in the accumulation of
intracellular adenosine (1).
Adenosine may be transported to the extracellular environment by
type 1 equilibrative nucleoside transporter (ENT1) (31); as such, the ADK-deficiency-induced
accumulation of intracellular adenosine may result in adenosine
being transported to the extracellular environment by ENT to
interact with adenosine receptors, including A1R (32,33) and A2BR (34,35), to promote osteoclast
differentiation. However, mice lacking ENT1 have been reported to
have reduced weight and lower bone density in the spine and femur
associated with increased osteoclast activity (36). This suggests that intracellular
adenosine may also participate in the functional regulation of
osteoclasts via a receptor independent pathway, although this
requires further investigation.
Metabolic analysis of liver tissue homogenates from
homozygous ADK deficient mice revealed a 40% decrease in AMP and
ADP and a 35% decrease in ATP (17). A previous study utilizing
osteoclast-specific conditional knockout Tfam mice, in which ATP
production is inhibited in osteoclasts, reported reduced growth and
increased bone-resorption (37).
An in vitro study demonstrated that ATP inhibits the
differentiation of osteoclast-like cells from RANKL-induced RAW
cells (38). A number of studies
have reported that metformin is able to effectively improve
osteoporosis in patients with long-term type 2 diabetes mellitus by
inhibiting osteoclast differentiation (39–41). The mechanism by this is achieved
is associated with the activation of AMP-activated protein kinase
(AMPK), a 'metabolic master switch' downstream of ADK (42–44). It was therefore speculated that
prolonged ADK deficiency in myeloid monocyte cells may result in
disordered purinergic signaling, which may contribute to bone
growth retardation and excessive osteoclast differentiation and
activation in ADKMMC-KO mice.
In summary, the results of the present study suggest
that ADK deficient myeloid monocyte cells contribute to reduced
bone formation in mice and this is associated with
osteoclastogenesis and bone resorption in ADKMMC-KO
mice.
Acknowledgments
The authors would like to thank Professor Yuqing Huo
for providing the ADK conditional knockout mice. The present study
was supported by the Guangdong Provincial Science & Technology
Project (grant nos. 2013B060300025, 2015A030303006 and
2016A030313790 to G.L.) and the Natural Science Foundation of China
(grant nos. 31702074 to G.L. and 31472061 to Y.Z.).
Notes
[1] Competing
interests
The authors declare that they have no competing
interests.
References
|
1
|
Boison D: Adenosine kinase: Exploitation
for therapeutic gain. Pharmacol Rev. 65:906–943. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Andres CM and Fox IH: Purification and
properties of human placental adenosine kinase. J Biol Chem.
254:11388–11393. 1979.PubMed/NCBI
|
|
3
|
Antonioli L, Blandizzi C, Pacher P and
Haskó G: Immunity, inflammation and cancer: A leading role for
adenosine. Nat Rev Cancer. 13:842–857. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Boison D: The adenosine kinase hypothesis
of epileptogenesis. Prog Neurobiol. 84:249–262. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Lusardi TA, Lytle NK, Szybala C and Boison
D: Caffeine prevents acute mortality after TBI in rats without
increased morbidity. Exp Neurol. 234:161–168. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Pignataro G, Simon RP and Boison D:
Transgenic overexpression of adenosine kinase aggravates cell death
in ischemia. J Cereb Blood Flow Metab. 27:1–5. 2007. View Article : Google Scholar
|
|
7
|
Pawelczyk T, Sakowicz M,
Szczepanska-Konkel M and Angielski S: Decreased expression of
adenosine kinase in streptozotocin-induced diabetes mellitus rats.
Arch Biochem Biophys. 375:1–6. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Giglioni S, Leoncini R, Aceto E, Chessa A,
Civitelli S, Bernini A, Tanzini G, Carraro F, Pucci A and Vannoni
D: Adenosine kinase gene expression in human colorectal cancer.
Nucleosides Nucleotides Nucleic Acids. 27:750–754. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Raggatt LJ and Partridge NC: Cellular and
molecular mechanisms of bone remodeling. J Biol Chem.
285:25103–25108. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Mediero A and Cronstein BN: Adenosine and
bone metabolism. Trends Endocrinol Metab. 24:290–300. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
He W, Mazumder A, Wilder T and Cronstein
BN: Adenosine regulates bone metabolism via A1, A2A, and A2B
receptors in bone marrow cells from normal humans and patients with
multiple myeloma. FASEB J. 27:3446–3454. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
He W and Cronstein BN: Adenosine A1
receptor regulates osteoclast formation by altering TRAF6/TAK1
signaling. Purinergic Signal. 8:327–337. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Mediero A, Kara FM, Wilder T and Cronstein
BN: Adenosine A2A receptor ligation inhibits osteoclast formation.
Am J Pathol. 180:775–786. 2012. View Article : Google Scholar :
|
|
14
|
Corciulo C, Wilder T and Cronstein BN:
Adenosine A2B receptors play an important role in bone homeostasis.
Purinergic Signal. 12:537–547. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Rath-Wolfson L, Bar-Yehuda S, Madi L,
Ochaion A, Cohen S, Zabutti A and Fishman P: IB-MECA, an A3
adenosine receptor agonist prevents bone resorption in rats with
adjuvant induced arthritis. Clin Exp Rheumatol. 24:400–406.
2006.PubMed/NCBI
|
|
16
|
Staufner C, Lindner M, Dionisi-Vici C,
Freisinger P, Dobbelaere D, Douillard C, Makhseed N, Straub BK,
Kahrizi K, Ballhausen D, et al: Adenosine kinase deficiency:
Expanding the clinical spectrum and evaluating therapeutic options.
J Inherit Metab Dis. 39:273–283. 2016. View Article : Google Scholar
|
|
17
|
Boison D, Scheurer L, Zumsteg V, Rülicke
T, Litynski P, Fowler B, Brandner S and Mohler H: Neonatal hepatic
steatosis by disruption of the adenosine kinase gene. Proc Natl
Acad Sci USA. 99:6985–6990. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Massaro EJ and Rogers JM: The Skeleton:
Biochemical, Genetic, and Molecular Interactions in Development and
Homeostasis. Humana Press; Totowa, NJ: 2004, View Article : Google Scholar
|
|
19
|
Sandau US, Colino-Oliveira M, Jones A,
Saleumvong B, Coffman SQ, Liu L, Miranda-Lourenço C, Palminha C,
Batalha VL, Xu Y, et al: Adenosine kinase deficiency in the brain
results in maladaptive synaptic plasticity. J Neurosci.
36:12117–12128. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Clausen BE, Burkhardt C, Reith W,
Renkawitz R and Förster I: Conditional gene targeting in
macrophages and granulocytes using LysMcre mice. Transgenic Res.
8:265–277. 1999. View Article : Google Scholar
|
|
21
|
Takahashi N, Udagawa N, Tanaka S and Suda
T: Generating murine osteoclasts from bone marrow. Methods Mol Med.
80:129–144. 2003.PubMed/NCBI
|
|
22
|
Tevlin R, McArdle A, Chan CK, Pluvinage J,
Walmsley GG, Wearda T, Marecic O, Hu MS, Paik KJ, Senarath-Yapa K,
et al: Osteoclast derivation from mouse bone marrow. J Vis Exp.
6:e520562014.
|
|
23
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
24
|
Dempster DW, Compston JE, Drezner MK,
Glorieux FH, Kanis JA, Malluche H, Meunier PJ, Ott SM, Recker RR
and Parfitt AM: Standardized nomenclature, symbols, and units for
bone histomorphometry: A 2012 update of the report of the ASBMR
Histomorphometry nomenclature committee. J Bone Miner Res. 28:2–17.
2013. View Article : Google Scholar :
|
|
25
|
Bjursell MK, Blom HJ, Cayuela JA, Engvall
ML, Lesko N, Balasubramaniam S, Brandberg G, Halldin M, Falkenberg
M, Jakobs C, et al: Adenosine kinase deficiency disrupts the
methionine cycle and causes hypermethioninemia, encephalopathy, and
abnormal liver function. Am J Hum Genet. 89:507–515. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Boyle WJ, Simonet WS and Lacey DL:
Osteoclast differentiation and activation. Nature. 423:337–342.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Albers J, Keller J, Baranowsky A, Beil FT,
Catala-Lehnen P, Schulze J, Amling M and Schinke T: Canonical Wnt
signaling inhibits osteoclastogenesis independent of
osteoprotegerin. J Cell Biol. 200:537–549. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Bartell SM, Kim HN, Ambrogini E, Han L,
Iyer S, Serra Ucer S, Rabinovitch P, Jilka RL, Weinstein RS, Zhao
H, et al: FoxO proteins restrain osteoclastogenesis and bone
resorption by attenuating H2O2 accumulation.
Nat Commun. 5:37732014. View Article : Google Scholar
|
|
29
|
Kim HN, Han L, Iyer S, de Cabo R, Zhao H,
O'Brien CA, Manolagas SC and Almeida M: Sirtuin1 suppresses
osteoclastogenesis by deacetylating FoxOs. Mol Endocrinol.
29:1498–1509. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Sun W, Zhao C, Li Y, Wang L, Nie G, Peng
J, Wang A, Zhang P, Tian W, Li Q, et al: Osteoclast-derived
microRNA-containing exosomes selectively inhibit osteoblast
activity. Cell Discov. 2:160152016. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Young JD, Yao SY, Sun L, Cass CE and
Baldwin SA: Human equilibrative nucleoside transporter (ENT) family
of nucleoside and nucleobase transporter proteins. Xenobiotica.
38:995–1021. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Kara FM, Chitu V, Sloane J, Axelrod M,
Fredholm BB, Stanley ER and Cronstein BN: Adenosine A1 receptors
(A1Rs) play a critical role in osteoclast formation and function.
FASEB J. 24:2325–2333. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Kara FM, Doty SB, Boskey A, Goldring S,
Zaidi M, Fredholm BB and Cronstein BN: Adenosine A(1) receptors
regulate bone resorption in mice: Adenosine A(1) receptor blockade
or deletion increases bone density and prevents ovariectomy-induced
bone loss in adenosine A(1) receptor-knockout mice. Arthritis
Rheum. 62:534–541. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Carroll SH, Wigner NA, Kulkarni N,
Johnston-Cox H, Gerstenfeld LC and Ravid K: A2B adenosine receptor
promotes mesenchymal stem cell differentiation to osteoblasts and
bone formation in vivo. J Biol Chem. 287:15718–15727. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Gharibi B, Abraham AA, Ham J and Evans BA:
Adenosine receptor subtype expression and activation influence the
differentiation of mesenchymal stem cells to osteoblasts and
adipocytes. J Bone Miner Res. 26:2112–2124. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Hinton DJ, McGee-Lawrence ME, Lee MR,
Kwong HK, Westendorf JJ and Choi DS: Aberrant bone density in aging
mice lacking the adenosine transporter ENT1. PLoS One.
9:e888182014. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Tsuchiya A, Kanno T, Saito M, Miyoshi Y,
Gotoh A, Nakano T and Nishizaki T: Intracellularly transported
adenosine induces apoptosis in (corrected) MCF-7 human breast
cancer cells by accumulating AMID in the nucleus. Cancer Lett.
321:65–72. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Hiken JF and Steinberg TH: ATP
downregulates P2X7 and inhibits osteoclast formation in RAW cells.
Am J Physiol Cell Physiol. 287:C403–C412. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Paschou SA, Dede AD, Anagnostis PG,
Vryonidou A, Morganstein D and Goulis DG: Type 2 diabetes and
osteoporosis: A guide to optimal management. J Clin Endocrinol
Metab. 102:3621–3634. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Hegazy SK: Evaluation of the
anti-osteoporotic effects of metformin and sitagliptin in
postmenopausal diabetic women. J Bone Miner Metab. 33:207–212.
2015. View Article : Google Scholar
|
|
41
|
Montagnani A, Gonnelli S, Alessandri M and
Nuti R: Osteoporosis and risk of fracture in patients with
diabetes: An update. Aging Clin Exp Res. 23:84–90. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Lee YS, Kim YS, Lee SY, Kim GH, Kim BJ,
Lee SH, Lee KU, Kim GS, Kim SW and Koh JM: AMP kinase acts as a
negative regulator of RANKL in the differentiation of osteoclasts.
Bone. 47:926–937. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Mai QG, Zhang ZM, Xu S, Lu M, Zhou RP,
Zhao L, Jia CH, Wen ZH, Jin DD and Bai XC: Metformin stimulates
osteoprotegerin and reduces RANKL expression in osteoblasts and
ovariectomized rats. J Cell Biochem. 112:2902–2909. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
McCarthy AD, Cortizo AM and Sedlinsky C:
Metformin revisited: Does this regulator of AMP-activated protein
kinase secondarily affect bone metabolism and prevent diabetic
osteopathy. World J Diabetes. 7:122–133. 2016. View Article : Google Scholar : PubMed/NCBI
|