Introduction
Atherosclerosis is a chronic inflammatory vascular
disease characterized by infiltration of lipid particles into the
arterial wall, leading to inflammatory responses accompanied by
endothelial cell dysfunction and recruitment of inflammatory and
immune cells (1). Previous
studies have reported that epigenetic mechanisms may be associated
with the pathogenesis of atherosclerosis and may account for some
of the missing heritability in atherosclerotic cardiovascular
disease (2,3). Epigenetic control of transcription
results in a heritable change in gene expression without a change
in DNA sequence. DNA methylation and post-translational
modifications of histone tails, including lysine methylation and
acetylation, are the most common mechanisms that cause changes in
DNA accessibility (3).
DNA methylation is a vital epigenetic modification
that has been implicated in the pathogenesis of a number of common
complex diseases, including atherosclerosis and cardiovascular
disease (4–12). DNA methylation serves a role in a
variety of cellular processes (5,13),
is affected by environmental factors and is influenced by age, sex
and genetic variants (4,6). As such, elucidating the differences
in DNA methylation patterns between atherosclerotic plaque lesions
and plaque-free intima tissue may provide an insight into the
underlying molecular mechanisms of atherosclerotic cardiovascular
disease. Although previous analyses of DNA methylation have
identified various CpG sites and genes associated with
atherosclerosis in European-ancestry (14–17) or Mexican (18) populations, the pattern of DNA
methylation in the atherosclerotic human aorta at the genome-wide
level has remained relatively uncharacterized in Japanese
individuals.
A previous study examined DNA methylation at
~450,000 CpG sites (Human Methylation 450 BeadChip; Illumina, Inc.,
San Diego, CA, USA) in 48 human aortic intima specimens obtained
from 24 autopsy cases (19); it
was demonstrated that DNA methylation was significantly
(P<1.03×10−7) increased at 30 CpG sites and reduced
at 15 CpG sites in atheromatous plaque tissues compared with
plaque-free intima (19). In the
present study, to further assess the association between DNA
methylation and the development of atherosclerosis, a genome-wide
analysis of DNA methylation at ~853,000 CpG sites (Infinium
MethylationEPIC BeadChip) was performed in 128 human aortic intima
specimens obtained from 64 autopsy cases. Compared with the Human
Methylation 450 BeadChip array, the newly developed Infinium
MethylationEPIC BeadChip array is a more reliable tool for
comprehensive DNA methylation analyses (20). A total of 16 significantly hyper-
or hypomethylated novel genes in atheromatous plaque lesions were
identified.
Materials and methods
Study specimens
Characteristics of the 64 deceased patients from
whom tissues were harvested for use in the present study are
presented in Table I.
Inter-individual variation in DNA methylation was detected for the
same cell and tissue type of unrelated individuals (21,22). To avoid the effects of such
variation, intra-individual paired comparisons of DNA methylation
were performed between atheromatous plaque lesions and
corresponding plaque-free intima. A total of 128 postmortem
specimens of the aortic intima were obtained from 64 deceased
Japanese patients for analysis. The specimens were collected
specifically for this study in participating hospitals (Gifu
Prefectural Tajimi Hospital, Tajimi; Japanese Red Cross Nagoya
First Hospital, Nagoya; Kasugai Municipal Hospital, Kasugai; Tokyo
Metropolitan Geriatric Hospital, Tokyo, Japan) between August 2012
and August 2017. A total of 48 of these specimens obtained from 24
subjects were also analyzed in a previous study (19).
 | Table IPatient characteristics (n=64). |
Table I
Patient characteristics (n=64).
| Characteristic | Value |
|---|
| Mean age (years) ±
SD (range) | 75.8±12.9
(41–95) |
| Sex (male/female,
%) | 73.4/26.6 |
| Mean body mass
index (kg/m2) ± SD (range) | 19.8±4.3
(12.2–32.2) |
| Current or former
smoker (%) | 57.5 |
| Hypertension
(%) | 40.6 |
| Diabetes mellitus
(%) | 20.3 |
| Dyslipidemia
(%) | 9.4 |
| Chronic kidney
disease (%) | 32.8 |
| Myocardial
infarction (%) | 21.9 |
| Ischemic stroke
(%) | 9.4 |
| Cause of death
(n) | |
| Pneumonia | 13 |
| Myocardial
infarction | 12 |
| Dissecting aortic
aneurysm | 4 |
| Amyloidosis | 2 |
| Dilated
cardiomyopathy | 2 |
| Hypertrophic
cardiomyopathy | 2 |
| Interstitial
pneumonia | 2 |
| Lung cancer | 2 |
| Necrotizing
enterocolitis | 2 |
| Amyotrophic
lateral sclerosis | 1 |
| Arrhythmogenic
right ventricular cardiomyopathy | 1 |
| Aspergillosis | 1 |
| Bacterial
meningitis | 1 |
| Cardiac
sarcoidosis | 1 |
| Chronic
obstructive pulmonary disease | 1 |
| Embolic
stroke | 1 |
| Gastric
cancer | 1 |
| Heatstroke | 1 |
| Huntingdon's
disease | 1 |
| Intestinal
obstruction | 1 |
| Ischemic heart
disease | 1 |
| Malignant
mesothelioma | 1 |
| Malnutrition | 1 |
| Multiple
myeloma | 1 |
| Parkinson's
disease | 1 |
| Primary biliary
cirrhosis | 1 |
| Pulmonary
hypertension | 1 |
| Relapsing
polychondritis | 1 |
| Renal failure | 1 |
| Superior
mesenteric artery thrombosis | 1 |
| Traumatic lung
contusion | 1 |
| Valvular heart
disease | 1 |
Immunohistochemical analysis of
atheromatous plaque lesions and plaque-free intima
Specimens of atheromatous plaque lesions and
plaque-free intima were subjected to immunohistochemical analysis
as described previously (19).
Formalin (20%)-fixed (6 h at room temperature) and
paraffin-embedded sections (3 µm) were deparaffinized,
hydrated, immersed in 0.01 mol/l citrate buffer (pH 6.0), and
heated for 10 min in a pressure cooker. Endogenous peroxidase was
blocked with 3% hydrogen peroxide for 5 min at room temperature and
sections were incubated for 30 min at room temperature with mouse
monoclonal antibodies against human α-smooth muscle actin (1:100;
M0851; Dako; Agilent Technologies, Inc., Santa Clara, CA, USA),
CD68 (1:100; N1576; Dako; Agilent Technologies, Inc.) and CD45
(1:100; 722071; Nichirei Bioscience, Inc., Tokyo, Japan).
Proteinase K (0.1%) pre-treatment (5 min at room temperature) was
used for CD68 and CD45. Sections were subsequently incubated for 30
min at room temperature with horseradish peroxidase
(HRP)-conjugated goat poly-clonal antibody to rabbit and mouse
immunoglobulin (1:100; K5007; Dako; Agilent Technologies, Inc.).
Sections were stained with diaminobenzidine for 10 min at room
temperature (ChemMate Envision/HRP kit; K5007; Dako; Agilent
Technologies, Inc.).
The present study was approved by the Committees on
the Ethics of Human Research of: Mie University Graduate School of
Medicine, Tsu; Tokyo Metropolitan Institute of Gerontology, Tokyo;
Japanese Red Cross Nagoya First Hospital, Nagoya; Gifu Prefectural
Tajimi Hospital, Tajimi; and Kasugai Municipal Hospital, Kasugai
(all Japan). Written informed consent was obtained from the
families of the deceased patients.
Genome-wide analysis of DNA
methylation
The intima tissue samples were frozen at −80°C
immediately following dissection from the aorta. The finely minced
(cut to ~1 mm3 with a surgical blade) tissue was
subsequently mixed with 250 µl phenol-chloroform and
centrifuged at 12,000 × g for 5 min at room temperature. The upper
aqueous phase was collected for the precipitation of genomic DNA
and 100% ethanol containing 0.3 mol/l sodium acetate was added and
incubated at −30°C for 30 min. The mixture was then centrifuged at
12,000 × g for 20 min at 4°C and the DNA pellet was dissolved in
Tris-EDTA buffer (pH 7.4; Takara Bio, Inc., Otsu, Japan). Bisulfite
conversion of genomic DNA was performed using an EZ DNA Methylation
kit (Zymo Research Corp., Irvine, CA, USA).
The bisulfite-modified genomic DNA was analyzed for
DNA methylation with a DNA methylation-specific microarray
(Infinium MethylationEPIC BeadChip, Illumina, Inc.) that included
853,307 CpG sitets distributed throughout the entire genome. A
total of 439,562 (91.1%) of these sites were assessed in a previous
study (19) using the Human
Methylation 450 BeadChip. Furthermore, 413,745 additional CpG
sites, including 333,265 located in enhancer regions, were
identified by the Encyclopedia of DNA elements (23) and FANTOM5 (24) projects. The Infinium Methylation
EPIC BeadChip microarray also interrogates 2,880 CNG (C, cytosine;
N, any nucleotide; G, guanine) sites (20).
Methylation at CpG sites in genomic DNA isolated
from atheromatous plaque lesions or plaque-free intima was assessed
using a GenomeStudio Methylation Module (Illumina, Inc.). Call rate
values for the 128 specimens were all >99.3%, with a mean value
of 99.7%. The DNA methylation level at each CpG site was calculated
as the β value, where β = (intensity of the methylated
allele)/(intensity of the methylated allele + intensity of the
unmethylated allele + 100) (25).
Statistical analysis
The levels of DNA methylation at 853,307 CpG sites
(β values) were compared between atheromatous plaque lesions and
plaque-free intima using the unpaired Student's t-test. To
compensate for multiple comparisons, Bonferroni's correction for
the statistical significance of associations was used. The
significance level was thus P<5.86×10−8
(0.05/853,307) for the genome-wide analysis of DNA methylation.
Statistical tests were performed using JMP Genomics 6.0 software
(SAS Institute, Inc., Cary, NC, USA).
Analysis of public databases
The potential association between CpG sites and
genes identified in the present study with atherosclerosis was
assessed by searching public databases between January 2007 and
October 2017 [Google Scholar (http://scholar.google.co.jp); PubMed (National Center
for Biotechnology Information, Bethesda, MD, USA; https://www.ncbi.nlm.nih.gov/pubmed), GWAS
Catalog (National Human Genome Research Institute, Bethesda, MD,
USA and European Bioinformatics Institute, Hinxton, UK; http://www.ebi.ac.uk/gwas) and GWAS Central
(http://www.gwascentral.org)] for
previously associated phenotypes. Genome-wide analyses of DNA
methylation or genome-wide association studies for atherosclerosis
or cardiovascular disease were included in the results. DNA
methylation analyses or association studies of candidate genes were
excluded.
Results
Study specimens
In the present study, the methylation status of
853,307 CpG sites of genomic DNA purified from atheromatous plaque
lesions and corresponding plaque-free intima were compared.
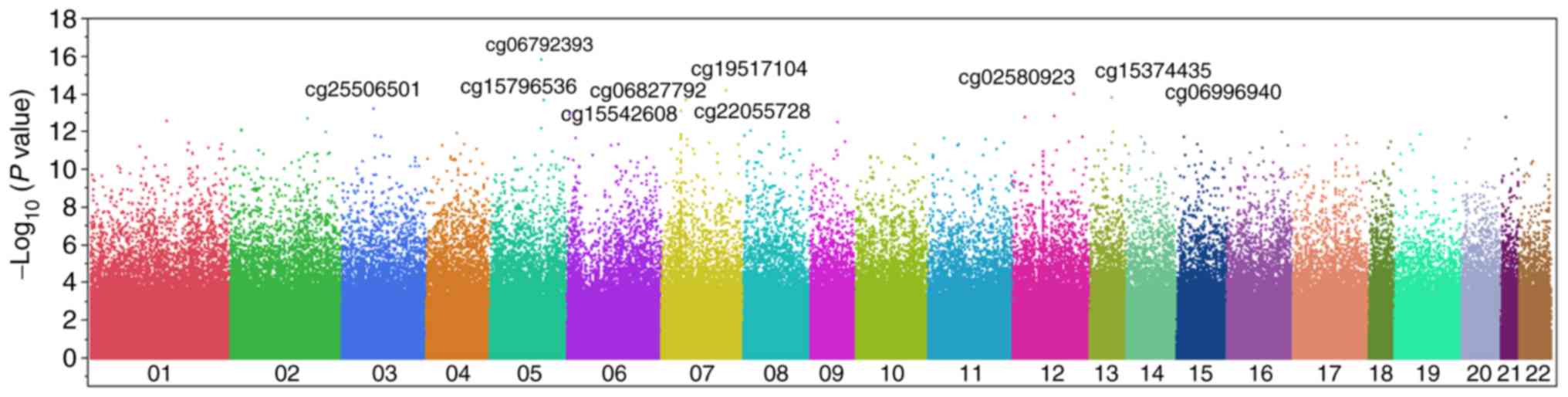
Manhattan and volcano plots for the genome-wide analysis of
differences in methylation status at these sites are presented in
Figs. 1 and 2, respectively. Following Bonferroni's
correction, the methylation of 2,679 CpG sites was revealed to
differ significantly (P<5.86×10−8) between
atheromatous plaque lesions and corresponding plaque-free intima.
The 50 CpG sites with the lowest P-values (P≤3.48×10−12)
are presented in Table II; to
the best of our knowledge, none of these sites have previously been
reported to be associated with atherosclerosis.
| Table IIA total of 50 CpG sites with the
lowest P-values (P≤3.48×10−12) for the comparison of
methylation status (β values) between atheromatous plaque lesions
and plaque-free intima by a genome-wide analysis of DNA
methylation. |
Table II
A total of 50 CpG sites with the
lowest P-values (P≤3.48×10−12) for the comparison of
methylation status (β values) between atheromatous plaque lesions
and plaque-free intima by a genome-wide analysis of DNA
methylation.
| CpG | Chromosome:
position | Gene | Methylation
site | Mean β value
(plaque) | Mean β value
(plaque-free) | β ratio
(plaque/plaque-free) | P-value |
|---|
| cg06792393 | 5:139242953 | NRG2 | Body | 0.7906 | 0.6985 | 1.13 |
1.11×10−16 |
| cg19517104 | 7:134204895 | | | 0.8458 | 0.7535 | 1.12 |
5.60×10−15 |
| cg02580923 | 12:117470541 | | | 0.4173 | 0.5093 | 0.82 |
9.21×10−15 |
| cg15374435 | 13:80911692 | SPRY2 | Body | 0.7301 | 0.5885 | 1.24 |
1.42×10−14 |
| cg15796536 | 5:140873082 | PCDHGA8 | Body | 0.8437 | 0.7766 | 1.09 |
1.82×10−14 |
| cg06827792 | 7:36723794 | AOAH | Body | 0.8015 | 0.7180 | 1.12 |
1.92×10−14 |
| cg06996940 | 15:28197405 | OCA2 | Body | 0.8065 | 0.7492 | 1.08 |
3.82×10−14 |
| cg25506501 | 3:59521337 | | | 0.7693 | 0.6500 | 1.18 |
5.56×10−14 |
| cg22055728 | 7:27205658 | HOXA9 | TSS1500 | 0.1162 | 0.2026 | 0.57 |
7.80×10−14 |
| cg15542608 | 6:12827379 | PHACTR1 | Body | 0.8312 | 0.7371 | 1.13 |
7.86×10−14 |
| cg07145664 | 6:2579591 | | | 0.6933 | 0.5966 | 1.16 |
1.21×10−13 |
| cg26968433 | 12:76397292 | | | 0.7965 | 0.7363 | 1.08 |
1.28×10−13 |
| cg24082440 | 21:33672186 | MRAP | Body | 0.8245 | 0.7598 | 1.09 |
1.53×10−13 |
| cg27554156 | 12:13248725 | GSG1 | Body, TSS200 | 0.8345 | 0.7781 | 1.07 |
1.64×10−13 |
| cg12433228 | 2:177486421 | | | 0.6642 | 0.5898 | 1.13 |
1.83×10−13 |
| cg12336358 | 1:114000078 | MAGI3 | Body | 0.7586 | 0.6597 | 1.15 |
2.34×10−13 |
| cg09723384 | 9:122217063 | | | 0.5751 | 0.6710 | 0.86 |
2.89×10−13 |
| cg05454595 | 5:139243335 | NRG2 | Body | 0.6737 | 0.5909 | 1.14 |
5.69×10−13 |
| cg06019613 | 2:18717524 | | | 0.4711 | 0.6044 | 0.78 |
7.60×10−13 |
| cg22651416 | 8:10643634 | PINX1 | Body | 0.8567 | 0.7935 | 1.08 |
8.42×10−13 |
| cg17882587 | 2:18933408 | | | 0.6727 | 0.5779 | 1.16 |
8.56×10−13 |
| cg19691778 | 8:97157756 | GDF6 | Body | 0.3900 | 0.5395 | 0.72 |
9.03×10−13 |
| cg17820365 | 8:97157856 | GDF6 | Body | 0.2811 | 0.4348 | 0.65 |
9.07×10−13 |
| cg15086256 | 2:222631916 | | | 0.7039 | 0.6146 | 1.15 |
9.39×10−13 |
| cg18141318 | 13:96350690 | DNAJC3 | Body | 0.8180 | 0.7723 | 1.06 |
9.67×10−13 |
| cg01754709 | 16:84961336 | | | 0.7191 | 0.6133 | 1.17 |
9.74×10−13 |
| cg17128320 | 4:78633646 | | | 0.8804 | 0.8235 | 1.07 |
1.14×10−12 |
| cg16597993 | 19:16578633 | EPS15L1 | Body | 0.8226 | 0.7540 | 1.09 |
1.20×10−12 |
| cg10224937 | 7:27208594 | HOXA10-AS,
HOXA10, HOXA9 | Body | 0.5089 | 0.6669 | 0.76 |
1.32×10−12 |
| cg12786452 | 17:62309293 | TEX2 | 5′UTR | 0.8392 | 0.7737 | 1.08 |
1.44×10−12 |
| cg05069228 | 3:61793623 | PTPRG | Body | 0.7403 | 0.6545 | 1.13 |
1.48×10−12 |
| cg17741799 | 8:885656 | | | 0.7770 | 0.7158 | 1.09 |
1.49×10−12 |
| cg23854860 | 7:27208590 | HOXA10-AS,
HOXA10, HOXA9 | Body | 0.5480 | 0.6949 | 0.79 |
1.67×10−12 |
| cg03735712 | 15:35263065 | AQR | TSS1500 | 0.7572 | 0.6263 | 1.21 |
1.69×10−12 |
| cg09164580 | 8:97157878 | GDF6 | Body | 0.4534 | 0.6044 | 0.75 |
1.73×10−12 |
| cg10166915 | 14:54260975 | | | 0.7714 | 0.6711 | 1.15 |
1.73×10−12 |
| cg17868595 | 3:90112204 | | | 0.7352 | 0.8183 | 0.90 |
1.74×10−12 |
| cg00246590 | 12:126968300 | | | 0.8422 | 0.6984 | 1.21 |
1.84×10−12 |
| cg19183743 | 7:27188020 | HOXA6 | TSS1500 | 0.3144 | 0.4401 | 0.71 |
1.86×10−12 |
| cg27584713 | 6:12717016 | PHACTR1 | TSS1500 | 0.6815 | 0.5733 | 1.19 |
1.98×10−12 |
| cg02750391 | 11:14274978 | SPON1 | Body | 0.7800 | 0.7205 | 1.08 |
2.04×10−12 |
| cg21310745 | 7:27208454 | HOXA10-AS,
HOXA10, HOXA9 | TSS200, body | 0.6244 | 0.7815 | 0.80 |
2.22×10−12 |
| cg16739092 | 20:17519424 | BFSP1 | Body, 5′UTR | 0.3781 | 0.4932 | 0.77 |
2.23×10−12 |
| cg03804397 | 11:76925617 | MYO7A | Body | 0.7302 | 0.6427 | 1.14 |
2.31×10−12 |
| cg22790931 | 7:39017455 | POU6F2 | TSS200 | 0.9070 | 0.8683 | 1.04 |
2.36×10−12 |
| cg23080761 | 12:111857575 | SH2B3 | Body | 0.8194 | 0.7611 | 1.08 |
3.04×10−12 |
| cg13913011 | 9:132711808 | FNBP1 | Body | 0.7628 | 0.6517 | 1.17 |
3.18×10−12 |
| cg08272913 | 18:68079266 | | | 0.6156 | 0.4664 | 1.32 |
3.33×10−12 |
| cg20337028 | 17:75181836 | SEC14L1 | Body, 5′UTR | 0.7624 | 0.6871 | 1.11 |
3.44×10−12 |
| cg12873661 | 1:166277235 | | | 0.6713 | 0.5187 | 1.29 |
3.48×10−12 |
Genome-wide analysis of gene
methylation
Of the 2,679 CpG sites significantly associated with
atherosclerosis, 2,272 and 407 sites were hyper- or hypomethylated,
respectively, in atheromatous plaque lesions compared with
plaque-free intima. Among the 2,272 CpG sites that were
hypermethylated in atheromatous plaque lesions, 5 had a β value
difference (plaque lesion-plaque-free intima) >0.15 (Table III) and 11 had a β ratio (plaque
lesion/plaque-free intima) >1.50 (Table IV). Among these CpG sites,
cg15648389 of homeobox (HOX) C4 (15), cg17466857 of
HOXA11-HOXA11-AS (14,15), cg15700739 of
HOXC4/HOXC5 (17)
and cg02384661 of HOXC11 (15) have previously been demonstrated to
be associated with atherosclerosis.
| Table IIIFive CpG sites whose methylation
status differed significantly (P<5.86×10−8) between
atheromatous plaque lesions and plaque-free intima with a
difference in β values (plaque lesion-plaque-free intima) of
>0.15. |
Table III
Five CpG sites whose methylation
status differed significantly (P<5.86×10−8) between
atheromatous plaque lesions and plaque-free intima with a
difference in β values (plaque lesion-plaque-free intima) of
>0.15.
| CpG | Chromosome:
position | Gene | Mean β value
(plaque) | Mean β value
(plaque-free) | β ratio
(plaque/plaque-free) | Difference in β
values (plaque-plaque-free) | P-value |
|---|
| cg26809635 | 12:54355087 | | 0.2669 | 0.0835 | 3.1957 | 0.1834 |
6.35×10−10 |
| cg23786812 | 7:156296516 | | 0.5526 | 0.3922 | 1.4090 | 0.1604 |
4.12×10−12 |
| cg15648389 | 12:54448769 | HOXC4 | 0.5564 | 0.4005 | 1.3893 | 0.1559 |
3.60×10−11 |
| cg27178293 | 12:54371246 | | 0.4042 | 0.2484 | 1.6270 | 0.1558 |
1.24×10−8 |
| cg12873661 | 1:166277235 | | 0.6713 | 0.5187 | 1.2941 | 0.1526 |
3.48×10−12 |
| Table IVEleven CpG sites whose methylation
status differed significantly (P<5.86×10−8) between
atheromatous plaque lesions and plaque-free intima with a β ratio
(plaque/plaque-free) of >1.50. |
Table IV
Eleven CpG sites whose methylation
status differed significantly (P<5.86×10−8) between
atheromatous plaque lesions and plaque-free intima with a β ratio
(plaque/plaque-free) of >1.50.
| CpG | Chromosome:
position | Gene | Methylation
site | Mean β value
(plaque) | Mean β value
(plaque-free) | β ratio
(plaque/plaque-free) | P-value |
|---|
| cg26809635 | 12:54355087 | | | 0.2669 | 0.0835 | 3.20 |
6.35×10−10 |
| cg18040901 | 12:54357530 | HOTAIR | Body | 0.1839 | 0.0979 | 1.88 |
6.12×10−11 |
| cg00576279 | 12:54427293 | HOXC4,
HOXC5 | 5′UTR, body | 0.2516 | 0.1419 | 1.77 |
5.04×10−9 |
| cg17466857 | 7:27225528 | HOXA11-AS,
HOXA11 | Body, TSS1500 | 0.2618 | 0.1479 | 1.77 |
1.16×10−8 |
| cg08857479 | 12:54369987 | HOXC11 | 3′UTR | 0.3479 | 0.2112 | 1.65 |
1.93×10−8 |
| cg27178293 | 12:54371246 | | | 0.4042 | 0.2484 | 1.63 |
1.24×10−8 |
| cg15700739 | 12:54427700 | HOXC4,
HOXC5 | 5′UTR, body | 0.3313 | 0.2050 | 1.62 |
1.15×10−9 |
| cg00862376 | 12:54343711 | | | 0.2745 | 0.1708 | 1.61 |
5.27×10−9 |
| cg00187380 | 12:54427384 | HOXC4,
HOXC5 | 5′UTR, body | 0.3373 | 0.2144 | 1.57 |
2.21×10−8 |
| cg02384661 | 12:54369638 | HOXC11 | 3′UTR | 0.3804 | 0.2450 | 1.55 |
5.36×10−9 |
| cg05951084 | 12:54343681 | | | 0.3480 | 0.2304 | 1.51 |
7.64×10−9 |
Among the 407 CpG sites hypomethylated in
atheromatous plaque lesions, 15 were observed to have a β value
difference <−0.15 (Table V)
and 17 sites had a β ratio <0.67 (Table VI). Of these CpG sites,
cg03217995 of HOXA9 (15)
was previously reported to be associated with atherosclerosis.
| Table VFifteen CpG sites whose methylation
status differed significantly (P<5.86×10−8) between
atheromatous plaque lesions and plaque-free intima with a
difference in β values (plaque lesion-plaque-free intima) of
<−0.15. |
Table V
Fifteen CpG sites whose methylation
status differed significantly (P<5.86×10−8) between
atheromatous plaque lesions and plaque-free intima with a
difference in β values (plaque lesion-plaque-free intima) of
<−0.15.
| CpG | Chromosome:
position | Gene | Mean β value
(plaque) | Mean β value
(plaque-free) | β ratio
(plaque/plaque-free) | Difference in β
values (plaque-plaque-free) | P-value |
|---|
| cg18461866 | 3:59996864 | FHIT | 0.3759 | 0.5558 | 0.6763 | −0.1799 |
1.78×10−10 |
| cg21007852 | 7:27203546 | HOXA9 | 0.5134 | 0.6904 | 0.7435 | −0.1771 |
2.96×10−11 |
| cg25227803 | 10:102239027 | WNT8B | 0.3980 | 0.5727 | 0.6951 | −0.1746 |
1.84×10−11 |
| cg03217995 | 7:27203430 | HOXA9 | 0.5377 | 0.7107 | 0.7565 | −0.1731 |
5.49×10−10 |
| cg02886033 | 7:27208114 | | 0.5367 | 0.7048 | 0.7615 | −0.1681 |
7.82×10−10 |
| cg11052578 | 21:39695801 | | 0.3391 | 0.5064 | 0.6697 | −0.1672 |
4.05×10−8 |
| cg13669152 | 6:130923610 | | 0.4987 | 0.6580 | 0.7579 | −0.1593 |
2.46×10−8 |
| cg10224937 | 7:27208594 | HOXA10-AS,
HOXA10, HOXA9 | 0.5089 | 0.6669 | 0.7631 | −0.1580 |
1.32×10−12 |
| cg09699744 | 12:54390705 |
HOXC-AS2 | 0.3719 | 0.5298 | 0.7020 | −0.1579 |
1.60×10−10 |
| cg21310745 | 7:27208454 | HOXA10-AS,
HOXA10, HOXA9 | 0.6244 | 0.7815 | 0.7990 | −0.1571 |
2.22×10−12 |
| cg01016793 | 15:64894722 | ZNF609 | 0.4212 | 0.5773 | 0.7297 | −0.1561 |
1.03×10−11 |
| cg10374314 | 7:27189610 |
HOXA-AS3 | 0.5017 | 0.6558 | 0.7650 | −0.1541 |
4.03×10−11 |
| cg17820365 | 8:97157856 | GDF6 | 0.2811 | 0.4348 | 0.6465 | −0.1537 |
9.07×10−13 |
| cg09164580 | 8:97157878 | GDF6 | 0.4534 | 0.6044 | 0.7502 | −0.1510 |
1.73×10−12 |
| cg06136628 | 7:35289993 | TBX20 | 0.2790 | 0.4293 | 0.6498 | −0.1503 |
4.81×10−10 |
| Table VISeventeen CpG sites whose methylation
status differed significantly (P<5.86×10−8) between
atheromatous plaque lesions and plaque-free intima with a β ratio
(plaque/plaque-free) of <0.67. |
Table VI
Seventeen CpG sites whose methylation
status differed significantly (P<5.86×10−8) between
atheromatous plaque lesions and plaque-free intima with a β ratio
(plaque/plaque-free) of <0.67.
| CpG | Chromosome:
position | Gene | Methylation
site | Mean β value
(plaque) | Mean β value
(plaque-free) | β ratio
(plaque/plaque-free) | P-value |
|---|
| cg22055728 | 7:27205658 | HOXA9 | TSS1500 | 0.1162 | 0.2026 | 0.57 |
7.80×10−14 |
| cg13335081 | 8:66894111 | | | 0.1758 | 0.2913 | 0.60 |
1.67×10−8 |
| cg23345300 | 2:177000621 | | | 0.1669 | 0.2752 | 0.61 |
3.81×10−8 |
| cg04122553 | 14:21250646 | | | 0.1587 | 0.2615 | 0.61 |
3.43×10−9 |
| cg24719020 | 7:27187943 | HOXA6 | TSS1500 | 0.2018 | 0.3324 | 0.61 |
1.51×10−9 |
| cg14554869 | 5:74231171 | | | 0.1656 | 0.2630 | 0.63 |
9.29×10−11 |
| cg11348442 | 2:220117784 | TUBA4B,
TUBA4A | TSS200, body | 0.1177 | 0.1869 | 0.63 |
5.22×10−8 |
| cg10893095 | 6:85478296 | | | 0.2545 | 0.4008 | 0.64 |
1.10×10−10 |
| cg26437522 | 7:27174169 | | | 0.2220 | 0.3460 | 0.64 |
4.59×10−9 |
| cg01428378 | 12:123259789 | CCDC62 | Body | 0.1784 | 0.2770 | 0.64 |
2.67×10−8 |
| cg17820365 | 8:97157856 | GDF6 | Body | 0.2811 | 0.4348 | 0.65 |
9.07×10−13 |
| cg25541958 | 8:1992965 | MYOM2 | TSS200 | 0.1679 | 0.2585 | 0.65 |
4.63×10−11 |
| cg06136628 | 7:35289993 | TBX20 | Body | 0.2790 | 0.4293 | 0.65 |
4.81×10−10 |
| cg19783626 | 7:98311436 | | | 0.2204 | 0.3366 | 0.65 |
3.72×10−12 |
| cg00980698 | 14:21250509 | RNASE6 | 3′UTR | 0.1554 | 0.2365 | 0.66 |
1.67×10−10 |
| cg12110087 | 7:27187691 | HOXA6 | TSS1500 | 0.2103 | 0.3168 | 0.66 |
8.08×10−9 |
| cg11052578 | 21:39695801 | | | 0.3391 | 0.5064 | 0.66 |
4.05×10−8 |
Discussion
Atherosclerosis occurs as a result of endothelial
damage and dysfunction that leads to the accumulation and oxidation
of low-density lipoprotein (LDL) cholesterol in the vessel wall.
Monocytes migrate from blood into the subendothelial intima and
transform into macrophages, which accumulate lipids as foam cells
in the lipid core of the atherosclerotic plaque (26,27). Inflammatory and thrombotic
processes serve primary roles in the formation of atherosclerotic
lesions and subsequent plaque rupture that causes acute coronary
syndrome (26,27). A number of mechanisms by which
changes in DNA methylation may affect the development of
atherosclerosis have been identified. These mechanisms include the
promotion of inflammation, endothelial dysfunction, proliferation
and migration of smooth muscle cells or monocyte-macrophages,
extracellular matrix production, homocysteine metabolism and
apoptosis of vascular cells (12,28,29). However, given the dynamic nature
and tissue heterogeneity of atherosclerosis, defining the precise
role of DNA methylation in the pathogenesis of this condition is
challenging (12). A marked
increase in DNA methylation in atherosclerotic lesions may warrant
the development of DNA demethylation agents, including DNA
methyltransferase inhibitors, for the treatment of atherosclerotic
cardiovascular disease (29).
Arteriosclerosis is classified into three types:
atherosclerosis, Mönckeberg medial sclerosis and arteriolosclerosis
(30). Given that atherosclerosis
is the most important pathological change in the development of
cardiovascular disease (1,26,27,30),
the aortic intima was examined in the present study. The results
revealed that 2,272 and 407 CpG sites were hyper- and
hypomethylated, respectively, in genomic DNA isolated from
atheromatous plaque lesions compared with matched plaque-free
intima. A total of 5 CpG sites had a >0.15 difference in β
values and 11 CpG sites had a β ratio of >1.50. Among these CpG
sites, cg15648389 of HOXC4 (15), cg17466857 of
HOXA11-HOXA11-AS (14,15), cg15700739 of
HOXC4/HOXC5 (17)
and cg02384661 of HOXC11 (15) have previously been reported to be
associated with atherosclerosis. A total of 10 novel CpG sites
(cg26809635, cg23786812, cg27178293, cg12873661, cg18040901,
cg00576279, cg08857479, cg00862376, cg00187380, cg05951084) that
were significantly hypermethylated in atheromatous plaque lesions
compared with plaque-free intima were identified in the present
study. Of these 10 CpG sites, cg18040901 is located in the HOX
transcript antisense RNA (HOTAIR) gene, whose
methylation status has not previously been associated with
atherosclerosis. The HOTAIR gene is located at chromosome
12q13.13 and encodes a protein that has been reported to promote
the proliferation and migration of vascular endothelial cells and
to protect these cells against oxidized LDL-induced injury and
apoptosis (31). Endothelial
damage and dysfunction are early key processes in the development
of atherosclerosis, resulting in the accumulation and oxidation of
LDL-cholesterol in the arterial wall (26,27), and so HOTAIR may protect
against this (31).
A total of 15 CpG sites with a <−0.15 difference
in β values and 17 CpG sites with a β ratio of <0.67 were
identified in the present study, including 2 CpG sites (cg13669152,
cg13335081) located in enhancer regions as described by the FANTOM5
project (24). Of these sites,
cg03217995 of HOXA9 (15)
was previously reported to be associated with atherosclerosis.
Additionally, 28 novel CpG sites that were significantly
hypomethylated in atheromatous plaque lesions compared with
plaque-free intima were identified in the present study:
cg18461866, cg21007852, cg25227803, cg02886033, cg11052578,
cg13669152, cg10224937, cg09699744, cg21310745, cg01016793,
cg10374314, cg17820365, cg09164580, cg06136628, cg22055728,
cg13335081, cg23345300, cg04122553, cg24719020, cg14554869,
cg11348442, cg10893095, cg26437522, cg01428378, cg25541958,
cg19783626, cg00980698 and cg12110087. Of these sites, 16 are
located in genes whose methylation status has not previously been
reported as associated with atherosclerosis, including fragile
histidine triad (FHIT; cg18461866), wnt family member 8B
(WNT8B; cg25227803), HOXA10-HOXA10-antisense RNA
(AS; cg10224937 and cg21310745), HOXC cluster antisense RNA
2 (HOXC-AS2; cg09699744), zinc finger protein 609
(ZNF609; cg01016793), HOXA-AS3 (cg10374314),
growth differentiation factor 6 (GDF6; cg17820365,
cg09164580), T-box 20 (TBX20; cg06136628), HOXA6
(cg24719020, cg12110087), tubulin alpha 4a and 4b
(TUBA4A/TUBA4B; cg11348442), coiled-coil domain
containing 62 (CCDC62; cg01428378), myomesin 2
(MYOM2; cg25541958) and ribonuclease A family member
k6 (RNASE6; cg00980698).
TBX20 is located at chromosome 7p14.2 and
encodes a protein that has been demonstrated to protect endothelial
cells against oxidized LDL-induced injury via upregulating
peroxisome proliferator-activated receptor γ, indicating that it
may protect against the development of atherosclerosis (32). It has also previously been
determined that a polymorphism (rs3206736) near TBX20 is
associated with a decrease in diastolic blood pressure (33). Several of the genes that were
demonstrated to be hypomethylated in atherosclerotic tissues in the
present study have been reported to be correlated with
atherosclerosis-related phenotypes: The FHIT gene located at
chromosome 3p14.2 is associated with body mass index (34); the HOXA10-HOXA10-AS
gene at 7p15.2 is expressed differentially in porcine coronary and
iliac artery endothelial cells (35); the HOXA-AS3 gene at 7p15.2
is associated with chronic venous disease (36) and monocyte count (37); HOXA6 at 7p15.2 is
associated with chronic venous disease (36); and the TUBA4A/TUBA4B
gene at 2q35 is associated with the size distribution of platelets
(37). The remaining novel genes
identified in the present study (WNT8B located at chromosome
10q24.31; HOXC-AS2 at 12q13.13; ZNF609 at 15q22.31;
GDF6 at 8q22.1; CCDC62 at 12q24.31; MYOM2 at
8p23.3; and the RNASE6 gene at 14q11.2) have not previously
been reported as associated with atherosclerosis-related
phenotypes.
It was previously demonstrated that the methylation
at 45 CpG sites differed significantly (P<1.03×10−7)
between atheromatous plaque lesions and plaque-free intima
(19). The associations between
23 of these CpG sites [cg02240539 (P=0.0499), cg04304054
(P=0.0071), cg14521421 (P=0.0033), cg14477581 (P=0.0302),
cg00909706 (P=3.49×10−6), cg00716848 (P=0.0009),
cg08466030 (P=8.08×10−6), cg22046201 (P=0.0002),
cg18516609 (P=0.0188), cg24634746 (P=8.43×10−6),
cg26619894 (P=1.20×10−5), cg03962451
(P=5.52×10−5), cg12556802 (P=4.90×10−7),
cg10586883 (P=0.0309), cg06208382 (P=1.06×10−6),
cg18177814 (P=0.0013), cg20556639 (P=1.07×10−6),
cg09349128 (P=0.0112), cg26724841 (P=0.0379), cg02196592
(P=0.0235), cg16906765 (P=0.0032), cg27647755
(P=1.16×10−6), cg01473038 (P=0.0044)] to atherosclerosis
were replicated in the present study.
There are several limitations to the present study:
i) The aortic intima specimens comprised heterogeneous cell types,
as described previously (19).
ii) Although DNA methylation status may differ among
atherosclerosis, Mönckeberg medial sclerosis and
arteriolosclerosis, only the aortic intima was examined. iii) The
association between atherosclerosis grade and DNA methylation
status was not assessed. iv) The effects of hyper- or
hypomethylation of CpG sites on the expression of genes were not
investigated. v) Given the small sample size of the current study,
the statistical power of the genome-wide analysis of DNA
methylation was not optimal. vi) The molecular mechanisms
underlying the effects of DNA methylation identified in the present
study have not been determined definitively. vii) The validation of
the results of the present study will require replication with
other independent subject panels.
In conclusion, 16 novel genes that were
significantly hyper-or hypomethylated in atheromatous plaque
lesions compared with plaque-free intima were identified in the
present study. These results suggest that the methylation status of
these genes may contribute to the pathogenesis of atherosclerosis.
The determination of DNA methylation status for the identified CpG
sites may prove informative for the assessment of epigenetic risks
associated with atherosclerotic cardiovascular disease in the
Japanese population.
Acknowledgments
The present study was supported by CREST (grant no.
JPMJCR1302), Japan Science and Technology Agency (Kawaguchi,
Japan).
Notes
[1] Competing
interests
The authors declare that they have no competing
interests.
References
|
1
|
Lusis AJ: Atherosclerosis. Nature.
407:233–241. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
2
|
Baccarelli A, Rienstra M and Benjamin EJ:
Cardiovascular epigenetics: Basic concepts and results from animal
and human studies. Circ Cardiovasc Genet. 3:567–573. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Neele AE, Van den Bossche J, Hoeksema MA
and de Winther MP: Epigenetic pathways in macrophages emerge as
novel targets in atherosclerosis. Eur J Pharmacol. 763:79–89. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Rakyan VK, Down TA, Balding DJ and Beck S:
Epigenome-wide association studies for common human diseases. Nat
Rev Genet. 12:529–541. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
Handy DE, Castro R and Loscalzo J:
Epigenetic modifications: Basic mechanisms and role in
cardiovascular disease. Circulation. 123:2145–2156. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Tsai PC, Spector TD and Bell JT: Using
epigenome-wide association scans of DNA methylation in age-related
complex human traits. Epigenomics. 4:511–526. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Pfeiffer L, Wahl S, Pilling LC, Reischl E,
Sandling JK, Kunze S, Holdt LM, Kretschmer A, Schramm K, Adamski J,
et al: DNA methylation of lipid-related genes affects blood lipid
levels. Circ Cardiovasc Genet. 8:334–342. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Rask-Andersen M, Martinsson D, Ahsan M,
Enroth S, Ek WE, Gyllensten U and Johansson Å: Epigenome-wide
association study reveals differential DNA methylation in
individuals with a history of myocardial infarction. Hum Mol Genet.
25:4739–4748. 2016.
|
|
9
|
Wahl S, Drong A, Lehne B, Loh M, Scott WR,
Kunze S, Tsai PC, Ried JS, Zhang W, Yang Y, et al: Epigenome-wide
association study of body mass index, and the adverse outcomes of
adiposity. Nature. 541:81–86. 2017. View Article : Google Scholar :
|
|
10
|
Li J, Zhu X, Yu K, Jiang H, Zhang Y, Deng
S, Cheng L, Liu X, Zhong J, Zhang X, et al: Genome-wide analysis of
DNA methylation and acute coronary syndrome. Circ Res.
120:1754–1767. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Fernández-Sanlés A, Sayols-Baixeras S,
Subirana I, Degano IR and Elosua R: Association between DNA
methylation and coronary heart disease or other atherosclerotic
events: A systematic review. Atherosclerosis. 263:325–333. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Khyzha N, Alizada A, Wilson MD and Fish
JE: Epigenetics of atherosclerosis: Emerging mechanisms and
methods. Trends Mol Med. 23:332–347. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Deaton AM and Bird A: CpG islands and the
regulation of transcription. Genes Dev. 25:1010–1022. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Nazarenko MS, Puzyreva VP, Lebedev IN,
Frolov AV, Barbarash OL and Barbarash LS: Methylation profiling of
DNA in the area of atherosclerotic plaque in humans. Mol Biol.
45:5612011. View Article : Google Scholar
|
|
15
|
Zaina S, Heyn H, Carmona FJ, Varol N,
Sayols S, Condom E, Ramírez-Ruz J, Gomez A, Gonçalves I, Moran S
and Esteller M: DNA methylation map of human atherosclerosis. Circ
Cardiovasc Genet. 7:692–700. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Aavik E, Lumivuori H, Leppänen O, Wirth T,
Häkkinen SK, Bräsen JH, Beschorner U, Zeller T, Braspenning M, van
Criekinge W, et al: Global DNA methylation analysis of human
atherosclerotic plaques reveals extensive genomic hypomethylation
and reactivation at imprinted locus 14q32 involving induction of a
miRNA cluster. Eur Heart J. 36:993–1000. 2015. View Article : Google Scholar
|
|
17
|
Kucher AN, Nazarenko MS, Markov AV,
Koroleva IA and Barbarash OL: Variability of methylation profiles
of CpG sites in microRNA genes in leukocytes and vascular tissues
of patients with atherosclerosis. Biochemistry. 82:698–706.
2017.PubMed/NCBI
|
|
18
|
Castillo-Díaz SA, Garay-Sevilla ME,
Hernández-González MA, Solís-Martínez MO and Zaina S: Extensive
demethylation of normally hypermethylated CpG islands occurs in
human atherosclerotic arteries. Int J Mol Med. 26:691–700.
2010.PubMed/NCBI
|
|
19
|
Yamada Y, Nishida T, Horibe H, Oguri M,
Kato K and Sawabe M: Identification of hypo- and hypermethylated
genes related to atherosclerosis by a genome-wide analysis of DNA
methylation. Int J Mol Med. 33:1355–1363. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Moran S, Arribas C and Esteller M:
Validation of a DNA meth-ylation microarray for 850,000 CpG sites
of the human genome enriched in enhancer sequences. Epigenomics.
8:389–399. 2016. View Article : Google Scholar
|
|
21
|
Christensen BC, Houseman EA, Marsit CJ,
Zheng S, Wrensch MR, Wiemels JL, Nelson HH, Karagas MR, Padbury JF,
Bueno R, et al: Aging and environmental exposures alter
tissue-specific DNA methylation dependent upon CpG island context.
PLoS Genet. 5:e10006022009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Bell JT, Pai AA, Pickrell JK, Gaffney DJ,
Pique-Regi R, Degner JF, Gilad Y and Pritchard JK: DNA methylation
patterns associate with genetic and gene expression variation in
HapMap cell lines. Genome Biol. 12:R102011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
ENCODE Project Consortium: An integrated
encyclopedia of DNA elements in the human genome. Nature.
489:57–74. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Lizio M, Harshbarger J, Shimoji H, Severin
J, Kasukawa T, Sahin S, Abugessaisa I, Fukuda S, Hori F,
Ishikawa-Kato S, et al: Gateways to the FANTOM5 promoter level
mammalian expression atlas. Genome Biol. 16:222015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Bibikova M, Barnes B, Tsan C, Ho V,
Klotzle B, Le JM, Delano D, Zhang L, Schroth GP, Gunderson KL, et
al: High density DNA methylation array with single CpG site
resolution. Genomics. 98:288–295. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Libby P: Inflammation in atherosclerosis.
Nature. 420:868–874. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Libby P: Mechanisms of acute coronary
syndromes and their implications for therapy. N Engl J Med.
368:2004–2013. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Turunen MP, Aavik E and Ylä-Herttuala S:
Epigenetics and atherosclerosis. Biochim Biophys Acta.
1790:886–891. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Hai Z and Zuo W: Aberrant DNA methylation
in the pathogenesis of atherosclerosis. Clin Chim Acta. 456:69–74.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Fishbein GA and Fishbein MC:
Arteriosclerosis: Rethinking the current classification. Arch
Pathol Lab Med. 133:1309–1316. 2009.PubMed/NCBI
|
|
31
|
Peng Y, Meng K, Jiang L, Zhong Y, Yang Y,
Lan Y, Zeng Q and Cheng L: Thymic stromal lymphopoietin-induced
HOTAIR activation promotes endothelial cell proliferation and
migration in atherosclerosis. Biosci Rep. 37:pii: BSR201703512017.
View Article : Google Scholar
|
|
32
|
Shen T, Zhu Y, Patel J, Ruan Y, Chen B,
Zhao G, Cao Y, Pang J, Guo H, Li H, et al: T-box20 suppresses
oxidized low-density lipoprotein-induced human vascular endothelial
cell injury by upregulation of PPAR-γ. Cell Physiol Biochem.
32:1137–1150. 2013. View Article : Google Scholar
|
|
33
|
Warren HR, Evangelou E, Cabrera CP, Gao H,
Ren M, Mifsud B, Ntalla I, Surendran P, Liu C, Cook JP, et al:
Genome-wide association analysis identifies novel blood pressure
loci and offers biological insights into cardiovascular risk. Nat
Genet. 49:403–415. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Graff M, Scott RA, Justice AE, Young KL,
Feitosa MF, Barata L, Winkler TW, Chu AY, Mahajan A, Hadley D, et
al: Genome-wide physical activity interactions in adiposity-a
meta-analysis of 200,452 adults. PLoS Genet. 13:e10065282017.
View Article : Google Scholar
|
|
35
|
Zhang J, Burridge KA and Friedman MH: In
vivo differences between endothelial transcriptional profiles of
coronary and iliac arteries revealed by microarray analysis. Am J
Physiol Heart Circ Physiol. 295:H1556–H1561. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Ellinghaus E, Ellinghaus D, Krusche P,
Greiner A, Schreiber C, Nikolaus S, Gieger C, Strauch K, Lieb W,
Rosenstiel P, et al: Genome-wide association analysis for chronic
venous disease identifies EFEMP1 and KCNH8 as susceptibility loci.
Sci Rep. 7:456522017. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Astle WJ, Elding H, Jiang T, Allen D,
Ruklisa D, Mann AL, Mead D, Bouman H, Riveros-Mckay F, Kostadima
MA, et al: The allelic landscape of human blood cell trait
variation and links to common complex disease. Cell.
167:1415–1429.e19. 2016. View Article : Google Scholar : PubMed/NCBI
|