Introduction
Aging is among the most prominent risk factors for
human diseases, including Alzheimer's disease, cataract, glaucoma,
Parkinson's disease, arteriosclerosis, and heart failure (1). At present, the biological basis of
aging is largely unknown, although several hypotheses have been
proposed, such as the genetic theory of aging, radical theory, and
decreased immune function theory (2). A growing body of evidence supports a
critical role of mitochondrial homeostasis in the aging process
(3–7). Damage and dysfunction in
mitochondria are important factors in a range of human disorders
because these organelles serve an irreplaceable role in energy
production and cell metabolism (6,8–10).
Mitochondria are highly dynamic, and the regulation of
mitochondrial quality control is subtle. Mitochondrial homeostasis
is tightly modulated by two pathways: Biogenesis and mitochondrial
autophagy, termed mitophagy (11). Mitophagy is a selective
degradation process of mitochondria that are damaged or stressed
(12,13). Several studies have reported that
mitophagy occurs in senescent myocardium and may have an important
role in cardioprotection (14,15).
Sirtuins is a protein family of nicotinamide adenine
dinucleotide (NAD)-dependent deacetylases, which serve important
roles in regulating cell stress, metabolism, growth, aging and
apoptosis (16). Recently,
several members of the family have been reported to be associated
with autophagy for their deacetylation function (17). Sirtuin 3 (SIRT3) is the only
family member that is highly expressed in the population of
longevity (18). As a typical
mitochondrial sirtuin family member, SIRT3 counteracts oxidative
stress, defends against cell apoptosis, and prevents cell aging and
tumorigenesis (19–22). Some studies have reported that
SIRT3 reduces the level of reactive oxygen species (ROS) in the
myocardium by deacetylating the transcription factor forkhead box
(Fox)O3a, thus increasing the expression of its target genes,
manganese superoxide dismutase (MnSOD) and catalase (CAT) (23,24). Other reports have also
demonstrated that SIRT3 increases the activity of these antioxidant
enzymes through nuclear factor κB and protects tissue from
ROS-induced injury (25). SIRT3
levels in the murine heart can be increased by hypertrophic
agonists in response to pressure overload and exercise.
Furthermore, a recent study demonstrated that mouse mitochondrial
dysfunction caused by loss of SIRT3 strongly contributed to
obesity-related heart failure (26). However, the role of SIRT3 in
regulating mitochondrial homeostasis in the myocardium remains
largely unknown.
The present study aimed to explore the role of SIRT3
on mitochondrial homeostasis in the aged myocardium. The results
demonstrated that SIRT3 knockout (KO) greatly inhibited
p53/Parkin-mediated mitophagy. This inhibition disrupted
mitochondrial homeostasis and resulted in irreversible
mitochondrial dysfunction.
Materials and methods
Animals and ethics statements
Heart-specific SIRT3 KO (SIRT3−/−) weaned
mice (n=54) and wild-type (WT) C57BL mice (n=96) were purchased
from the Jackson Laboratory (Ben Harbor, ME, USA) in the present
study. The age of the male WT or KO mice used in the present study
were as follows: Young (4 months), aged (20 months), and aged +
aerobic intermittent training (AIT) (20 month old mice that
underwent 13 weeks of AIT). To determine the effect of SIRT3 on
Parkin-mediated cardiac mitophagy, specific mitophagy agonist CCCP
(5 mg CCCP/kg body weight, dissolved in DMSO; cat no. C2759;
Sigma-Aldrich), alone or with the autophagy antagonist
bafilomycin-A1 (12 µg/kg body weight, dissolved in DMSO,
cat. no. ab120497; Abcam, Cambridge, UK), was injected
intraperitoneally into the WT young, WT aged, and SIRT3 KO mice 12
h (n=8 for each group) prior to subsequent detections. As a
control, an equivalent dose of DMSO was injected into the
littermates of the CCCP injection group. The mice were housed in a
humidity- and temperature-controlled institutional laboratory
animal facility where they had access to food and water ad
libitum under a 12-h light/dark cycle. For sampling, 1%
pentobarbital sodium was used to anesthetize the mice (50 mg/kg).
The mice were euthanized immediately following the operation in a
Mobile Anesthetic Workstation (Harvard Apparatus; Harvard
Bioscience Inc., Cambridge, MA). All animal handling and
experimental procedures described in the present study were
approved by the Institutional Animal Care and Use Committee of the
Fourth Military Medical University (Xi'an, China), and in
compliance with the Guidelines for the Care and Use of Laboratory
Animals (27).
Aerobic intermittent training (AIT)
AIT was performed on aged or SIRT3 KO mice to
evaluate the impacts of aerobic excise on SIRT3 activity and
myocardial mitochondrial functions. The procedure was performed as
follows: 1 week adaptive treadmill training at a speed of 15 m/min
(30 min/day, 5 days/week), followed by 12 weeks formal intermittent
aerobic exercise at regularly changing speed (1 h/day, 5
days/week). For each formal exercise section, the mouse first did
warm-up exercise (40–50% of maximum oxygen uptake) for 10 min,
followed by seven cycles of 4 min high-intensity (80–85% VO2 max,
speed of 23 m/min) and 3 min low-intensity intermittent aerobic
exercise (65–75% VO2 max, speed of 12 m/min). At the end of each
section, the mouse had a 1 min cool-down period.
Mitochondria/cytosol fractionation
Mitochondria and cytosol protein compartments were
fractionated with Mitochondria/Cytosol Fractionation Kit
(BioVision, Inc., Milpitas, CA, USA) according to the
manufacturer's instructions.
Co-Immunoprecipitation assay (co-IP)
Myocardial samples were collected from each group
(n=8), separately homogenized in Nonidet P-40 (NP-40) IP lysis
buffer (50 mM Tris, 0.1% NP-40, 150 mM NaCl and 2 mM EDTA; pH 7.5;
Beyotime Institute of Biotechnology, Shanghai, China) containing
protease inhibitor (Invitrogen; Thermo Fisher Scientific, Inc.,
Waltham, MA, USA), and centrifuged at 12,000 × g for 10 min at 4°C.
Supernatants were collected, incubated with the primary antibody
overnight at 4°C, and then mixed with protein G-Sepharose Fast Flow
beads (Merck KGaA, Darmstadt, Germany) that were pre-equilibrated
in lysis buffer. The primary antibodies used for IP were as
follows: MnSOD (1:200; cat. no. S5069) and PGC-1α (1:100; cat. no.
AV40129) obtained from Sigma-Aldrich (St. Louis, MO, USA); p53
(1:100; cat. no. ab26) and Parkin (1:100; cat. no. ab15954) were
obtained from Abcam (Cambridge, UK). The beads were collected by
centrifugation (3,000 × g for 5 min), washed and resuspended in an
equal volume of 5xSDS loading buffer. Immunoprecipitated proteins
were separated by 10% SDS-PAGE, and immunoblotting was performed as
described below.
Western blotting
Myocardial proteins were extracted using
radioimmunopreciptation assay buffer (50 mM Tris-HCl, pH7.5, 2 mM
EDTA, 0.5% deoxycholate, 150 mM NaCl, 1% Triton X-100, 0.1% SDS, 1
mM Na3VO4 and 1 mM PMSF). A total of 60 mg of proteins from each
sample was separated by 12% SDS-PAGE and transferred to
polyvinylidene difluoride membranes at 120 V for 2 h. The membranes
were briefly washed with methanol and left to dry for 15 min to
enhance the protein binding. The membranes were blocked with 5%
bovine serum albumin (BSA; Invitrogen; Thermo Fisher Scientific,
Inc.) in TBS-T and then incubated with primary antibodies overnight
at 4°C. For loading control, the membranes were probed with
anti-GAPDH or tubulin (for non-mitochondrial proteins; 1:600; cat.
no. sc-25778 and sc-69971, respectively, Santa Cruz Biotechnology,
Inc., Dallas, TX, USA) or anti-cytocchrome c oxidase complex
(COX) IV (for mitochondrial proteins; 1:600; cat. no. HPA002485;
Sigma-Aldrich) antibodies in TBS-T for 1 h. The membranes were next
incubated with horseradish peroxidase-conjugated goat anti-rabbit
secondary antibody (1:2,000 dilution) in TBS-T for 1 h. Finally,
the membranes were incubated with enhanced chemiluminescence
solution (Boehringer Mannheim; Roche Diagnostics GmbH, Mannheim,
Germany), exposed in a ChemiDoc XRS imaging system, and analyzed
with the Quantity One software (Bio-Rad Laboratories, Inc.,
Hercules, CA, USA). Rabbit/mouse monoclonal or poly-clonal primary
antibodies purchased from Sigma-Aldrich were used at dilutions:
SIRT3 (1:500; cat. no. S4072), MnSOD (1:300), PGC-1α (1:300),
cyclin-dependent kinase inhibitor 2A (p16; 1:800; cat. no.
SAB4500072), acetyl-p53 (K317; 1:200; cat. no. SAB4503014), SIRT1
(1:500; cat. no. AV32386, SIRT1 served as a control to demonstrate
the specificity of SIRT3 KO), glucose-regulated protein (GRP) 75
(1:400; cat. no. SAB4501454), NADH:Ubiquinone oxidoreductase
subunit A9 (NDUFA9; 1:400; cat. no. WH0004704M1), and anti-acetyl
lysine (1:200; cat. no. SAB5200090). In addition, rabbit polyclonal
primary antibody against sequestosome 1 (p62; 1:400; cat. no.
23214) was obtained from Cell Signaling Technology, Inc. (Danvers,
MA, USA). Antibodies against general control of amino acid
synthesis 5-like 1 (GCN5L1; 1:500; cat. no. ab18381), p53 (1:800)
and Parkin (1:400) were obtained from Abcam.
Measurement of protein content by UV
spectrophotometry
A standard BSA solution at a concentration of 1
mg/ml was prepared, and aliquots of 0, 1, 2, 3, 4 and 5 ml of the
standard solution were placed in six tubes, respectively. Each tube
was filled with distilled water up to 5 ml. The first one was set
as a blank control, and the absorption values of the standard
solutions were successively measured under 280 nm UV light. The
concentration of each tube was plotted to create a standard curve,
which was then used to determine protein content of samples based
on their A280 values.
Detection of enzyme activity and
measurement of substrate content
Mice were euthanized by cervical dislocation. The
hearts were rapidly removed, and a piece of full-thickness left
ventricular myocardium was immediately clipped from each heart. The
samples were snap-frozen in liquid nitrogen, and 0.3 g of each
sample was placed in an ice-cold glass homogenizer. MnSOD and
citrate synthase activity were detected with Manganese Superoxide
Dismutase Activity Assay kit (Cayman Chemical Company, Ann Arbor,
MI, USA) and Citrate Synthase Activity Assay kit (cat. no.
ab119692; Abcam), respectively. ROS and lipofuscin were measured by
Cellular Reactive Oxygen Species Detection Assay kit (cat. no.
ab113851; Abcam) and Mouse Lipofuscin ELISA kit (Sigma-Aldrich).
ATP and malondialdehyde (MDA) were detected with ATP Determination
kit (Invitrogen; Thermo Fisher Scientific, Inc.) and OxiSelect
TBARS Assay kit (Cell Biolabs, Inc., San Diego, CA, USA),
respectively. Each detection or measurement was performed according
to the manufacturer's instructions. The aforementioned measurements
were performed in 12 mice per group to minimize the variation
between individual animals and to reduce the
false-positive/positive effect of the detection methods.
β-galactosidase+ cell staining
and counting
Myocardium (0.1 g) was sampled from the WT young, WT
aged, and SIRT3 KO mice. The tissue samples were shredded and then
digested with 200 U/ml collagenase I for ~2 h to obtain dissociated
cardiomyocytes. The cardiomyocytes were stained with Senescence
β-Galactosidase Staining kit (Cell Signaling Technology, Inc.)
according to the manufacturer's instructions and analyzed with a
flow cytometer (BD Biosciences, Franklin Lakes, NJ, USA).
Transmission electron microscopy
For transmission electron microscopy examination,
the full-thickness left ventricular myocardium was mechanically
dissected. The tissues were fixed in 3% glutaraldehyde in 0.1 M
cacodylate buffer (pH 7.4) at 4°C for 24 h, post-fixed in 1%
OsO4 in the same buffer for 1 h, dehydrated in graded
ethanols, and embedded in Epon 812. Thin (60–90 nm) sections were
used for ultrastructural evaluation using a 100 SX transmission
electron microscope (JEOL Inc., Peabody, MA, USA) operating at 80
kV.
Statistical analysis
Data are expressed as mean ± standard error of the
mean. One-way analysis of variance followed by the LSD test was
used for multiple comparisons. All statistical tests were performed
using SPSS v. 19.0 (IBM Corp., Armonk NY, USA). P<0.05 was
considered to indicate a statistically significant difference.
Results
SIRT3-mediated mitochondrial protein
deacetylation is attenuated by aging but partially mitigated by
AIT
The expression levels of SIRT3 protein in aged mouse
myocardium were significantly lower compared with young mouse
myocardium (Fig. 1A and B).
Consistent with the change in SIRT3 protein levels, the overall
acetylation of mitochondrial proteins was markedly elevated in the
senile myocardium (Fig. 1C).
However, the levels of the conserved mitochondrial
acetyltransferase GCN5L1 did not differ significantly among the
young, the aged and AIT groups (F=0.175, P=0.844; Fig. 1A and B). These results suggested
that increase of overall acetylation in senile myocardium might be
associated with downregulation of SIRT3; therefore, elevation of
SIRT3 level might enhance the deacetylation of mitochondrial
proteins in senile myocardium.
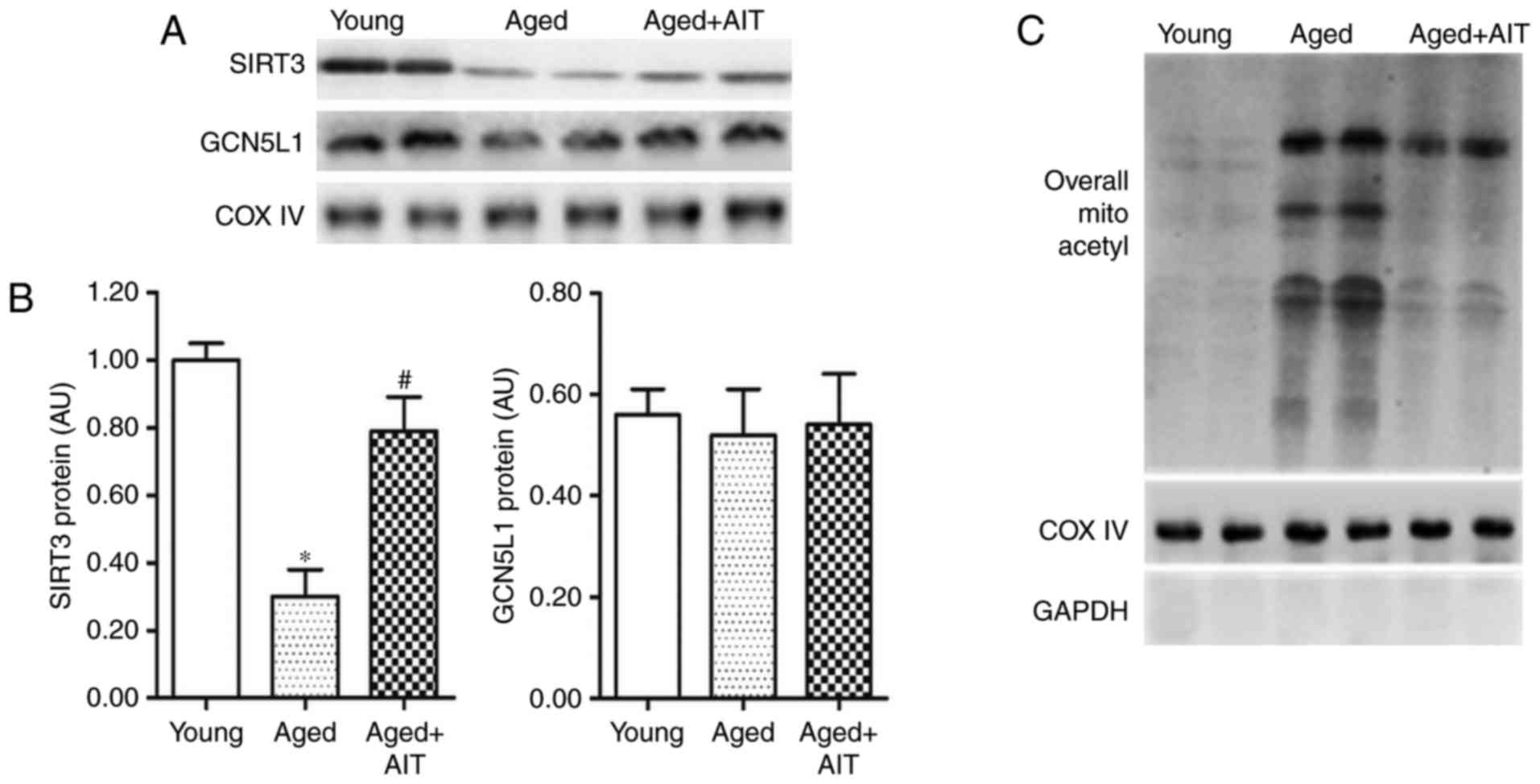 | Figure 1SIRT3 and SIRT3-mediated
mitochondrial protein deacetylation are reduced in the aged
myocardium. (A) Representative blots and (B) quantification of
western blot analysis for the levels of SIRT3 and acetyltransferase
GCN5L1 in the myocardial mitochondria of young, aged, and aged+AIT
mice. Myocardial mitochondrial samples were separately isolated
from WT mice aged as follows: Young (4 months), aged (20 months),
and aged+AIT (20 month old mice that underwent 13 weeks of AIT).
(C) The overall acetylation levels of mitochondrial protein in the
myocardia of young, aged and aged+AIT mice. n=8 per group.
*P<0.05 compared with the young;
#P<0.05 compared with both the young and the aged.
SIRT3, sirtuin 3; GCN5L1, general control of amino acid synthesis
5-like 1; AIT, aerobic intermittent training; WT, wild-type; COX
IV, cytochrome c oxidase complex IV. |
Previous studies have demonstrated that AIT is
beneficial for the expression of mammalian SIRT3 in adult
myocardium (28). In the present
study, the effects of AIT on myocardial SIRT3 expression and
acetylation of mitochondrial proteins in aged mice were
investigated. The results demonstrated that myocardial SIRT3
expression could be partially rescued by AIT (Fig. 1A and B). Likewise, the
deacetylation of mitochondrial proteins was also partially
increased by AIT (Fig. 1C).
Oxidative stress and energy metabolism
dysfunction are increased in aged myocardium
Mitochondrial MnSOD is important in mitigating
oxidative stress (29), which is
one of the main manifestations of aging. Mitochondrial proteins
were isolated from young and aged WT myocardia, and the expression
and activity of MnSOD were detected. Western blot and fluorescence
spectrophotometric analyses revealed that the levels of MnSOD in
aged myocardium were significantly lower compared with young
myocardium (F=13.261, P<0.001; Fig. 2A and B). Co-IP analysis
demonstrated that the acetylation of MnSOD in the aged myocardium
was markedly elevated (Fig. 2C).
In addition, the activity of MnSOD in aged myocardium was ~60% of
that in the young myocardium (Fig.
2D). In the aged+AIT group, the expression levels, activity and
deacetylation of MnSOD were less affected (Fig. 2A–D). As key factors in oxidative
stress, MDA and ROS were upregulated by 30 and 70%, respectively,
in the aged myocardium compared with the young (Fig. 2E and F). This upregulation was
partially eliminated by AIT (Fig. 2E
and F).
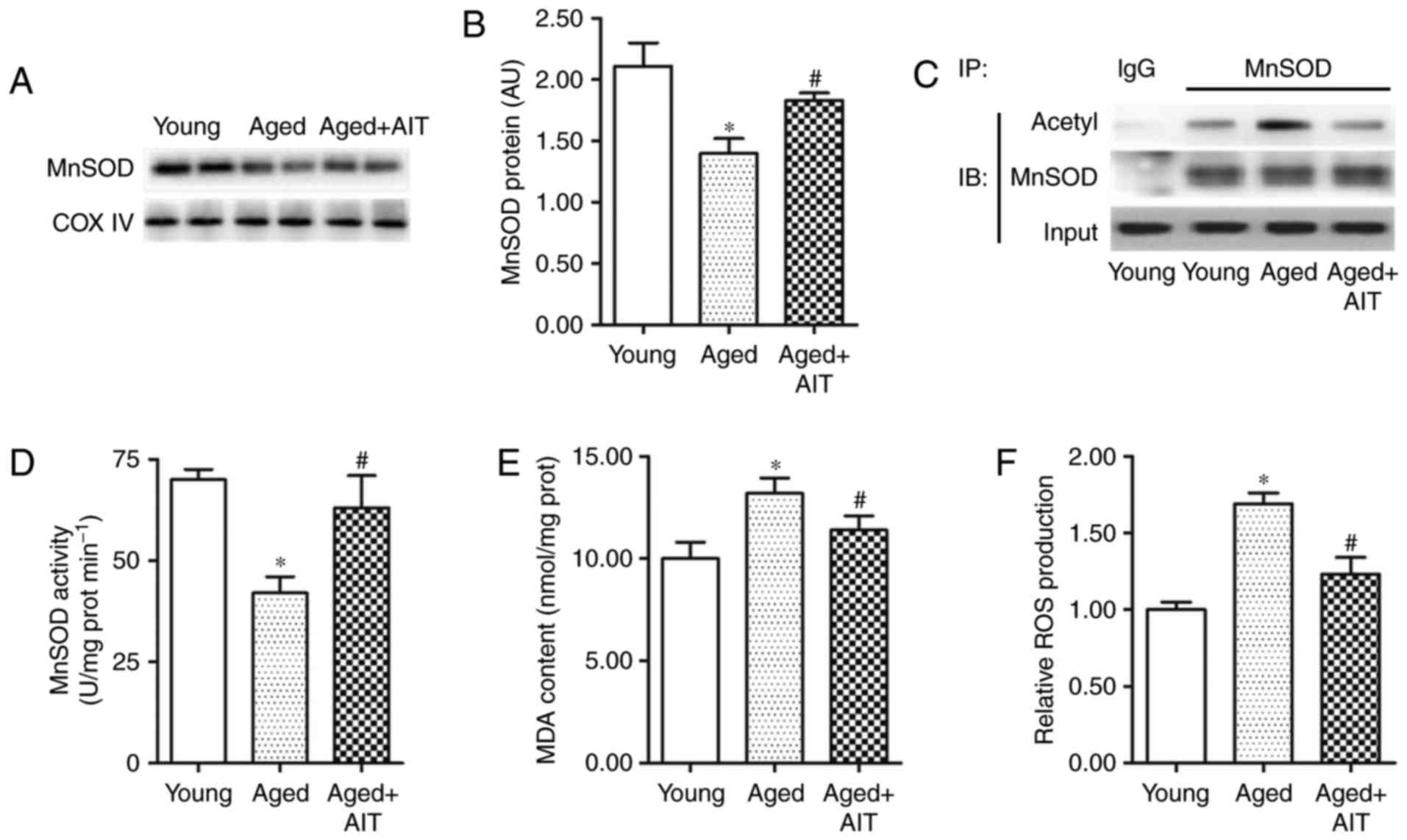 | Figure 2Expression and activity of MnSOD are
reduced in the aged myocardium. Expression of MnSOD in the
myocardial mitochondria of young, aged, and aged+AIT groups were
detected with (A) western blot analysis and (B) UV
spectrophotometry. (C) The acetylation levels of mitochondrial
MnSOD were assayed by IP analysis. (D) Activity of MnSOD in the
young, aged, and aged+AIT myocardia was analyzed with a MnSOD
activity assay kit. (E) MDA content and (F) ROS production were
detected with a MDA Quantitation Assay kit and a Cellular Detection
Assay kit, respectively. n=8 per group for (A–C), and n=12 per
group for (D–F). *P<0.05 compared with the young;
#P<0.05 compared with both the young and the aged.
MnSOD, manganese superoxide dismutase; AIT, aerobic intermittent
training; IP, immunoprecipitation; MDA, malondialdehyde; ROS,
reactive oxygen species; COX IV, cytochrome c oxidase
complex IV; IB, immunoblotting. |
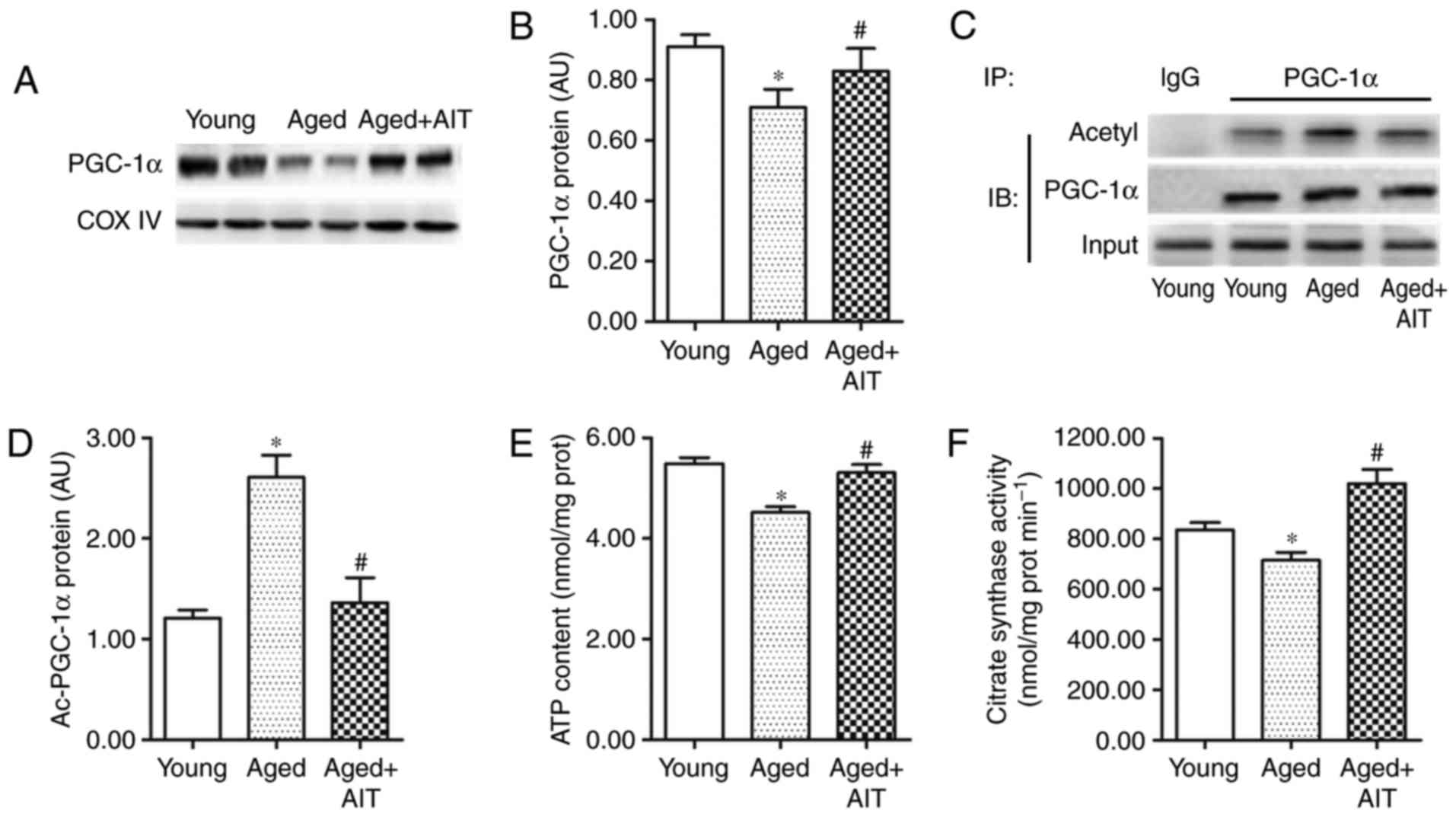
Energy metabolism dysfunction is also a main
manifestation of aging. PGC-1α is a key regulator of energy
metabolism (30), and its
expression and activity are commonly measured as a method to
evaluate the energy metabolism of young and aged myocardia. PGC-1α
had lower expression, deacetylation and activity in aged myocardium
compared with young (Fig. 3A–D).
In addition, the ATP content and citrate synthase activity were
demonstrated to be reduced in aged myocardium compared with the
young (Fig. 3E and F), suggesting
a decrease in the metabolic ability of aged myocardium. Similar to
the changes of MnSOD, the reduced PGC-1α expression, deacetylation,
and activity, as well as the energy metabolism, were improved by
AIT (Fig. 3). These results and
the expression profiles of SIRT3 in aged myocardium suggested that
SIRT3 reduction was closely related to acetylation and
downregulation of MnSOD and PGC-1α-induced aging in mitochondrial
homeostasis.
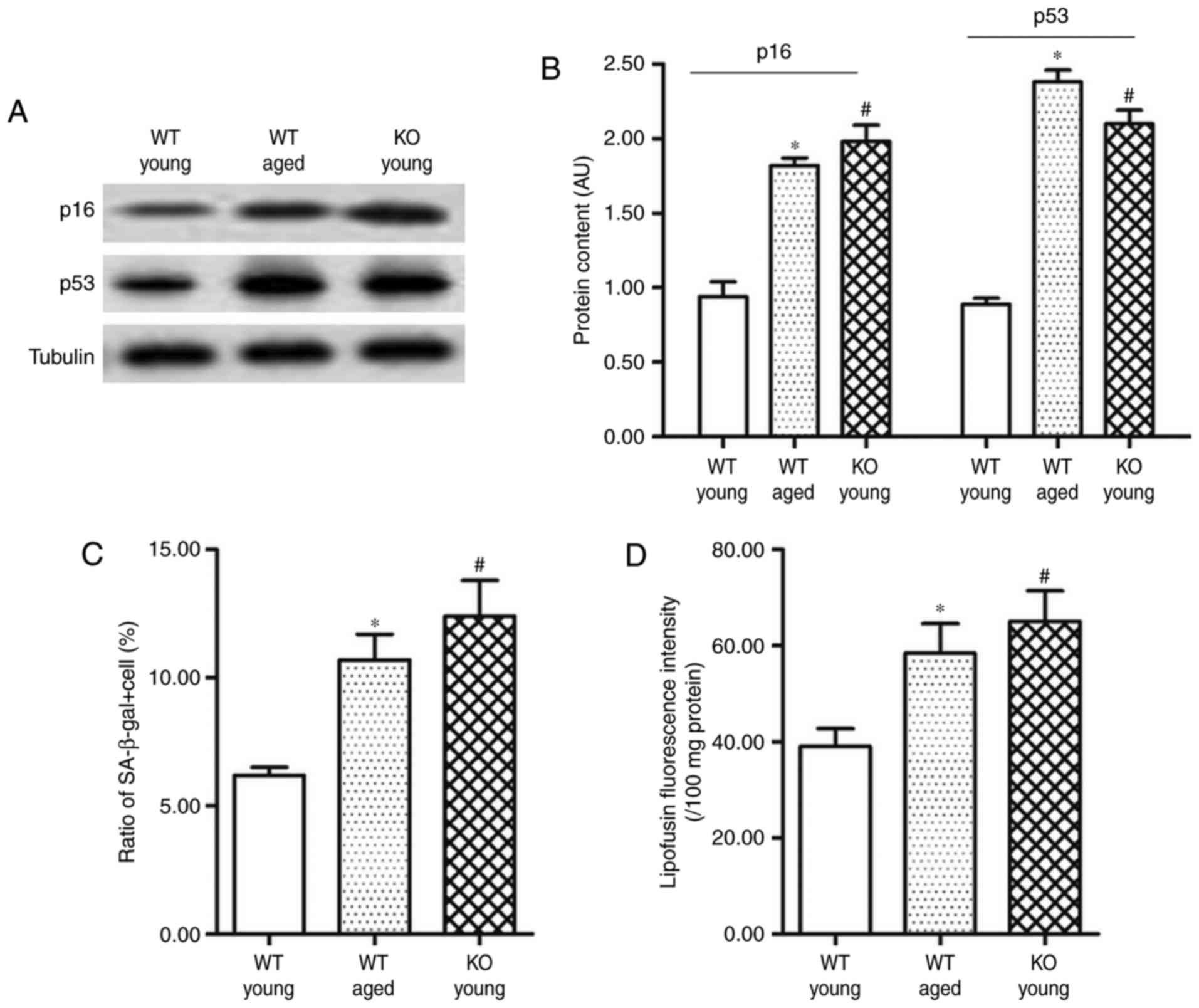
Myocardium of SIRT3−/− mice
displays obvious features of aging, including mitochondrial protein
dysfunction, enhanced oxidative stress and energy metabolism
dysfunction
To explore the association between SIRT3 and
myocardial senescence, SIRT3 KO (SIRT3−/−) mice were
used in subsequent experiments. Compared with the young WT mice
(aged 4 months), the levels of senescence marker genes p16 and p53
in the adult SIRT3−/− mice were upregulated by ~80 and
140%, respectively (Fig. 4A and
B). In addition, both the proportion of
β-galactosidase+ cells and the lipofuscin content in the
SIRT3−/− mice were increased by ~50% (Fig. 4C and D).
As in the analyses for senescent myocardium, the
overall acetylation of mitochondrial proteins and the expression
and acetylation of MnSOD and PGC-1α were then analyzed in the
SIRT3−/− myocardium. Western blot analysis revealed that
the overall acetylation was robustly enhanced in the absence of
SIRT3 (Fig. 5A). A strong
increase in the acetylation of MnSOD and PGC-1α accompanied by a
sharp decrease in their activity was also detected (Fig. 5B, C and F). Consistent with the
alteration of the MnSOD and PGC-1α levels, production of MDA and
ROS was greatly elevated, while ATP content was reduced and citrate
synthase activity was inhibited (Fig.
5D, E, G and H). However, in the absence of SIRT3, AIT had no
effect on the deacetylation of mitochondrial proteins or energy
metabolism (Fig. 5A, B and F-H)
and it even exacerbated oxidative stress in myocardial mitochondria
(Fig. 5D and E). These data
indicated that myocardial mitochondrial protein damage and
dysfunction caused by SIRT3 deletion could not be mitigated by
AIT.
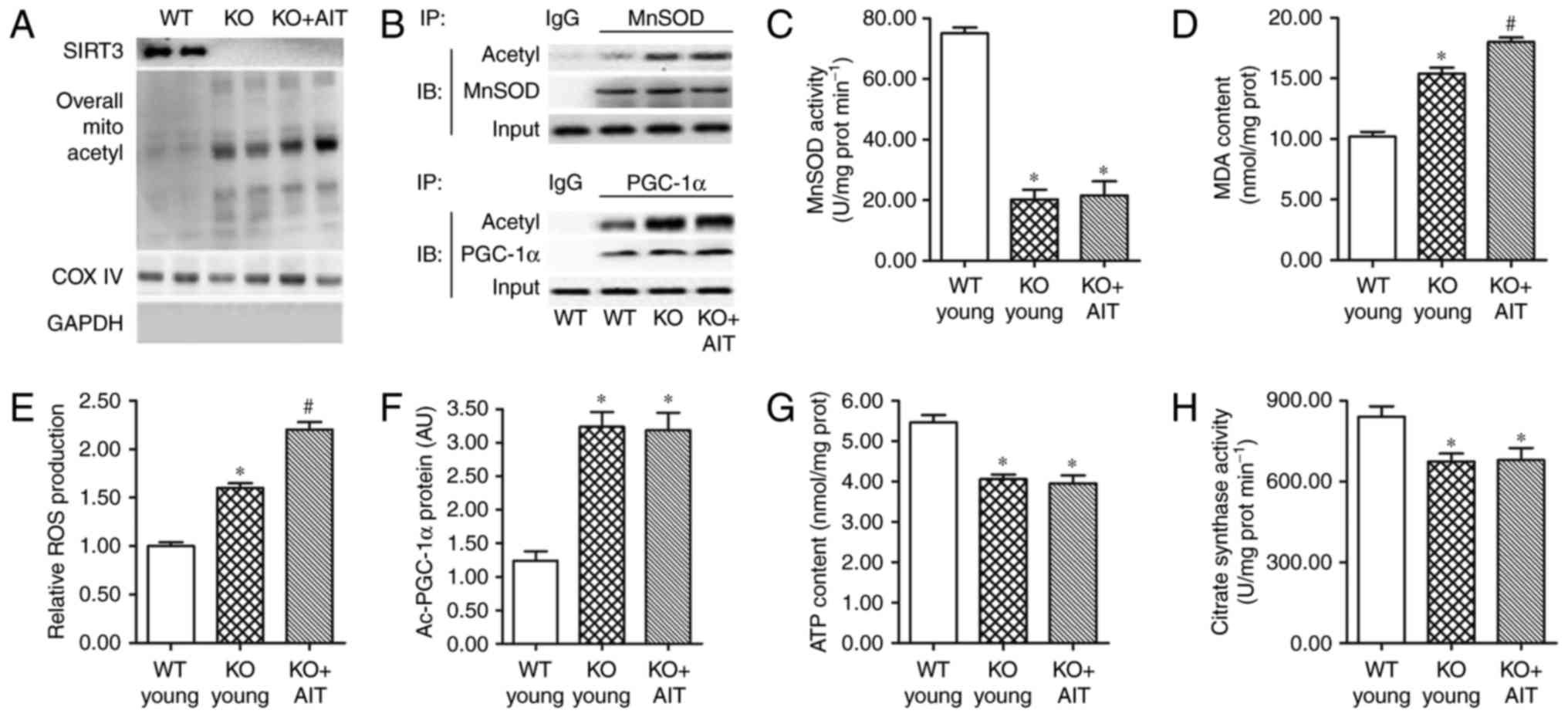 | Figure 5The myocardium of SIRT3−/−
mice displays features of enhanced oxidative stress and energy
metabolism dysfunction. (A) Detection of SIRT3 and mitochondrial
protein acetylation levels in the WT young, SIRT3−/−
(KO), and KO+AIT myocardia by western blotting. (B) Detection of
acetyl/total MnSOD and acetyl/total PGC-1α by IP/western blot
analyses. (C) MnSOD activity, (D) MDA content and (E) ROS
production in the myocardia of WT young, KO young and KO+AIT mice.
(F) Acetylation levels of PGC-1α, (G) ATP content and (H) citrate
synthase activity in the myocardia of WT young, KO young, and
KO+AIT mice. n=8 per group. *P<0.05 compared with the
WT group; #P<0.05 compared with both the WT and the
KO groups. SIRT3, sirtuin 3; WT, wild-type; KO, knockout; AIT,
aerobic intermittent training; MnSOD, manganese superoxide
dismutase; PGC-1α, peroxisome proliferator-activated receptor γ
coactivator-1α; IP, immunoprecipitation; MDA, malondialdehyde; ROS,
reactive oxygen species; COX IV, cytochrome c oxidase
complex IV; IB, immunoblotting; Ac, acetylated. |
SIRT3 deficiency impairs Parkin-mediated
mitophagy by increasing p53-Parkin binding
Mitophagy serves important roles in mitochondrial
self-renewal and inhibition of mitochondrial dysfunction during
aging or post-injury. It was speculated that SIRT3 deficiency may
have caused abnormal mitophagy, therefore, further experiments
explored autophagosome morphology and signal transduction in
mitophagy regulation. Electron microscopy revealed that autophagic
vacuoles were reduced in aged and SIRT3−/− myocardium
compared with adult WT myocardium (Fig. 6A).
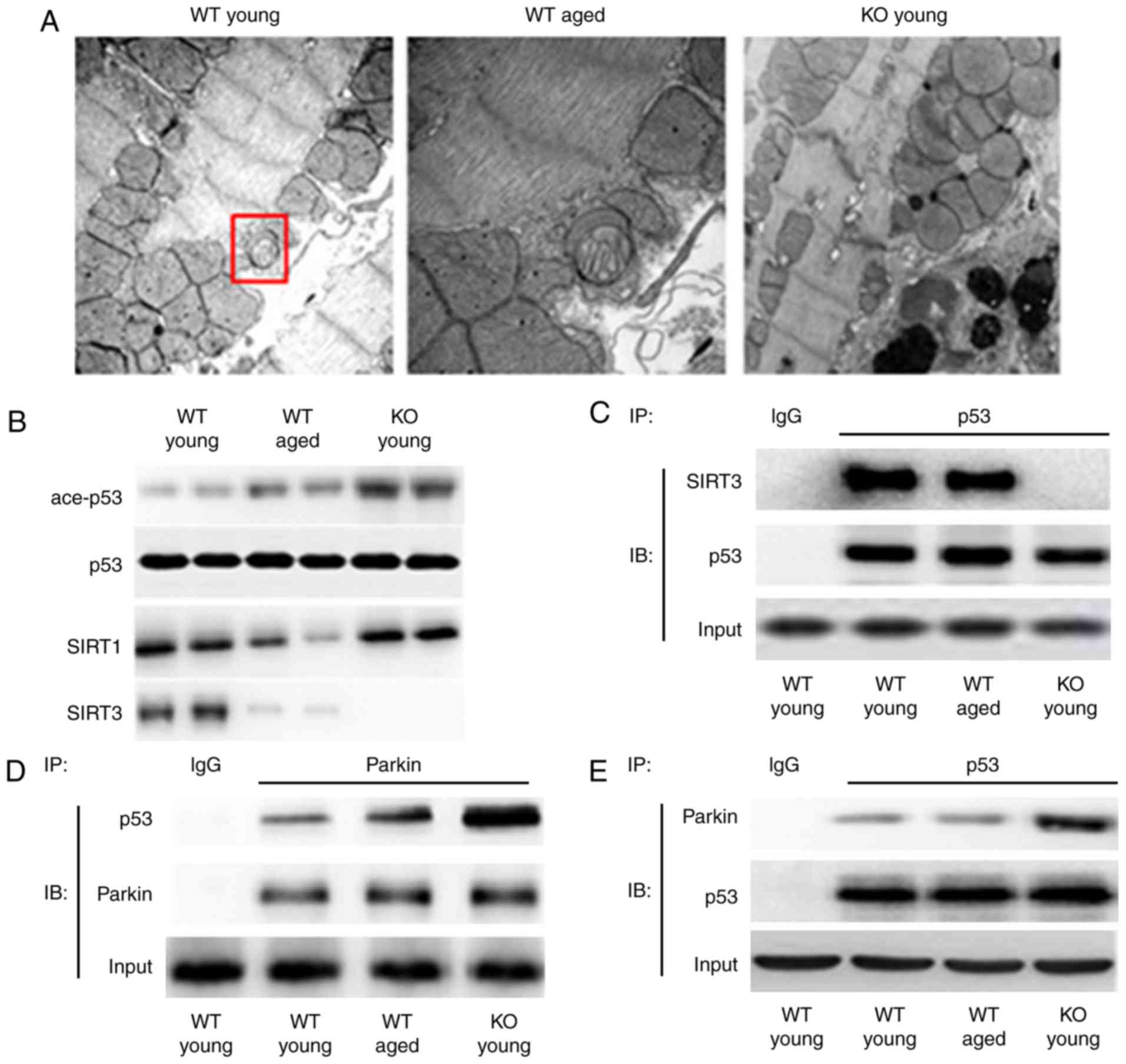 | Figure 6The myocardium of SIRT3−/−
mice displays features of enhanced oxidative stress and energy
metabolism dysfunction. (A) Representative images from transmission
electron microscopy analysis for autophagic vacuoles in the WT
young, WT aged, and SIRT−/− (KO) myocardia. Original
magnification, ×8,000. The red box demonstrated a representative
autophagic vacuole. (B) Detection of acetyl/total p53 by western
blot analysis. SIRT1 herein served as a control to demonstrate the
specificity of SIRT3 KO. (C) The interaction between p53 and SIRT3
in WT young, WT aged, and KO young myocardial mitochondria was
determined by co-IP assay. p53 primary antibody was incubated with
the mitochondrial proteins for IP; IgG was the negative control.
Then a SIRT3 or p53 secondary antibody was applied in the following
IB experiment. (D and E) The interaction between p53 and Parkin in
WT young, WT aged and KO young myocardial mitochondria was
determined by co-IP assay. n=8 per group. SIRT3, sirtuin 3; WT,
wild-type; KO, knockout; p53, tumor protein p53; IP,
immunoprecipitation; Ig, immunoglobulin; IB, immunoblotting; ace,
acetylated. |
Several previous studies have suggested that p53 is
likely to be involved in aging and myocardial mitophagy (31,32). Therefore, in the present study
western blot analysis was used to detect the impact of aged SIRT3
and SIRT3 deficiency on the level and activity of p53. The results
demonstrated that aged SIRT3 raised the acetylation level of p53,
and SIRT3 deficiency further increased the acetylation level
(Fig. 6B). The co-IP analysis
revealed that in aged myocardium the interaction between SIRT3 and
p53 was reduced, and in SIRT3−/− myocardium the
interaction was hardly detectable (Fig. 6C). These results indicated that
deacetylation of p53 was regulated by SIRT3 and the deacetylating
function on p53 was attenuated by aging or SIRT3 deficiency.
Parkin is a key factor in mitophagy, and its
translocation into mitochondria is essential for inducing mitophagy
(33,34). Parkin can be bound by activated
p53 (acetylated), which then blocks its translocation (35). The co-IP analysis for p53 and
SIRT3 revealed that p53-Parkin binding increased in aged myocardium
and even further elevated in the SIRT3−/− myocardium
(Fig. 6D and E). These results
indicated that p53 was activated and bound to Parkin, which was
likely an important reason for impaired Parkin-mediated mitophagy
in the aged and SIRT3−/− myocardia.
SIRT3 is required to maintain
Parkin-mediated mitophagy
To further validate that SIRT3 deficiency was
involved in p53 re-acetylation, binding to Parkin and inhibition of
Parkin-mediated mitophagy, a specific mitophagy agonist CCCP was
intraperitoneally injected into young, aged and SIRT3−/−
mice (5 mg CCCP/kg body weight). Twelve hours following injection,
changes in the levels of the myocardial mitophagy markers Parkin
and its translocation blocker p62 were detected. Western blot
analyses demonstrated that the levels of Parkin and p62 did not
differ among young, aged, and SIRT3−/− mice in cytosol
or mitochondria compared with controls (Fig. 7A). CCCP administration resulted in
a significant increase in the mitochondrial levels of Parkin and
p62 in the young because it induced their translocation into
mitochondria. Notably, the translocation was quite marked in the
young mice, but not so obvious in the aged mice and even less in
SIRT3−/− mice (Fig.
7A). These data suggested that CCCP-induced Parkin/p62
translocation was reduced in aged and SIRT3−/−
hearts.
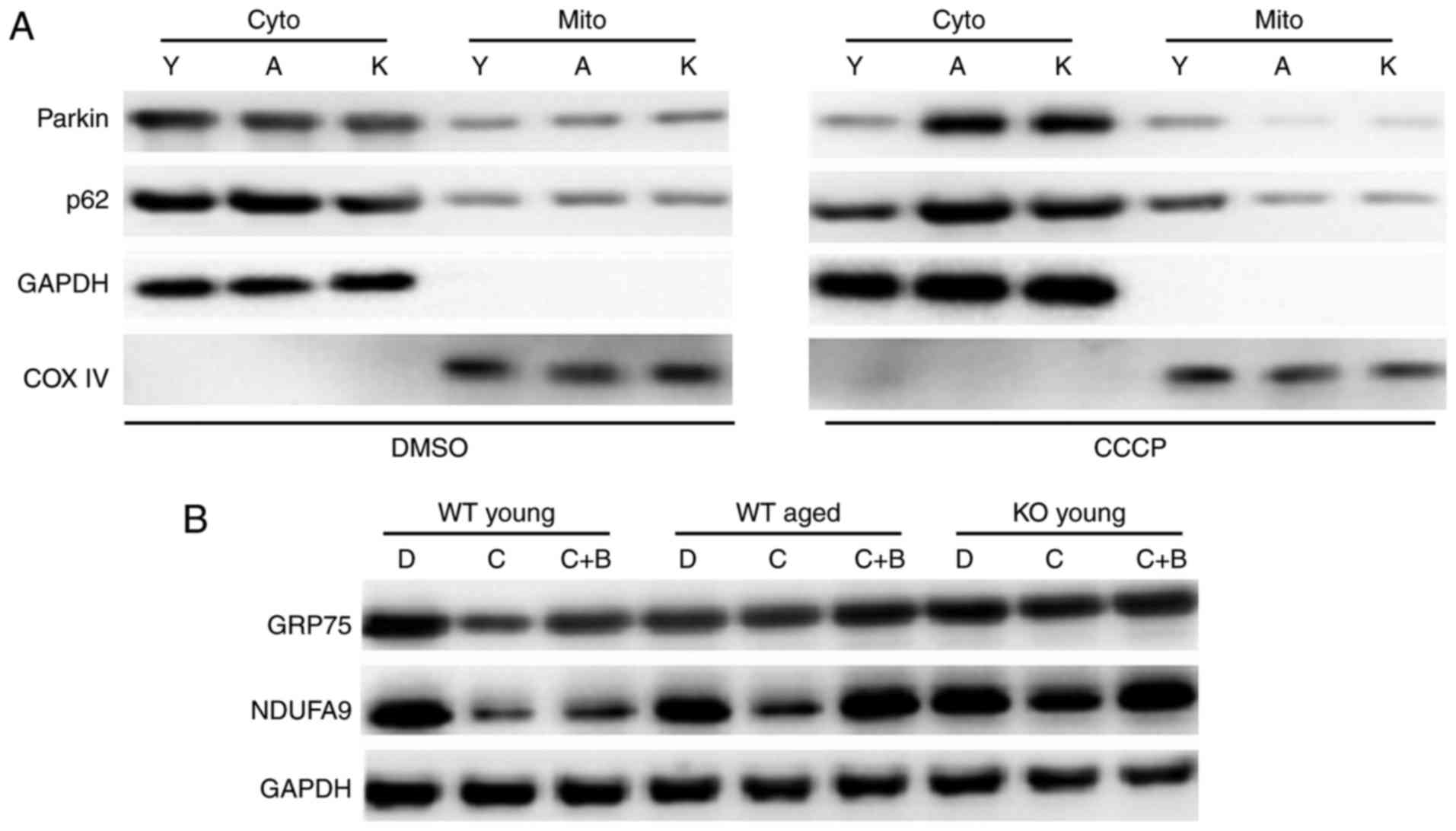 | Figure 7SIRT3 is required to maintain Parkin
translocation and Parkin-mediated mitophagy. (A) Parkin
mitochondrial translocation was blocked in WT aged and
SIRT3−/− (KO) myocardia when mitophagy was stimulated by
the agonist CCCP. CCCP was injected (5 mg/kg body weight)
intraperitoneally into WT young, WT aged, and KO mice (n=5). As a
control, an equivalent dose of DMSO was injected into the
littermates of the CCCP injection group (n=4). Following 12 h, the
heart tissues were sampled, and the levels of Parkin and of its
translocation blocker p62 were detected by western blot analysis.
(B) CCCP-induced mitochondrial clearance was markedly weakened in
WT aged and KO myocardia. CCCP, alone or with the autophagy
antagonist bafilomycin-A1, was injected intraperitoneally into the
WT young, WT aged, and KO mice (n=8 per group). As a control, an
equivalent dose of DMSO was injected into the littermates of the
CCCP injection group (n=8). Following 12 h, the heart tissues were
sampled, and the levels of mitochondrial chaperone GRP75 and
mitochondrial ubiquinone NDUFA9 were detected by western blot
analysis. n=8 per group. SIRT3, sirtuin 3; WT, wild-type; KO,
knockout; DMSO, dimethyl sulfoxide; p62, sequestosome 1; GRP75,
glucose-regulated protein 75; NDUFA9, NADH dehydrogenase 1 alpha
subcomplex 9; COX IV, cytochrome c oxidase complex IV; cyto,
cytoplasmic; mito, mitochondrial; Y, WT young group; A, WT aged
group; K, SIRT3 knockout group; D, DMSO-treated group; C,
CCCP-treated group; C+B, CCCP+ bafilomycin-A1-treated group. |
Finally, autophagy antagonist bafilomycin-A1
(Baf-A1), CCCP, or both were applied to young, aged, and
SIRT3−/− mice. In the young cardiomyocytes, levels of
mitochondrial chaperone GRP75 and mitochondrial ubiquinone NDUFA9,
typical markers of mitochondrial clearance, were significantly
downregulated following CCCP administration, but could be rescued
by Baf-A1 administration (Fig.
7B). CCCP-induced mitochondrial clearance was markedly
attenuated in aged and SIRT3−/− hearts compared with the
young (Fig. 7B). Thus
mitochondria were impaired by blockage of autophagy in aged and
SIRT3−/− hearts, and SIRT3 decline or deficiency was
responsible for impaired Parkin-mediated mitophagy in senescent or
SIRT3−/− hearts.
Discussion
Cardiac aging is characterized by hypertrophy and
fibrosis of the heart, which results in increased susceptibility to
stress, such as ischemia and hemodynamic overload. It has been
demonstrated that the incidence of heart failure dramatically
increases with aging (36).
However, the exact causes of heart aging remain unknown. The heart
is an organ in which mitochondria are enriched in high density in
order to meet the high energy-demand and to maintain redox stress
homeostasis (37). Therefore,
mitochondrial dysfunction caused by oxidative stress is believed to
be an important cause of cardiac aging and failure. Several lines
of evidence have revealed that SIRT3 is involved in cardiac aging
related to oxidative stress and mitochondrial dysfunction. However,
the role and mechanisms of SIRT3 in modulating heart aging and
mitochondrial function require further study. In the present study,
SIRT3 deficiency was demonstrated to be associated with cardiac
aging due to, at least in part, its inhibition of mitophagy and its
effect on mitochondrial functions related to oxidative stress and
energy metabolism.
SIRT3, an aging-related deacetylase, has been
previously reported to protect cardiomyocytes from stress-mediated
cell death and improve mitochondrial respiration (38,39). In fact, its roles in many
important biological processes are mostly based on its
deacetylating function. In the present study, the role of SIRT3 in
aged myocardium was investigated. Acetylation of p53 and other
mitochondrial proteins was demonstrated to be upregulated in aged
and SIRT3−/− myocardia. Strikingly, Kim et al
(40) also demonstrated that
acetyl-lysine was present in >20% of mitochondrial proteins,
including many oxidative stress-related proteins, longevity
regulators and metabolism enzymes. SIRT3−/− mice are
well-suited for experiments to investigate the impact of SIRT3
deficiency on the biological characteristics of the myocardium.
Several reports have revealed that SIRT3 knockdown/knockout
exacerbates ischemia-reperfusion injury, myocardium failure, and
nutrient- or exercise-induced stress in the heart (20,41,42). In the present study, it was
demonstrated that protein deacetylation disorder, enhanced
oxidative stress and energy metabolism dysfunction, which occurred
in aged hearts, were also present in SIRT3−/− hearts.
These data indicated that SIRT3 deficiency likely had a strong
impact on the regulation of myocardial mitochondrial function under
the aging condition.
In the present study, SIRT3 deficiency was confirmed
to be associated with the elevated oxidative stress and disturbed
energy metabolism, which were recognized as typical features of
cardiac aging. Mitochondria are involved in a range of other
processes besides energy supply, such as signaling, cellular ion
homeostasis, oxidative stress, apoptotic and necrotic cell death.
In the present study, the impacts of SIRT3 on oxidative stress and
energy metabolism in mitochondria were explored. Mitochondrial
MnSOD is very important in resisting mitochondrial ROS-induced
oxidative stress (43), which is
an important part of radical theory. In aged myocardial
mitochondria, the level and activity of MnSOD were sharply reduced
by SIRT3. The mechanism for SIRT3 influencing ROS-induced oxidative
stress was previously demonstrated to involve SIRT3 deacetylating
FoxO3a and then upregulating its target genes, MnSOD and
CAT (23,24). Moderate intensity AIT is often
recommended for weight loss and prevention of chronic disorders,
including aging-related diseases (44–46). In the present study, it was
demonstrated that MnSOD suppression could be attenuated by aging
but partially maintained by AIT. Similar results were obtained for
PGC-1α, the key regulator of energy metabolism. Hence, protein
deacetylation disorder, enhanced oxidative stress and energy
metabolism dysfunction caused by aging could be alleviated, but not
eliminated, by physical training. Additionally, an important role
of SIRT3 in these processes may be speculated.
Furthermore, SIRT3 deficiency suppressed p53/Parkin-
mediated mitophagy, in turn leading to the inhibition of damaged
mitochondrial clearance. Mitophagy is vital to maintaining
mitochondrial homeostasis, and its inhibition can lead to
mitochondrial dysfunction and abnormal cell functions. Parkin, an
E3 ligase originally discovered as mutated in monogenic forms of
Parkinson's disease, was recently found to translocate specifically
to uncoupled mitochondria and to induce autophagy of damaged
mitochondria (47). Present in
the cytosol, Parkin can be targeted by activated cytosolic p53,
which disrupts its translocation to damaged mitochondria and
subsequent mitophagy (35). In
the present study, Parkin-mediated mitophagy was significantly
inhibited in aged or SIRT3 deficient animals. It is likely that,
compared with normal conditions, SIRT3 in aged or
SIRT3−/− myocardium cannot maintain p53 deacetylation,
leading to higher amounts of activated p53, more binding with
Parkin, and consequent inhibition of Parkin translocation. The
results from the co-IP analysis demonstrated that the interaction
between SIRT3 and p53 was reduced in aged or SIRT3−/−
myocardium, which supported the aforementioned hypothesis.
Overall, SIRT3 deficiency raised the acetylation
levels of mitochondrial proteins and disrupted mitochondrial
homeostasis. On one hand, proteins such as MnSOD and PGC-1α are
important to resist the redox stress and maintain normal
mitochondrial biogenesis; increased acetylation suppressed MnSOD
and PGC-1α activity and exacerbated mitochondrial dysfunction in
aged hearts. On the other hand, with a lack of SIRT3, p53-Parkin
binding was enhanced, the translocation of Parkin was blocked, and
Parkin-mediated mitophagy was inhibited; mitochondria could not
renew through mitophagy. Of note, the functions of mitochondria in
a range of biological processes, as well as a comprehensive
understanding of the role of SIRT3 in regulating mitochondria
dynamics, will require further studies in the future.
In conclusion, decreased SIRT3 could disrupt
mitochondrial homeostasis and increase the susceptibility of the
aged heart to cardiac injury. The present results suggest that
therapeutic activation of SIRT3 and improved mitochondrial function
may ameliorate the symptoms of cardiac aging.
Acknowledgments
Not applicable.
Notes
[1]
Funding
This study was supported by research grants from the
National Natural Science Foundation of China (81570252; 81500195;
81170184) and Military Foundation (CWS14J065). Availability of data
and materials. The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
[2] Availability
of data and materials
The analyzed data sets generated during the study
are available from the corresponding author upon reasonable
request.
[3] Authors'
contributions
YL and LQS and HM conceived and designed the study.
YL and YM drafted the manuscript. YM and LY conducted the
experiments. LZ, YX and YY analyzed and interpreted the data; LQS
and YMZ critical revised the manuscript for important intellectual
content. YL and HM final approval of the drafted manuscript. All
authors have read and approved the final manuscript.
[4] Ethics
approval and consent to participate
All animal handling and experimental procedures
described in the present study were approved by the Institutional
Animal Care and Use Committee of the Fourth Military Medical
University (Xi'an, China), and in compliance with the Guidelines
for the Care and Use of Laboratory Animals (27).
[5] Consent for
publication
Not applicable.
[6] Competing
interests
The authors declare that they have no competing
interests.
References
|
1
|
Niccoli T and Partridge L: Ageing as a
risk factor for disease. Current Biol. 22:R741–R752. 2012.
View Article : Google Scholar
|
|
2
|
Rattan SI: Rationale and methods of
discovering hormetins as drugs for healthy ageing. Expert Opin Drug
Discov. 7:439–448. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Schmitt K, Grimm A, Kazmierczak A,
Strosznajder JB, Götz J and Eckert A: Insights into mitochondrial
dysfunction: Aging, amyloid-β, and tau-A deleterious trio. Antioxid
Redox Signal. 16:1456–1466. 2012. View Article : Google Scholar
|
|
4
|
Biala AK, Dhingra R and Kirshenbaum LA:
Mitochondrial dynamics: Orchestrating the journey to advanced age.
J Mol Cell Cardiol. 83:37–43. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Nasrallah CM and Horvath TL: Mitochondrial
dynamics in the central regulation of metabolism. Nat Rev
Endocrinol. 10:650–658. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Bullon P, Newman HN and Battino M:
Obesity, diabetes mellitus, atherosclerosis and chronic
periodontitis: A shared pathology via oxidative stress and
mitochondrial dysfunction? Periodontol 2000. 64:139–153. 2014.
View Article : Google Scholar
|
|
7
|
Schiavi A and Ventura N: The interplay
between mitochondria and autophagy and its role in the aging
process. Expe Gerontol. 56:147–153. 2014. View Article : Google Scholar
|
|
8
|
Currais A: Ageing and inflammation-A
central role for mitochondria in brain health and disease. Ageing
Res Rev. 21:30–42. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Sorrentino G, Comel A, Mantovani F and Del
Sal G: Regulation of mitochondrial apoptosis by Pin1 in cancer and
neurodegeneration. Mitochondrion. 19:88–96. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Paradies G, Paradies V, Ruggiero FM and
Petrosillo G: Cardiolipin and mitochondrial function in health and
disease. Antioxid Redox Signal. 20:1925–1953. 2014. View Article : Google Scholar
|
|
11
|
Palikaras K and Tavernarakis N:
Mitochondrial homeostasis: The interplay between mitophagy and
mitochondrial biogenesis. Exp Gerontol. 56:182–188. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Chu CT, Bayır H and Kagan VE: LC3 binds
externalized cardiolipin on injured mitochondria to signal
mitophagy in neurons: Implications for Parkinson disease.
Autophagy. 10:376–378. 2014. View Article : Google Scholar
|
|
13
|
Youle RJ and Narendra DP: Mechanisms of
mitophagy. Nat Rev Mol Cell Biol. 12:9–14. 2011. View Article : Google Scholar
|
|
14
|
Thomas RL and Gustafsson AB: Mitochondrial
autophagy: An essential quality control mechanism for myocardial
homeostasis. Circ J. 77:2449–2454. 2013. View Article : Google Scholar
|
|
15
|
Moyzis AG, Sadoshima J and Gustafsson AB:
Mending a broken heart: The role of mitophagy in cardioprotection.
Am J Physiol Heart and Circ Physiol. 308:H183–H192. 2015.
View Article : Google Scholar
|
|
16
|
Houtkooper RH, Pirinen E and Auwerx J:
Sirtuins as regulators of metabolism and healthspan. Nat Rev Mol
Cell Biol. 13:225–238. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
17
|
Lee IH, Yun J and Finkel T: The emerging
links between sirtuins and autophagy. Methods Mol Biol.
1077:259–271. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zhang B, Cui S, Bai X, Zhuo L, Sun X, Hong
Q, Fu B, Wang J, Chen X and Cai G: SIRT3 overexpression antagonizes
high glucose accelerated cellular senescence in human diploid
fibroblasts via the SIRT3-FOXO1 signaling pathway. Age (Dordr).
35:2237–2253. 2013. View Article : Google Scholar
|
|
19
|
Ahn BH, Kim HS, Song S, Lee IH, Liu J,
Vassilopoulos A, Deng CX and Finkel T: A role for the mitochondrial
deacetylase Sirt3 in regulating energy homeostasis. Proc Nat Acad
Sci USA. 105:14447–14452. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Jing E, Emanuelli B, Hirschey MD, Boucher
J, Lee KY, Lombard D, Verdin EM and Kahn CR: Sirtuin-3 (Sirt3)
regulates skeletal muscle metabolism and insulin signaling via
altered mitochondrial oxidation and reactive oxygen species
production. Proc Nat Acad Sci. 108:14608–14613. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Haigis MC, Deng CX, Finley LW, Kim HS and
Gius D: SIRT3 is a mitochondrial tumor suppressor: A scientific
tale that connects aberrant cellular ROS, the Warburg effect, and
carcinogenesis. Cancer Res. 72:2468–2472. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Alhazzazi TY, Kamarajan P, Verdin E and
Kapila YL: SIRT3 and cancer: Tumor promoter or suppressor? Biochim
Biophys Acta. 1816:80–88. 2011.PubMed/NCBI
|
|
23
|
Aldakkak M, Stowe DF, Chen Q, Lesnefsky EJ
and Camara AK: Inhibited mitochondrial respiration by amobarbital
during cardiac ischaemia improves redox state and reduces matrix
Ca2+ overload and ROS release. Cardiovasc Res.
77:406–415. 2008.
|
|
24
|
Sundaresan NR, Gupta M, Kim G, Rajamohan
SB, Isbatan A and Gupta MP: Sirt3 blocks the cardiac hypertrophic
response by augmenting Foxo3a-dependent antioxidant defense
mechanisms in mice. J Clin Invest. 119:2758–2771. 2009.PubMed/NCBI
|
|
25
|
Chen CJ, Fu YC, Yu W and Wang W: SIRT3
protects cardiomyocytes from oxidative stress-mediated cell death
by activating NF-κB. Biochem Biophys Res Commun. 430:798–803. 2013.
View Article : Google Scholar
|
|
26
|
Sack MN: Emerging characterization of the
role of SIRT3-mediated mitochondrial protein deacetylation in the
heart. Am J Physiol-Heart Circ Physiol. 301:H2191–H2197. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Garber JC, Barbee RW, Bielitzki JT,
Clayton L, Donovan J, Hendriksen C, Kohn DF, Lipman NS, Locke PA,
Melcher J, et al: Guide for the care and use of laboratory animals.
Washington DC: The National Academic Press; pp. 2202011
|
|
28
|
Jiang HK, Miao Y, Wang YH, Zhao M, Feng
ZH, Yu XJ, Liu JK and Zang WJ: Aerobic interval training protects
against myocardial infarction-induced oxidative injury by enhancing
antioxidase system and mitochondrial biosynthesis. Clin Exp
Pharmacol Physiol. 41:192–201. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Merksamer PI, Liu Y, He W, Hirschey MD,
Chen D and Verdin E: The sirtuins, oxidative stress and aging: An
emerging link. Aging (Albany NY). 5:144–150. 2013. View Article : Google Scholar
|
|
30
|
Adamovich Y, Shlomai A, Tsvetkov P,
Umansky KB, Reuven N, Estall JL, Spiegelman BM and Shaul Y: The
protein level of PGC-1α, a key metabolic regulator, is controlled
by NADH-NQO1. Mol Cell Biol. 33:2603–2613. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Rufini A, Tucci P, Celardo I and Melino G:
Senescence and aging: The critical roles of p53. Oncogene.
32:5129–5143. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Hoshino A, Matoba S, Iwai-Kanai E,
Nakamura H, Kimata M, Nakaoka M, Katamura M, Okawa Y, Ariyoshi M,
Mita Y, et al: p53-TIGAR axis attenuates mitophagy to exacerbate
cardiac damage after ischemia. J Mole Cell Cardiol. 52:175–184.
2012. View Article : Google Scholar
|
|
33
|
Saito T and Sadoshima J: Molecular
mechanisms of mitochondrial Autophagy/mitophagy in the heart. Circ
Res. 116:1477–1490. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Herman AM and Moussa CE: The ubiquitin
ligase parkin modulates the execution of autophagy. Autophagy.
7:919–921. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Hoshino A, Mita Y, Okawa Y, Ariyoshi M,
Iwai-Kanai E, Ueyama T, Ikeda K, Ogata T and Matoba S: Cytosolic
p53 inhibits Parkin-mediated mitophagy and promotes mitochondrial
dysfunction in the mouse heart. Nat Commun. 4:23082013. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Sadoshima J: Sirt3 targets mPTP and
prevents aging in the heart. Aging. 3:12–13. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Sack MN: The role of SIRT3 in
mitochondrial homeostasis and cardiac adaptation to hypertrophy and
aging. J Mol Cell Cardiol. 52:520–525. 2012. View Article : Google Scholar :
|
|
38
|
Samant S, Pillai V, Wolfgeher D and Gupta
M: SIRT3 protects cardiomyocytes from Doxorubicin-induced
mitochondrial damage and Cell-death by Activating Opa1.
Circulation. 130(Suppl 2): A146642014.
|
|
39
|
Cheung KG, Cole LK, Xiang B, Chen K, Ma X,
Myal Y, Hatch GM, Tong Q and Dolinsky VW: Sirtuin-3 (SIRT3) protein
attenuates Doxorubicin-induced Oxidative Stress and improves
mitochondrial respiration in H9c2 cardiomyocytes. J Biol Chem.
290:10981–10993. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Kim SC, Sprung R, Chen Y, Xu Y, Ball H,
Pei J, Cheng T, Kho Y, Xiao H, Xiao L, et al: Substrate and
functional diversity of lysine acetylation revealed by a proteomics
survey. Mol Cell. 23:607–618. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Sol EM, Wagner SA, Weinert BT, Kumar A,
Kim HS, Deng CX and Choudhary C: Proteomic investigations of lysine
acetylation identify diverse substrates of mitochondrial
deacetylase sirt3. PLoS One. 7:e505452012. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Kawamura Y, Uchijima Y, Horike N, Tonami
K, Nishiyama K, Amano T, Asano T, Kurihara Y and Kurihara H: Sirt3
protects in vitro-fertilized mouse preimplantation embryos against
oxidative stress-induced p53-mediated developmental arrest. J Clin
Invest. 120:2817–2828. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
James AM, Collins Y, Logan A and Murphy
MP: Mitochondrial oxidative stress and the metabolic syndrome.
Trend Endocrinol Metab. 23:429–434. 2012. View Article : Google Scholar
|
|
44
|
Hosseinzadeh S, Dabidi Roshan V and
Pourasghar M: Effects of intermittent aerobic training on passive
avoidance test (shuttle box) and stress markers in the dorsal
hippocampus of wistar rats exposed to administration of
homocysteine. Iran J Psychiatry Behav Sci. 7:37–44. 2013.
|
|
45
|
Cardoso AM, Bagatini MD, Roth MA, Martins
CC, Rezer JF, Mello FF, Lopes LF, Morsch VM and Schetinger MR:
Acute effects of resistance exercise and intermittent intense
aerobic exercise on blood cell count and oxidative stress in
trained middle-aged women. Braz J Med Biol Res. 45:1172–1182. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Tanaka M, Sugawara M, Ogasawara Y, Izumi
T, Niki K and Kajiya F: Intermittent, moderate-intensity aerobic
exercise for only eight weeks reduces arterial stiffness:
Evaluation by measurement of stiffness parameter and
pressure-strain elastic modulus by use of ultrasonic echo tracking.
J Med Ultrason (2001). 40:119–124. 2013. View Article : Google Scholar
|
|
47
|
Narendra D, Tanaka A, Suen DF and Youle
RJ: Parkin is recruited selectively to impaired mitochondria and
promotes their autophagy. J Cell Biol. 183:795–803. 2008.
View Article : Google Scholar : PubMed/NCBI
|