Introduction
Biliary tract cancer (BTC) refers to a group of
cancers of the biliary tract, including gallbladder cancer,
cholangiocarcinoma of intrahepatic and extrahepatic bile ducts, and
cancers of the ampulla and papilla of Vater (1,2).
The incidence rate of BTC differs among geographic areas: It is
high in Asia, Latin America and eastern European countries, while
it is low in the US and certain western European countries
(2). Complete resection has been
considered the best treatment for BTC; however, most patients are
ineligible for surgery due to its rapid progression and
non-specific symptoms (3–5). Even patients who are treated with
surgery have a poor prognosis.
Although surgery remains the only curative treatment
option, chemotherapy prolongs the survival of patients with BTC
(6,7). Among the chemotherapeutic drugs,
gemcitabine and cisplatin have proven to be the most effective
first-line drugs (8–10). However, drug resistance to
gemcitabine limits its effect, and the median overall survival of
patients with advanced BTC who receive chemotherapy is only ~1 year
(11). Therefore, it is essential
to explore the potential mechanism underlying the resistance of BTC
to gemcitabine in order to enhance its effect and prolong patient
survival.
Midkine (MDK), a heparin-binding growth factor, was
first identified as a highly expressed factor involved in embryonic
development (12). MDK has been
reported to have important roles in the survival, growth and
migration of cells, which may contribute to oncogenesis and tumor
progression in numerous types of cancer (13–18). Several studies have demonstrated
that MDK mediates drug resistance. Mirkin et al (19) employed a cytokine complementary
DNA array to identify putative survival molecules in human
neuroblastoma and osteosarcoma cells and identified MDK as a lead
candidate responsible for doxorubicin resistance via regulation of
the AKT pathway. Furthermore, Lorente et al (20) identified MDK as a pivotal factor
involved in the resistance of glioma cells to the pro-autophagic
and anti-tumoral action of tetrahydrocannabinol by regulation of
the anaplastic lymphoma kinase receptor. Xu et al (21) proved that MDK, which activates AKT
and extracellular signal-regulated kinase by phosphorylation,
induced doxorubicin resistance in gastric cancer cells. Hu et
al (22) indicated that MDK
increased the drug-efflux ability in lymphoblastic leukemia,
thereby having an important role in multidrug resistance. These
results highlighted the fact that MDK may have an essential role in
cancer chemotherapy resistance. However, the role of MDK in the
drug resistance of BTC has remained largely elusive.
Epithelial to mesenchymal transition (EMT) is the
process wherein epithelial cells lose their apical-basal polarity
and cell-cell adhesion and transit to invasive mesenchymal cells.
EMT cells exhibit decreased expression of epithelial genes (e.g.,
E-cadherin) and increased expression of mesenchymal genes (e.g.,
vimentin) (23). The link between
EMT and drug resistance of cancer cells has been suggested in a
previous study. Furthermore, increasing evidence has indicated that
drug resistance of several cancer types, including lung (24), pancreatic (25), liver (26) and breast cancer (27), is frequently accompanied by EMT.
In BTC, EMT involves the invasion and migration of BTC cells.
However, evidence supporting the role of EMT in drug resistance of
BTC has remained insufficient.
Therefore, the present study aimed to determine the
association between MDK, EMT and gemcitabine resistance in BTC and
explore the potential mechanisms underlying gemcitabine
resistance.
Materials and methods
Cell culture and reagents
Two human BTC cell lines, RBE and GBC-SD, were
purchased from the American Type Culture Collection (Manassas, VA,
USA) and cultured according to the supplier's recommendation in
RPMI-1640 medium (Gibco; Thermo Fisher Scientific, Inc., Waltham,
MA, USA) supplemented with 10% fetal bovine serum (Gibco; Thermo
Fisher Scientific, Inc.) and maintained at 37°C in a humidified
atmosphere of air with 5% CO2. For hypoxia culture,
cells were maintained at 37°C in a humidified atmosphere with 5%
CO2, 1% O2 and 94% N2. MDK was
from Sigma-Aldrich (cat. no. SRP3114; Merck KGaA, Darmstadt,
Germany) and was used at a concentration of 50 ng/ml. Gemcitabine
(cat. no. S1714) was purchased from Selleck Chemicals (Houston, TX,
USA).
Cell viability assay
RBE and GBC-SD cells were seeded into 96-well
microplates at a density of 5,000 cells/well and cultured with
different concentrations of gemcitabine ranging from 0.00 to 0.06
µg/ml, MDK (50 ng/ml) or a combination of the two drugs for
48 h. Cell viability was detected using the Cell Counting Kit-8
(CCK-8) assay (Dojindo, Kumamoto, Japan) according to the
manufacturer's protocol. In brief, 100 µl medium and 10
µl CCK-8 solution were added to microplates, and the cells
were incubated for 2 h. The optical density at 450 nm was
determined using a MRX II microplate reader (Dynex, Chantilly, VA,
USA). Cell viability in each group was determined by comparison
with untreated control cells.
Cell proliferation assay
Cell proliferation was analyzed by
5-ethynyl-2′-deoxyuridine (EdU) staining using the Click-iTEdU
Imaging kit (Invitrogen; Thermo Fisher Scientific, Inc.) following
the manufacturer's protocol. RBE or GBC-SD cells were treated with
gemcitabine alone or a combination of gemcitabine (0.03136
µg/ml for RBE; 0.1433 µg/ml for GBC-SD) and MDK (50
ng/ml) for 48 h, and then exposed to 10 µMEdU for 2 h at
37°C. The cells were then fixed with 3.7% formaldehyde for 15 min
at room temperature and treated with 0.5% Triton X-100 (Sangon
Biotech, Shanghai, China) for 20 min at room temperature for
permeabilization. After washing twice with PBS containing 3% bovine
serum albumin, the cells were treated with 0.5 ml of
Click-iT® reaction cocktail (Invitrogen; Thermo Fisher
Scientific, Inc.) for 30 min in the dark. Subsequently, the cell
DNA was stained with 1 ml 1X Hoechst 33342 (1:2,000 dilution) for
30 min. Finally, three random fields of view per slide were
selected under a fluorescence microscope (Olympus, Tokyo, Japan),
and the number of proliferative (EdU-positive) cells was
counted.
Cell transfection for RNA
interference
Human Twist small interfering RNA (siRNA) was
synthesized by Shanghai GeneChem Co., Ltd. (Shanghai, China). The
human Twist siRNA sequence was as follows: Twist1,
5′-GGUGUCUAAAUGCAUUCAUTT-3′ and 5′-AUGAAUGCAUUUAGACACCTT-3′;
Notch1, 5′-CCAACCCUGUCAAUGGCAATT-3′ and
5′-UUGCCAUUGACAGGGUUGGTT-3′; Scrambled siRNA,
5′-UUCUCCGAACGUGUCACGUTT-3′ and 5′-ACGUGACACGUUCGGAGAATT-3′. The
transfection was performed by using Lipofectamine 2000 (Invitrogen;
Thermo Fisher Scientific, Inc.) according to the manufacturer's
protocol.
Western blot analysis
The interference efficiency of Twist siRNA and its
effect on the expression of various proteins was determined by
western blot analysis (28). The
following antibodies were used: Anti-E-cadherin (cat. no. 3195),
anti-vimentin (cat. no. 5741), anti-Twist (cat. no. 46702),
anti-β-actin (cat. no. 8457) (all at 1:1,000 dilution; Cell
Signaling Technology, Inc., Danvers, MA, USA) and anti-Notch-1
(cat. no. ab8925; 1:1,000 dilution; Abcam, Cambridge, UK). The
corresponding secondary antibodies conjugated to horseradish
peroxidase (cat. no. ab98489; 1:2,000 dilution) were obtained from
Abcam. The grey value was analyzed by QuantityOnev. 4.62 software
(Bio-Rad Laboratories, Inc., Hercules, CA, USA).
Statistical analysis
Three independent experiments were performed for
each experiment. Experimental data were expressed as the mean ±
standard deviation. Analyses were performed using GraphPad Prism 5
(GraphPad Software, Inc., La Jolla, CA, USA). Comparisons among
datasets were performed by using one-way analysis of variance
followed by Tukey's post hoc test or a unpaired Student's t-test.
P<0.05 was considered to indicate a statistically significant
difference.
Results
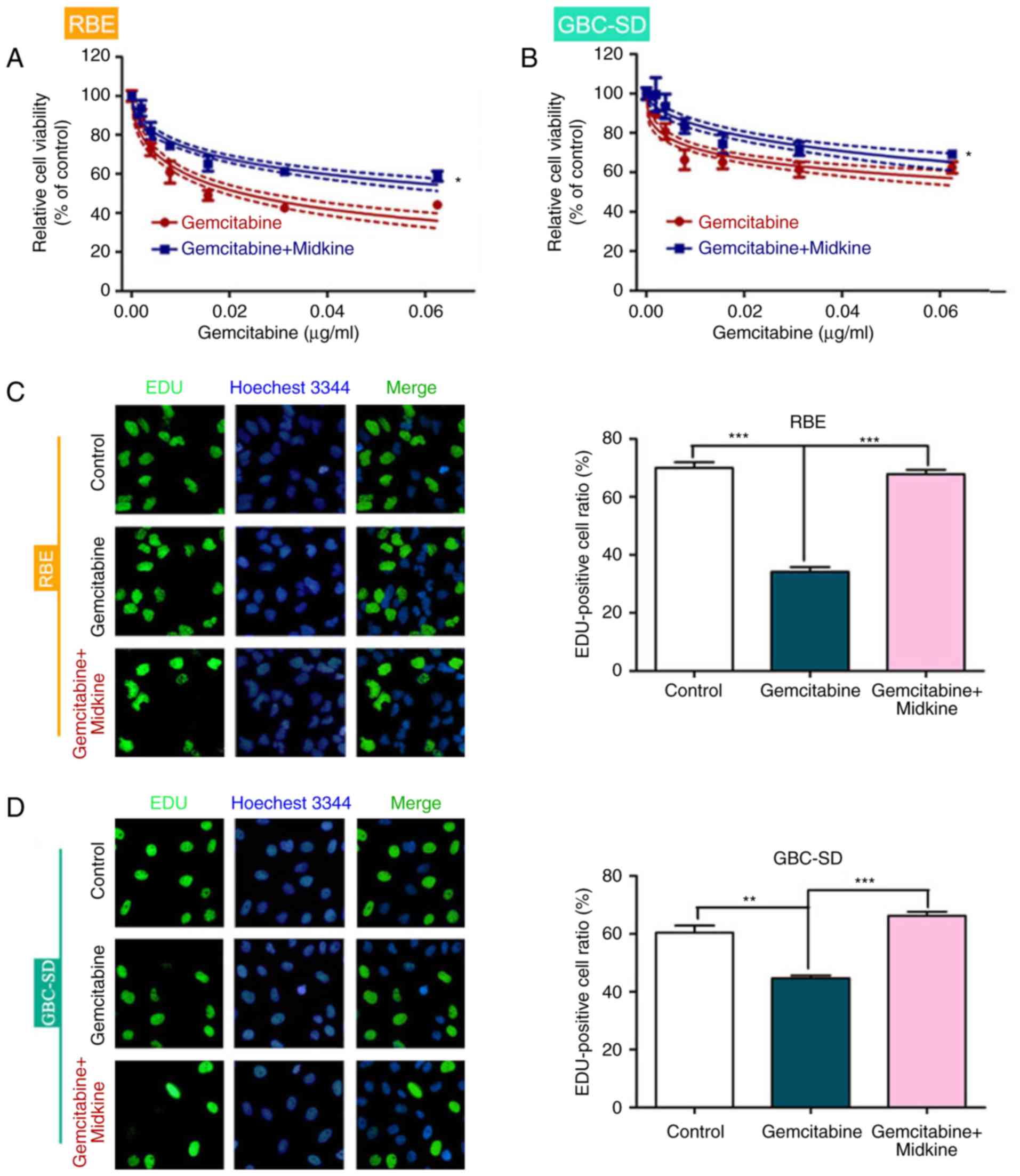
MDK induces gemcitabine resistance in BTC
cells
To determine whether MDK is involved in gemcitabine
resistance of BTC, the effect of MDK on BTC cell viability was
evaluated in the presence of different concentrations of
gemcitabine. BTC cells exhibited higher cell viability after
gemcitabine + MDK treatment than cells treated with gemcitabine
alone (Fig. 1A and B). In
addition, the EdU assay indicated that BTC cells had increased
proliferation in the presence of gemcitabine and MDK compared with
gemcitabine alone, which suggests that MDK induces gemcitabine
resistance in BTC (Fig. 1C and
D).
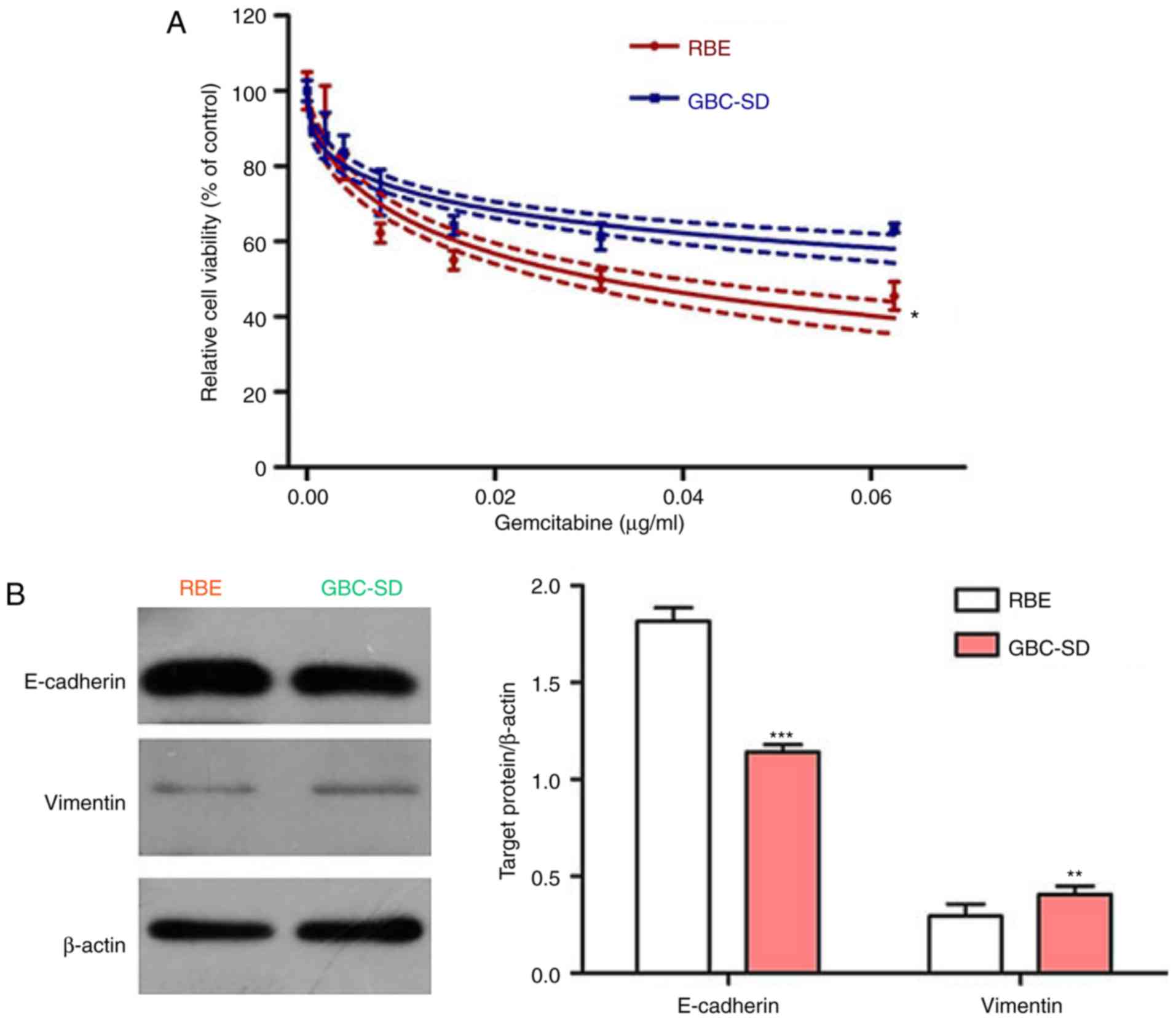
EMT is involved in gemcitabine resistance
of BTC cells
The EMT is known to be associated with
chemoresistance of cancers, and MDK was reported to induce EMT in
several cancer types (29–31).
Thus, the present study hypothesized that EMT may be involved in
gemcitabine resistance in BTC and that MDK may promote gemcitabine
resistance in BTC by regulating the EMT pathway. To prove this
hypothesis, the efficiency of gemcitabine on the two BTC cell lines
was first examined. The CCK-8 assay indicated that gemcitabine
effectively inhibited the viability of BTC cells in a
concentration-dependent manner. Of note, the BTC cell line RBE was
more sensitive to gemcitabine than the GBC-SD cell line (Fig. 2A). Subsequently, the expression of
the epithelial marker E-cadherin and the mesenchymal marker
vimentin was assessed in these two cell lines, revealing that the
cell line RBE had high E-cadherin levels, but low vimentin levels,
whereas the opposite results were observed in the GBC-SD cell line
(Fig. 2B). Accordingly, the
present results indicate that EMT may be involved in gemcitabine
resistance in BTC.
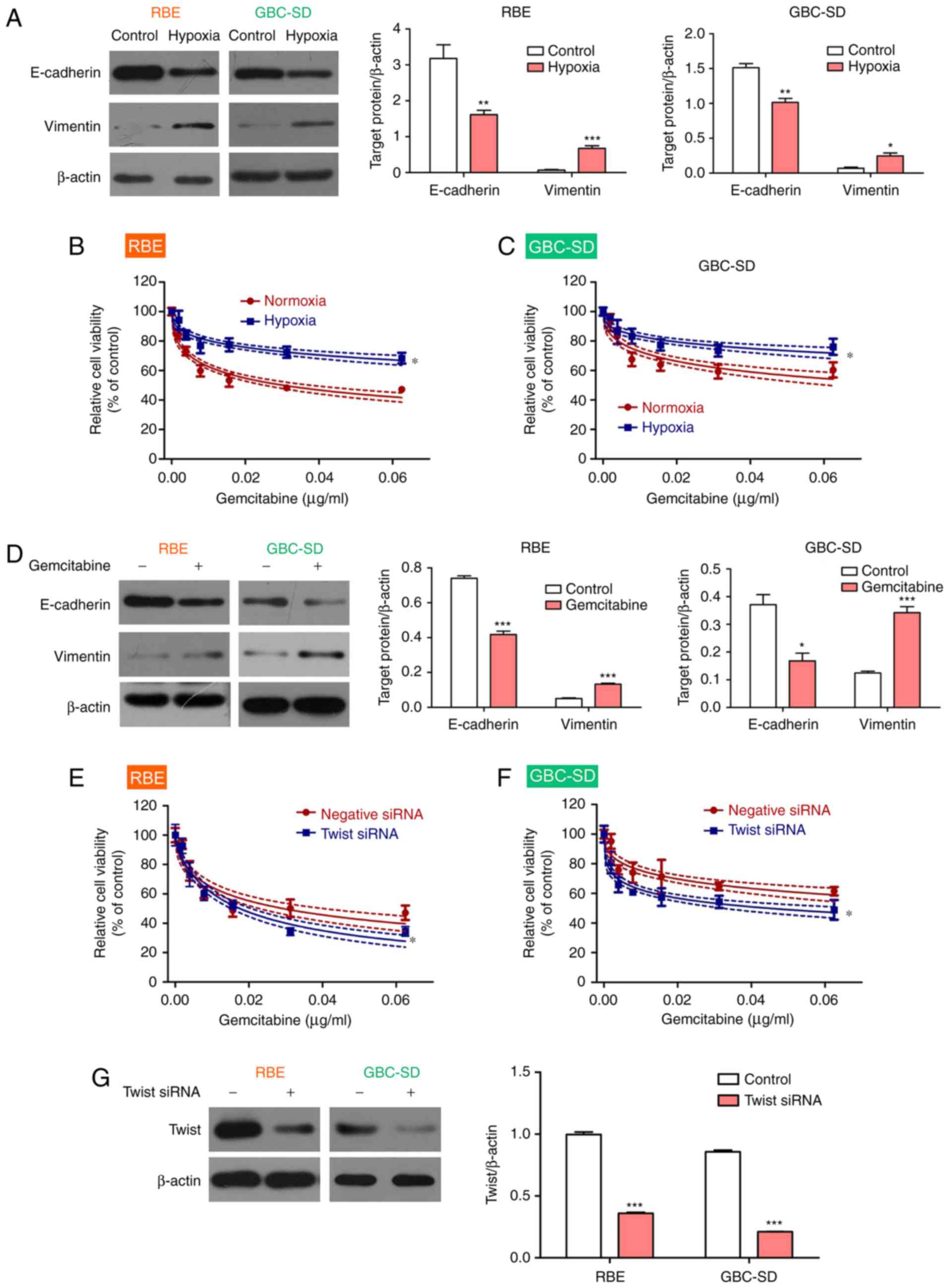
To confirm this result, the BTC cell lines were
exposed to hypoxic conditions to induce EMT, as reported previously
(32,33). The results demonstrated that
hypoxia upregulated the expression of vimentin and downregulated
the expression of E-cadherin in BTC cell lines, thereby promoting
EMT (Fig. 3A). As expected, BTC
cell viability increased in the presence of gemcitabine under
hypoxic conditions compared with that under normoxic conditions
(Fig. 3B and C). In addition,
gemcitabine treatment led to an upregulation of the expression of
vimentin and a downregulation of the expression of E-cadherin in
the BTC cell lines under normoxia conditions (Fig. 3D). To confirm the role of the EMT
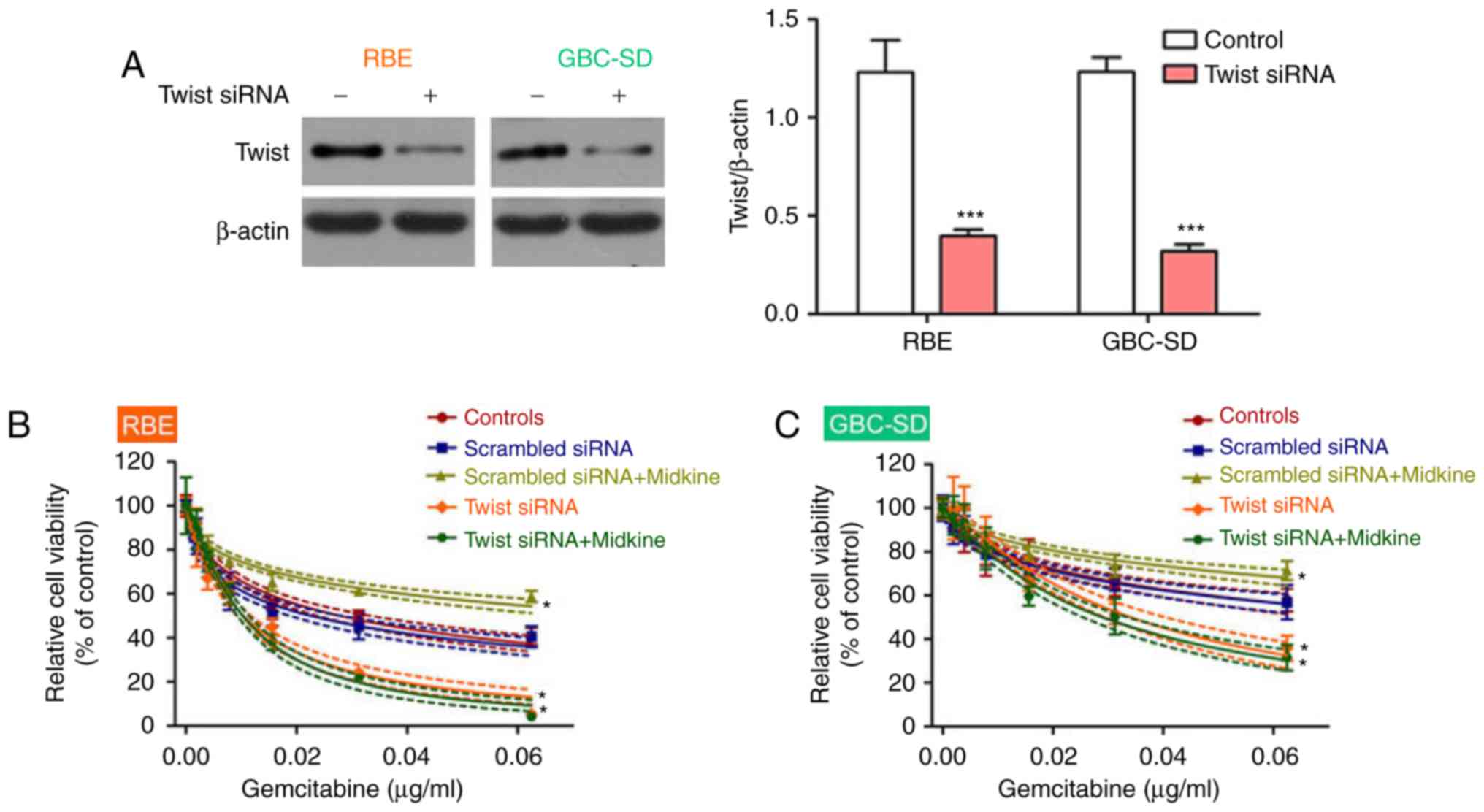
in gemcitabine resistance, Twist, a key regulator of EMT (34), was knocked down to block EMT in
BTC cell lines. The results demonstrated that the BTC cell lines
became more sensitive to gemcitabine after Twist inhibition
(Fig. 3E and F). Interference
efficiency of Twist1 was measured using western blot analysis
(Fig. 3G). Therefore, the present
results indicated that gemcitabine resistance in BTC may be
mediated via the EMT.
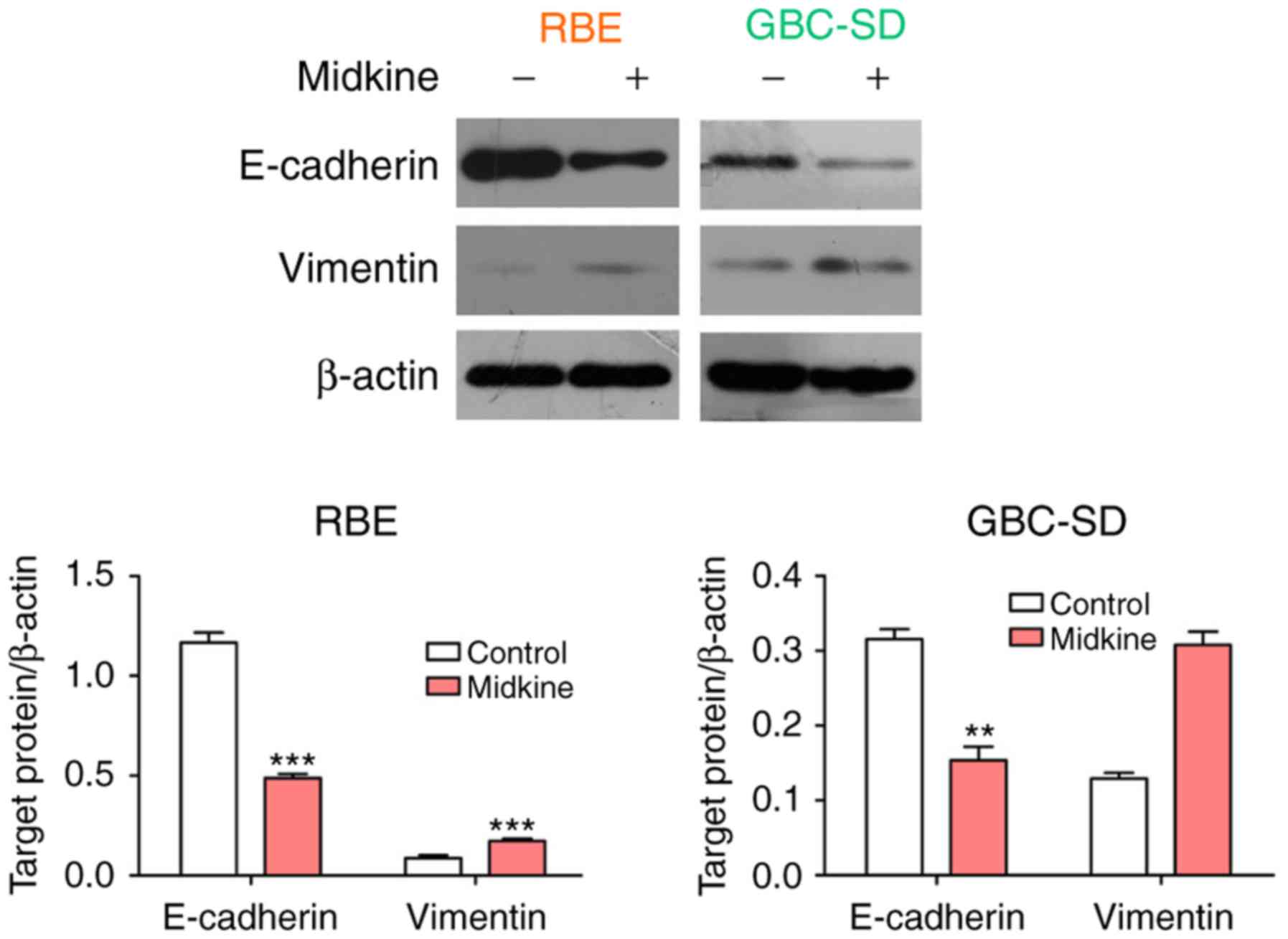
MDK mediates gemcitabine resistance in
BTC cells by regulating EMT
To further prove the abovementioned hypothesis, the
association between MDK and EMT in gemcitabine resistance was
examined. First, the expression of E-cadherin and vimentin was
detected in the two BTC cell lines cultured in the presence of MDK.
The results indicated that MDK treatment led to a significant
upregulation of vimentin expression and a downregulation of
E-cadherin expression in BTC cell lines (Fig. 4). Thereafter, the efficiency of
MDK to induce gemcitabine resistance was examined after EMT
blockage. To block EMT, Twist, the key molecule in the EMT pathway,
was inhibited using siRNA. The knockdown efficiency of Twist siRNA
was confirmed by western blot (Fig.
5A). As expected, cells transfected with scrambled siRNA were
more sensitive to treatment with gemcitabine compared with those in
the MDK+scrambled siRNA group, and after EMT inhibition with Twist
siRNA, the effect of MDK to induce gemcitabine resistance in BTC
cells was lost (Fig. 5B and C).
Hence, the present results proved the hypothesis that MDK promotes
gemcitabine resistance in BTC by regulating EMT.
Midkine induces EMT by upregulating
Notch-1 expression
The Notch pathway has a significant role in EMT of
cancer cells (35,36), and Notch-1 activation has been
reported to be linked to acquired chemoresistance in several cancer
types (37–40). Therefore, the role of MDK in
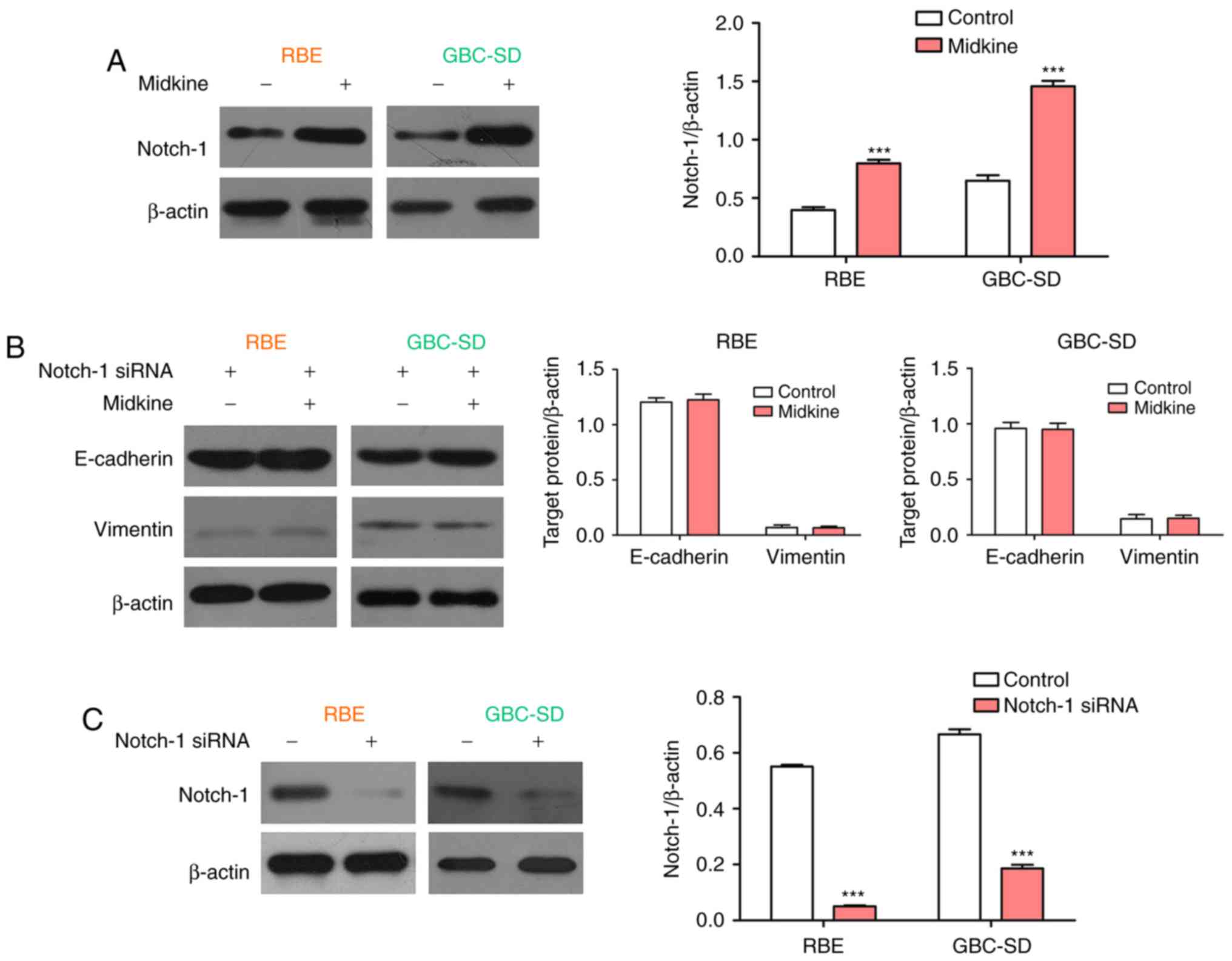
Notch-1 expression was then examined in the present study. The
western blot results demonstrated that MDK significantly promoted
the expression of Notch-1 in BTC cell lines (Fig. 6A). Subsequently, the expression of
E-cadherin and vimentin was examined in BTC cells treated with
Notch-1 siRNA or a combination of Notch-1 siRNA and MDK. The
results indicated that siRNA-mediated knockdown of Notch-1
completely abolished the regulatory effect of MDK on the expression
of E-cadherin and vimentin in BTC cells (Fig. 6B). The interference efficiency of
Notch1 was detected by western blot analysis (Fig. 6C). These results indicate that
Notch-1 is a mediator in MDK-induced EMT.
Discussion
Chemotherapy has been considered an effective
adjuvant therapy for BTC; however, drug resistance limits the
efficiency of chemotherapy (7,41).
Therefore, further studies are required to determine the potential
mechanism of drug resistance in BTC. In the present study, MDK was
demonstrated to induce drug resistance in BTC via induction of the
EMT through regulating the expression of the Notch-1 protein.
MDK is a growth factor that was first identified as
a mediator of retinoic acid-induced differentiation (12). Further studies indicated that it
was associated with drug resistance. Mirkin et al (19) proved that MDK was secreted from
drug-resistant cells and protected the neighboring drug-sensitive
cells from the toxicity of doxorubicin. Kang et al (42) identified >250 differentially
expressed genes in 5-fluorouracil-, cisplatin- or
doxorubicin-resistant gastric cancer cell lines by microarray
analysis and determined that MDK was overexpressed in all
drug-resistant cell lines. Qi et al (43) reported that MDK protected murine
kidney cells and cultured Wilms' tumor cells from cisplatin-induced
apoptotic cell death by upregulating the expression of B-cell
lymphoma 2. Regarding BTC, MDK was upregulated in intrahepatic
cholangiocarcinoma. However, little is known regarding the effect
of MDK on the drug resistance of BTC. Therefore, the present study
assessed this aspect and proved that MDK induced gemcitabine
resistance in BTC.
The EMT is known to be involved in cancer drug
resistance via various functions, including regulation of cancer
cell stemness, overexpression of ATP binding cassette transporters,
inhibition of epithelial growth factor receptor tyrosine kinase
inhibitor-induced apoptosis and alteration of the tumor
microenvironment (44). Previous
studies have reported that replication stress-induced MDK
expression activates Notch-2, which drives EMT and chemoresistance
in pancreatic cancer (30). The
MDK-induced crosstalk of Notch2/Janus kinase 2/signal transducer
and activator of transcription 3 signaling pathways regulates cell
plasticity and motility, thereby contributing to EMT in human
keratinocytes (31). In lung
adenocarcinoma, estrogen receptor β-mediated estradiol enhanced MDK
expression and increased EMT (29). Considering these previous studies,
it was hypothesized that MDK may mediate gemcitabine resistance in
BTC cells by regulating EMT. To the best of our knowledge, the
present study was the first to provide in vitro evidence to
prove this hypothesis.
The Notch signaling pathway has critical roles in
the development and progression of human cancers, as this pathway
is critically involved in numerous cellular processes, including
proliferation, survival, apoptosis, migration, invasion,
angiogenesis and metastasis. Emerging evidence suggests that Notch
regulates EMT, leading to tumor invasion and metastasis (35,36,45–49). Notch-1 has been reported to
promote EMT in several cancer types (50–52). Although the association between
MDK and Notch-2 is well known, the association between MDK and
Notch-1 has remained elusive. In the present study, MDK was
demonstrated to upregulate Notch-1 expression and it was revealed
that MDK-induced EMT was mediated by Notch-1. These results
highlight the role of Notch-1 in MDK-induced EMT.
To the best of our knowledge, the present study was
the first to provide evidence that MDK enhances gemcitabine
resistance in BTC cells via the Notch-1/EMT axis. Therefore,
targeting MDK or blocking/reversing EMT prior to or during
chemotherapy may force chemoresistant cells to revert to sensitive
cells and may thus provide a tremendous benefit to patients with
advanced chemoresistant cancers. Further study is required to
understand the precise molecular mechanisms underlying gemcitabine
resistance in BTC.
Acknowledgments
This study was financially supported by the National
Natural Science Foundation of China (grant no. 81501830), the
Zhejiang Province Natural Science Foundation of China (grant nos.
LY16H160041 and LY16H160041) and the Huzhou Science and Technology
Project (grant nos. 2014GZ11 and 2015GZ16).
Notes
[1] Competing
interests
The authors declare that they have no competing
interests.
References
|
1
|
Hennedige TP, Neo WT and Venkatesh SK:
Imaging of malignancies of the biliary tract-an update. Cancer
Imaging. 14:142014.
|
|
2
|
Randi G, Malvezzi M, Levi F, Ferlay J,
Negri E, Franceschi S and La Vecchia C: Epidemiology of biliary
tract cancers: An update. Ann Oncol. 20:146–159. 2009. View Article : Google Scholar
|
|
3
|
Horgan AM, Amir E, Walter T and Knox JJ:
Adjuvant therapy in the treatment of biliary tract cancer: A
systematic review and meta-analysis. J Clin Oncol. 30:1934–1940.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hezel AF and Zhu AX: Systemic therapy for
biliary tract cancers. Oncologist. 13:415–423. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Khan SA, Davidson BR, Goldin RD, Heaton N,
Karani J, Pereira SP, Rosenberg WM, Tait P, Taylor-Robinson SD,
Thillainayagam AV, et al: Guidelines for the diagnosis and
treatment of cholangiocarcinoma: An update. Gut. 61:1657–1669.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Goyal L, Chong DQ, Duda DG and Zhu AX:
Chemotherapy and antiangiogenics in biliary tract cancer. Lancet
Oncol. 16:882–883. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ghosn M, Kourie HR, El Rassy E, Chebib R,
El Karak F, Hanna C and Nasr D: Optimum chemotherapy for the
management of advanced biliary tract cancer. World J Gastroenterol.
21:4121–4125. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Park K, Kim KP, Park S and Chang HM:
Comparison of gemcitabine plus cisplatin versus capecitabine plus
cisplatin as first-line chemotherapy for advanced biliary tract
cancer. Asia Pac J Clin Oncol. 13:13–20. 2017. View Article : Google Scholar
|
|
9
|
Stein A, Arnold D, Bridgewater J,
Goldstein D, Jensen LH, Klümpen HJ, Lohse AW, Nashan B, Primrose J,
Schrum S, et al: Adjuvant chemotherapy with gemcitabine and
cisplatin compared to observation after curative intent resection
of cholangiocarcinoma and muscle invasive gallbladder carcinoma
(ACTICCA-1 trial)-a randomized, multidisciplinary, multinational
phase III trial. BMC Cancer. 15:5642015. View Article : Google Scholar
|
|
10
|
Lamarca A, Benafif S, Ross P, Bridgewater
J and Valle JW: Cisplatin and gemcitabine in patients with advanced
biliary tract cancer (ABC) and persistent jaundice despite optimal
stenting: Effective intervention in patients with luminal disease.
Eur J Cancer. 51:1694–1703. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Yonemoto N, Furuse J, Okusaka T, Yamao K,
Funakoshi A, Ohkawa S, Boku N, Tanaka K, Nagase M, Saisho H and
Sato T: A multi-center retrospective analysis of survival benefits
of chemotherapy for unresectable biliary tract cancer. Jpn J Clin
Oncol. 37:843–851. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Böhlen P and Kovesdi I: HBNF and MK
members of a novel gene family of heparin-binding proteins with
potential roles in embryogenesis and brain function. Prog Growth
Factor Res. 3:143–157. 1991. View Article : Google Scholar
|
|
13
|
Vu Van D, Heberling U, Wirth MP and
Fuessel S: Validation of the diagnostic utility of urinary midkine
for the detection of bladder cancer. Oncol Lett. 12:3143–3152.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Edfeldt K, Daskalakis K, Bäcklin C, Norlén
O, Tiensuu Janson E, Westin G, Hellman P and Stålberg P: DcR3, TFF3
and midkine are novel serum biomarkers in small intestinal
neuroendocrine tumors. Neuroendocrinology. 105:170–181. 2017.
View Article : Google Scholar
|
|
15
|
Krzystek-Korpacka M, Gorska S, Diakowska
D, Kapturkiewicz B, Podkowik M, Gamian A and Bednarz-Misa I:
Midkine is up-regulated in both cancerous and inflamed bowel,
reflecting lymph node metastasis in colorectal cancer and clinical
activity of ulcerative colitis. Cytokine. 89:68–75. 2017.
View Article : Google Scholar
|
|
16
|
Vongsuvanh R, van der Poorten D, Iseli T,
Strasser SI, McCaughan GW and George J: Midkine increases
diagnostic yield in AFP negative and NASH-related hepatocellular
carcinoma. PLoS One. 11:e01558002016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Yamashita T, Shimada H, Tanaka S, Araki K,
Tomifuji M, Mizokami D, Tanaka N, Kamide D, Miyagawa Y, Suzuki H,
et al: Serum midkine as a biomarker for malignancy, prognosis, and
chemosensitivity in head and neck squamous cell carcinoma. Cancer
Med. 5:415–425. 2016. View
Article : Google Scholar : PubMed/NCBI
|
|
18
|
Yao J, Li WY and Gao SG: The advances of
Midkine with peripheral invasion in pancreatic cancer. Am J Cancer
Res. 5:2912–2917. 2015.PubMed/NCBI
|
|
19
|
Mirkin BL, Clark S, Zheng X, Chu F, White
BD, Greene M and Rebbaa A: Identification of midkine as a mediator
for intercellular transfer of drug resistance. Oncogene.
24:4965–4974. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lorente M, Torres S, Salazar M, Carracedo
A, Hernández-Tiedra S, Rodríguez-Fornés F, García-Taboada E,
Meléndez B, Mollejo M, Campos-Martín Y, et al: Stimulation of ALK
by the growth factor midkine renders glioma cells resistant to
autophagy-mediated cell death. Autophagy. 7:1071–1073. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Xu YY, Mao XY, Song YX, Zhao F, Wang ZN,
Zhang WX, Xu HM and Jin F: Midkine confers Adriamycin resistance in
human gastric cancer cells. Tumor Biol. 33:1543–1548. 2012.
View Article : Google Scholar
|
|
22
|
Hu R, Yan Y, Li Q, Lin Y, Jin W, Li H, Lu
Y and Pang T: Increased drug efflux along with midkine gene high
expression in childhood B-lineage acute lymphoblastic leukemia
cells. Int J Hematol. 92:105–110. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Vaquero J, Guedj N, Clapéron A, Nguyen
Ho-Bouldoires TH, Paradis V and Fouassier L: Epithelial-mesenchymal
transition in cholangiocarcinoma: From clinical evidence to
regulatory networks. J Hepatol. 66:424–441. 2017. View Article : Google Scholar
|
|
24
|
Sung WJ, Kim H and Park KK: The biological
role of epithelial-mesenchymal transition in lung cancer (Review).
Oncol Rep. 36:1199–1206. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Nomura A, Majumder K, Giri B, Dauer P,
Dudeja V, Roy S, Banerjee S and Saluja AK: Inhibition of NF-kappa B
pathway leads to deregulation of epithelial-mesenchymal transition
and neural invasion in pancreatic cancer. Lab Invest. 96:1268–1278.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kim HS, Lee KS, Bae HJ, Eun JW, Shen Q,
Park SJ, Shin WC, Yang HD, Park M, Park WS, et al: MicroRNA-31
functions as a tumor suppressor by regulating cell cycle and
epithelial-mesenchymal transition regulatory proteins in liver
cancer. Oncotarget. 6:8089–8102. 2015.PubMed/NCBI
|
|
27
|
Zhang X, Liu X, Luo J, Xiao W, Ye X, Chen
M, Li Y and Zhang GJ: Notch3 inhibits epithelial-mesenchymal
transition by activating Kibra-mediated Hippo/YAP signaling in
breast cancer epithelial cells. Oncogenesis. 5:e2692016. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Xu G, Shao G, Pan Q, Sun L, Zheng D and Li
M: MicroRNA-9 regulates non-small cell lung cancer cell invasion
and migration by targeting eukaryotic translation initiation factor
5A2. Am J Transl Res. 9:478–488. 2017.PubMed/NCBI
|
|
29
|
Zhao G, Nie Y, Lv M, He L, Wang T and Hou
Y: ERβ-mediated estradiol enhances epithelial mesenchymal
transition of lung adenocarcinoma through increasing transcription
of midkine. Mol Endocrinol. 26:1304–1315. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Güngör C, Zander H, Effenberger KE,
Vashist YK, Kalinina T, Izbicki JR, Yekebas E and Bockhorn M: Notch
signaling activated by replication stress-induced expression of
midkine drives epithelial-mesenchymal transition and
chemoresistance in pancreatic cancer. Cancer Res. 71:5009–5019.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Huang Y, Hoque MO, Wu F, Trink B,
Sidransky D and Ratovitski EA: Midkine induces
epithelial-mesenchymal transition through Notch2/Jak2-Stat3
signaling in human keratinocytes. Cell Cycle. 7:1613–1622. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Chen S, Chen JZ, Zhang JQ, Chen HX, Yan
ML, Huang L, Tian YF, Chen YL and Wang YD: Hypoxia induces
TWIST-activated epithelial-mesenchymal transition and proliferation
of pancreatic cancer cells in vitro and in nude mice. Cancer Lett.
383:73–84. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Yang J, Zhang X, Zhang Y, Zhu D, Zhang L,
Li Y, Zhu Y, Li D and Zhou J: HIF-2α promotes
epithelial-mesenchymal transition through regulating Twist2 binding
to the promoter of E-cadherin in pancreatic cancer. J Exp Clin
Cancer Res. 35:262016. View Article : Google Scholar
|
|
34
|
Lee JY and Kong G: Roles and epigenetic
regulation of epithelial-mesenchymal transition and its
transcription factors in cancer initiation and progression. Cell
Mol Life Sci. 73:4643–4660. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Espinoza I, Pochampally R, Xing F, Watabe
K and Miele L: Notch signaling: Targeting cancer stem cells and
epithelial-to-mesenchymal transition. Onco Targets Ther.
6:1249–1259. 2013.PubMed/NCBI
|
|
36
|
Ma J, Xia J, Miele L, Sarkar FH and Wang
Z: Notch signaling pathway in pancreatic cancer progression.
Pancreat Disord Ther. 3(pii): 10001142013. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Liu H, Yin Y, Hu Y, Feng Y, Bian Z, Yao S,
Li M, You Q and Huang Z: miR-139 5p sensitizes colorectal cancer
cells to 5-fluorouracil by targeting NOTCH-1. Pathol Res Pract.
212:643–649. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Mirone G, Perna S, Shukla A and Marfe G:
Involvement of Notch-1 in resistance to regorafenib in colon cancer
cells. J Cell Physiol. 231:1097–1105. 2016. View Article : Google Scholar
|
|
39
|
Xie M, He CS, Wei SH and Zhang L: Notch-1
contributes to epidermal growth factor receptor tyrosine kinase
inhibitor acquired resistance in non-small cell lung cancer in
vitro and in vivo. Eur J Cancer. 49:3559–3572. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Osipo C, Patel P, Rizzo P, Clementz AG,
Hao L, Golde TE and Miele L: ErbB-2 inhibition activates Notch-1
and sensitizes breast cancer cells to a gamma-secretase inhibitor.
Oncogene. 27:5019–5032. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Oyasiji T, Zhang J, Kuvshinoff B, Iyer R
and Hochwald SN: Molecular targets in biliary carcinogenesis and
implications for therapy. Oncologist. 20:742–751. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Kang HC, Kim IJ, Park JH, Shin Y, Ku JL,
Jung MS, Yoo BC, Kim HK and Park JG: Identification of genes with
differential expression in acquired drug-resistant gastric cancer
cells using high-density oligonucleotide microarrays. Clin Cancer
Res. 10:272–284. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Qi M, Ikematsu S, Ichihara-Tanaka K,
Sakuma S, Muramatsu T and Kadomatsu K: Midkine rescues Wilms' tumor
cells from cisplatin-induced apoptosis: Regulation of Bcl-2
expression by Midkine. J Biochem. 127:269–277. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Du B and Shim JS: Targeting
epithelial-mesenchymal transition (EMT) to overcome drug resistance
in cancer. Molecules. 21(pii): E9652016. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Brabletz S, Bajdak K, Meidhof S, Burk U,
Niedermann G, Firat E, Wellner U, Dimmler A, Faller G, Schubert J
and Brabletz T: The ZEB1/miR-200 feedback loop controls Notch
signalling in cancer cells. EMBO J. 30:770–782. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Espinoza I and Miele L: Deadly crosstalk:
Notch signaling at the intersection of EMT and cancer stem cells.
Cancer Lett. 341:41–45. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Hu YY, Zheng MH, Zhang R, Liang YM and Han
H: Notch signaling pathway and cancer metastasis. Adv Exp Med Biol.
727:186–198. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Vinson KE, George DC, Fender AW, Bertrand
FE and Sigounas G: The Notch pathway in colorectal cancer. Int J
Cancer. 138:1835–1842. 2016. View Article : Google Scholar
|
|
49
|
Wang Z, Li Y, Banerjee S and Sarkar FH:
Emerging role of Notch in stem cells and cancer. Cancer Lett.
279:8–12. 2009. View Article : Google Scholar :
|
|
50
|
Bao B, Wang Z, Ali S, Kong D, Li Y, Ahmad
A, Banerjee S, Azmi AS, Miele L and Sarkar FH: Notch-1 induces
epithelial-mesenchymal transition consistent with cancer stem cell
phenotype in pancreatic cancer cells. Cancer Lett. 307:26–36. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Fender AW, Nutter JM, Fitzgerald TL,
Bertrand FE and Sigounas G: Notch-1 promotes stemness and
epithelial to mesenchymal transition in colorectal cancer. J Cell
Biochem. 116:2517–2527. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Hao L, Rizzo P, Osipo C, Pannuti A, Wyatt
D, Cheung LW, Sonenshein G, Osborne BA and Miele L: Notch-1
activates estrogen receptor-alpha-dependent transcription via IKK
alpha in breast cancer cells. Oncogene. 29:201–213. 2010.
View Article : Google Scholar
|