Introduction
Acute lymphoblastic leukemia (ALL) is the most
common malignancy in pediatric patients, whose malignant white
blood cells continuously multiply and lead to an excess of
lymphoblasts in peripheral blood and bone marrow (1). During the past decade, several
genetic and transcriptional factors have been identified as
aberrant in B-ALL, which is a common subtype of ALL. Among these
factors, overexpression of c-Myc is the main characteristic of
B-ALL (2,3).
The c-Myc protein belongs to a larger family of the
helix-loop-helix leucine zipper (HLHzip) transcription factors. The
C-terminal basic region HLHzip domain and the N-terminal
transcriptional activation domain (TAD) are essential for the
activation and repression of target genes (4). c-Myc forms a heterodimer with Max to
bind target DNA sequences through the canonical E box DNA binding
motif (CACGTG) (5).
Transcription/transformation-associated protein (TRRAP), a subunit
of different histone acetyltransferases, is a coactivator with the
TAD domain of c-Myc (6). Most
estimates suggest that c-Myc-regulated genes are generally involved
in cell-cycle progression and metabolism, which includes cyclin
(CCN) D kinases (CDKs), cyclins and ribosomal RNAs. The target
genes of c-Myc affect the multiple cellular processes by direct or
indirect pathways in a diversity of cancer cell types. When
directly activating the oncogenic pathway, c-Myc upregulates a
series of transcriptional programs, which influences physiological
processes including metabolic adaptation, cell division and
survival (5,7). When acting indirectly, c-Myc alters
microRNA and long non-coding RNA expression patterns or histone
modifications, which impacts target gene expression in various
carcinoma types (8,9).
In contrast to the tightly regulated expression of
c-Myc in normal cells, it is frequently dysregulated in human
cancers. Abundant evidence has indicated that c-Myc is
pathologically activated in numerous types of human malignancy
(5,9,10,11). c-Myc mRNA has been detected to be
5- to 40-fold overexpressed in 60–80% of colon carcinomas (12), 29% of prostate cancers (13), 40% of ovarian cancers (14), 23% of lung carcinomas (15), 61% of nodular melanomas and in 30%
of metastases (10). In virtually
all Burkitt's lymphoma, the c-Myc gene is translocated to one of
the immunoglobulin loci enhancers that drive the high expression
levels of c-Myc mRNA and protein. A typical translocation of c-Myc
into the immunoglobulin heavy chain locus is observed in ~80% of
Burkitt's lymphomas. Variant translocations of c-Myc into either
the κ or λ light chain locus each occur at a frequency of ~10%
(16,17). However, the association between
c-Myc overexpression and tumorigenesis has remained to be fully
elucidated. In this light, the present study on the function of
c-Myc aimed to contribute to the current knowledge on the molecular
mechanisms underlying the genesis of B-ALL.
Materials and methods
Mice
Severe combined immunodeficient (SCID) mice were
purchase from Beijing Huafu Kang Company (Beijing, China) and
maintained in a specific-pathogen-free environment at 24–26°C with
a relative humidity of 60–65%. They were provided with water and
food ad libitum throughout the experimental period. The
procedures for handling animals complied with the Current
Laboratory Animal Laws and Regulations, Policies and Administration
in China. All experiments were approved by the Animal Ethics
Committee of Dalian Medical University (no. AEE17013).
Antibodies
Mouse monoclonal anti-c-Myc (9E10; cat. no. E1809)
antibody was purchased from Santa Cruz Biotechnology, Inc. (Dallas,
TX, USA); mouse monoclonal anti-cyclin E1 (HE12; cat. no. 4129S),
anti-cyclin B1 (V152; cat. no. 4135S), anti-caspase-3 (3G2; cat.
no. 9668), anti-caspase-8 (1C12; cat. no. 9746), anti-caspase-9
(C9; cat. no. 9508) antibodies, and rabbit polyclonal anti-cyclin D
kinase (CDK)1 (cat. no. 9112) and anti-phospho-(p)CDK1 (Tyr15)
(cat. no. 9111S) antibodies were purchased from Cell Signaling
Technology, Inc. (Danvers, MA, USA). Rabbit polyclonal
anti-acetyl-histone H4 (cat. no. 06-866) was purchased from EMD
Millipore (Billerica, MA, USA). Mouse monoclonal anti-c-Myc (9E10
cat. no. ab32)-chromatin immunoprecipitation (ChIP) grade antibody
and rabbit polyclonal anti-GAPDH antibody were purchased Abcam
(Cambridge, UK). Fluorescein isothiocyanate (FITC)-labeled
immunoglobulin (Ig)G (cat. no. 11-4011-85) was obtained from
e-Bioscience (Thermo Fisher Scientific, Inc., Waltham, MA, USA).
Horseradish peroxidase (HRP)-conjugated rabbit (cat. no. A0208) and
mouse (cat. no. A0216) IgG antibodies were from Beyotime Institute
of Biotechnology (Haimen, China).
Patient samples
The clinical manifestations and examination results
of B-ALL patients newly diagnosed and treated at Dalian Municipal
Central Hospital (Dalian, China) from May 2015 to January 2016 were
retrospectively analyzed. Bone marrow aspiration and biopsy were
obtained from all B-ALL patients (n=12; 7 women and 5 men; mean
age, 23 years; range, 18–35 years) and individuals with anemia
(n=4; 2 women and 2 men; mean age, 22 years; range, 18–30 years).
All B-ALL patients newly diagnosed and have not received any
medication prior to sample collection. All investigations were
performed either for diagnostic purposes or with residual material
obtained through diagnostic procedures. All experiments were
approved by the Ethics Committee of Dalian Municipal Central
Hospital (approval no. YN2016-019-01).
Cells and culture conditions
Raji cells were purchased from the American Type
Culture Collection (Manassas, VA, USA). Cells were cultured in
RPMI-1640 tissue culture medium supplemented with 2 mM glutamine
(both from Thermo Fisher Scientific, Inc.), 50 mM 2-mercaptoethanol
(Fluka, Buchs, Switzerland), 10% fetal bovine serum (FBS; Gibco;
Thermo Fisher Scientific, Inc.), 100 U/ml penicillin and 100 mg/ml
streptomycin.
Transient transfection of c-Myc small
interfering (si)RNA
Cells (5×104/well) were seeded into
six-well plates and allowed to grow to 90% confluence. Transient
transfections of c-Myc siRNAs were performed for 6 h with
TransIT-TKO transfection reagent (Takara Bio, Inc., Otsu, Japan)
according to the manufacturer's instructions. The siRNAs were
designed to form 19-bp double-stranded RNA with 2 thymine overhangs
at each 3′ end of RNA. The following 3 targeting sequences of c-Myc
siRNA were used: siRNA 1 sense, 5′-GCUUCACCAACAGGAACUAUU-3′ and
antisense, AACGAAGUGGUUGUCCUUGAU (region: 586–605 bp); siRNA 2
sense, 5′-GGCGAACACACAACGUCUUUU-3′ and antisense,
5′-AACCGCUUGUGUGUUGCUGUU (region: 1,636–1,655 bp); and siRNA 3
sense, 5′-GGAAACGACGAGAACAGUUUU-3′, and antisense,
5′-AACCUUUGCUGCUCUUGUCAA-3′ (region: 1,831–1,850 bp). Alexa
488-conjugated siRNA duplex (Qiagen, Hilden, Germany) was used to
determine the transfection efficiency.
Establishment of Raji cells with c-Myc
knockdown (Raji-KD cells) and those with c-Myc knockdown and
subsequent restoration of c-Myc expression (Raji-KD-Re cells)
A retroviral vector carrying siRNA targeting c-Myc
was constructed as follows. A 21-nucleotide sequence (siRNA 3
region: 1,831–1,850) of the c-Myc complementary (c)DNA was inserted
in the sense and antisense directions into the pSINsi-mU6 cassette
vector (recombinant retroviral vector; Takara Bio, Inc.),
containing the mouse U6 promoter. The recombinant retroviruses were
generated by co-transfection of the vector mixture into 293 cells
(Genomeditech, Shanghai, China), including recombinant retroviral
vector, pE-eco vector (ecotropic env) and pGP vector
(gag-pol; cat. no. 6161; Takara Bio, Inc.). Recombinant
retroviral particles containing the target sequence or a mock
control were transfected into the parental Raji cells, and the
geneticin (G418)-resistant clones were selected as transfected
cells. The Raji-derived cells, transfected with the plasmid
expressing siRNA that targeted c-Myc, are referred to hereafter as
'Raji-KD'. Next, pseudotyped retroviral vectors (cat. no. 6161;
Takara Bio, Inc.) were generated for transfection of the designated
mock cells.
For c-Myc reintroduction, the open reading frame
(ORF) of c-Myc was cloned into the ClaI site of the
pLHCXsi-mU6-c-Myc expression vector, which is resistant to the
siRNA expressed in the c-Myc-knockdown cells. For this purpose,
c-Myc shRNA containing multiple mutations were introduced into
pLHCX vector (cat. no. 631511; Clontech Laboratories, Inc.,
Mountainview, CA, USA; five point mutations in the 1,831–1,850
region) that did not alter the original amino acid residues.
Raji-KD cells with c-Myc restoration, referred to as 'Raji-KD-Re
cells', were established by the transfection of pLHCXsi-mU6-c-Myc
into Raji-KD cells.
Western blot analysis
Cell lysates were prepared with ice-cold buffer (50
mM Tris-HCl, 150 mM of NaCl, 1% Triton X-100, 5 mM EDTA, 10 mM NaF,
0.1 mM Na3VO4, supplemented with 0.1 mM
phenylmethylsulfonylflouride, 1 mM dithiothreitol and a mixture of
protease and phosphatase inhibitors (Pierce; Thermo Fisher
Scientific, Inc.). The cell lysate was cleared by centrifugation at
12,000 × g for 10 min at 4°C. The proteins were determined using a
bicinchoninic acid protein assay kit (Thermo Fisher Scientific,
Inc.). A total of 10 µg protein per lane in 4X Laemmli
loading buffer was resolved by 10% SDS-PAGE gel and immunostained
with. The protein samples were subjected to 10% SDS-PAGE and
transferred onto polyvinylidene difluoride membranes (Immobilon-P;
0.45 µm; EMD Millipore) at 240 mA for 60 min. Blots were
blocked at 37°C for 2 h with 5% skimmed milk in 10 mM Tris-HCl (pH
7.5) with 150 mM NaCl and 0.1% Tween-20 (TBST) for caspase
antibodies or with 5% bovine serum albumin (BSA; Sigma-Aldrich;
Merck KGaA, Darmstadt, Germany) in TBS-T for other antibodies.
Following incubation with the appropriate primary antibodies
overnight at 4°C (dilution, 1:4,000 dilution or 1:3,000 for caspase
antibodies), the slides were washed. After washing, the blots were
incubated at 37°C for 1 h with the HRP-conjugated anti-rabbit IgG
(1:8,000 dilution) or anti-mouse IgG (1:8,000 dilution) secondary
antibodies. Finally, specific proteins were visualized using an
enhanced chemiluminescence system (GE Healthcare, Little Chalfont,
UK).
Semi-quantitative reverse transcription
polymerase chain reaction (RT-PCR) and RT-quantitative (q)PCR
RNA was isolated from each cell population using
TRIzol reagent (Thermo Fisher Scientific, Inc.). First-strand cDNA
was synthesized using SuperScript II reverse transcriptase
(Invitrogen; Thermo Fisher Scientific, Inc.) and the oligo(dT) 18
primer. The mixture without SuperScript II and RNasin was heated to
70°C for 5 min to unfold the secondary structures of the RNA. After
adding SuperScript II and RNasin, reactions were performed for 70
min at 42°C and for 15 min at 70°C in a 20-µl reaction
volume. Prepared cDNAs were stored at −20°C.
For PCR, GAPDH served as an internal control gene.
The thermocycling conditions were as follows: 95°C for 5 min,
followed by 30 cycles of 95°C for 30 sec, 60°C for 30 sec and 72°C
for 30 sec. The PCR products were separated by electrophoresis on
2% agarose gels and images were captured under ultraviolet
light.
Real-time qPCR was performed in triplicate in a
reaction volume of 20 µl (20 µmol/l of primers and 10
µl of Master Mix (Takara Bio, Inc.), which was adapted from
the standard protocol provided by SYBR-Green PCR Master Mix. The
PCR procedure was performed on an Applied Biosystems Prism 7000
Sequence Detection System (Applied Biosystems; Thermo Fisher
Scientific, Inc.). The thermocycling conditions were as follows:
95°C for 10 min, followed by 40 cycles of 95°C for 15 sec and 60°C
for 1 min, with a subsequent standard dissociation run to obtain
melting curve profiles of the amplicons. Using the
2−∆∆Cq (18) method,
relative internal mRNA expression of target genes was normalized to
GAPDH.
Primer sequences used in the PCR assays were as
follows (5′-3′): CDK1 (NG_029877.1) forward, GAAATTGAGCGGAGAGCGAC
and reverse, CCGTTCCTCAATACTCGCCC; CDKN1A (NG_009364.1) forward,
GCGGAGTGGAGTAAGTTCGT and reverse, TGGCGTAAAGGACCTGAACC; CDKN1B
(NG_016341.1) forward, CGCTCGCCAGTCCATTTG and reverse,
AAAGACACAGACCCCGACG; CCNG1 (XM_011534685) forward,
GATCAGGGCCGAGTTGTCTC and reverse, GAGGAGAGGGGACTCGTAGG;
retinoblastoma 1 (RB1; NG_009009.1) forward, CCCTGTTTCAATTTATCAGGC
and reverse, TCACCCCAGATTAGTTTAGGC; tumor protein (TP)53
(NG_017013.2) forward, CTCAGACACTGGCATGGTGT and reverse,
GTGGGGATCCAGCATGAGAC; CCNG2 (NM_004354.2) forward,
CCTCCTGTGCCATTCAACCA and reverse, CCAAGTCAACGGGGGTAAGG; CCNB1
(NM_031966.3) forward, TGAGAGCCATCCTAATTGACT and reverse,
CAATTATTCTGCATGAACCGAT; c-Myc (NM_002467.4) forward,
AATGTCAAGAGGCGAACACAC and reverse, ATTGTTTTCCAACTCCGGGAT; histone
acetyl transferase (HAT) [K(Lysine) acetyltransferase 5 (kat5);
NM_001206833.1] forward, CATTGCCTGTCCTCTACCTG and reverse,
ACTCTTGTTCTTACGTCCATC; HAT (kat8; NM_032188.2) forward,
GCAAGATCACTCGCAACCAA and reverse, ATCAATTTCGTAGTTCCCGAT; GAPDH
(NM_001289746.1) forward, AGATCATCAGCAATGCCTCCTG and reverse,
ATGGCATGGACTGTGGTCATG.
Proliferation assay
An MTT assay was performed to assess viability of
Raji, Raji-KD and Raji-KD-Re cells. The tetrazo-lium salt MTT is
taken up by viable cells and reduced to a formazan residue by
functional mitochondria of living cells. For the MTT assay, cells
were seeded into a 96-well flat bottom microtiter plate. A total of
10 µl per well of a 5 mg/ml solution of MTT (Sigma-Aldrich;
Merck KGaA) in PBS was added for the last 4 h of incubation.
Subsequently, the plate was centrifuged 2,500 × g, at 4°C for 10
min, the media were removed and dimethylsulfoxide was added to each
well to dissolve the precipitate. The absorbance of each well was
read at 490 nm with a Benchmark microplate reader (Bio-Rad
Laboratories, Hercules, CA, USA).
Bromodeoxyuridine (BrdU) assay
BrdU labelling was performed using a BrdU assay kit
(CycLex Co., Ltd., Nagano, Japan) according to the manufacturer's
protocol. Briefly, cells were seeded in 96-well plates and exposed
to 10 µm of BrdU for 8 h at 37°C. The cells were fixed with
denaturing solution for 30 min at room temperature and incubated
with 50 µl anti-BrdU monoclonal antibody for 1 h at room
temperature. Following washing, the plates were incubated with 50
µl HRP conjugated anti-mouse IgG at 37°C for 1 h. The plates
were visualized by incubation with 50 µl substrate reagent
for 15 min. The absorbance was measured at a wavelength of 450
nm.
Cell-cycle and apoptosis analysis
The cells were harvested, fixed with ice-cold 70%
ethanol for 2 h, and single-cell suspensions were prepared. After
extensive washing, the cells were resuspended in PBS containing
propidium iodide (PI) and RNase A (both from Sigma-Aldrich; Merck
KGaA), incubated for 1 h at room temperature and analyzed using a
FACSCalibur flow cytometer (BD Biosciences, San Jose, CA, USA). The
cell-cycle-phase distribution was analyzed with CellQuest software
3.3 (BD Biosciences).
For analysis of apoptosis, 5×105 cells
were harvested. Subsequently, PI and Annexin V-FITC double staining
were performed at room temperature for 15 min. Finally, cells were
washed three times with PBS and analyzed by flow cytometry (BD
Biosciences).
PCR array
Total RNA was extracted from cells with TRIzol
reagent (Invitrogen; Thermo Fisher Scientific, Inc.). A Cell Cycle
PCR Array (PAHS-020) was used to simultaneously examine the mRNA
levels of 96 genes, including 6 'housekeeping genes' for
noralization, in 96-well plates, according to the protocol of the
manufacturer (cat. no. OHS-020; Zhejiang Kangchen Biotech Co.,
Ltd., Wuhan, China). The Cq value was calculated for each sample as
the difference in gene expression between Raji and Raji-KD
cells.
ChIP assay
During the culturing process of Raji cells and their
derivatives, formaldehyde was added to the medium at a final
concentration of 1%. Cross-linking was allowed to proceed for 10
min at room temperature and stopped by addition of glycine to a
final concentration of 125 mM, followed by an additional incubation
for 5 min at 25°C. Fixed cells were washed twice with PBS and
harvested in SDS buffer [50 mM Tris-HCl (pH=8.0), 0.5% SDS, 100 mM
NaCl, 5 mM EDTA and aforementioned protease inhibitors]. Cells were
pelleted by 1,500 × g centrifugation for 5 min at 4°C and suspended
in 4 ml IP buffer [100 mM Tris (pH 8.6), 0.3% SDS, 1.7% Triton
X-100 and 5 mM EDTA]. The cells were then disrupted by sonication
for 10 sec in a Branson 250 sonicator (Branson, Danbury, CT, USA)
at a power setting of 3 and duty cycle of 100%, yielding genomic
DNA fragments with a bulk size of 100–400 bp. The lysate was then
diluted with IP buffer to a final volume of 1 ml. For each
immunoprecipitation, 1 ml diluted lysate was precleared by adding
30 µl blocked protein G beads [50% slurry protein
A-Sepharose (GE Healthcare); 0.5 mg/ml fatty acid-free BSA and 0.2
mg/ml salmon sperm DNA (both from Sigma-Aldrich; Merck KGaA) in
Tris-EDTA (TE) buffer]. Samples were immunoprecipitated overnight
at 4°C with 1:500 anti-c-Myc antibody or 1:200 anti-AcH4 antibody).
Immune complexes were recovered by adding 30 µl blocked
protein G beads and incubated for 6 h at 4°C. Beads were washed and
eluted, and cross links were reversed. This included successive
washes in 1 ml Mixed Micelle Buffer [20 mM Tris (pH 8.1), 150 mM
NaCl, 5 mM EDTA, 5% w/v sucrose, 1% Triton X-100 and 0.2% SDS],
buffer 500 [50 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic
acid at pH 7.5, 0.1% w/v deoxycholic acid, 1% Triton X-100, 500 mM
NaCl, and 1 mM EDTA], LiCl Detergent Wash Buffer [10 mM Tris-HCl
(pH 8.0), 0.5% deoxycholic acid, 0.5% Nonidet P-40, 250 mM LiCl and
1 mM EDTA] and TE (pH 7.5). The eluted material was
phenol/chloroform-extracted and ethanol-precipitated. DNA was
re-suspended in 100 µl water (19). PCR was performed as described
above using 1 µl DNA and 800 nM aforementioned primers
diluted to a final volume of 20 µl in SYBR-Green Reaction
Mix (Takara Bio Inc.). Using the 2−∆∆Cq (18) method, relative internal mRNA
expression of target genes was normalized to GAPDH.
Immunostaining and immunofluorescence
analysis
Cells were grown on glass coverslips for 24 h. For
cell immunostaining, cells were fixed in 4% neutral-buffered
paraformaldehyde at 4°C for 20 min. Subsequently, the cells were
blocked with the 5% FBS in PBS for 1 h at room temperature.
Anti-acetyl-histone H4 antibody (1:250) staining was performed
overnight at 4°C, whereas the FITC-labeled IgG (1:250) staining was
performed for 1 h at room temperature. Finally, the slides were
visualized with 3,3′-diaminobenzidine. All the slides were observed
under a fluorescence microscope (Olympus, Tokyo, Japan) and images
were captured.
In vivo tumor formation assays (Xenograft
model)
Male SCID mice (age, 6 weeks, average weight 25 g;
n=11 mice/group) were allowed to acclimatize to the laboratory
environment for 1 week. To establish the xenograft tumor formation
model, 1×107 Raji cells and Raji-KD cells were injected
into the right and left sides axilla of a site in each mouse,
respectively. Over 30 days, tumor formation at the site of
injection and at distant tissue sites was monitored. Tumor size was
measured using a caliper and tumor volume was estimated using the
following equation: V=π/6xaxbxc (V, volume of the tumor; a, the
longest radial line; b, the shortest radial line; c,
thickness).
To assess the efficiency of the retroviral
vector-mediated c-Myc-siRNA in suppressing lymphoblastic tumor
growth in vivo, 2×106 of Raji cells were
inoculated into the bilateral hind axilla of 7-week-old SCID mice
(n=3 mice/group). A total of 50 µl retroviral vector (0.5
µg/µl) was injected once after 24 h inoculation. Mice
were injected with c-Myc shRNA retrovirus on the right axilla or
with the pseudotyped retrovirus on the left. Over 45 days, the
tumor weight and the date at which a palpable tumor first arose was
recorded.
Immunohistochemistry
For immunohistochemical detection of protein in the
xenograft tumors, 5-mm sections taken from formalin-fixed paraffin
blocks were mounted on glass slides and incubated at 56°C
overnight. The slides were then de-paraffinized with xylene and
dehydrated with alcohol. After washing with tap water, the slides
were incubated in 0.3% hydrogen peroxide to block endogenous
peroxidase activity. After washing with PBS, the slides were
blocked with 5% skimmed milk in PBS, followed by incubation with
the primary antibodies (c-Myc, 1:250; cyclin B1, 1:250; p-CDK1,
1:250; CDK1, 1:250; cyclin E1, 1:250) for 1 h at room temperature.
Next, the slides were washed in PBS and incubated with
HRP-conjugated rabbit or mouse IgG antibodies (1:500) for 30 min at
room temperature. Finally, the antibody binding was visualized
using a diaminobenzidine system and counterstained with Giemsa
staining at 37°C for 5 mins. Bone marrow from B-ALL patients was
smeared and stained as described above.
Semi-quantitative analysis of the
immunohistochemical staining in tissues
The intensity of positive staining in tissue
sections was analyzed via the integrated optical density (IOD)
using the Image-Pro Plus 5.1 software (Media Cybernetics, Inc.,
Rockville, MD, USA). In brief, 4 images for each of 4 individual
tumor samples were analyzed in a blinded manner. All of the images
were captured using the same microscope and camera settings.
Image-Pro Plus 5.1 software was used to calculate the average IOD
per stained area (IOD/µm2) to quantify the
staining intensity.
Statistical analysis
Each experiment was performed at least three times.
Values are expressed as the mean ± standard error mean. Statistical
analyses were performed with the statistical software package
GraphPad Prism 5 (GraphPad Inc., La Jolla, CA, USA). The
statistical significance was assessed by one-way analysis of
variance with Student-Newman-Keuls post-hoc test. The level of
differences between two groups was analyzed by Student's test.
P<0.05 was considered to indicate a statistically significant
difference.
Results
Loss of the c-Myc gene significantly
reduces the growth rate of Raji cells
To explore the function of c-Myc, specific
c-Myc-siRNA fragments corresponding to nucleotides 586–605,
1,636–1,655 and 1,831–1,850 of a human c-Myc ORF (NM_002467) were
designed. In the RT-PCR analysis, the siRNA fragment (1,831–1,850)
exhibited a stronger inhibitory effect than other siRNA fragments
(Fig. 1A). However, this effect
was short-lived (about 3 days by transient transfection; data not
shown). It is important to assess the role of c-Myc under
experimental conditions where the effect of endogenous c-Myc is
completely eliminated. To stably silence c-Myc gene expression,
c-Myc knockdown cells were established, which were referred to as
'Raji-KD cells', by transfection with a replication-defective
retrovirus encoding a c-Myc small hairpin (sh)RNA. For c-Myc
reintroduction, an ORF of the c-Myc gene was cloned into the
ClaI site of a pLHCXsi-mU6-c-Myc expression vector that is
resistant to the siRNA expressed in Raji-KD cells, and cells
transfected with the two were referred to as 'Raji-KD-Re cells'. As
presented in Fig. 1B, the mRNA
expression of the c-Myc gene was significantly downregulated in the
Raji-KD cells after c-Myc shRNA transduction. The loss of mRNA
expression was then restored in the Raji-KD-Re cells via the
introduction of the c-Myc gene. Again, the c-Myc protein expression
was significantly down-regulated from the Raji-KD cells as
evidenced by western blot analysis, and partly restored in the
Raji-KD-Re cells (Fig. 1C).
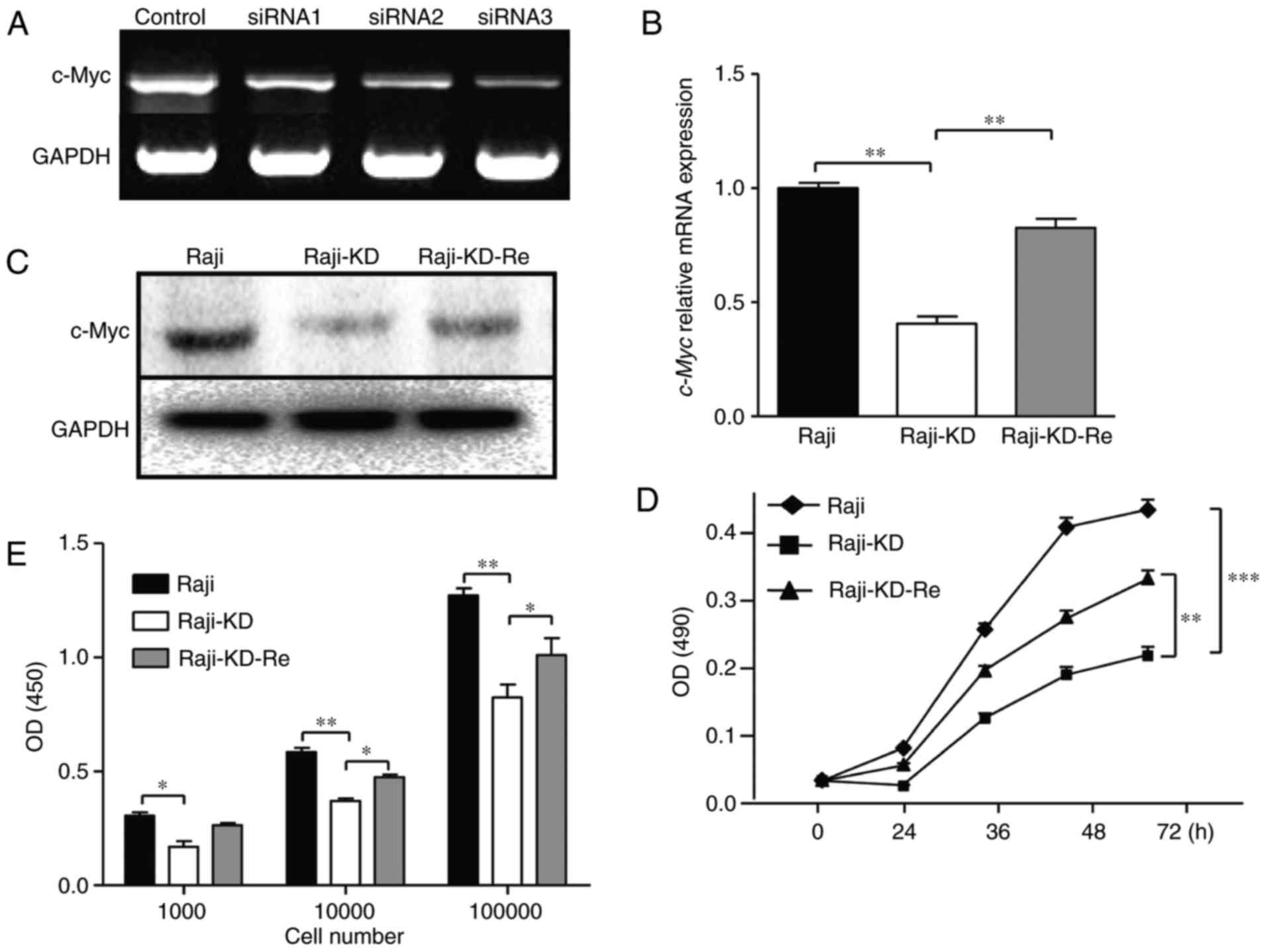 | Figure 1Knockdown of c-Myc significantly
reduces the proliferation rate of Raji cells. (A) Three different
c-Myc siRNAs were transiently transfected into Raji cells. mRNA
expression of c-Myc was determined by RT-PCR. GAPDH was used as an
internal loading control. (B) mRNA expression of c-Myc was
determined by RT-qPCR with normalization to GAPDH expression. (C)
Western blot analysis was used to determine c-Myc expression in
Raji, Raji-KD and Raji-KD-Re cells. GAPDH served as a loading
control. (D) MTT assays for Raji, Raji-KD and Raji-KD-Re cells.
OD(490) values were measured using a microplate reader. (E) Effect
of c-Myc-knockdown on cell proliferation was measured using BrdU
assays. Values are expressed as the mean ± standard error of the
mean. **P<0.01, ***P<0.001. RT-PCR,
semi-quantitative reverse transcription PCR; RT-qPCR,
RT-quantitative PCR; PCR, polymerase chain reaction; siRNA, small
interfering RNA; OD(490), optical density at 490 nm; Raji-KD, Raji
cells with c-Myc knockdown; Raji-KD-Re, Raji-KD with re-expression
of c-Myc. |
Previous studies have demonstrated that c-Myc has a
critical role in regulating cell proliferation and growth (20). Removal of c-Myc results in a
marked prolongation of the doubling time. In the case of Rat1
fibroblasts, the doubling time increases from ~16 to ~50 h without
c-Myc, indicating that the loss of c-Myc expression has an impact
the proliferation of cells. From the MTT assay, it was indicated
that in contrast to the mock-transfected cells, the growth rates of
the Raji-KD cells were significantly reduced (>50% inhibition)
after 72 h, and were restored by c-Myc re-expression in the
Raji-KD-Re cells (Fig. 1D).
Furthermore, the populations of Raji, Raji-KD and Raji-KD-Re cells
were detected immunochemically using a bromodeoxyuridine cell
proliferation assay, which mirrored the results of the MTT assay,
suggesting that c-Myc affected the cell proliferation by inhibiting
DNA synthesis (Fig. 1E).
Loss of c-Myc decreases the expression of
cell cycle-associated genes and induces G2/M-phase arrest
Previous studies suggest that the regulation of cell
proliferation by c-Myc involves several key components of
cell-cycle regulatory molecules (21,22). To determine whether c-Myc
influences cell proliferation via inducing cell-cycle arrest, the
three types of cells were processed for cell-cycle analysis by flow
cytometry. The cell-cycle profiles of Raji-KD cells were obviously
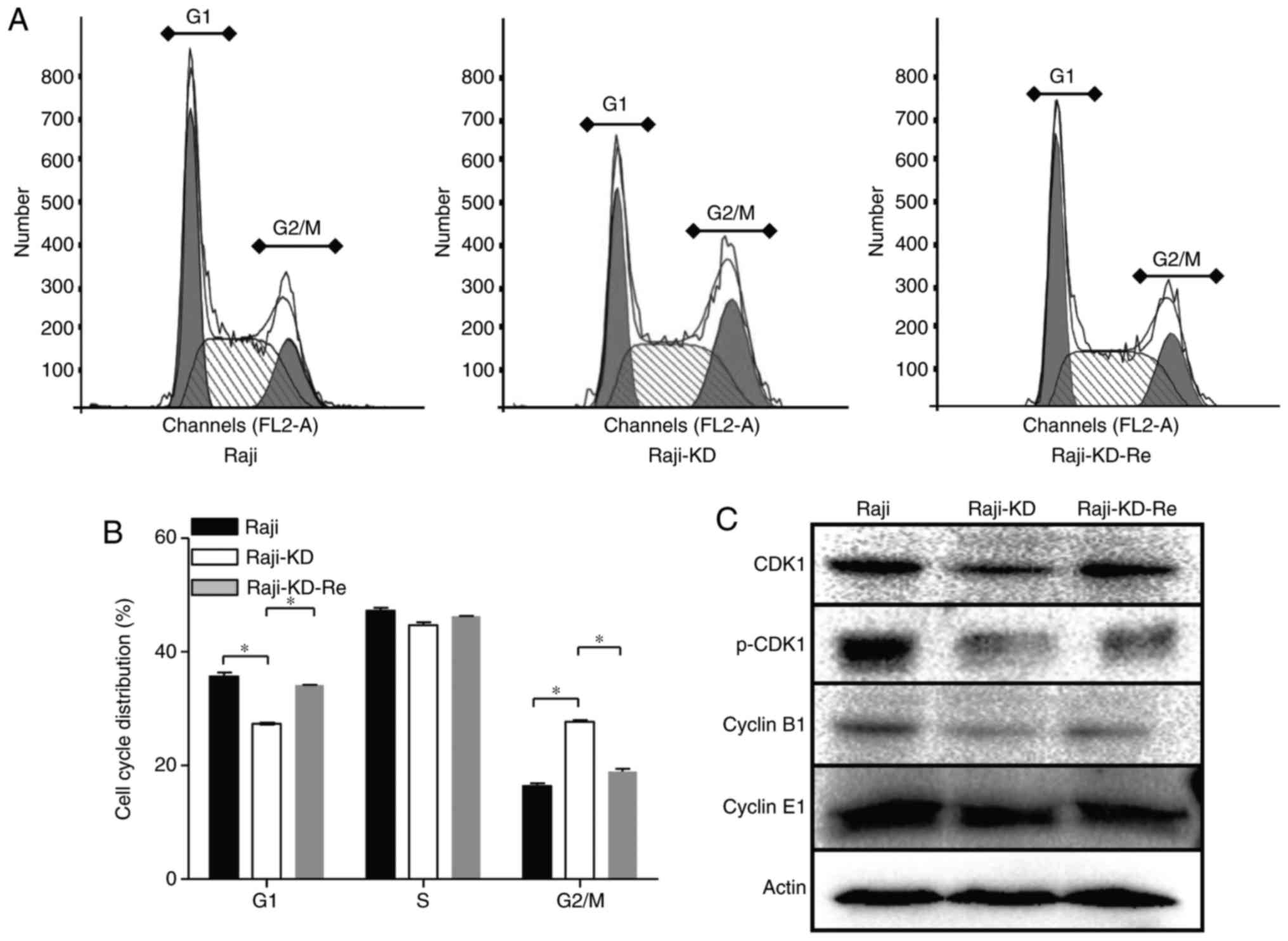
different from those of Raji and Raji-KD-Re cells (Fig. 2A and B). Loss of c-Myc expression
resulted in a significant G2/M-phase arrest, while the
G1-phase population was decreased (Fig. 2A and B).
Activation of CDK1 and cyclin B1 is a major
checkpoint of transition from the G2-phase to mitosis
(G2/M), which governs most of the processes involved in
mitotic initiation and progression (23,24). To assess why knockdown of the
c-Myc gene induced G2/M arrest, the expression of CDK1
and cyclin B1 in the Raji-derived cells was examined. The loss of
c-Myc caused a reduction in the levels of CDK1, p-CDK1 and cyclin
B1 in Raji-KD cells, while no significant difference was identified
in cyclin E1 expression. Of note, the c-Myc shRNA-associated
reduction in the expression of CDK1, p-CDK1 and cyclin B1 was
attenuated by re-expression of c-Myc (Fig. 2C), suggesting that the cell-cycle
arrest in G2/M-phase may be attributed to the
downregulation of CDK1, p-CDK1 and cyclin B1 (Fig. 2A and B). To further elucidate the
underlying mechanisms of the cell-cycle arrest caused by the
suppression of c-Myc, cell cycle-associated gene expression was
examined between Raji cells and Raji-KD cells via a PCR array. A
total of 28 genes with a 2-fold difference in their expression
levels were identified. Among them, 24 genes [CCNG1, CCNG2, HUS1
checkpoint clamp component, RB1, RB-like 2, BRCA2 and -1 DNA repair
associated, CDK1, S-phase kinase-associated protein 2, CDKN1B,
CDK6, caspase-3, growth arrest and DNA-damage-inducible α, meiotic
recombination 11 homolog A (S. cerevisiae), MRE11 homolog
double strand break repair nuclease, MDM2 protooncogene, CCNF,
CDK8, WEE1 G2 checkpoint kinase, cell division cycle 6,
hypoxanthine phosphoribosyl transferase 1, checkpoint kinase 1,
transcription factor Dp-2 and TP53], which are involved in cell
cycle regulation, were downregulated in Raji-KD cells, whereas 4
genes (CDKN1A, CCND2, CCND1 and baculoviral IAP repeat containing
5), which function in cell differentiation, were upregulated
(Table I). CDKN1A (also known as
p21) serves as an inhibitor of cellular proliferation in response
to DNA damage (25). CCND1 and
CCND2 are components of the cyclinD/CDK4/CDK6 complexes, and the
latter phosphorylates and inhibits members of the retinoblastoma
protein family and regulates the cell cycle during G1/S transition
(26). These results demonstrated
that downregulation of c-Myc induces G2/M arrest by
regulating the expression of CDK1 and cyclin B1, which are capable
of accelerating cell-cycle progression.
 | Table IUp- and downregulated genes in
Raji-KD cells. |
Table I
Up- and downregulated genes in
Raji-KD cells.
| Gene name | Definition | GenBank ID | Fold change
(Raji-KD/Raji) |
|---|
| CDKN1A | Cyclin-dependent
kinase inhibitor 1A | NM_000389 | 6.10 |
| CCND2 | Cyclin D2 | NM_001759 | 3.31 |
| CCND1 | Cyclin D1 | NM_053056 | 3.03 |
| BIRC5 | Baculoviral IAP
repeat containing 5 | NM_001012270 | 2.12 |
| CCNG1 | Cyclin G1 | NM_004060 | −6.90 |
| CCNG2 | Cyclin G2 | NM_004354 | −5.22 |
| HUS1 | HUS1 checkpoint
clamp component | NM_004507 | −3.75 |
| RB1 | RB transcriptional
corepressor 1 | NM_000321 | −3.69 |
| RBL2 | RB transcriptional
corepressor like 2 | NM_005611 | −3.44 |
| BRCA2 | BRCA2, DNA repair
associated | NM_000059 | −3.40 |
| CDK1 | Cyclin dependent
kinase 1 | NM_001170406 | −3.34 |
| SKP2 | S-phase
kinase-associated protein 2 | NM_001243120 | −2.90 |
| CDKN1B | Cyclin dependent
kinase inhibitor 1B | NM_004064 | −2.79 |
| CDK6 | Cyclin dependent
kinase 6 | NM_001145306 | −2.78 |
| CASP3 | Caspase-3 | NM_004346 | −2.66 |
| GADD45A | Growth arrest and
DNA damage inducible α | NM_001199741 | −2.60 |
| MRE11A | MRE11 homolog,
double strand break repair nuclease | NM_005590 | −2.54 |
| BRCA1 | BRCA1, DNA repair
associated | NM_007294 | −2.40 |
| MAD2L1 | Mitotic arrest
deficient 2 like 1 | NM_002358 | −2.24 |
| MDM2 | MDM2
proto-oncogene | NM_001145337 | −2.13 |
| CCNF | Cyclin F | NM_001761 | −2.03 |
| CDK8 | Cyclin dependent
kinase 8 | NM_001260 | −2.00 |
| WEE1 | WEE1 G2 checkpoint
kinase | NM_001143976 | −2.00 |
| CDC6 | Cell division cycle
6 | NM_001254 | −2.00 |
| HPRT1 | Hypoxanthine
phosphoribosyl transferase 1 | NM_000194 | −2.00 |
| CHEK1 | Checkpoint kinase
1 | NM_001114121 | −2.00 |
| TFDP2 | Transcription
factor Dp-2 | NM_001178138 | −2.00 |
| TP53 | Tumor protein
p53 | NM_000546 | −2.00 |
c-Myc induces cell cycle-associated gene
expression by transcriptionally regulating HAT
As a transcription factor, c-Myc usually binds to
specific sites in the target gene promoters, acts as a general
amplifier of transcription, and is known to alter histone
modifications, thereby having a role in remodeling target chromatin
(19,27). To assess whether c-Myc directly
impacts gene transcription in Raji cells, a ChIP assay was used to
evaluate the c-Myc-dependent regulation of cell cycle-associated
genes. Based on a PCR array and western blot analysis of
cell-cycle-associated genes (Fig.
2C and Table I), cyclin B1
and CDK1 were selected as the target genes. The real-time
PCR-amplified cyclin B1 and CDK1 signals from the c-Myc-ChIP assay
exhibited no significant differences between Raji, Raji-KD and
Raji-KD-Re cells; however, the PCR-amplified gene signals of TIP60
immunoprecipitated with c-Myc antibodies were reduced in the
Raji-KD cells compared with those in Raji cells, and were
significantly increased in Raji-KD-Re cells (Fig. 3A). In addition, the mRNA levels of
TIP60 and MOF were assessed in the three groups of cells,
indicating that knockdown of the c-Myc gene resulted in decreased
expression of TIP60 and MOF (Fig.
3B). Of note, the level of AcH4 was markedly decreased in the
Raji-KD cells, as evidenced by western blot analysis (Fig. 3C) and immunostaining assays
(Fig. 3D). These results suggest
that c-Myc regulates the transcription of cell cycle-associated
genes through an indirect AcH4-associated mechanism in Raji cells.
To further evaluate AcH4-dependent changes in cell cycle-associated
gene expression, ChIP assays using AcH4 antibodies and PCR analysis
were performed. As presented in Fig.
3E, the gene signals of cyclin B1, CDK1, RB1, CCNG2 and cyclin
dependent kinase inhibitor 1B (CDKN1B) from the amplification of
AcH4-ChIPed sequences were reduced in the Raji-KD cells compared
with Raji cells and restored in Raji-KD-Re cells, while the gene
signals for CCNG1 were not affected. It is conceivable that c-Myc
regulates the expression of cell cycle-associated genes, including
cyclin B1, CDK1, RB1 and CCNG2, by controlling the HAT-mediated
acetylation of histone H4 in target chromatin.
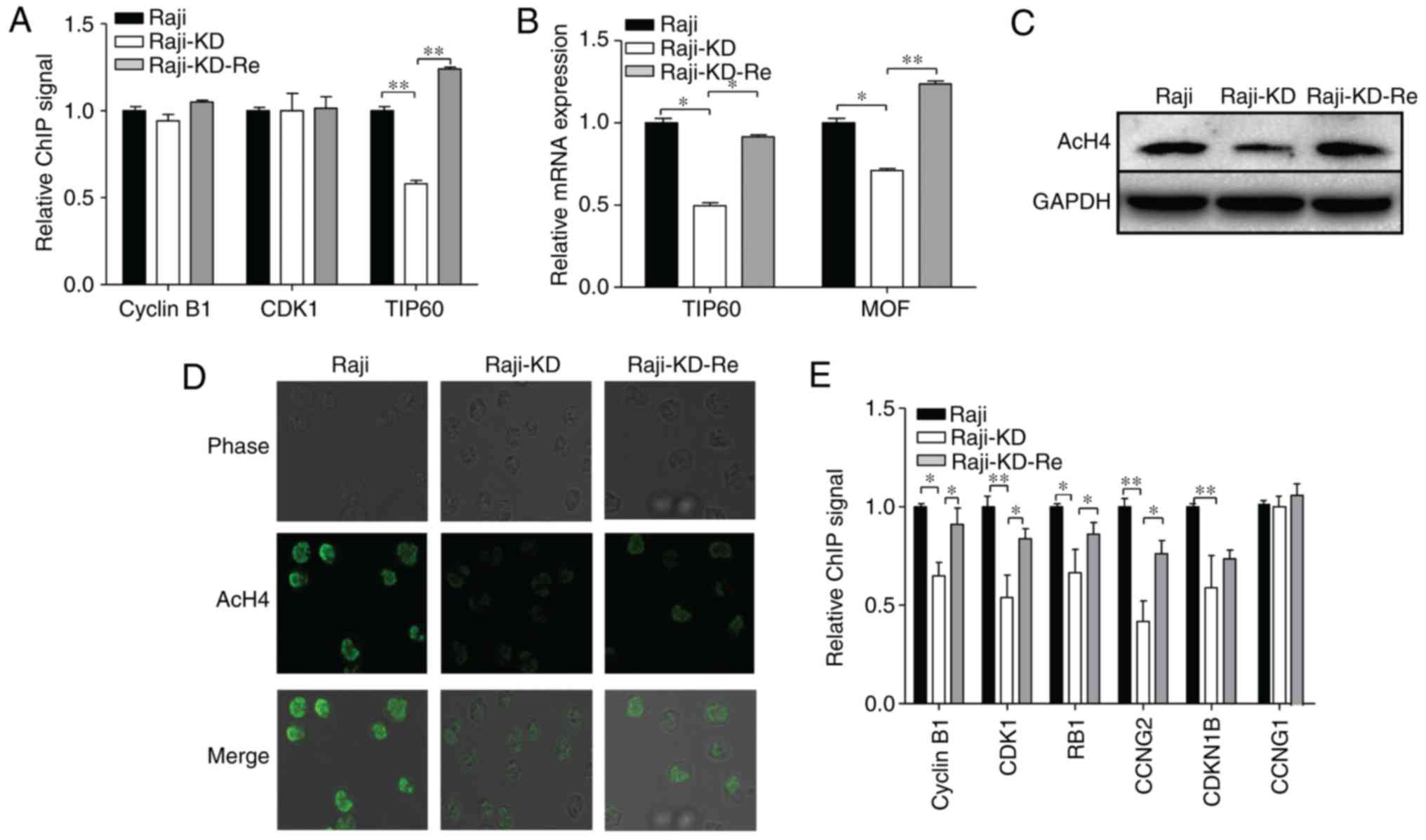 | Figure 3Loss of c-Myc suppresses the
expression of CDK by transcriptionally regulating HAT that targets
histone H4. (A) c-Myc-ChIP assay with Raji, Raji-KD and Raji-KD-Re
cells. Following formaldehyde cross-linking and DNA fragmentation,
samples were immunoprecipitated with antibody specific for c-Myc.
The c-Myc binding signal for TIP60 was reduced in Raji-KD cells,
while that for cyclin B1 and CDK1 did not change. (B) mRNA
expression of TIP60 and MOF was determined by reverse
transcription-quantitative polymerase chain reaction. GAPDH
expression was used for normalization. (C) Western blot analysis of
AcH4 expression in Raji, Raji-KD and Raji-KD-Re cells. GAPDH served
as a loading control. (D) Confocal microscopy analysis of Raji,
Raji-KD and Raji-KD-Re cells after staining with anti-AcH4 antibody
(magnification, ×400). (E) AcH4-ChIP assay of Raji, Raji-KD and
Raji-KD-Re cells. The AcH4 binding signals of cell cycle-associated
genes were reduced by the knockdown of c-Myc gene. Values are
expressed as the mean ± standard error of the mean.
*P<0.05, **P<0.01. Raji-KD, Raji cells
with c-Myc knockdown; Raji-KD-Re, Raji-KD with re-expression of
c-Myc; CDK, cyclin D kinase; ChIP, chromatin immunoprecipitation;
AcH4, acetylated histone H4; TIP60, 60 kDa Tat-interactive protein;
MOF, males absent on the first; RB, retinoblastoma; CCN, cyclin.
CDKN1B, cyclin dependent kinase inhibitor 1B. |
Knockdown of c-Myc suppresses apoptosis
by regulating caspase-3 expression
In addition to the function of c-Myc in cell growth
and proliferation, numerous studies have reported that c-Myc
induces cell apoptosis (4,5,28).
To investigate the effect of c-Myc on cell apoptosis, the three
cell groups of the present study were analyzed by flow cytometry
after Annexin V and PI staining. As presented in Fig. 4A and B, the percentage of early
apoptotic cells (PI− Annexin V+ cells) in
Raji, Raji-KD and Raji-KD-Re cells was 2.56, 2.39 and 3.7%,
respectively, and the percentage of late apoptotic cells
(PI+ Annexin V+ cells) was 6.91, 2.42 and
3.16%, respectively (Fig. 4B).
The apoptotic rate in the Raji-KD cells was significantly lower
than that in Raji cells and was partially restored in the
Raji-KD-Re cells, which indicated that c-Myc is involved in cell
apoptosis. To explore the underlying mechanism, the expression of
caspase-3, -8 and -9 was examined. In contrast to the Raji cells,
knockdown of the c-Myc gene led to a significant decrease in
pro-caspase-3 expression in Raji-KD cells (Fig. 4C), while the expression of
caspase-8 or 9 exhibited no difference among the Raji, Raji-KD and
Raji-KD-Re cells. These results indicate that c-Myc promotes the
apoptosis of Raji cells by regulating the expression of activated
caspase-3, which has a key role in the execution of multiple
apoptotic pathways.
c-Myc expression is essential for the
Raji cells-derived tumor formation
It is well known that c-Myc has a significant role
in promoting tumor formation in vivo. To examine the
influence of c-Myc knockdown in Raji cells on xenograft tumor
formation, a xenograft model of B-ALL was established in SCID mice.
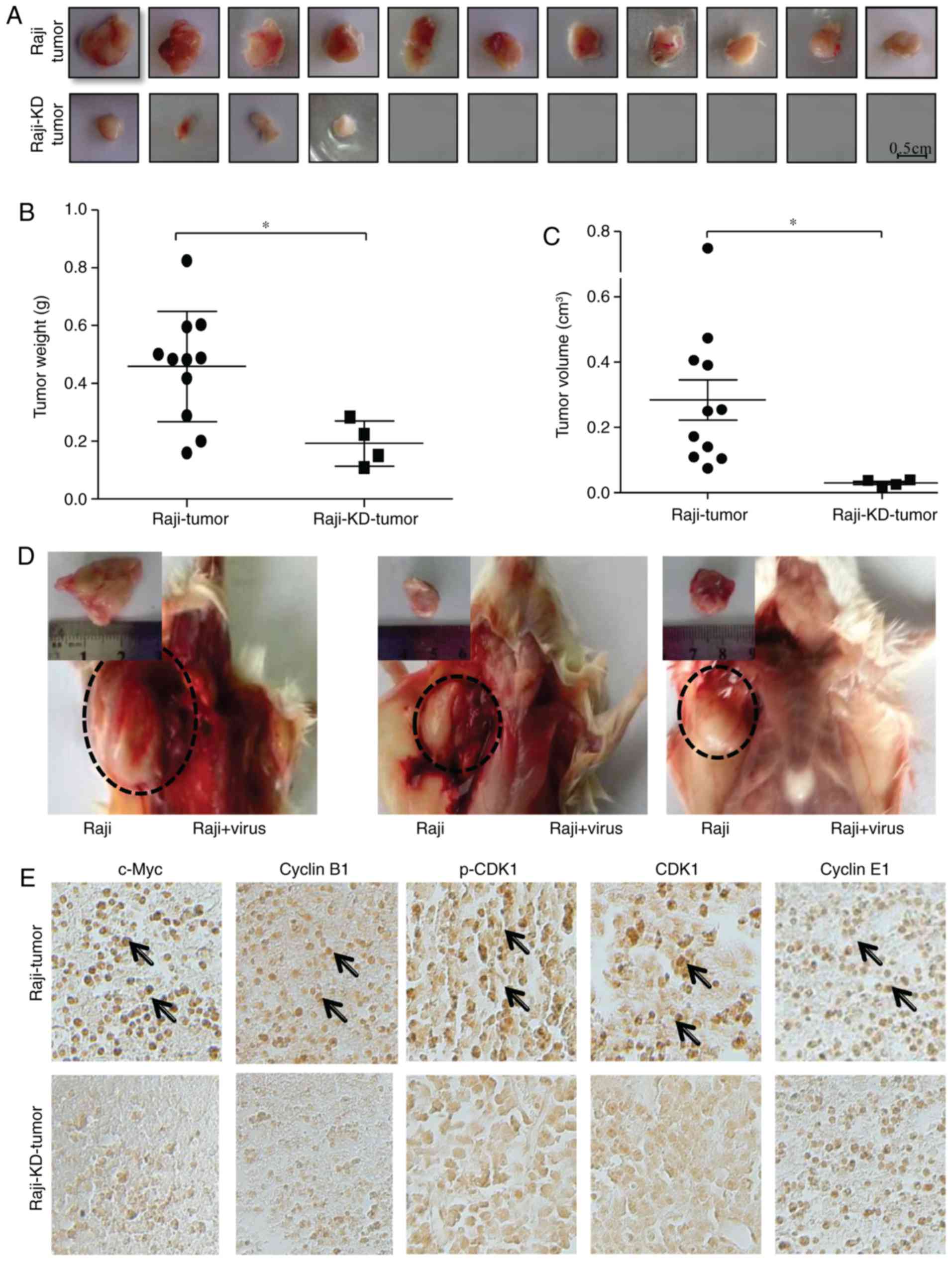
As presented in Fig. 5A, Raji
cells rapidly formed tumors with no notable rejection following
subcutaneous injection into the left axilla of mice, whereas the
incidence of tumor formation was significantly suppressed in mice
that were injected with Raji-KD cells over a period of 30 days.
Only 4 small tumors (4 tumors/11 mice) were observed in the mice
that were transplanted with Raji-KD cells (Fig. 5A). No adverse events were noted in
mice injected with Raji cells expressing c-Myc shRNA. After 30
days, the volume and weight of the tumors were measured. It was
apparent that the tumors of the Raji-KD cell group were smaller and
lighter than those of the Raji cells group (Fig. 5B and C), suggesting that the
tumor-forming ability of Raji-KD was severely reduced by knockdown
of c-Myc. Since the abovementioned results indicated that c-Myc
promotes tumor growth in vitro by modulating the cell cycle,
the efficiency of injection of the c-Myc shRNA retrovirus in
inhibiting tumor formation was then assessed in vivo. During
the first two days after inoculation of Raji cells into the
bilateral hind axilla, mice were injected with c-Myc shRNA
retrovirus on the right axilla or with a pseudotyped retrovirus on
the left over. As presented in Fig.
5D, injection of the c-Myc shRNA retrovirus markedly inhibited
the tumor formation in vivo. The weights and volumes of the
tumors were 1.27±0.85 g and 1.23±1.14 (cm3) in mice
injected with Raji cells, while the tumor was not generated
following injection with c-Myc shRNA retroviruses (Table II). Based on the
immunohistochemical analysis, the expressions of cyclin B1, CDK1
and p-CDK1 were suppressed in Raji cell-derived tumors following
the retroviral vector-mediated c-Myc-siRNA injection, while no
significant difference was identified in cyclin E1 expression
(Fig. 5E). These results are
consistent with the observation that the expression of these
proteins was significantly suppressed in Raji cells
afterc-Myc-siRNA injection vs. Raji cells (Fig. 2C). The present results suggested
that loss of c-Myc suppressed tumorigenesis, probably and at least
in part via downregulation of the expression of G2/M
checkpoint-associated genes, including CDK1 and cyclin B1.
| Table IIInjection of c-Myc shRNA retrovirus
completely inhibits tumor formation in vivo. |
Table II
Injection of c-Myc shRNA retrovirus
completely inhibits tumor formation in vivo.
| Cells | Tumor formation
time (days) | Tumor weight
(g) | Tumor volume
(cm3) |
|---|
| Raji
(2×106) | 45 | 1.27±0.85 | 1.23±1.14 |
| Raji
(2×106) + shRNA | 45 | 0.00 | 0.00 |
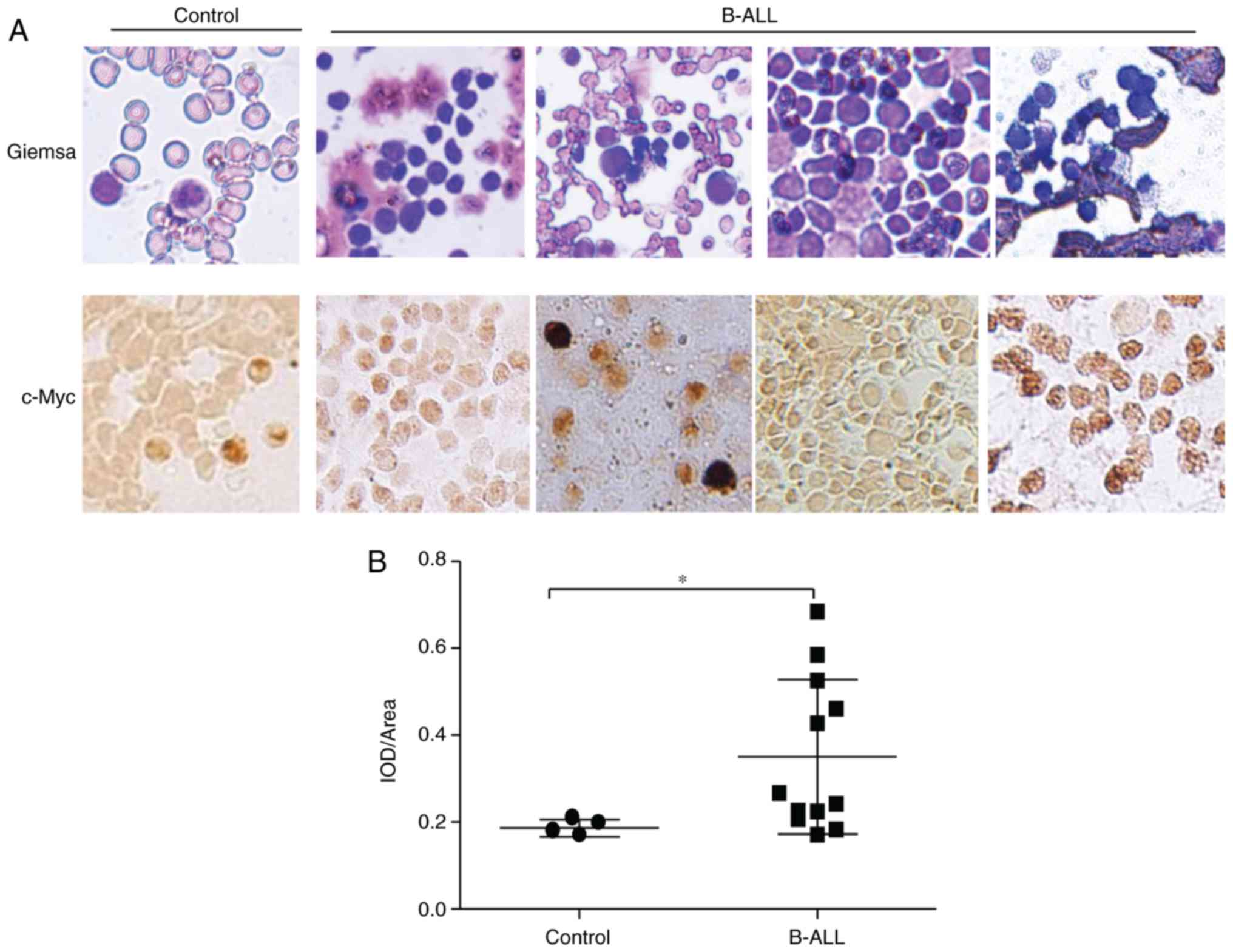
As reported previously, overexpression of c-Myc is
the major characteristic of B-ALL (2). To further confirm the tumor promoter
effect of c-Myc in B cell lymphoma tumorigenesis, c-Myc expression
in the bone marrow aspirations and biopsies of B-ALL patients was
examined. The immunohistochemistry assays using anti-c-Myc antibody
indicated that c-Myc was overexpressed in the B-ALL samples,
compared with that in the samples from individuals with anemia
(Fig. 6A). Semi-quantitative IOD
analysis also confirmed significantly enhanced anti-c-Myc antibody
staining in the patient group (Fig.
6B). These results indicated that the expression of c-Myc is
constitutively activated in the process of B-ALL tumorigenesis, and
that c-Myc RNA interference may serve as a potential therapeutic
strategy for B-ALL.
Discussion
The c-Myc protein has multiple functions in
tumorigenesis and progression, and c-Myc transgenic mice, carrying
c-Myc linked to the intron enhancer of the immunoglobulin heavy
chain gene, developed clonal B-cell malignancies (29). Furthermore, when lymphoblastoid
cells were transfected with a constitutively expressed c-Myc gene,
the cells became tumorigenic in nude mice (30). Understanding c-Myc tumorigenic
activity requires experimentally tractable models. To date, the
study of c-Myc target genes has not been standardized as to the
cell types used or the expression systems employed. With this
regard, the present study established a novel B-ALL cell model
(Raji-KD) with knockdown of the c-Myc gene, and demonstrated that
c-Myc regulates the CDK1/cyclin B1-dependent G2/M cell cycle
progression via controlling histone H4 acetylation.
A deletion of c-Myc in mice was lethal in
homozygotes between 9.5 and 10.5 days of gestation, and the embryos
were generally smaller and retarded in their development compared
with their littermates (31),
indicating that c-Myc is involved in the regulation of the cell
cycle and cell proliferation. Prior study into the mechanisms
underlying the effects of c-Myc have revealed that c-Myc-null rat
fibroblasts display a prolonged G2-phase (32). Song et al (33) reported that knockdown of c-Myc led
to a growth inhibition and cell cycle arrest at
G2/M-phase in Jijoye cells, but the role of c-Myc in the
G2/M-phase has remained elusive. The present study
indicated that loss of c-Myc controlled the expression of genes
associated with the G2/M-phase transition, including
cyclin B1 and CDK1, by modulating TIP60-mediated histone H4
acetylation in specific chromosomal regions, which resulted in a
marked G2/M-phase arrest in Raji-KD cells. Other studies
have reported that loss of c-Myc function impedes
G1-phase progression (28,34). Felsher et al (35) has reported that overexpression of
c-Myc causes TP53-dependent G2-phase arrest in normal
fibroblasts. These observations suggest that c-Myc has different
functions in diverse tumor cell types. Furthermore, the CDK
inhibitors p27 and p21 appear to be critical targets of c-Myc.
c-Myc cooperates with Zn-finger transcription factor Miz-1 to
repress the transcription of cell-cycle inhibitors, including p15,
p21 and p27 (5,36). The present study reported that
suppression of c-Myc induced the upregulation of p21 and the
downregulation of p27 in Raji-KD cells. However, further study is
required to determine whether G2/M arrest induced by the
loss of c-Myc is associated with changes in p27/p21 expression.
Distinct HATs, including TIP60, MOF, Kat2B and
Kat2A, associate with c-Myc TAD as the TRRAP, and form the
different HAT complexes that regulate histone H3 and H4 acetylation
(6,27,37). Recruitment of TIP60 to c-Myc is
significant for gene regulation by c-Myc (27,37). In the present study, a c-Myc-ChIP
assay indicated that c-Myc directly regulates TIP60 expression.
Reduction of MOF and AcH4 are known to correlate with the reduced
transcription of certain genes and with G2/M-phase
arrest (38). However, the
universal mechanism underlying c-Myc-mediated G2/M-phase
arrest remains to be elucidated. In the present study, the
AcH4-ChIP assay indicated that the transcription of cyclin B1,
CDK1, RB1, CCNG2 and CDKN1B was controlled by TIP60 and/or
MOF-mediated histone H4 acetylation, which was regulated by c-Myc.
The unique means by which c-Myc was demonstrated to regulate cell
cycle-associated factors is indicative of previously unsuspected
links between c-Myc, HATs, AcH4 and G2/M-phase
arrest.
Cell-cycle arrest in G2/M-phase usually
reflects a requirement for repairing cell damage; if not repaired,
apoptotic mechanisms are often activated (39). The present study indicated that
downregulation of c-Myc in Raji-KD cells induced decreases in cell
apoptosis. c-Myc induces apoptosis through various mechanisms; for
instance, it activates upstream events in the apoptotic cascade,
including death receptor Fas and Fas ligand (40). Furthermore, numerous target genes
of c-Myc that regulate mitochondrial function have been implicated
in c-Myc-induced apoptosis (41).
In addition, various target genes of c-Myc that regulate the cell
cycles, including CDC25A, have been implicated in cell apoptosis
(42). In the present study, TP53
expression was also downregulated in the Raji-KD cells.
Furthermore, knockdown of c-Myc decreased the expression of
pro-caspase-3, but did not affect the expression of caspase-8 and 9
in Raji cells. Although the detailed molecular mechanisms of how
c-Myc causes a decrease in caspase-3 levels remains to be
determined, elucidation of the links between c-Myc and the caspase
family may clarify the mechanisms underlying c-Myc-induced
apoptosis.
B-ALL is an aggressive form of Non-Hodgkin lymphoma
and is the most common type of childhood cancer (43). The survival rates of B-ALL
patients require to be improved via development of novel,
innovative therapies. To elucidated the biological functions of
c-Myc in B-ALL, an experimentally tractable model is required. In
this regard, to determine the role of c-Myc in modulating lymphoma
cell behavior, the present study used Raji cells to generate
Raji-KD and Raji-KD-Re cells. It was revealed that c-Myc recruited
HAT that induced histone H4 acetylation in specific chromatin
regions, thereby opening chromatin structures and amplifying the
expression of target genes. Knockdown of the c-Myc gene led to the
downregulation of the expression of CDK1 and cyclin B1 and resulted
in G2/M-phase arrest by histone H4 acetylation. Of note,
loss of c-Myc function by anti-sense suppression significantly
inhibited the tumorigenesis of Raji cells in vivo. Indeed,
c-Myc was overexpressed in the B-ALL samples, compared to the
samples from individuals with anemia (Fig. 6). It is consistent with the
reports that c-Myc contributes to the lymphoma tumorgenesis
(44). The therapeutic strategies
of c-Myc knockdown may be valuable for establishing novel methods
for B-ALL therapy.
Acknowledgments
The authors gratefully acknowledge Professor
Jianzhong Qin (Department of Biological Sciences, Dalian
University, Dalian, China) for supportive comments regarding the
manuscript.
Notes
[1]
Funding
This study is supported by National Nature Science
Foundation of China (grant nos. 31570797, 31270864, 30972675 and
31101717), the Science and Technology Planning Project of Dalian
City (grant no. 2010J21DW011) and the Natural Science Foundation of
Liaoning Province (grant no. 2015020253).
[2] Availability
of data and materials
The analyzed data sets generated during the study
are available from the corresponding author upon reasonable
request.
[3] Authors'
contributions
WL was desponsible for conception and designed of
the present study. YY, KX, ZL, WD, WZ, ZS, SS and TM performed
experiments; WL wrote the manuscript, which was reviewed by all
authors.
[4] Ethics
approval and consent to participate
The procedures for handling animals complied with
the Current Laboratory Animal Las and Regulations, Policies and
Administration in China. All experiments were approved by the
Animal Ethics Committee of Dalian Medical University (no.
AEE17013).
[5] Consent for
publication
Not applicable.
[6] Competing
interests
The authors declare that they have no competing
interests.
References
|
1
|
Villarreal-Martinez L, Jaime-Pérez JC,
Rodríguez-Martínez M, González-Llano O and Gómez-Almaguer D: Acute
lymphoblastic leukemia of childhood presenting as aplastic anemia:
Report of two cases. Rev Bras Hematol Hemoter. 34:165–167. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Liew M, Rowe L, Clement PW, Miles RR and
Salama ME: Validation of break-apart and fusion MYC probes using a
digital fluorescence in situ hybridization capture and imaging
system. J Pathol Inform. 7:202016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Zeller KI, Zhao X, Lee CW, Chiu KP, Yao F,
Yustein JT, Ooi HS, Orlov YL, Shahab A, Yong HC, et al: Global
mapping of c-Myc binding sites and target gene networks in human B
cells. Proc Natl Acad Sci USA. 103:17834–17839. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Wang H, Teriete P, Hu A,
Raveendra-Panickar D, Pendelton K, Lazo JS, Eiseman J, Holien T,
Misund K, Oliynyk G, et al: Direct inhibition of c-Myc-Max
heterodimers by celastrol and celastrol-inspired triterpenoids.
Oncotarget. 6:32380–32395. 2015.PubMed/NCBI
|
|
5
|
Dang CV: MYC on the path to cancer. Cell.
149:22–35. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Zhang N, Ichikawa W, Faiola F, Lo SY, Liu
X and Martinez E: MYC interacts with the human STAGA coactivator
complex via multivalent contacts with the GCN5 and TRRAP subunits.
Biochim Biophys Acta. 1839:395–405. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Dang CV: MYC, metabolism, cell growth, and
tumorigenesis. Cold Spring Harb Perspect Med. 3:a0142172013.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Cao X, Bennett RL and May WS: c-Myc and
caspase-2 are involved in activating Bax during cytotoxic
drug-induced apoptosis. J Biol Chem. 283:14490–14496. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wang O, Yang F, Liu Y, Lv L, Ma R, Chen C,
Wang J, Tan Q, Cheng Y, Xia E, et al: C-MYC-induced upregulation of
lncRNA SNHG12 regulates cell proliferation, apoptosis and migration
in triple-negative breast cancer. Am J Transl Res. 9:533–545.
2017.PubMed/NCBI
|
|
10
|
Treszl A, Adány R, Rákosy Z, Kardos L,
Bégány A, Gilde K and Balázs M: Extra copies of c-myc are more
pronounced in nodular melanomas than in superficial spreading
melanomas as revealed by fluorescence in situ hybridisation.
Cytometry B Clin Cytom. 60:37–46. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ruiz-Pérez MV, Henley AB and
Arsenian-Henriksson M: The MYCN protein in health and disease.
Genes (Basel). 8. pp. E1132017, View Article : Google Scholar
|
|
12
|
Yang Y, Yoo HM, Choi I, Pyun KH, Byun SM
and Ha H: Interleukin 4-induced proliferation in normal human
keratinocytes is associated with c-myc gene expression and
inhibited by genistein. J Invest Dermatol. 107:367–372. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Nupponen NN, Kakkola L, Koivisto P and
Visakorpi T: Genetic alterations in hormone-refractory recurrent
prostate carcinomas. Am J Pathol. 153:141–148. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Nupponen NN, Hyytinen ER, Kallioniemi AH
and Visakorpi T: Genetic alterations in prostate cancer cell lines
detected by comparative genomic hybridization. Cancer Genet
Cytogenet. 101:53–57. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Gugger M, Burckhardt E, Kappeler A,
Hirsiger H, Laissue JA and Mazzucchelli L: Quantitative expansion
of structural genomic alterations in the spectrum of neuroendocrine
lung carcinomas. J Pathol. 196:408–415. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Hecht JL and Aster JC: Molecular biology
of Burkitt's lymphoma. J Clin Oncol. 18:3707–3721. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Neri A, Barriga F, Knowles DM, Magrath IT
and Dalla-Favera R: Different regions of the immunoglobulin
heavy-chain locus are involved in chromosomal translocations in
distinct pathogenetic forms of Burkitt lymphoma. Proc Natl Acad Sci
USA. 85:2748–2752. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Schmittgen TD and Livak KJ: Analyzing
real-time PCR data by the comparative C(T) method. Nat Protoc.
3:1101–1108. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Frank SR, Schroeder M, Fernandez P,
Taubert S and Amati B: Binding of c-Myc to chromatin mediates
mitogen-induced acetylation of histone H4 and gene activation.
Genes Dev. 15:2069–2082. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Pfeifer D, Chung YM and Hu MC: Effects of
low-dose bisphenol A on DNA damage and proliferation of breast
cells: The role of c-Myc. Environ Health Perspect. 123:1271–1279.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Lane AN and Fan TW: Regulation of
mammalian nucleotide metabolism and biosynthesis. Nucleic Acids
Res. 43:2466–2485. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Yap CS, Peterson AL, Castellani G, Sedivy
JM and Neretti N: Kinetic profiling of the c-Myc transcriptome and
bioinformatic analysis of repressed gene promoters. Cell Cycle.
10:2184–2196. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Seo HR, Kim J, Bae S, Soh JW and Lee YS:
Cdk5-mediated phosphorylation of c-Myc on Ser-62 is essential in
transcriptional activation of cyclin B1 by cyclin G1. J Biol Chem.
283:15601–15610. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Xu YX and Manley JL: New insights into
mitotic chromosome condensation: A role for the prolyl isomerase
Pin1. Cell Cycle. 6:2896–2901. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ehedego H, Boekschoten MV, Hu W, Doler C,
Haybaeck J, Gaβler N, Müller M, Liedtke C and Trautwein C: p21
ablation in liver enhances DNA damage, cholestasis, and
carcinogenesis. Cancer Res. 75:1144–1155. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Huang X, Di Liberto M, Jayabalan D, Liang
J, Ely S, Bretz J, Shaffer AL III, Louie T, Chen I, Randolph S, et
al: Prolonged early G(1) arrest by selective CDK4/CDK6 inhibition
sensitizes myeloma cells to cytotoxic killing through cell
cycle-coupled loss of IRF4. Blood. 120:1095–1106. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Ravens S, Yu C, Ye T, Stierle M and Tora
L: Tip60 complex binds to active Pol II promoters and a subset of
enhancers and co-regulates the c-Myc network in mouse embryonic
stem cells. Epigenetics Chromatin. 8:452015. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Bretones G, Delgado MD and León J: Myc and
cell cycle control. Biochim Biophys Acta. 1849:506–516. 2015.
View Article : Google Scholar
|
|
29
|
Nussenzweig MC, Schmidt EV, Shaw AC, Sinn
E, Campos-Torres J, Mathey-Prevot B, Pattengale PK and Leder P: A
human immunoglobulin gene reduces the incidence of lymphomas in
c-Myc-bearing transgenic mice. Nature. 336:446–450. 1988.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Miller DM, Thomas SD, Islam A, Muench D
and Sedoris K: c-Myc and cancer metabolism. Clin Cancer Res.
18:5546–5553. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Davis AC, Wims M, Spotts GD, Hann SR and
Bradley A: A null c-myc mutation causes lethality before 10.5 days
of gestation in homozygotes and reduced fertility in heterozygous
female mice. Genes Dev. 7:671–682. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Mateyak MK, Obaya AJ and Sedivy JM: c-Myc
regulates cyclin D-Cdk4 and -Cdk6 activity but affects cell cycle
progression at multiple independent points. Mol Cell Biol.
19:4672–4683. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Song A, Ye J, Zhang K, Sun L, Zhao Y and
Yu H: Lentiviral vector-mediated siRNA knockdown of c-MYC: Cell
growth inhibition and cell cycle arrest at G2/M phase in Jijoye
cells. Biochem Genet. 51:603–617. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Schorl C and Sedivy JM: Loss of
protooncogene c-Myc function impedes G1 phase progression both
before and after the restriction point. Mol Biol Cell. 14:823–835.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Felsher DW, Zetterberg A, Zhu J, Tlsty T
and Bishop JM: Overexpression of MYC causes 53-dependent G2 arrest
of normal fibroblasts. Proc Natl Acad Sci USA. 97:10544–10548.
2000. View Article : Google Scholar
|
|
36
|
Vo BT, Wolf E, Kawauchi D, Gebhardt A,
Rehg JE, Finkelstein D, Walz S, Murphy BL, Youn YH, Han YG, et al:
The interaction of Myc with Miz1 defines medulloblastoma subgroup
identity. Cancer Cell. 29:5–16. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Wolf E, Lin CY, Eilers M and Levens DL:
Taming of the beast: Shaping Myc-dependent amplification. Trends
Cell Biol. 25:241–248. 2015. View Article : Google Scholar :
|
|
38
|
Smith ER, Cayrou C, Huang R, Lane WS, Côté
J and Lucchesi JC: A human protein complex homologous to the
Drosophila MSL complex is responsible for the majority of histone
H4 acetylation at lysine 16. Mol Cell Biol. 25:9175–9188. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Rozenblat S, Grossman S, Bergman M,
Gottlieb H, Cohen Y and Dovrat S: Induction of G2/M arrest and
apoptosis by sesquiterpene lactones in human melanoma cell lines.
Biochem Pharmacol. 75:369–382. 2008. View Article : Google Scholar
|
|
40
|
Wiese KE, Haikala HM, von Eyss B, Wolf E,
Esnault C, Rosenwald A, Treisman R, Klefström J and Eilers M:
Repression of SRF target genes is critical for Myc-dependent
apoptosis of epithelial cells. EMBO J. 34:1554–1571. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Hoffman B and Liebermann DA: Apoptotic
signaling by c-MYC. Oncogene. 27:6462–6472. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Shen T and Huang S: The role of Cdc25A in
the regulation of cell proliferation and apoptosis. Anticancer
Agents Med Chem. 12:631–639. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Shah S, Schrader KA, Waanders E, Timms AE,
Vijai J, Miething C, Wechsler J, Yang J, Hayes J, Klein RJ, et al:
A recurrent germline PAX5 mutation confers susceptibility to pre-B
cell acute lymphoblastic leukemia. Nat Genet. 45:1226–1231. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Shou Y, Martelli ML, Gabrea A, Qi Y,
Brents LA, Roschke A, Dewald G, Kirsch IR, Bergsagel PL and Kuehl
WM: Diverse karyotypic abnormalities of the c-myc locus associated
with c-myc dysregulation and tumor progression in multiple myeloma.
Proc Natl Acad Sci USA. 97:228–233. 2000. View Article : Google Scholar : PubMed/NCBI
|