Introduction
Acute lung injury (ALI) and acute respiratory
distress syndrome (ARDS) are life-threatening conditions, the
clinical manifestations of which are refractory hypoxemia and
progressive dyspnea (1). Current
studies indicate that the release of proinflammatory cytokines and
chemokines are key factors in ALI. Inflammation is a protective
defense system that acts against the invasion of pathogens
(2,3); however, the excessive inflammation
induces tissue and organ injury (3). Excessive inflammation induced by
bacterial infection performs a major role in the progression of
ALI, although the exact mechanisms remain unclear (4). Blocking the progression of
inflammatory cascades has been demonstrated as a valid method of
mitigating ALI (5,6), indicating that inflammatory blocking
agents may be used to prevent and treat ALI.
In 2000, triggering receptor expressed on myeloid
cells-1 (TREM-1) was first reported by Bouchon et al
(7). TREM-1, a member of the
immunoglobulin superfamily, is expressed on monocytes/macrophages
and neutrophils (7,8). TREM-1 is deemed to be an amplifier
of inflammation (8). TREM-1
triggers neutrophil degranulation and oxidative burst (7,9–11),
which is one of the most upregulated signaling pathways in
inflammation (12). Engagement of
TREM-1 with agonist monoclonal antibodies increases the secretion
of tumor necrosis factor (TNF)-α, keratinocyte chemokine (KC) and
monocyte chemoattractant protein-1 (MCP-1), and inhibits the
release of interleukin (IL)-10 by monocytes (13–15). Studies have demonstrated that
TREM-1 is a necessary regulator of immunity and a potential
therapeutic target in septic shock (16). Blocking TREM-1 signaling using a
fusion protein or a short inhibitory peptide shows a protective
effect in bacterial sepsis caused by live Escherichia coli
and Pseudomonas aeruginosa, and in lipopolysaccharide
(LPS)-induced shock, as well as in cecal ligation and puncture
models of sepsis (9,16–19). In addition, TREM-1 deficiency
reduced local release of cytokines and chemokines, and delayed the
influx of neutrophils (20).
Furthermore, TREM-1 deficiency decreased the transepithelial
migration of neutrophils into the lung (21). In other acute or chronic
inflammatory diseases, the protective effects of blocking TREM-1
have been confirmed (22–25). Recently, Liu's group (26) reported the regulation of NLR
family pyrin domain containing 3 (NLRP3) inflammasome activation by
TREM-1, which relieves LPS-induced ALI and improves survival rate
via pretreatment methods. Hence, the role of TREM-1 in LPS-induced
ALI requires further investigation.
Crystallographic studies reveal that the structure
between TREM-like transcript-1 (TLT-1) and TREM-1 are similar,
which indicates the existence of interactions between TLT-1 and
TREM-1 (27). Previous studies
demonstrate that TLT-1 and LR12 behave like natural TREM-1
inhibitors, which exhibit anti-inflammatory properties by
inhibiting TREM-1 signaling (28,29). It has been identified that LR12
exerts protective effects in hypodynamic septic shock in pigs, and
mediates inflammatory injury and cardiac remodeling following
myocardial infarction in mice (30,31). Furthermore, LR12 weakens
endotoxin-associated clinical and biological alterations, and no
obvious side effects were identified in nonhuman primates (29).
Hence, by using an LPS-induced ALI model in C57BL/6
mice, the roles and potential underlying mechanisms of TREM-1 in
LPS-induced ALI were investigated in the present study.
Materials and methods
Animals
All animal experiments were approved by the Animal
Care and Use Committee of Tongji Medical College of Huazhong
University of Science and Technology (Wuhan, China; certification
no. 2015 S621). Six to eight-week old male C57BL/6 mice (weight,
20–25 g) were purchased from the animal experimental center of
Wuhan University (Wuhan, China). Mice were housed (4 mice/cage) in
a specific-pathogen-free room at a temperature of 22–24°C, humidity
of 60–65% and under a 12-h light/dark cycle. The animals were
allowed free access to standard laboratory chow and water, and
acclimatized to the environment for 5 days before the
experiments.
Introduction of LR12 and LR12
scramble
LR12 is a 12-amino acid peptide, which is derived
from TLT-1 (LQEEDAGEYGCM), and the placebo, LR12 scramble (a
COOH-terminal amidated peptide) is the corresponding scramble
peptide (YQMGELCAGEED). These peptides were synthesized by
Bioyeargene Biosciences Co., Ltd. (Wuhan, China). The peptides were
homogeneous and with a purity of >99%, which was confirmed by
mass spectrometry and analytic reversed-phase high-performance
liquid chromatography. These peptides were free of endotoxin.
Experimental procedures
All the mice were divided randomly into three groups
as follows: Sham, LPS + scramble and LPS + LR12 (n=5 per group).
The preliminary experiment identified that there was no difference
in ALI between model groups treated with normal saline (NS) and
LR12 scramble (data not shown). The mice were anaesthetized
intraperitoneally with 90 mg/kg of 1% sodium pentobarbital
(Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) prior to surgery.
Following successful endotracheal intubation, mice were instilled
with LPS (Escherichia coli serotype O55:B5; Sigma-Aldrich,
Merck KGaA) at a dosage of 3 mg/kg (LPS + scramble group and LPS +
LR12 group) or NS (1.5 ml/kg; sham group) (2). LR12 (5 mg/kg LPS + LR12 group), LR12
scramble (5 mg/kg; LPS + scramble group) or NS (5 ml/kg; sham
group) was administrated intraperitoneally 30 min after
intratracheal instillation with LPS or NS (30). The treatment was repeated every 3
h three times.
Twenty-four hours after intratracheal instillation
with LPS or NS, mice were anesthetized with an overdose of sodium
pentobarbital and 1 ml blood samples were collected from the
eyeball. The blood was centrifuged at 1,000 × g for 10 min at room
temperature and the supernatant was stored at −80°C for further
experiments. The right lung was clamped at the level of the main
stem bronchus. Lung tissue samples were excised and rinsed with
cold phosphate-buffered saline to obtain the lung wet/dry (W/D)
weight ratio and for histologic analysis, or frozen in liquid
nitrogen and stored at −80°C for further investigation.
Histological analysis of lung tissue
samples
The right lower lobes of the lungs were embedded in
paraffin wax and the sections (~6-mm thick) were stained with
hematoxylin for 10 min at room temperature and then with eosin for
3 min at room temperature. The lung injury scores were evaluated by
an investigator who was blinded to the experimental design
according to the published criteria (Table I):
Score=(20xA+14xB+7xC+7xD+2xE)/(number of fields x100) (32).
 | Table ILung injury scoring system. |
Table I
Lung injury scoring system.
| Parameter | Score per field
|
|---|
| 0 | 1 | 2 |
|---|
| A. Neutrophils in
the alveolar space | None | 1–5 | >5 |
| B. Neutrophils in
the interstitial space | None | 1–5 | >5 |
| C. Hyaline
membranes | None | 1 | >1 |
| D. Proteinaceous
debris filling the airspaces | None | 1 | >1 |
| E. Alveolar septal
thickening | <2x | 2–4x | >4x |
Differential leukocyte counts and
pulmonary edema
Bronchoalveolar lavage fluid (BALF) was collected by
bronchoalveolar lavaging with 0.4 ml NS from the left lung three
times. The BALF was spun at 4°C for 10 min at 250 × g. Supernatant
was collected and stored at −80°C for protein and cytokine
detection. The cell aggregate underwent differential leukocyte
counting. Total BALF cells were measured using a hematocytometer
and stained with Giemsa stain solution for 30 min at room
temperature. A total of 200 cells/slide were randomly selected to
calculate the percentage of neutrophils and monocytes/macrophages
in the sample under the microscope.
Pulmonary W/D weight ratio and the BALF protein
concentrations were detected for the assessment of pulmonary edema.
For the W/D weight ratio, the right-middle lobes were weighed as
soon as they were removed. The lobe samples were placed in an oven
and weighted again 24 h later when they were dried. BALF protein
concentration was estimated using a Bicinchoninic acid Protein
Assay kit (cat. no. 23227; Thermo Fisher Scientific, Inc., Waltham,
MA, USA) and the procedure was performed according to the
manufacturer's instructions.
Pulmonary MPO activity
Lung tissue samples were homogenized with isotonic
sodium chloride to detect the MPO activity. Lung tissue MPO
activity was measured using the MPO kits (cat. no. A044; Nanjing
Jiancheng Bioengineering Institute, Nanjing, China). Detection was
conducted according to the manufacturer's instructions.
Immunohistochemistry
Following deparaffinization and repair, the sections
were incubated in 3% hydrogen peroxide for 30 min at room
temperature and blocked using 5% goat serum albumin (cat. no.
SL038; Beijing Solarbio Science and Technology Co., Ltd., Beijing,
China) for 20 min at room temperature. The sections were incubated
with MPO antibody (cat. no. ab208670; 1:100; Abcam, Cambridge, MA,
USA) overnight at 4°C. Then the secondary antibody (cat. no.
A25112; 1:100; Abbkine Scientific Co., Ltd., Wuhan, China) was
added to the sections for 50 min at 4°C. The sections were stained
with 3,3′-diaminobenzidine and re-dyed with hematoxylin at room
temperature. The integrated optical density was analyzed using the
Image-pro Plus 6.0 software (Media Cybernetics, Inc., Rockville,
MD, USA).
Immunofluorescence
Following deparaffinization, microwave-treated
antigen retrieval was conducted in sodium citrate solution.
Subsequently, nonspecific binding was blocked using goat serum for
30 min. The sections were incubated with the MPO antibody (1:100)
or p65 antibody (cat. no. ab16502; 1:100; Abcam) overnight at 4°C.
The secondary antibody was added for 2 h after washing three times
with PBS. The coverslips were counterstained at room temperature
with 4′,6-diamidino-2-phenylindole for 5 min and images were
acquired using a fluorescence microscope in a dark room.
ELISA
The expression levels of TNF-α, IL-1β, IL-6, IL-10
and KC in BALF were detected by using ELISA kits (cat. nos.
EMC102a, EMC001b, EMC004, EMC005 and EMC104, respectively;
NeoBioscience Technology Co., Shanghai, China). The expression
levels of MCP-1 in BALF were evaluated using the ELISA kit (cat.
no. ELM-MCP1-001; RayBiotech, Inc., Norcross, GA, USA) and sTREM-1
was analyzed with an ELISA kit (cat. no. CSB-E13848m; Cusabio
Biotech Co., Ltd., Hubei, China). The intensity of the color was
measured at 450 nm using a microplate reader (Bio-Rad Laboratories,
Inc., Hercules, CA, USA) and the procedure was performed according
to the manufacturer's instructions.
Western blotting
Protein was extracted from the lung tissue sample
homogenates according to the manual provided by the Protein
Extraction kit (cat. no. KGP150; Nanjing KeyGen Biotech Co., Ltd.,
Nanjing, China). The nuclear protein and cytosolic protein were
separated during the procedure. Proteins were separated by
electrophoresis on 10% polyacrylamide SDS gels and transferred to a
polyvinylidene difluoride membrane by wet transfer (200 mA, 1 h).
The membranes were blocked with 5% non-fat milk for 1 h, and
incubated with p65 (cat. no. ab32536; 1:2,000), inhibitor of NF-κB
(IκB) (cat. no. ab32518; 1:1,000), TREM-1(cat. no. ab104413;
1:500), β-actin (cat. no. ab8226; 1:1,000) (all Abcam) and histone
H3 (cat. no. 4243; 1:500; Cell Signaling Technology, Inc., Danvers,
MA, USA) individually, overnight at 4°C. After washing three times
with TBS containing 0.05% Tween-20, horse radish
peroxidase-conjugated goat anti-rabbit Immunoglobulin G (IgG)
antibodies (cat. no. SA00001-2; 1:2,000) or anti-mouse IgG
antibodies (cat. no. SA00001-1; 1:2,000) (both Proteintech Group,
Inc., Wuhan, China) was added at room temperature for 1 h.
Chemiluminescent detection was performed using Western Lighting
Chemiluminescence Reagent (Beyotime Institute of Biotechnology,
Shanghai, China). Images were scanned using a UVP imaging system
and analyzed using ImageJ software (version 1.45s; National
Institutes of Health, Bethesda, MD, USA).
Electrophoretic mobility shift assay (EMSA) for
p65. The protein extract was prepared using a protein
extraction kit (cat. no. KGP150; Nanjing KeyGen Biotech Co., Ltd.).
Each sample contained an equivalent magnitude of nuclear extract
protein (10 µg), and the 50-fmol biotin-labeled, double-strand
probe was incubated for 15 min at room temperature. The
oligonucleotide probe sequence of the p65 binding site was 5′-AGT
TGA GGG GAC TTT CCA GGC-3′. The DNA-protein complexes were
electrophoresed on a 6.5% nondenaturing polyacrylamide gel and
electrotransferred for detection.
Statistical analysis
All data are presented as the mean ± standard error
of the mean. GraphPad Prism version 5.0 (GraphPad Software, Inc.,
La Jolla, CA, USA) was used to analyze the results. Following
testing for their normal distribution (using the Kolmogorov-Smirnov
test), the significance of the difference between the groups was
tested by one-way ANOVA, and pairwise comparisons were performed
between groups using the Newman-Keuls method. P<0.05 was
considered to indicate a statistically significant difference.
Results
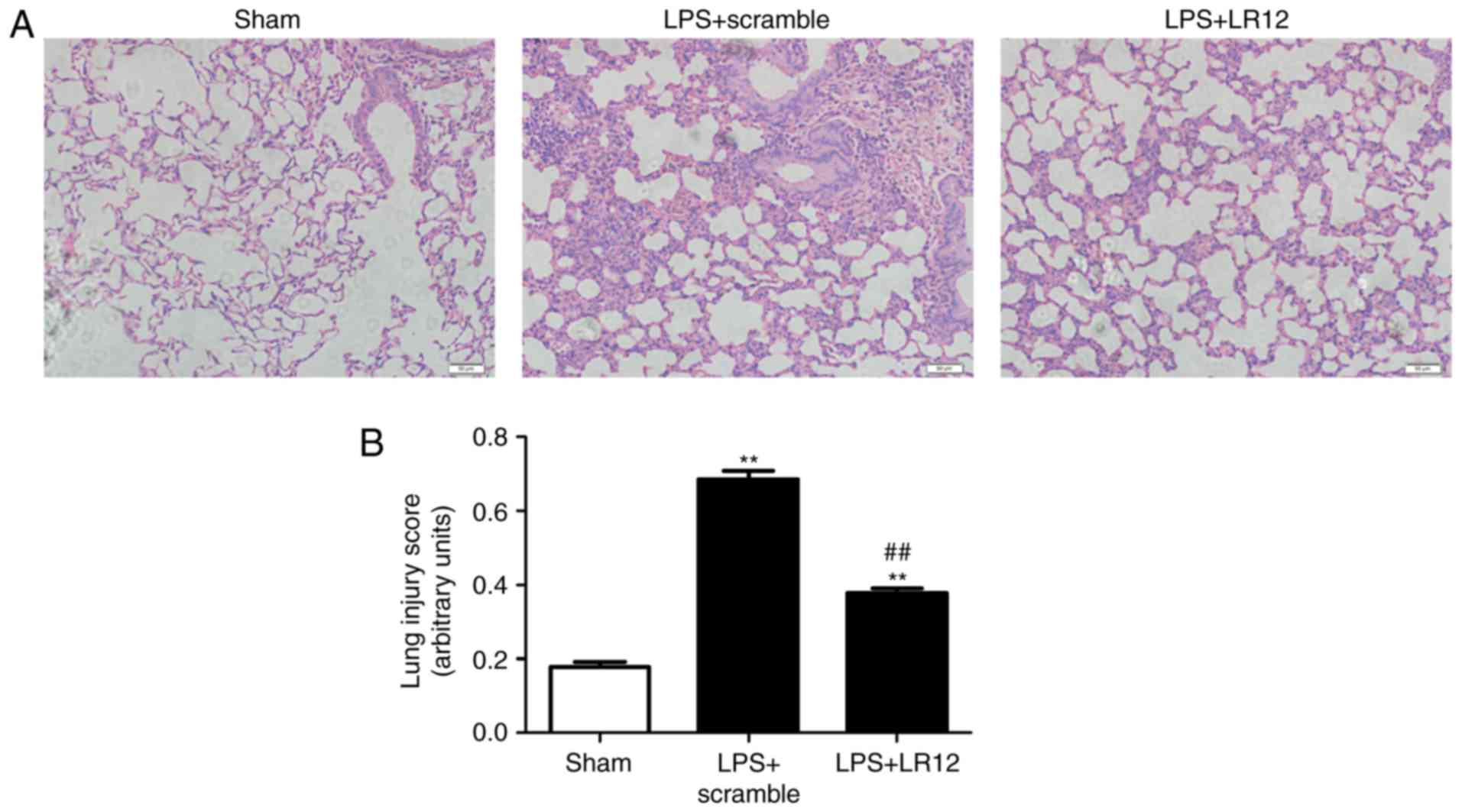
LR12 attenuates pathological changes and
lung injury scores in mice with LPS-induced ALI
The pulmonary construction was normal and, under
light microscopy, fewer macrophages were observed in the alveolar
space in the sham group (Fig.
1A). LPS treatment significantly increased the damage and
hemorrhaging in the lung tissue samples. The thickness of the
alveolar septal interstitium and neutrophil infiltration were
significantly increased following treatment with LPS (Fig. 1A). LR12 significantly alleviated
lung tissue injury when compared with the LPS + scramble group
(Fig. 1A). Furthermore, the lung
injury scores were assessed in parallel with the pathohistological
changes (Fig. 1B).
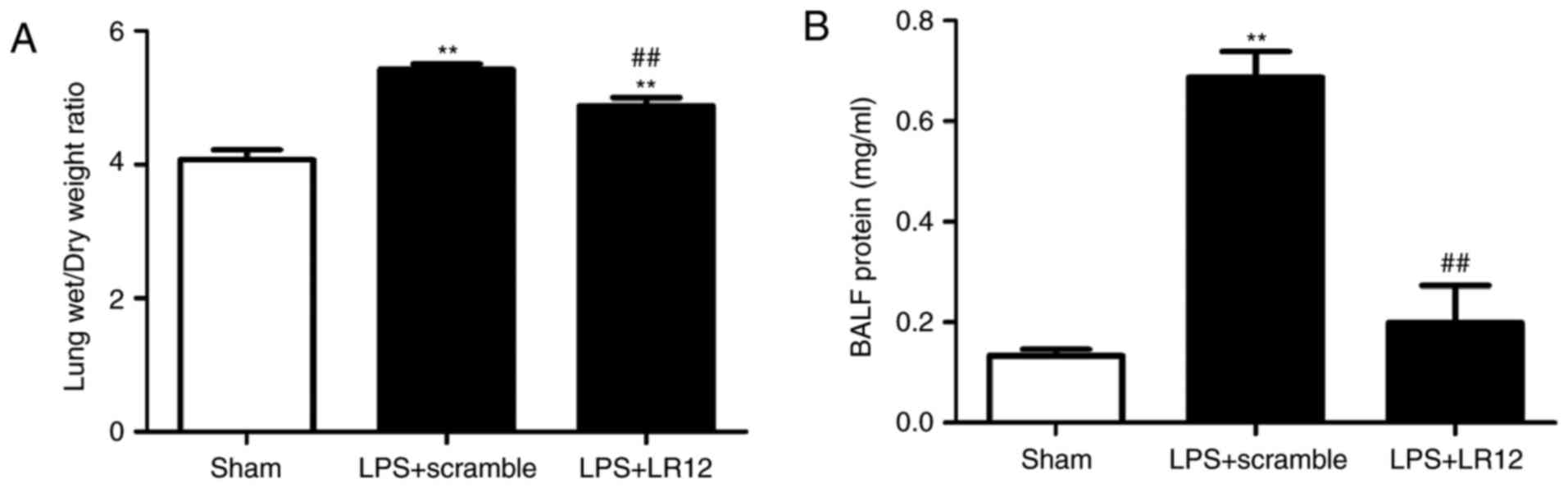
LR12 attenuates the changes of lung
tissue permeability in mice with LPS-induced ALI
The lung tissue sample W/D weight ratio and BALF
protein concentration reflect the changes in lung tissue
permeability. Compared with the sham group, the lung tissue sample
W/D weight ratio and BALF protein concentration were significantly
elevated in the LPS + scramble group, which indicated an obvious
disruption in the microvascular permeability of the lung tissues.
The lung tissue sample W/D weight ratio and BALF protein
concentration in the LPS + LR12 group were significantly decreased
compared with that in the LPS + scramble group (Fig. 2).
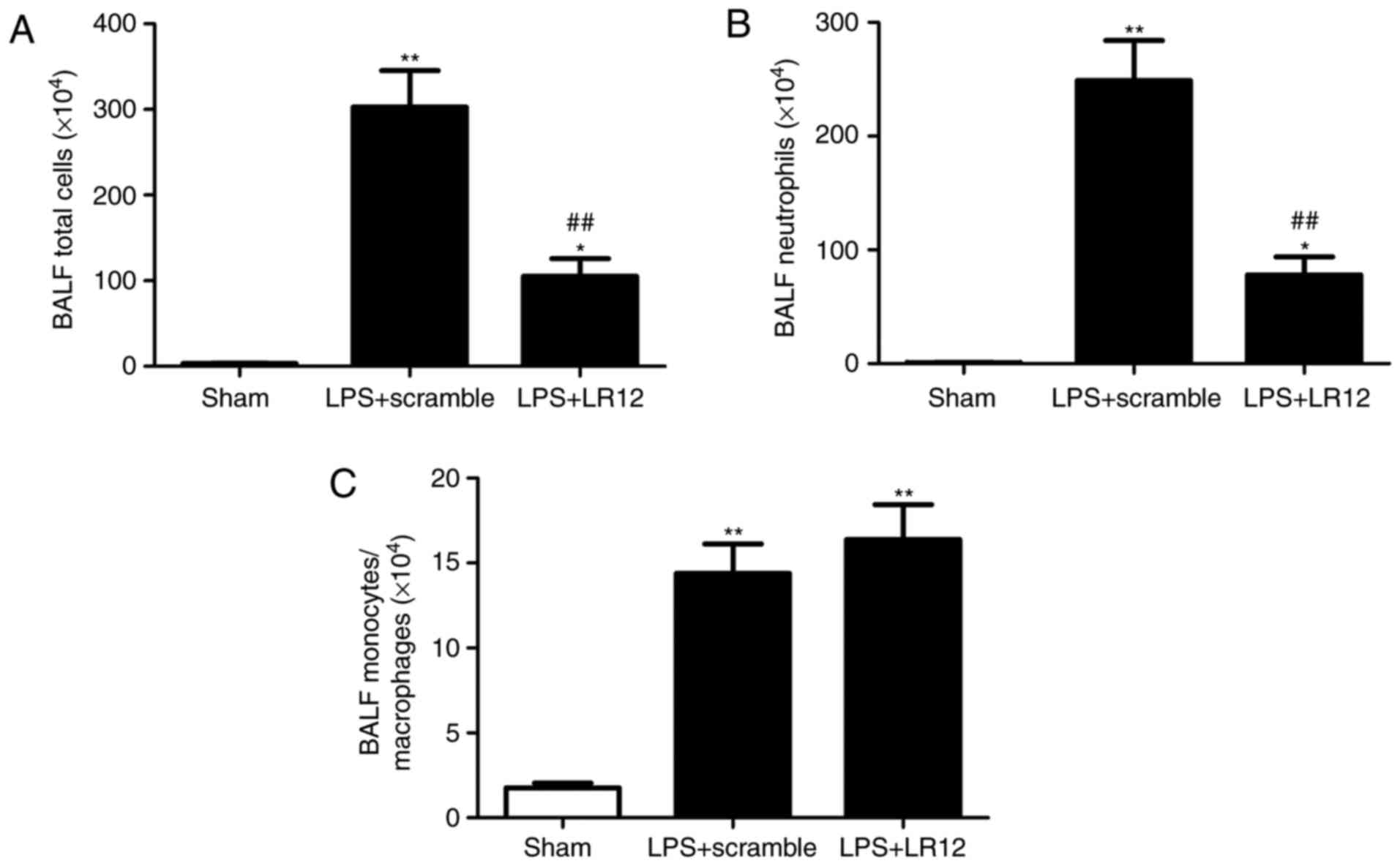
LR12 suppresses the variation of lung
leukocyte recruitment in mice with LPS-induced ALI
Following LPS treatment, the BALF cytology changed
markedly in the alveoli. Compared with that in the sham group, the
numbers of total cells, macrophages and neutrophils were
significantly increased in the LPS + scramble group (Fig. 3A–C).
The number of total cells and neutrophils, but not
macrophages in the BALF were significantly decreased following
treatment with LR12 (Fig.
3A–C).
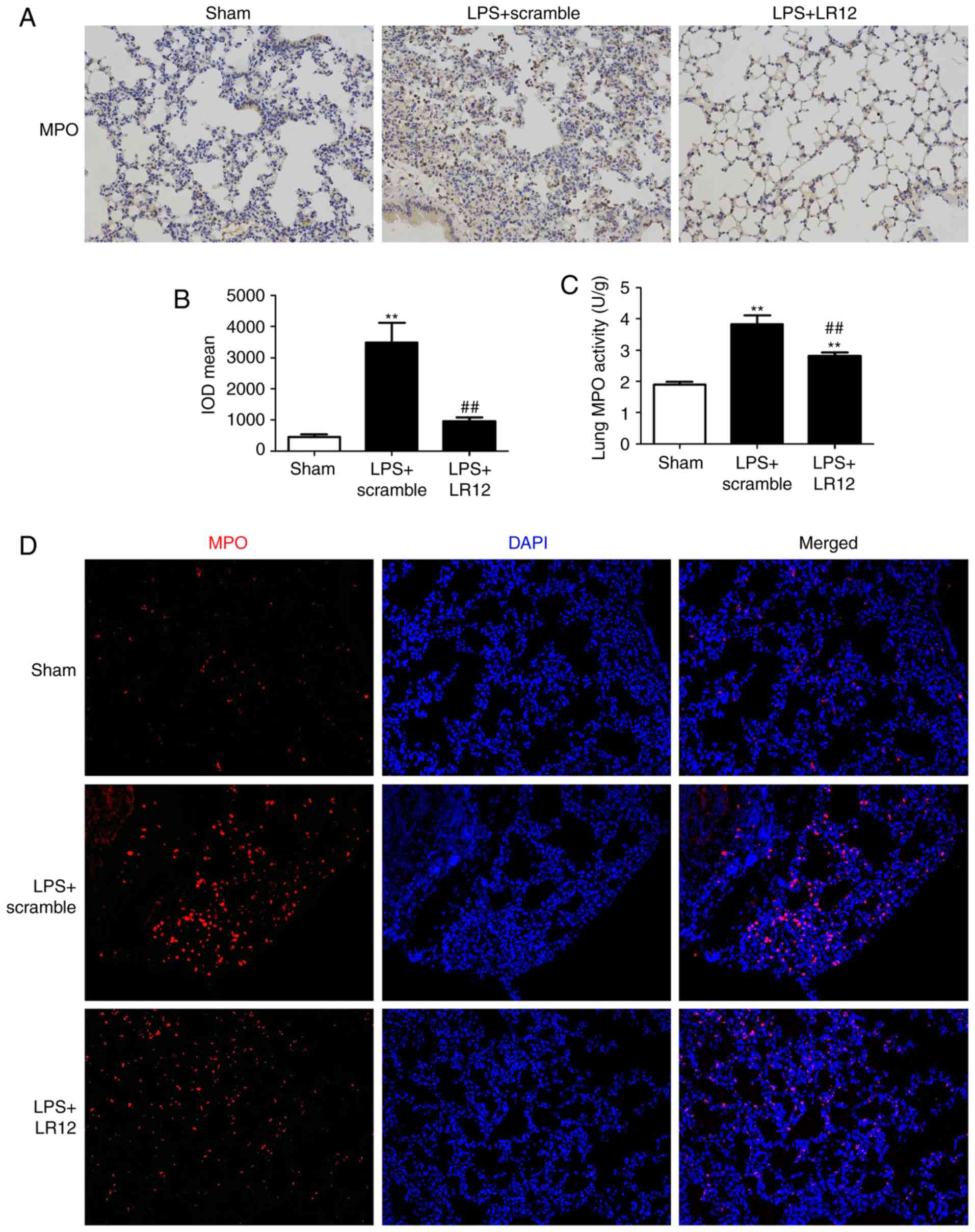
LR12 suppresses neutrophil infiltration
of lung tissues in mice with LPS-induced ALI
Immunohistochemical staining indicated that LPS
treatment significantly increased the MPO-positive cells in the
lung tissue samples (Fig. 4A and
B). Treatment with LR12 significantly attenuated the
LPS-induced significant increase of the MPO-positive cells in the
lung tissue samples when compared with that in the LPS + scramble
group (Fig. 4A and B). MPO
activity of the lung tissue samples is an important indicator of
neutrophil infiltration. The MPO activity of lung tissue samples
from the LPS + LR12 group was significantly decreased when compared
with that in the LPS + scramble group (Fig. 4C). The immunofluorescence staining
for MPO confirmed the results (Fig.
4D).
LR12 suppresses the expression of
proinflammatory cytokines and promotes the expression of
anti-inflammatory cytokines in mice with LPS-induced ALI
Inflammatory cytokines are important in neutrophil
recruitment and propagation of the inflammatory response. These
particular inflammatory cytokines were detected in BALF using ELISA
in order to investigate their effects. Compared with that in the
sham group, the expression levels of proinflammatory cytokines
(TNF-α, IL-1β, IL-6, MCP-1 and KC) were significantly increased in
the LPS + scramble group (Fig.
5). Treatment with LR12 significantly attenuated the
LPS-induced significant increase in proinflammatory cytokines
(Fig. 5). Furthermore, the
expression level of anti-inflammatory cytokine, IL-10 in the LPS +
LR12 group was significantly increased when compared with that in
the LPS + scramble group (Fig.
5).
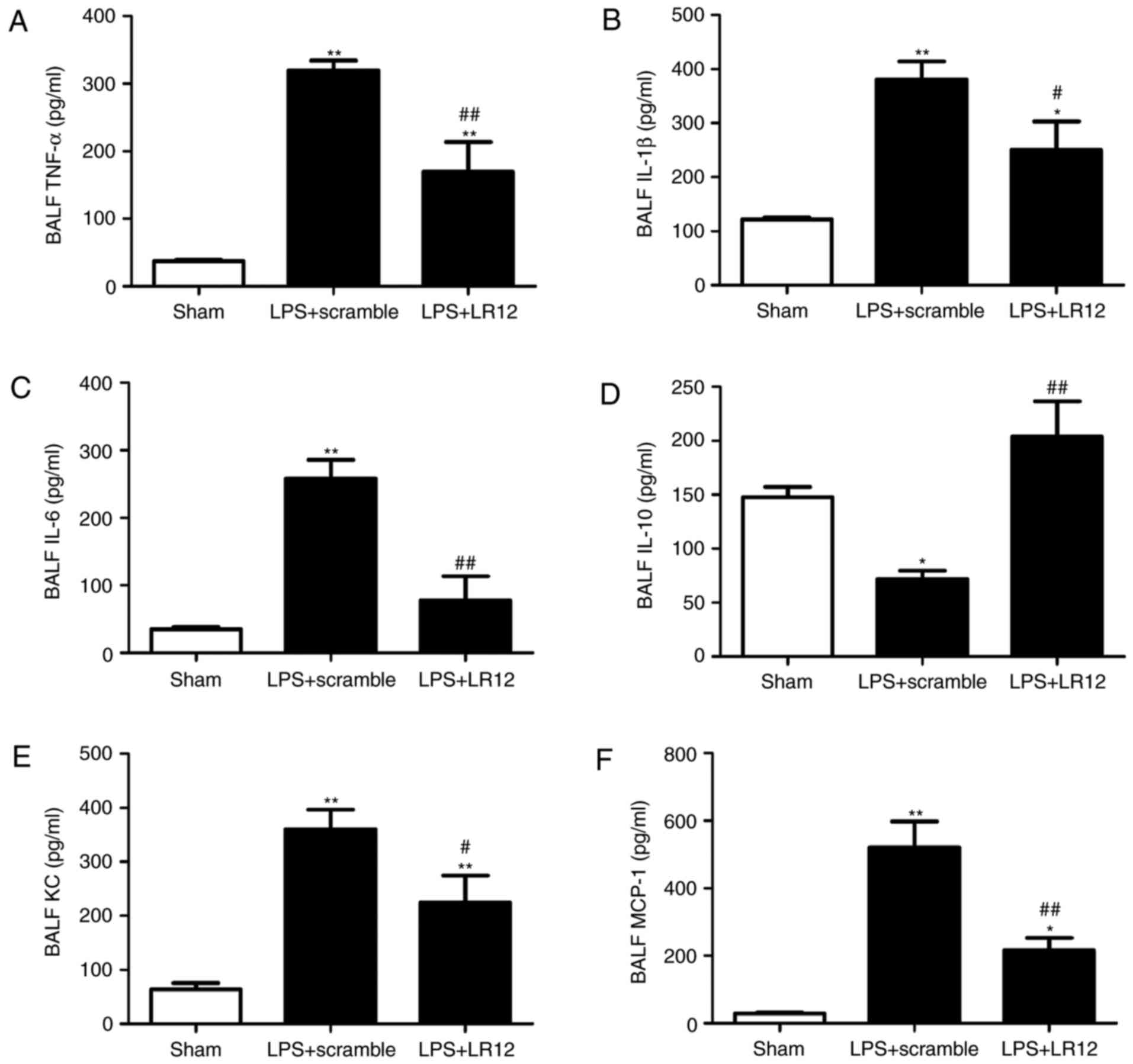 | Figure 5Treatment with LR12 significantly
attenuated the LPS-induced significant increase in proinflammatory
cytokines. (A) The changes of inflammatory cytokines in BALF (A)
TNF-α, (B) IL-1β, (C) IL-6, (D) IL-10, (E) KC and (F) MCP-1. Data
are presented as means ± standard error of the mean (n=5).
*P<0.05 and **P<0.01 vs. sham group;
#P<0.05 and ##P<0.01 vs. LPS + scramble
group. LPS, lipopolysaccharide; BALF, bronchoalveolar lavage fluid;
TNF-α, tumor necrosis factor-α; IL, interleukin; KC, keratinocyte
chemokine; MCP-1, monocyte chemoattractant protein-1. |
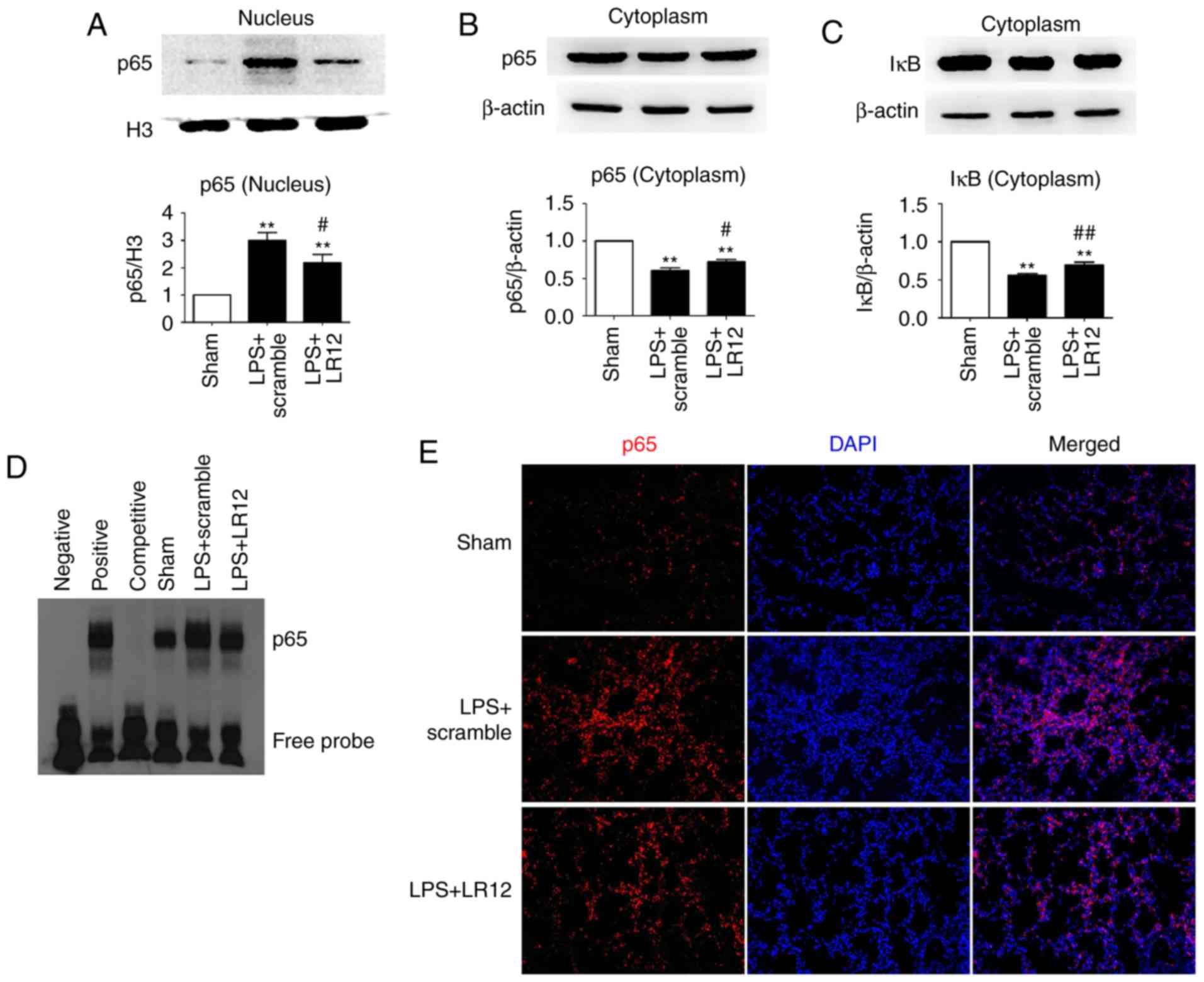
LR12 inhibits NF-κB activation in mice
with LPS-induced ALI
NF-κB existed as an inactive form combined with its
inhibitor, IκB when cells were in the resting state. The expression
level of p65 in the nucleus was significantly increased with the
treatment of LPS compared with that in the sham group, and
administration of LR12 significantly decreased the expression level
of p65 in the nucleus (Fig. 6A).
The expression level of p65 and IκB in the cytoplasm was
significantly increased following treatment with LR12 compared with
the LPS + scramble group (Fig. 6B and
C). The EMSA and immunofluorescence staining of p65 confirmed
the results (Fig. 6D and E).
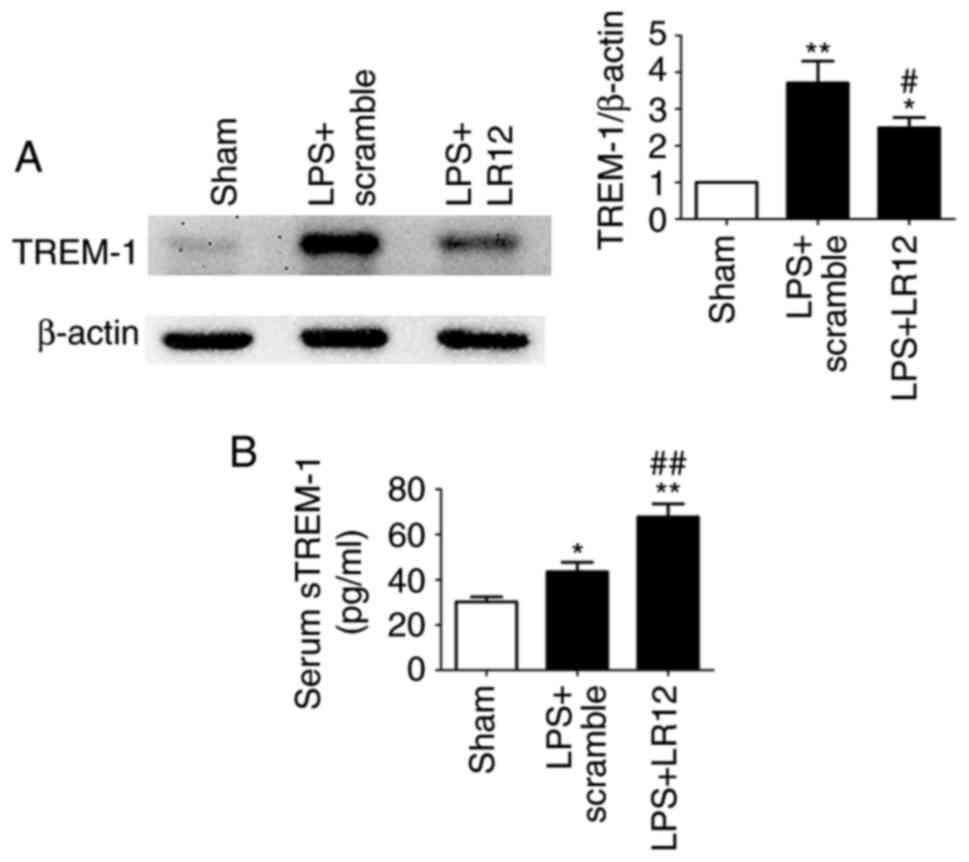
LR12 suppresses the expression of TREM-1
and promotes the release of sTREM-1 in mice with LPS-induced
ALI
The expression level of TREM-1 was significantly
increased in the LPS + scramble group compared with the sham group
(Fig. 7A). Following treatment
with LR12, the expression level of TREM-1 in the lung tissue
samples was significantly decreased (Fig. 7A). The expression levels of
anti-inflammatory mediator sTREM-1 in the serum were significantly
increased following treatment with LR12 compared with the LPS +
scramble group (Fig. 7B).
Discussion
Inflammatory disorders, the activities and
inappropriate accumulation of leukocytes and platelets, activation
of uncontrollable coagulation pathways, and the changes of
permeability of alveolar epithelial and endothelial barriers are
the core pathophysiological characteristics of ALI/ARDS (33,34). Furthermore, the glycolipid of the
outer membrane of gram-negative bacteria, LPS, is a pathogenic
factor of ALI/ARDS (35).
Intratracheal instillation of LPS is a commendable model for ALI,
which mimics gram-negative pulmonary infection in the clinical
development of ALI (36).
First reported in the year 2000 (7), TREM-1 amplified the inflammatory
response and contributed to the innate and adaptive immune
responses (11,37–39). Under a variety of inflammatory
conditions, the TREM-1 expression level was upregulated (25,40) and it has been demonstrated that
the expression level of TREM-1 in ALI mice was increased (41). The present study demonstrated that
the expression level of TREM-1 in lung tissue samples was increased
and that TREM-1 contributed significantly to mediating inflammatory
cell recruitment following LPS-induced ALI. Its therapeutic
modulation achieved via LR12 administration conferred protection in
mice. LR12, a 12-amino acid sequence representative of residues
94–105, was one of the TLT-1 derived peptides (31). Previously, it was observed that
TLT-1 binds and impedes TREM-1 engagement (28). Intraperitoneal injection of LR12
(5 mg/kg) was used in a study by Boufenzer et al (30), which determined a reference dose
for the present study. The present study confirmed that LPS-induced
ALI may be alleviated by LR12. The present data indicated that LR12
treatment significantly alleviated pathological damage, reduced
pulmonary edema, decreased the infiltration and activity of
neutrophils, reduced LPS-induced production of proinflammatory
cytokines and chemokines (TNF-α, IL-6, IL-1β, MCP-1 and KC), and
increased the release of anti-inflammatory cytokines (IL-10 and
sTREM-1). Therefore, the present results indicate that LR12 confers
protection against LPS-induced ALI.
NF-κB, as a nuclear transcription factor,
participates in LPS-induced generation of proinflammatory
cytokines, chemokines and adhesion molecules (42–44). Cytokines, such as TNF-α, IL-1β,
IL-6 and IL-8, are transcriptionally regulated by NF-κB in
vitro (43). NF-κB activation
enhances the transcription of TNF-α and IL-1β, and these cytokines
activate NF-κB. Other mediators, such as IL-6 and IL-8, are
released later and lead to more sustained elevations. These later
mediators may depend largely on TNF-α and IL-1β to stimulate their
production. Inflammatory stimuli, such as endotoxin, TNF-α and
IL-1β stimulate the production of counterregulatory cytokines, such
as IL-10, that suppress the production of proinflammatory
cytokines. It was shown that IL-10 inhibits cytokine production in
monocytes by blocking endotoxin-induced NF-κB activation (45). When bound by the IκB family
proteins, NF-κB is inactivated and sequestered in the cytoplasmic
compartment. Once certain inflammatory stimuli appear, degradation
of IκB family proteins is induced, which leads to the release of
NF-κB, followed by its trans-location from the cytosol to the
nucleus, where it initiates transcription of proinflammatory genes.
It has been shown that NF-κB signaling is important in mediating
LPS-induced ALI (46,47). Evidence has indicated that the
NF-κB signaling pathway is involved in TREM-1-mediated inflammatory
responses (48). As TREM-1
contributed to LPS-induced ALI via promotion of neutrophil
infiltration, and production of proinflammatory cytokines and
chemokines in the present study, it is hypothesized that TREM-1 may
mediate LPS-induced ALI via activation of the NF-κB signaling
pathway. In addition, TREM-1 aggravates inflammation in ALI by
activating the NLRP3 inflammasome (26). NF-κB inhibition leads to a
dose-dependent reduction of NLRP3 protein induction by LPS, which
indicates a key role for NF-κB in priming the NLRP3 inflammasome
(49). However, it was not
verified by the administration of a TREM-1 inhibitor (LP17) or a
NF-κB inhibitor (Bay 11-7082) in the present study. Future studies
are required to confirm these findings.
sTREM-1, a 27-kDa glycosylated peptide that is
detected in biological fluids and tissues in response to infection
(18,50,51), is most likely produced by cleavage
of the extracellular domain from the membrane-bound form of matrix
metalloproteinases (52). sTREM-1
is unable to transfer signals, but acts as a decoy receptor to
prevent the binding of its ligand to membrane-bound TREM-1, inhibit
the effect of TREM-1 activation and attenuate inflammation
(53). The current results
revealed that the expression level of sTREM-1 increased following
treatment with LR12, and it acted as an anti-inflammatory mediator,
similar to IL-10 (54). The
present data indicate that an increase of sTREM-1 expression level
after LR12 treatment could inhibit TREM-1-mediated ALI for the
first time.
In conclusion, the current results clearly reveal a
previously uncharacterized property of LR12, that is, the
LR12-mediated anti-inflammatory effects in LPS-induced ALI.
Alleviating the expression level of TREM-1, increasing the release
of sTREM-1 and inhibiting activation of the NF-κB signaling pathway
may be involved in this process. The present study provides a
theoretical basis and a novel therapeutic approach for ALI/ARDS.
The NF-κB signaling pathway was exclusively investigated in the
present study, therefore, further studies are required to
investigate other signaling pathways involved in this process.
Acknowledgments
The present study was supported by the National
Natural Science Foundation of China (grant no. 81401568).
Notes
[1] Competing
interests
The authors declare there is no competing
interest.
References
|
1
|
Matthay MA, Ware LB and Zimmerman GA: The
acute respiratory distress syndrome. J Clin Invest. 122:2731–2740.
2012. View
Article : Google Scholar : PubMed/NCBI
|
|
2
|
Gong J, Guo S, Li HB, Yuan SY, Shang Y and
Yao SL: BML-111, a lipoxin receptor agonist, protects haemorrhagic
shock-induced acute lung injury in rats. Resuscitation. 83:907–912.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Serhan CN: Resolution phase of
inflammation: Novel endogenous anti-inflammatory and proresolving
lipid mediators and pathways. Annu Rev Immunol. 25:101–137. 2007.
View Article : Google Scholar
|
|
4
|
Tsushima K, King LS, Aggarwal NR, De
Gorordo A, D'Alessio FR and Kubo K: Acute lung injury review.
Intern Med. 48:621–630. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Shang Y, Jiang YX, Ding ZJ, Shen AL, Xu
SP, Yuan SY and Yao SL: Valproic acid attenuates the multiple-organ
dysfunction in a rat model of septic shock. Chin Med J (Engl).
123:2682–2687. 2010.
|
|
6
|
Maderna P and Godson C: Lipoxins:
Resolutionary road. Br J Pharmacol. 158:947–959. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Bouchon A, Dietrich J and Colonna M:
Cutting edge: Inflammatory responses can be triggered by TREM-1, a
novel receptor expressed on neutrophils and monocytes. J Immunol.
164:4991–4995. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Schenk M, Bouchon A, Birrer S, Colonna M
and Mueller C: Macrophages expressing triggering receptor expressed
on myeloid cells-1 are underrepresented in the human intestine. J
Immunol. 174:517–524. 2005. View Article : Google Scholar
|
|
9
|
Bouchon A, Facchetti F, Weigand MA and
Colonna M: TREM-1 amplifies inflammation and is a crucial mediator
of septic shock. Nature. 410:1103–1107. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Colonna M and Facchetti F: TREM-1
(triggering receptor expressed on myeloid cells): A new player in
acute inflammatory responses. J Infect Dis. 187(Suppl 2):
S397–S401. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
11
|
Dower K, Ellis DK, Saraf K, Jelinsky SA
and Lin LL: Innate immune responses to TREM-1 activation: Overlap,
divergence, and positive and negative cross-talk with bacterial
lipopolysaccharide. J Immunol. 180:3520–3534. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Xiao W, Mindrinos MN, Seok J, Cuschieri J,
Cuenca AG, Gao H, Hayden DL, Hennessy L, Moore EE, Minei JP, et al:
A genomic storm in critically injured humans. J Exp Med.
208:2581–2590. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Hara H, Ishihara C, Takeuchi A, Imanishi
T, Xue L, Morris SW, Inui M, Takai T, Shibuya A, Saijo S, et al:
The adaptor protein CARD9 is essential for the activation of
myeloid cells through ITAM-associated and Toll-like receptors. Nat
Immunol. 8:619–629. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
14
|
Fortin CF, Lesur O and Fulop T Jr: Effects
of TREM-1 activation in human neutrophils: Activation of signaling
pathways, recruitment into lipid rafts and association with TLR4.
Int Immunol. 19:41–50. 2007. View Article : Google Scholar
|
|
15
|
Ornatowska M, Azim AC, Wang X, Christman
JW, Xiao L, Joo M and Sadikot RT: Functional genomics of silencing
TREM-1 on TLR4 signaling in macrophages. Am J Physiol Lung Cell Mol
Physiol. 293:L1377–L1384. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Nathan C and Ding A: TREM-1: A new
regulator of innate immunity in sepsis syndrome. Nat Med.
7:530–532. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wang F, Liu S, Wu S, Zhu Q, Ou G, Liu C,
Wang Y, Liao Y and Sun Z: Blocking TREM-1 signaling prolongs
survival of mice with Pseudomonas aeruginosa induced sepsis. Cell
Immunol. 272:251–258. 2012. View Article : Google Scholar
|
|
18
|
Gibot S, Kolopp-Sarda MN, Béné MC,
Bollaert PE, Lozniewski A, Mory F, Levy B and Faure GC: A soluble
form of the triggering receptor expressed on myeloid cells-1
modulates the inflammatory response in murine sepsis. J Exp Med.
200:1419–1426. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Gibot S, Buonsanti C, Massin F, Romano M,
Kolopp-Sarda MN, Benigni F, Faure GC, Béné MC, Panina-Bordignon P,
Passini N and Lévy B: Modulation of the triggering receptor
expressed on the myeloid cell type 1 pathway in murine septic
shock. Infect Immun. 74:2823–2830. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Hommes TJ, Hoogendijk AJ, Dessing MC,
Van't Veer C, Florquin S, Colonna M, de Vos AF and van der Poll T:
Triggering receptor expressed on myeloid cells-1 (TREM-1) improves
host defence in pneumococcal pneumonia. J Pathol. 233:357–367.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Klesney-Tait J, Keck K, Li X, Gilfillan S,
Otero K, Baruah S, Meyerholz DK, Varga SM, Knudson CJ, Moninger TO,
et al: Transepithelial migration of neutrophils into the lung
requires TREM-1. J Clin Invest. 123:138–149. 2013. View Article : Google Scholar :
|
|
22
|
Gibot S, Massin F, Alauzet C, Montemont C,
Lozniewski A, Bollaert PE and Levy B: Effects of the TREM-1 pathway
modulation during mesenteric ischemia-reperfusion in rats. Crit
Care Med. 36:504–510. 2008. View Article : Google Scholar
|
|
23
|
Kamei K, Yasuda T, Ueda T, Qiang F,
Takeyama Y and Shiozaki H: Role of triggering receptor expressed on
myeloid cells-1 in experimental severe acute pancreatitis. J
Hepatobiliary Pancreat Sci. 17:305–312. 2010. View Article : Google Scholar
|
|
24
|
Gibot S, Massin F, Alauzet C, Derive M,
Montemont C, Collin S, Fremont S and Levy B: Effects of the TREM 1
pathway modulation during hemorrhagic shock in rats. Shock.
32:633–637. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Schenk M, Bouchon A, Seibold F and Mueller
C: TREM-1-expressing intestinal macrophages crucially amplify
chronic inflammation in experimental colitis and inflammatory bowel
diseases. J Clin Invest. 117:3097–3106. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Liu T, Zhou Y, Li P, Duan JX, Liu YP, Sun
JY, Wan L, Dong L, Fang X, Jiang JX and Guan CX: Blocking
triggering receptor expressed on myeloid cells-1 attenuates
lipopolysaccharide-induced acute lung injury via inhibiting NLRP3
inflammasome activation. Sci Rep. 6:394732016. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Gattis JL, Washington AV, Chisholm MM,
Quigley L, Szyk A, McVicar DW and Lubkowski J: The structure of the
extracellular domain of triggering receptor expressed on myeloid
cells like transcript-1 and evidence for a naturally occurring
soluble fragment. J Biol Chem. 281:13396–13403. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Derive M, Bouazza Y, Sennoun N, Marchionni
S, Quigley L, Washington V, Massin F, Max JP, Ford J, Alauzet C, et
al: Soluble TREM-like transcript-1 regulates leukocyte activation
and controls microbial sepsis. J Immunol. 188:5585–5592. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Derive M, Boufenzer A and Gibot S:
Attenuation of responses to endotoxin by the triggering receptor
expressed on myeloid cells-1 inhibitor LR12 in nonhuman primate.
Anesthesiology. 120:935–942. 2014. View Article : Google Scholar
|
|
30
|
Boufenzer A, Lemarié J, Simon T, Derive M,
Bouazza Y, Tran N, Maskali F, Groubatch F, Bonnin P, Bastien C, et
al: TREM-1 mediates inflammatory injury and cardiac remodeling
following myocardial infarction. Circ Res. 116:1772–1782. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Derive M, Boufenzer A, Bouazza Y,
Groubatch F, Alauzet C, Barraud D, Lozniewski A, Leroy P, Tran N
and Gibot S: Effects of a TREM-like transcript-1-derived peptide
during hypodynamic septic shock in pigs. Shock. 39:176–182. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Matute-Bello G, Downey G, Moore BB,
Groshong SD, Matthay MA, Slutsky AS and Kuebler WM; Acute
LungInjury in Animals Study Group: An official American Thoracic
Society workshop report: Features and measurements of experimental
acute lung injury in animals. Am J Respir Cell Mol Biol.
44:725–738. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Matthay MA and Zimmerman GA: Acute lung
injury and the acute respiratory distress syndrome: Four decades of
inquiry into pathogenesis and rational management. Am J Respir Cell
Mol Biol. 33:319–327. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Matthay MA, Zimmerman GA, Esmon C,
Bhattacharya J, Coller B, Doerschuk CM, Floros J, Gimbrone MA Jr,
Hoffman E, Hubmayr RD, et al: Future research directions in acute
lung injury: Summary of a National Heart, Lung, and Blood Institute
working group. Am J Respir Crit Care Med. 167:1027–1035. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Moon C, Han JR, Park HJ, Hah JS and Kang
JL: Synthetic RGDS peptide attenuates lipopolysaccharide-induced
pulmonary inflammation by inhibiting integrin signaled MAP kinase
pathways. Respir Res. 10:182009. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Wang B, Gong X, Wan JY, Zhang L, Zhang Z,
Li HZ and Min S: Resolvin D1 protects mice from LPS-induced acute
lung injury. Pulm Pharmacol Ther. 24:434–441. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Bleharski JR, Kiessler V, Buonsanti C,
Sieling PA, Stenger S, Colonna M and Modlin RL: A role for
triggering receptor expressed on myeloid cells-1 in host defense
during the early-induced and adaptive phases of the immune
response. J Immunol. 170:3812–3818. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Tessarz AS and Cerwenka A: The
TREM-1/DAP12 pathway. Immunol Lett. 116:111–116. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Klesney-Tait J, Turnbull IR and Colonna M:
The TREM receptor family and signal integration. Nat Immunol.
7:1266–1273. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
40
|
Wang DY, Qin RY, Liu ZR, Gupta MK and
Chang Q: Expression of TREM-1 mRNA in acute pancreatitis. World J
Gastroenterol. 10:2744–2746. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Liu N, Gu Q and Zheng YS: Expression of
triggering receptor-1 in myeloid cells of mice with acute lung
injury. World J Emerg Med. 1:144–148. 2010.PubMed/NCBI
|
|
42
|
Baig MS, Zaichick SV, Mao M, de Abreu AL,
Bakhshi FR, Hart PC, Saqib U, Deng J, Chatterjee S, Block ML, et
al: NOS1-derived nitric oxide promotes NF-kB transcriptional
activity through inhibition of suppressor of cytokine signaling-1.
J Exp Med. 212:1725–1738. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Blackwell TS, Lancaster LH, Blackwell TR,
Venkatakrishnan A and Christman JW: Differential NF-kappaB
activation after intratracheal endotoxin. Am J Physiol.
277:L823–L830. 1999.PubMed/NCBI
|
|
44
|
Kang JL, Lee HW, Lee HS, Pack IS, Chong Y,
Castranova V and Koh Y: Genistein prevents nuclear factor-kappa B
activation and acute lung injury induced by lipopolysaccharide. Am
J Respir Crit Care Med. 164:2206–2212. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Blackwell TS and Christman JW: The role of
nuclear factor-kappaB in cytokine gene regulation. Am J Respir Cell
Mol Biol. 17:3–9. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Zhang A, Wang S, Zhang J and Wu H: Genipin
alleviates LPS-induced acute lung injury by inhibiting NF-κB and
NLRP3 signaling pathways. Int Immunopharmacol. 38:115–119. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Yan C, Guan F, Shen Y, Tang H, Yuan D, Gao
H and Feng X: Bigelovii A protects against
lipopolysaccharide-induced acute lung injury by blocking NF-κB and
CCAAT/Enhancer-binding protein δ pathways. Mediators Inflamm.
2016:92016042016. View Article : Google Scholar
|
|
48
|
Gomez-Pina V, Martinez E, Fernández-Ruiz
I, Del Fresno C, Soares-Schanoski A, Jurado T, Siliceo M, Toledano
V, Fernández-Palomares R, Garcia-Rio F, et al: Role of MMPs in
orchestrating inflammatory response in human monocytes via a
TREM-PI3K-NF-κB pathway. J Leukoc Biol. 91:933–945. 2012.
View Article : Google Scholar
|
|
49
|
Bauernfeind FG, Horvath G, Stutz A,
Alnemri ES, MacDonald K, Speert D, Fernandes-Alnemri T, Wu J, Monks
BG, Fitzgerald KA, et al: Cutting Edge: NF-kappaB activating
pattern recognition and cytokine receptors license NLRP3
inflammasome activation by regulating NLRP3 expression. J Immunol.
183:787–791. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Knapp S, Gibot S, de Vos A, Versteeg HH,
Colonna M and van der Poll T: Cutting edge: Expression patterns of
surface and soluble triggering receptor expressed on myeloid
cells-1 in human endotoxemia. J Immunol. 173:7131–7134. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Mahdy AM, Lowes DA, Galley HF, Bruce JE
and Webster NR: Production of soluble triggering receptor expressed
on myeloid cells by lipopolysaccharide-stimulated human neutrophils
involves de novo protein synthesis. Clin Vaccine Immunol.
13:492–495. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Gomez-Piña V, Soares-Schanoski A,
Rodriguez-Rojas A, Del Fresno C, Garcia F, Vallejo-Cremades MT,
Fernández-Ruiz I, Arnalich F, Fuentes-Prior P and López-Collazo E:
Metalloproteinases shed TREM-1 ectodomain from
lipopolysaccharide-stimulated human monocytes. J Immunol.
179:4065–4073. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Palazzo SJ, Simpson T and Schnapp LM:
Triggering receptor expressed on myeloid cells type 1 as a
potential therapeutic target in sepsis. Dimens Crit Care Nurs.
31:1–6. 2012. View Article : Google Scholar
|
|
54
|
Giamarellos-Bourboulis EJ, Zakynthinos S,
Baziaka F, Papadomichelakis E, Virtzili S, Koutoukas P, Armaganidis
A, Giamarellou H and Roussos C: Soluble triggering receptor
expressed on myeloid cells 1 as an anti-inflammatory mediator in
sepsis. Intensive Care Med. 32:237–243. 2006. View Article : Google Scholar : PubMed/NCBI
|