Introduction
Chronic hypoxia is an important factor contributing
to the near-irreversibility of hypoxia-induced pulmonary
hypertension (HPH), a condition characterized by structural
remodeling of small pulmonary arteries (PAs). These important
morphological changes are characterized by an abnormal increase in
numbers of smooth muscle (SM)-like cells [cells expressing α-SM
actin (α-SMA)] in the PA wall (1,2),
which results in thickening of muscular and elastic vessels,
muscularization of distal vessels, and eventual development of
pulmonary hypertension. The small pulmonary arteries and veins may
be distinguished based on the relative abundance of SM cells or
SM-like cells in vessel walls. However, the origin of SM-like cells
has remained elusive.
Epithelial-mesenchymal transition (EMT) is a process
wherein epithelial cells undergo phenotypic changes and develop
into mesenchymal/SM-like cells (3). Similarly, endothelial cells may
acquire a mesenchymal or SM-like phenotype, a process referred to
as endothelial-mesenchymal transition (EndMT). Evidence of EndMT
has been reported in the context of cardiac and vascular
development, wound healing and various diseases, including
fibrosis, diabetic nephropathy, heterotopic ossification and cancer
(4–10). Transdifferentiated cells
co-express the endothelial marker CD31 and the SM-like cell type
marker α-SMA. Thus, EndMT is regarded as another important
mechanism for the generation of SM-like cells. The endothelial cell
appears to be one of the targets in hypoxia, and endothelial cell
dysfunction has a direct and indirect role in the process of
pulmonary vascular remodelling (11). Ranchoux et al (12) reported that EndMT may be a source
of α-SMA-expressing cells. The EndMT participates in vascular
remodeling as a characteristic of pulmonary hypertension; however,
the underlying mechanism has remained to be fully elucidated.
Hypoxia-inducible transcription factor-1α (HIF-1α)
is a critical regulator of the cellular response to hypoxia. HIF-1α
activity in the endothelium of PAs has been observed to
significantly increase in pulmonary hypertension (13). The HIF-1α-regulated gene Twist
(14,15) has an important role in EMT, cell
movement and proliferation (16–18). However, the role of HIF-1α/Twist
in EndMT has remained to be fully characterized. In the present
study, a significant upregulation of HIF-1α in hypoxic pulmonary
microvascular endothelial cells (PMVECs) was demonstrated, and
knockdown of HIF-1α and Twist1 effectively blocked hypoxia-induced
EndMT. It appeared that HIF-1α regulated the EndMT through binding
to the promoter of the Twist1 gene and subsequently activating
Twist1 transcription and expression.
Materials and methods
Animal model
A total of 36 male Sprague Dawley rats (age, 6–8
weeks; weight, 200–250 g) were obtained from the Fourth Military
Medical University. All experimental procedures were approved by
the Animal Use and Care Committee for Research and Education of The
Fourth Military Medical University (Xi'an, China). Control rats
were housed under a 12-h light/dark cycle at room temperature
(humidity: 50–60%) and were provided water and a standard
laboratory diet ad libitum. Rats were housed for 28 days in
a chamber containing 10% oxygen for exposure to continuous
hypobaric hypoxia. A gradual decrease in oxygen concentration was
employed in order to acclimatize the rats to the hypoxic conditions
(19). Over 30 min, the pressure
was slowly increased at the beginning of hypoxia by 0.5 atm, and
the oxygen concentration was reduced to 10%. The gradual decrease
in the oxygen concentration had no observable negative effect on
the rats. After 4 weeks, the HPH models were successfully
replicated. Rats in the normoxic group (control group) were housed
continuously in room air (n=6 per group). At the end of hypoxia
exposure, the right ventricular systolic pressure (RVSP) and the
ratio of ventricular weight [right ventricle/(left
ventricle+septum), denoted as RV/(LV+S)] were measured. Increases
in RVSP and RV/[LV+S] were considered as being indicative of
HPH.
Cell culture and chemical reagents
The lung was isolated from healthy adult rats under
sterile conditions. After washing repeatedly with sterile D-Hank's
buffer, tissues at the edge and surface of the lung (thickness, ~1
mm) were removed, dissected into small pieces and cultured in M200
medium (Gibco; Thermo Fisher Scientific, Inc., Waltham, MA, USA),
supplemented with a growth factor cocktail [fibroblast growth
factor, heparin, hydrocortisone and epidermal growth factor (Gibco;
Thermo Fisher Scientific, Inc.)] and 20% fetal bovine serum FBS
(HyClone; GE Healthcare, Little Chalfont, UK). Primary cultures
used were confirmed to be uncontaminated with SM cells by
immunostaining for α-SMA and CD31 or VIII factor. In the normoxic
group, PMVECs were cultured at 37°C in a humidified atmosphere of
5% CO2 in air. In the hypoxic groups, PMVECs were
incubated in a hypoxic chamber containing 1% O2, 5%
CO2 and 94% N2 for 7 days, and SM-like cells
were then obtained. In another experiment, PMVECs were also
stimulated with transforming growth factor (TGF)-β (10 ng/ml) in
normoxic conditions for 7 days. Antibodies against α-SMA (MA1-744)
were obtained from Thermo Fischer Scientific, Inc. (Waltham, MA,
USA). Monoclonal antibodies against CD31 (ab64543) and Twist1
(ab50581) were purchased from Abcam (Cambridge, MA, USA). collagen
(Col) 1A1 (sc-293182) and Col3A1 (sc-271249) were purchased from
Santa Cruz Biotechnology, Inc. (Dallas, TX, USA). Anti-HIF-1α
monoclonal antibody (MAB5382) was purchased from EMD Millipore
(Billerica, MA, USA). Secondary antibodies (anti-rabbit or
anti-mouse IgG antibody conjugated to horseradish peroxidase; 7074
and 7076; 1:2,000) were purchased from Cell Signaling Technology
(Danvers, MA, USA). Recombinant human TGF-β was purchased from
R&D Systems (Minneapolis, MN, USA).
Immunofluorescence
Frozen tissue sections or cultured cells grown on
coverslips were washed with PBS and fixed in 1% paraformaldehyde.
Cells were subsequently permeabilized with 0.2% Triton X-100 in PBS
at room temperature for 5 min. The cells and tissue sections were
blocked by 2% goat serum (OriGene Technologies, Beijing, China) at
37°C for 1 h. Then they were incubated with primary antibodies
(CD31, 1:500; α-SMA, 1:1,000) overnight at 4°C. Subsequently,
samples were incubated with secondary antibody (Alexa
Fluor® 594 donkey anti-mouse IgG; R37115; 1:500;
Molecular Probes; Thermo Fischer Scientific, Inc.) for 2 h at room
temperature. Fluorescence was examined by confocal laser scanning
microscopy.
Immunohistochemistry
Sagittal sections of the right lung were placed in
4% paraformaldehyde and processed for paraffin embedding. Sections
(5 μm) were prepared and mounted on glass slides prior to
overnight incubation at 4°C with anti-Twist1 antibody (1:500).
Slides were washed and incubated with corresponding secondary
antibodies conjugated with alkaline phosphatase (Expose Rabbit
specific HRP/DAB detection IHC kit; ab80437; Abcam). Sections were
evaluated using an Olympus BX50 optical microscope (Olympus
Corporation, Tokyo, Japan) equipped with an image analysis program
(Image Pro Plus version 6.0; Media Cybernetics, Inc., Rockville,
MD, USA).
Electron microscopy
Transmission electron microscopy (TEM) analysis was
performed using glutaraldehyde-fixed PMVECs embedded in resin (Epon
812 epoxy resin). The clean slide was placed in 0.25% Formvar
solution and then placed vertically on filter paper to air dry.
Then the slide was exposed vertically to the surface of the water.
The supporting membrane on the surface of the glass slide was
removed and the 75-mesh copper grids placed on the membrane,
removed from water and allowed to dry. Thin sections (50–80 nm)
were collected on the formvar-coated copper grids and stained with
3% uranyl acetate and lead citrate solutions for 20–30 min
respectively at room temperature. Following air drying at room
temperature, the sections were examined under an electron
microscope (JEM1200EX II; JEOL, Tokyo, Japan); images were
captured.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was extracted from lung tissues and PMVECs
with TRIzol (Invitrogen; Thermo Fisher Scientific, Inc.) according
to the manufacturer's instructions. Briefly, template RNA (1
μg/μl) was added to the individual tubes containing
the master mix, using the Omniscript RT kit (Qiagen, Inc.). These
were centrifuged briefly to collect residual liquid from the walls
of the tubes, which were then incubated for 2 h at 37°C. Then 2
μl RNase A (10 mg/ml) was added and the reaction mix
incubated for 10 min at 65°C, 5 min at 93°C and then cooled
immediately on ice. RT-qPCR was performed with a MyiQ (Bio-Rad
Laboratories, Inc., Hercules, CA, USA) using SYBR-Green I dye
(Applied Biosystems; Thermo Fisher Scientific, Inc.). Primer pairs
used in the present study are listed in Table I. Each PCR was performed in a 50
μl mixture containing 1 μl cDNA, 5 μl 10X
Qiagen PCR buffer (Qiagen, Inc.), 10 μl 5X Q-Solution
(Qiagen, Inc.), 1 μl each of deoxynucleotide triphosphate
mix (10 mM), 0.1 μM of each sense and antisense primer and
0.5 μl Taq DNA polymerase (Qiagen, Inc.), by using the Taq
DNA Polymerase kit (Qiagen, Inc.). The amplification reaction
consisted of initial denaturation at 95°C for 2 min, followed by
three-step cycling that consisted of denaturation at 95°C for 10
sec, annealing at 60°C for 30 sec and extension at 70°C for 30 sec
for 30 cycles. The comparative threshold cycle (Cq) method was
employed for quantification of transcripts (20).
 | Table IPrimer sequences for real-time
polymerase chain reaction. |
Table I
Primer sequences for real-time
polymerase chain reaction.
| Gene | Forward
(5′-3′) | Reverse
(5′-3′) |
|---|
| α-SMA |
TTGTGGATCAGCGCCTTCAGTT |
GACAGGCCAGGGCTAGAAGG |
| vWF |
CCAACAGCCAGTCTCCCGTT |
TGACGTAGGAGCAGTTGCCG |
| CD31 |
TTCCGAGGGTGGCTTGAGTG |
GAGGGAAGGCAATGGGGGTT |
| Vimentin |
TGCAGTCACTCACCTGCGAA |
CATTTCACGCATCTGGCGCT |
| Col1A1 |
ATCACCAGACGCAGAAGTCATA |
ACCAGGAGGACCAGGAAGTC |
| Col3A1 |
GGAACAACTGATGGTGCTACTG CC |
AAAATAAGAGGGGTGAAG |
| β-actin |
ATCATGTTTGAGACCTTCAACA C |
ATCTCTTGCTCGAAGTCCA |
Western blot assay
Lung tissues and cultured PMVECs were placed in
lysis buffer (50 mM NaCl, 50 mM NaF, 50 mM sodium pyrophosphate, 5
mM EDTA, 5 mM EGTA, 2 mM Na3VO4, 0.5 mM
phenylmethylsulfonyl fluoride and 10 mM
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid at pH 7.4,
together with 0.1% Triton X-100, 10 mg/l leupeptin and 5%
aprotinin) respectively. Protein concentrations were determined
with the bicinchoninic acid protein assay. Equivalent amounts of
protein (50 μg) were subjected to 10% SDS-PAGE and
transferred onto a nitrocellulose membrane (Bio-Rad Laboratories).
Membranes were blocked overnight at 4°C with PBS containing 5%
non-fat dry milk and 0.1% Tween-20. Following blocking, membranes
were incubated overnight at 4°C with primary antibodies, including
anti-HIF-1α (1:500), anti-CD31 (1:500), anti-Twist1 (1:500),
anti-α-SMA (1:1,000), anti-vimentin (ab92547; 1:200) and
anti-β-actin (A2228; 1:4,000; Sigma-Aldrich; Merck KGaA). Proteins
were incubated with secondary antibodies (anti-rabbit or anti-mouse
IgG antibody; Cat. 7074 and Cat. 7076; 1:2,000; Cell Signaling
Technology) for 1 h at 37°C in PBS containing 1% non-fat dry milk
and 0.1% Tween-20. The chemiluminescence signal was detected using
enhanced chemiluminescent substrate (SuperSignal™ West Femto Trial
Kit, Thermo Fisher Scientific, Inc.).
Cell transfection
Primary PMVECs were transduced with lentiviral
vector containing small interfering (si)RNA targeting either rat
HIF-1α or Twist1 (Genechemgene, Shanghai, China). For transduction,
cells were seeded in 6-well plates at a density of 5×105
cells per well. Virus particles were added at a multiplicity of
infection of 40, followed by incubation for 36 h. Cells were then
washed and incubated at 37°C for 60 h prior to PCR and western blot
analysis. The sequences of Twist1 siRNA and HIF-1α siRNA were
5′-GGCGGCCAGGTACATCGACTT-3′ and 5′-AACCAGTTGAATCTTCAGATA-3′,
respectively. The sequences of negative control was
TTCTCCGAACGTGTCACGT.
Cell proliferation
PMVECs or hypoxia-induced SM-like cells were seeded
on to 96-well plates and incubated for 24 and 48 h under normoxic
or hypoxic conditions. Subsequently, 10 μl MTT solution (5
mg/ml) was added into each well and following 4 h of incubation the
supernatant was gently removed by a pipette and discarded, and 150
μl dimethyl sulfoxide added. The optical density values were
detected at a wavelength of 490 nm using a spectrophotometer
(Bio-Tek Power Wave XS, Bio-Tek, Winooski, VT, USA).
Wound healing assay
PMVECs and SM-like cells were grown to confluence. A
linear wound was made by scraping the cell layer with a 0.5 ml
plastic Pasteur pipette. Following two washes with culture medium
to remove the detached cells and debris, the cells were exposed to
normoxic or hypoxic conditions for 24 and 48 h, and the size of the
wounds was measured.
Chromatin immunoprecipitation (ChIP)
The ChIP assay was performed using the EZ-Zyme™
Chromatin Prep Kit (EMD Millipore) according to the manufacturer's
instructions. In brief, cell lysates were incubated with 1
μg RNA polymerase II, IgG (1 μg/ml; Wanleibio Co.,
Ltd., Shenyang, China) or anti-HIF-1α antibody (1:500). The
immunoprecipitated DNA was amplified using Twist1 promoter-specific
primers (21). PCR generated a
196-bp product from the regulatory region of the Twist1 promoter
containing CCACGTGG. Primers for Twist1 were as follows: Forward
5′-TCGGATGAAAGCACAGTCG-3′ and reverse 5′-GCCACCGACTTCCTGAGA-3′.
Statistical analysis
All values are expressed as the mean ± standard
error of the mean. Statistical analysis was performed using
analysis of variance and multiple comparisons were made by Tukey's
method. Statistical analysis was processed by SPSS version 16.0
(SPSS, Inc., Chicago, IL, USA). Differences were considered to be
statistically significant at P<0.05.
Results
Hypoxia induces transdifferentiation of
endothelial cells into SM-like cells in small, but not in large
Pas
A rat model of chronic HPH was used to examine the
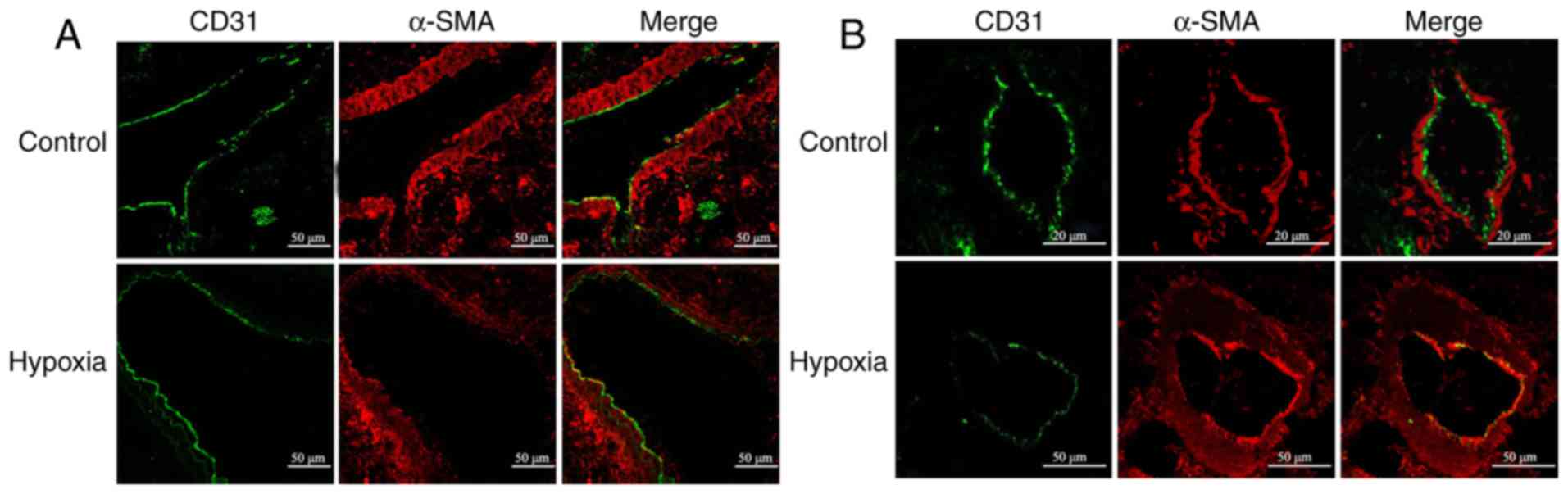
effect of hypoxia on EndMT in vivo. In large proximal PAs,
hypoxia did not appear to alter the expression of endothelial
marker CD31 and mesenchymal marker α-SMA (Fig. 1A). By contrast, a considerable
downregulation of CD31 along with significant upregulation of α-SMA
was observed in small distal PAs (Fig. 1B). In addition, hypoxia led to PA
remodeling, as evidenced by the appreciable intimal thickening
(Fig. 1B). Of note, CD31 was
co-localized with α-SMA (Fig.
1B), which suggested trans-differentiation of endothelial cells
into α-SMA-expressing mesenchymal-like cells (SM-like cells). These
results indicate that HPH is associated with remodeling of small
PAs and that hypoxia may induce EndMT in vivo.
Hypoxia induces transdifferentiation of
endothelial cells into SM-like cells in vitro
Next, the present study sought to determine the role
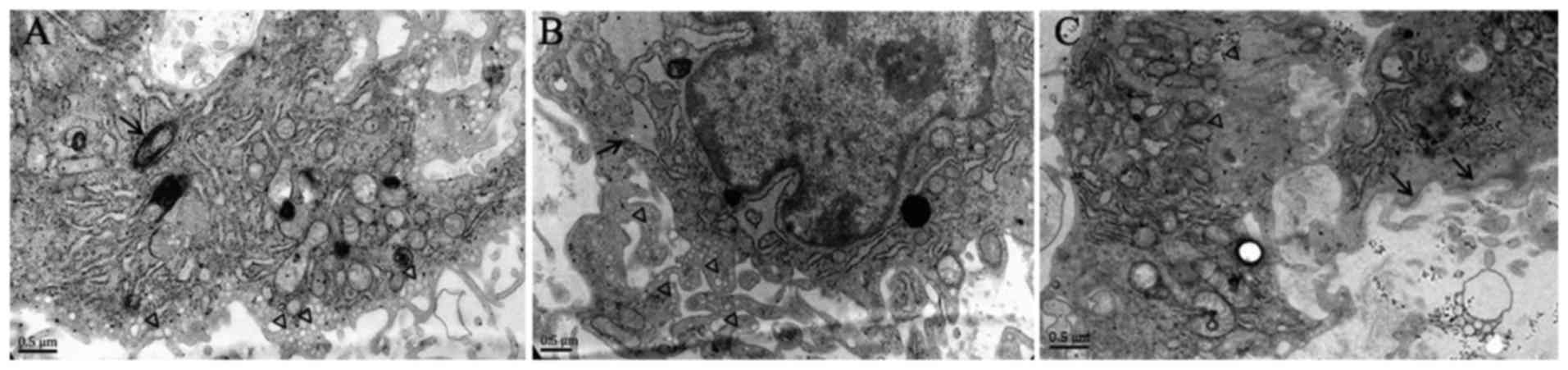
of hypoxia in EndMT in vitro. As presented in Fig. 2, Weibel-Palade bodies, as well as
abundant pinocytotic vesicles near the plasma membrane, were
observed in the cytoplasm of normal PMVECs, while α-SMA stress
fibers were undetectable, which is a typical phenotype of
endothelial cells. However, following hypoxic exposure, mixed cell
populations were observed in PMVECs. Certain cells exhibited
well-developed endoplasmic reticulum whose cisternae were expanded,
and increased protein secretion and abundant VEC-specific
pinocytotic vesicles were present in the cytoplasm (Fig. 2B), whereas other cells exhibited
SM-like features, including aggregation of α-SMA filaments in the
cytoplasm and presence of dense bodies abutting the cell membrane
(Fig. 2C). Pinocytotic vesicles
were not observed in these cells (Fig. 2C). These results suggest that
hypoxic PMVECs undergo a transition from endothelial cells to
SM-like cells.
To further confirm the role of hypoxia in EndMT
in vitro, endothelial and mesenchymal markers were examined
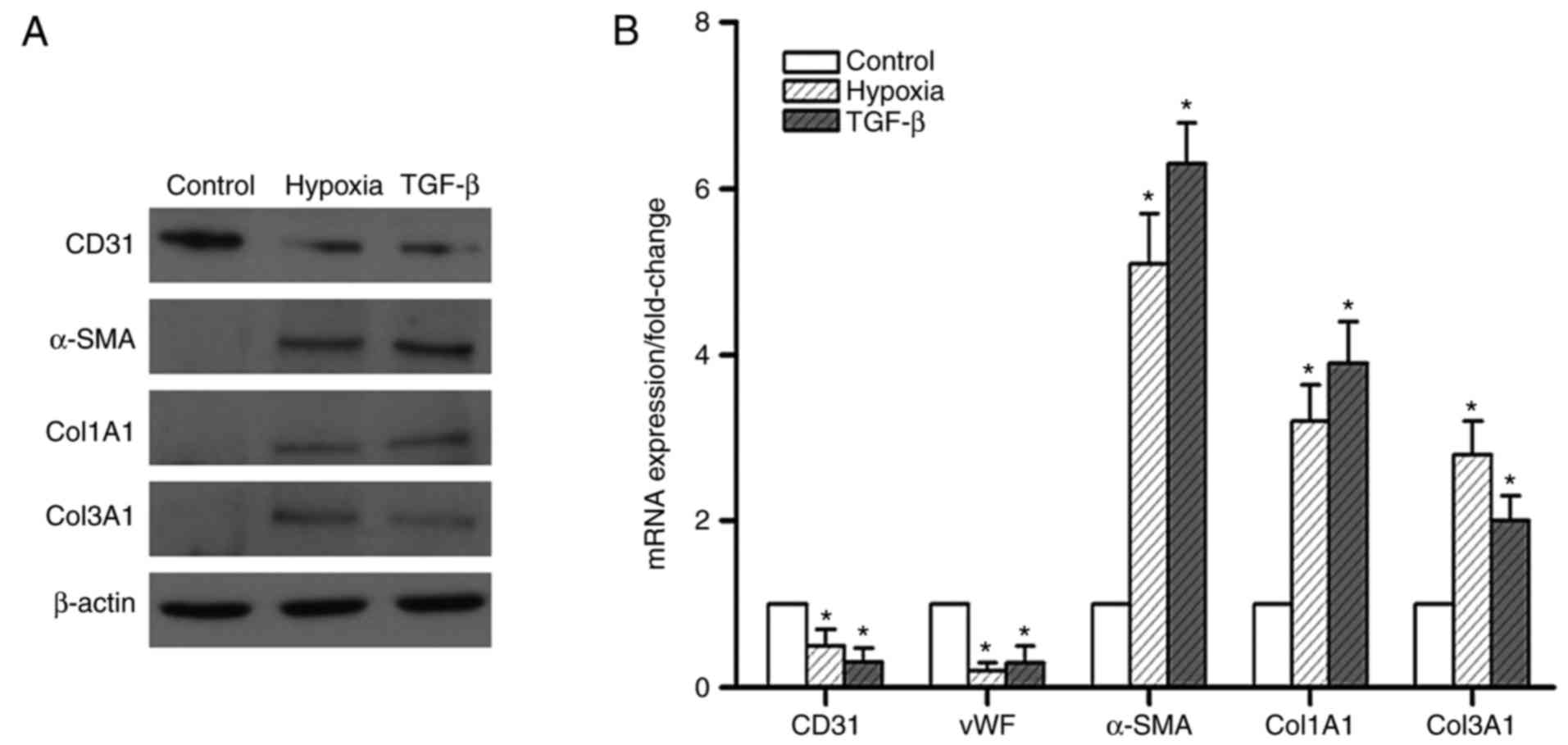
in hypoxic PMVECs. The results indicated that 7 days of hypoxia
exposure attenuated the protein expression of CD31, but markedly
enhanced the expression of α-SMA and the mesenchymal markers Col1A1
and Col3A1 (Fig. 3A). This effect
was comparable to that of TGF-β, a known potent inducer of EndMT.
The mRNA expression of these markers exhibited a similar trend
(Fig. 3B). These results suggest
that the effect of hypoxia in inducing EndMT was akin to that of
TGF-β.
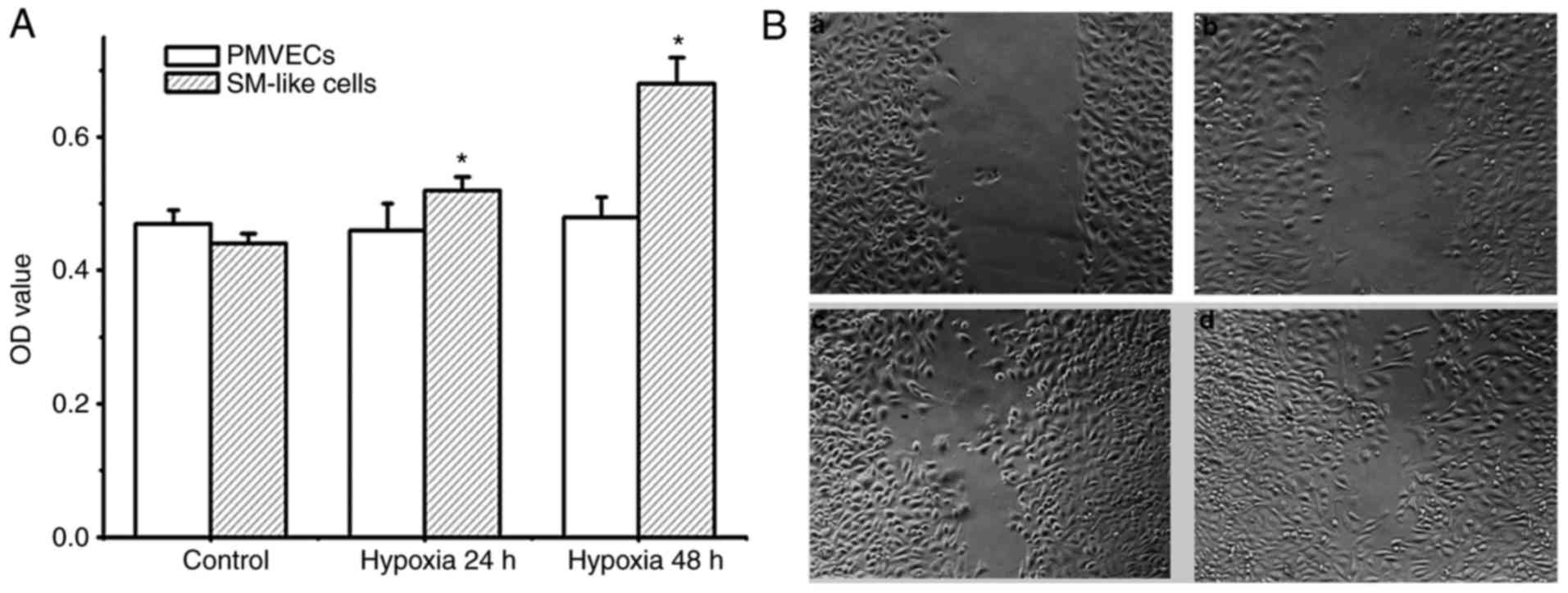
Increased cell proliferation and migration are
important features associated with EndMT. Therefore, the degree of
proliferation and migration of PMVECs and SM-like cells after EndMT
under hypoxic conditions was detected. As presented in Fig. 4A and B, PMVECs did not exhibit any
proliferation and migration potential under hypoxia over 48 h.
Conversely, SM-like cells exhibited time-dependent proliferation
and migration under hypoxia. As the SM-like cells were derived from
the PMVECs incubated in hypoxia over 7 days, these results
demonstrate that hypoxia induces EndMT, resulting in a phenotype
with increased cell proliferation and migration.
HIF-1α is essential for hypoxia-induced
EndMT
HIF-1α is known to be a master regulator of the
hypoxic response. Therefore, the present study examined the role of
HIF-1α in hypoxia-induced EndMT. The present results indicated that
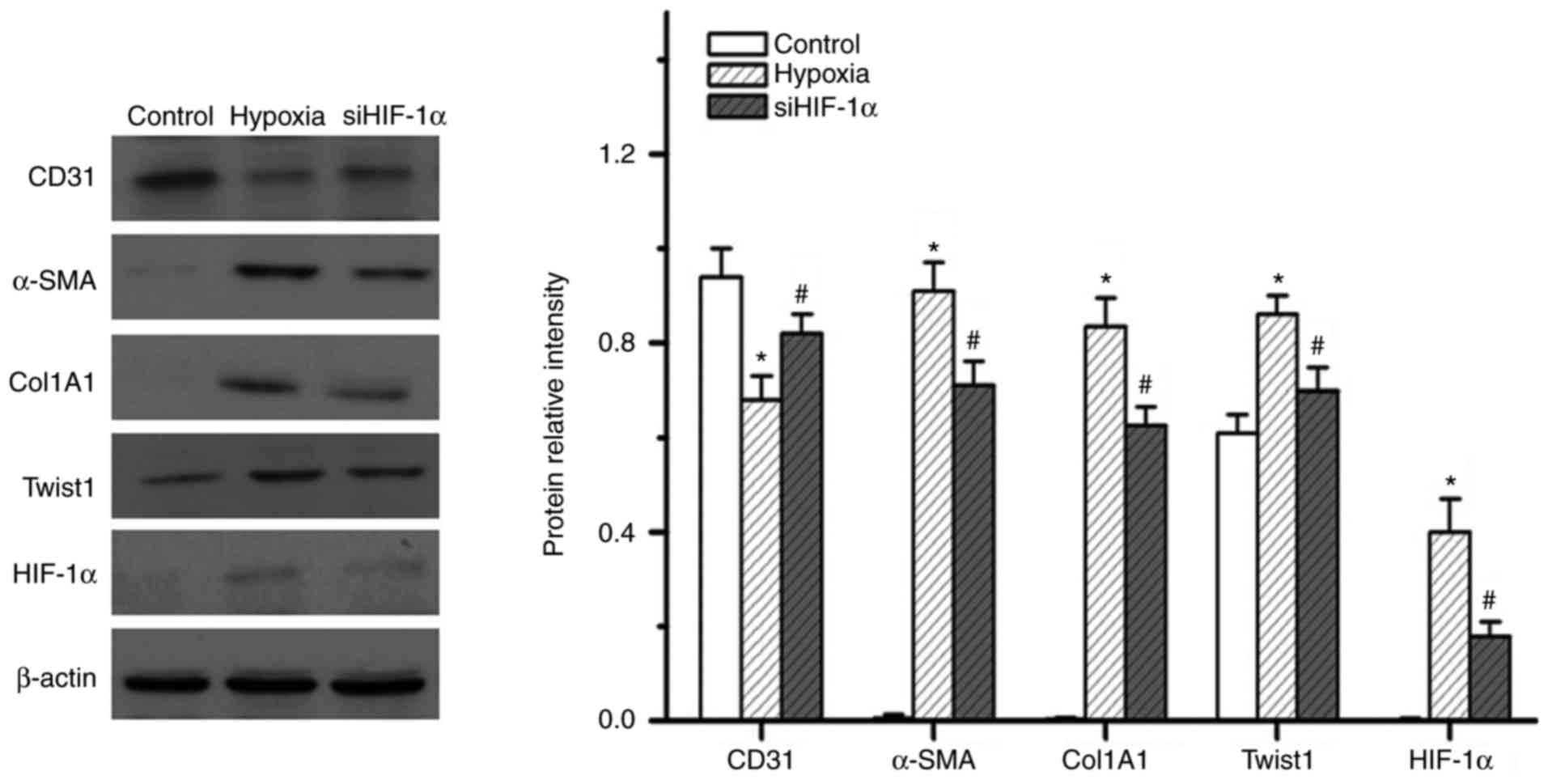
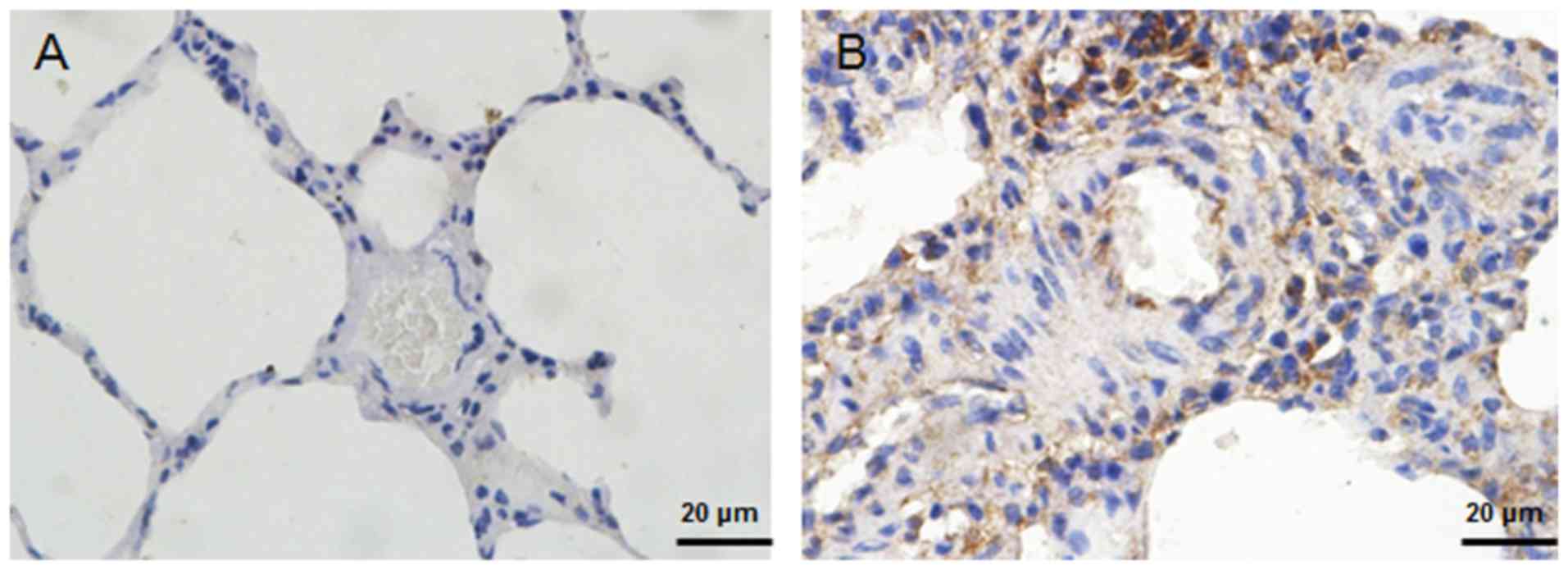
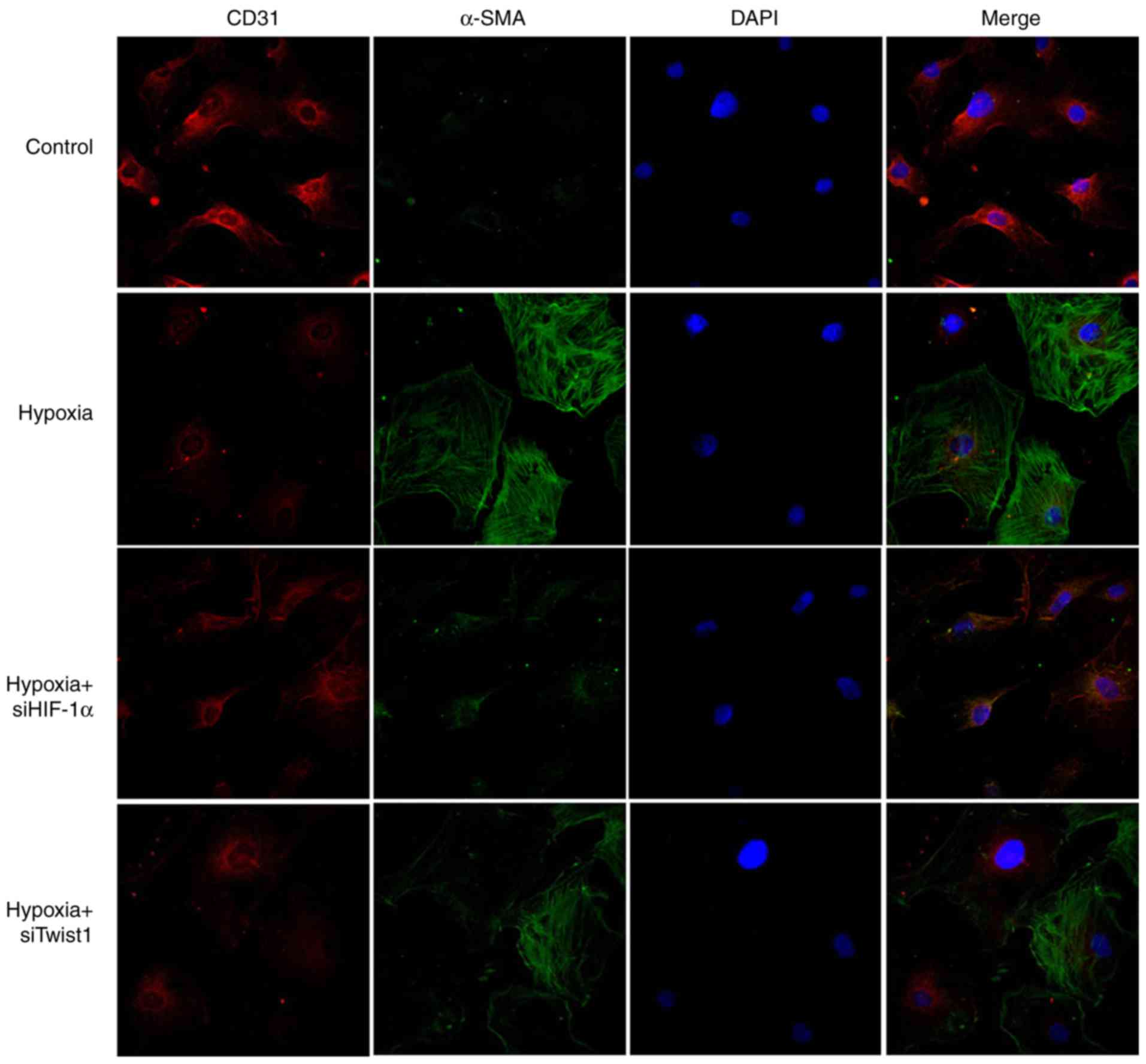
hypoxia induced the expression of HIF-1α (Fig. 5A). Upregulation of Twist1
expression was also detected in the small PA specimens from HPH
rats (Fig. 6). Of note, knockdown
of HIF-1α effectively inhibited 7 days of hypoxia exposure-induced
expression of Twist1, α-SMA and Col1A1, while reversing
hypoxia-induced suppression of CD31 expression (Fig. 5). These results indicate that
HIF-1α has an essential role in hypoxia-induced EndMT and that it
may act as an upstream regulator of Twist1.
Twist1 is important in hypoxia-induced
EndMT
Since hypoxia also upregulates Twist1, the present
study next sought to determine the function of Twist1 in
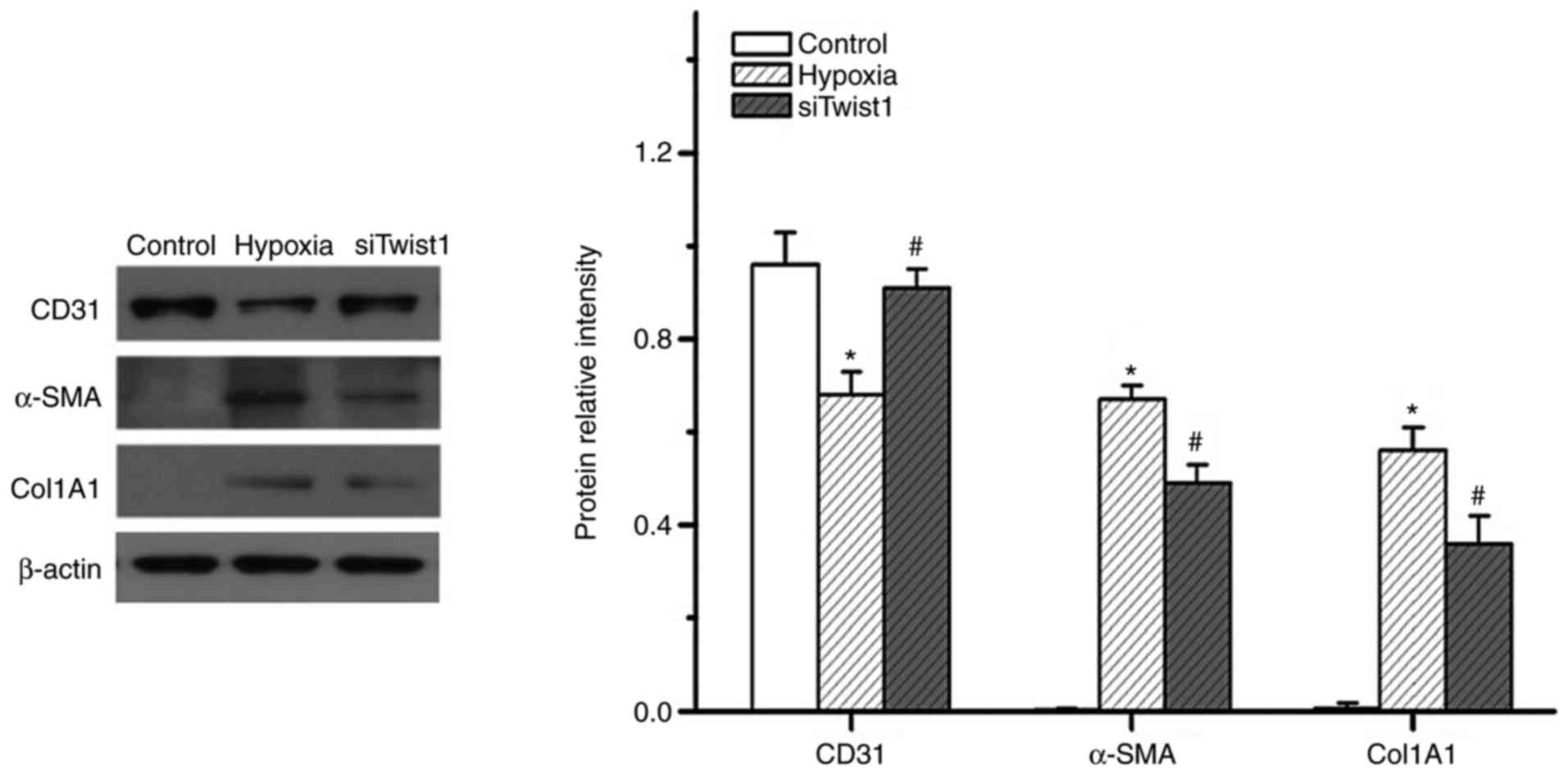
hypoxia-induced EndMT. Similar to HIF-1α, knockdown of Twist1 also
altered the expression of CD31, α-SMA and Col1A1 in hypoxic PMVECs
(Fig. 7). The knockdown
efficiency of the siRNAs was 80–85%. It appeared that knockdown of
HIF-1α exerted a more obvious effect on hypoxia-induced PMVEC
transdifferentiation to mesenchymal-like cells than that of Twist1.
This was evidenced in the obviously greater inhibition of
hypoxia-induced α-SMA expression by knockdown of HIF-1α, as
compared with the effect of Twist1 knockdown (Fig. 8). This observation is consistent
with the hypothesis that HIF-1α is an upstream regulator of Twist1.
These results suggest that Twist1 may be a mediator of
hypoxia/HIF-1α-induced EndMT in vitro.
HIF-1α binds with the promoter of the
Twist1 gene
The present study then investigated the mechanism by
which HIF-1α regulates Twist1.An 8-bp HIF-1α-binding sequence (CCA
CGT GG) located between 321 and 328 bp upstream of the first exon
of the Twist1 transcript was identified using JASPAR database
(http://jaspar2016.genereg.net/). A ChIP
assay was performed to determine whether HIF-1α indeed binds to
this site. As presented in Fig.
9, HIF-1α physically interacted with the binding sequence of
the Twist1 promoter in PMVECs, which suggests that HIF-1α regulates
Twist1 expression by binding to the Twist1 promoter, and that the
HIF-1α-Twist1 axis is an important pathway in hypoxia-induced
EndMT.
Discussion
The present study indicated that endothelial cells
in the pulmonary vasculature differentiate towards an SM phenotype
via EndM transdifferentiation upon exposure to hypoxia. The
endothelium may be a potential source of SM cells in the vascular
system, and the HIF-1α-Twist1 intracellular signaling pathway is
involved in this process.
Increased expression of α-SMA was observed in the
walls of small PAs in response to chronic exposure to hypoxia.
These SM-like cells not only lead to abnormal vasoconstriction, but
also have the ability to proliferate, migrate and produce
extracellular matrix, which contributes to structural remodeling
and results in pulmonary hypertension. These SM-like cells are
thought to be derived from the proliferation of resident vascular
SM cells, an SM-like subpopulation residing within the media
(22) or from adventitial
fibroblasts (23). Several
studies have indicated that circulating progenitor cells and
resident vascular progenitor cells may acquire an SM-like phenotype
at the site of vascular injury (19,24). However, endothelial cells were
observed to retain the capacity to transition into mesenchymal or
SM-like cells in vitro. With TGF-β treatment, isolated
endothelial cells from the bovine aorta were previously reported to
convert to spindle-shaped α-SMA-expressing cells (25). Zhu et al (26) reported that hypoxia induced
transdifferentiation of endothelial cells into SM-like cells and
that this process was regulated by myocardin. Similarly, Ranchoux
et al (12) reported that
EndMT may be a source of those α-SMA-expressing cells. EndMT
participates in vascular remodeling associated with pulmonary
hypertension. In the present study, cells with expression of α-SMA
were not observed in the intimal layer of large proximal PA
specimens from HPH rats, although certain α-SMA-expressing cells
were observed in distal PA specimens. Pulmonary hypertension is
characterized by cellular changes in the walls of PAs. These
changes include the appearance of SM-like cells in previously
non-muscularized vessels, and medial and adventitial thickening of
the muscular and elastic vessels. In addition, intimal changes have
been consistently observed in rat models of HPH; however, these
intimal changes are typically minimal, at least from a
morphological point of view (27). The magnitude of the changes
depends on the species studied for exposure to hypoxia. Since it is
hard to morphologically distinguish between small pulmonary
arteries and veins, small circular vessels that had a greater
expression of SM cells than pulmonary arteries were selected for
analysis in the present study. In vitro, it was observed
that hypoxia induced transdifferentiation of PMVECs into SM-like
cells. Therefore, endothelium appears to be a source of SM-like
cells.
Next, the present study explored whether HIF-1α may
modulate the transdifferentiation of endothelial cells. HIF-1α is a
transcriptional regulator that has an important role in the
cellular response to hypoxia. In addition, animals deficient in
HIF-1α have attenuated HPH (28).
HIF-1α transcriptionally controls a diverse number of genes,
including those involved in vascular remodeling. Higgins et
al (29) reported that
hypoxia promotes fibrogenesis through HIF-1α-mediated stimulation
of EMT. As an HIF-1α-regulated gene, Twist, a basic
helix-loop-helix transcription factor, was identified as a crucial
factor in the EMT (30,31). Ranchoux et al (12) reported that EndMT involves
neoexpression of Twist1. Sun et al (32) reported that Twist promoters
contain HIF-1α-binding sites, that HIF-1α induces Twist expression
in hypoxic tubular cells, and that this has a role in the EMT
during renal fibrogenesis. In the present study, when siRNA was
used to block the expression of HIF-1α, hypoxia-induced α-SMA
expression was markedly suppressed in PMVECs. Knockdown of HIF-1α
also resulted in a marked inhibition of Twist1 expression induced
by hypoxia. When Twist1 was knocked down by siRNA, the expression
of CD31, α-SMA and Col1A1 was also altered in hypoxic PMVECs, and
hypoxia-induced EndMT was effectively inhibited. This indicates
that Twist1 also mediates hypoxia-induced EndMT in PMVECs, and that
HIF-1α is an upstream regulator of Twist1. The process of EndMT is
therefore likely to be in part regulated by HIF-1α-Twist1. In
addition, the Twist1 gene promoter was identified to contain
HIF-1α-binding sites, and that HIF-1α induces Twist1 expression in
PMVECs, further confirming this interaction.
Although HPH is an important subtype of pulmonary
hypertension and has the features of pulmonary hypertension,
hypoxia is not defined as the major etiological factor. Thus, the
HPH model used in the present study may not entirely replicate the
actual pathogenesis of pulmonary hypertension. To better understand
the precise molecular mechanisms involved in the pathogenesis of
pulmonary hypertension, other factors, including inflammation,
should also be taken into consideration.
In conclusion, the present study provides evidence
of the EndMT process taking place in pulmonary vascular remodeling
under chronic hypoxic conditions. The HIF-1α-Twist1 pathway was
also suggested to be involved in EndMT. These observations may help
explain the appearance of SM-like cells in the intima of PAs in
HPH. Identification of EndMT in PAs and elucidation of associated
signaling pathways will confer novel insight into the pathogenesis
of pulmonary hypertension.
Acknowledgments
Not applicable.
References
|
1
|
Morrell NW, Adnot S, Archer SL, Dupuis J,
Jones PL, MacLean MR, McMurtry IF, Stenmark KR, Thistlethwaite PA,
Weissmann N, et al: Cellular and molecular basis of pulmonary
arterial hypertension. J Am Coll Cardiol. 54(1 Suppl): S20–S31.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Stenmark KR, Fagan KA and Frid MG:
Hypoxia-induced pulmonary vascular remodeling: Cellular and
molecular mechanisms. Circ Res. 99:675–691. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Hay ED: An overview of
epithelio-mesenchymal transformation. Acta Anat. 154:8–20. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Arciniegas E, Neves CY, Carrillo LM,
Zambrano EA and Ramirez R: Endothelial-mesenchymal transition
occurs during embryonic pulmonary artery development. Endothelium.
12:193–200. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Armstrong EJ and Bischoff J: Heart valve
development: Endothelial cell signaling and differentiation. Circ
Res. 95:459–470. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Hashimoto N, Phan SH, Imaizumi K, Matsuo
M, Nakashima H, Kawabe T, Shimokata K and Hasegawa Y:
Endothelial-mesenchymal transition in bleomycin-induced pulmonary
fibrosis. Am J Respir Cell Mol Biol. 43:161–172. 2010. View Article : Google Scholar
|
|
7
|
Li J, Qu X, Yao J, Caruana G, Ricardo SD,
Yamamoto Y, Yamamoto H and Bertram JF: Blockade of
endothelial-mesenchymal transition by a Smad3 inhibitor delays the
early development of streptozotocin-induced diabetic nephropathy.
Diabetes. 59:2612–2624. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Liebner S, Cattelino A, Gallini R, Rudini
N, Iurlaro M, Piccolo S and Dejana E: Beta-catenin is required for
endothelial-mesenchymal transformation during heart cushion
development in the mouse. J Cell Biol. 166:359–367. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Potenta S, Zeisberg E and Kalluri R: The
role of endothe-lial-to-mesenchymal transition in cancer
progression. Br J Cancer. 99:1375–1379. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zeisberg EM, Tarnavski O, Zeisberg M,
Dorfman AL, McMullen JR, Gustafsson E, Chandraker A, Yuan X, Pu WT,
Roberts AB, et al: Endothelial-to-mesenchymal transition
contributes to cardiac fibrosis. Nat Med. 13:952–961. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Humbert M, Montani D, Perros F, Dorfmüller
P, Adnot S and Eddahibi S: Endothelial cell dysfunction and cross
talk between endothelium and smooth muscle cells in pulmonary
arterial hypertension. Vascul Pharmacol. 49:113–118. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ranchoux B, Antigny F, Rucker-Martin C,
Hautefort A, Péchoux C, Bogaard HJ, Dorfmüller P, Remy S, Lecerf F,
Planté S, et al: Endothelial-to-mesenchymal transition in pulmonary
hypertension. Circulation. 131:1006–1018. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wojciak-Stothard B, Tsang LY and Haworth
SG: Rac and Rho play opposing roles in the regulation of
hypoxia/reoxygenation-induced permeability changes in pulmonary
artery endothelial cells. Am J Physiol Lung Cell Mol Physiol.
288:L749–L760. 2005. View Article : Google Scholar
|
|
14
|
Yang MH and Wu KJ: TWIST activation by
hypoxia inducible factor-1 (HIF-1): Implications in metastasis and
development. Cell Cycle. 7:2090–2096. 2008. View Article : Google Scholar
|
|
15
|
Yang MH, Wu MZ, Chiou SH, Chen PM, Chang
SY, Liu CJ, Teng SC and Wu KJ: Direct regulation of TWIST by
HIF-1alpha promotes metastasis. Nat Cell Biol. 10:295–305. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Desprez PY, Sumida T and Coppe JP:
Helix-loop-helix proteins in mammary gland development and breast
cancer. J Mammary Gland Biol Neoplasia. 8:225–239. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Lo HW, Hsu SC, Xia W, Cao X, Shih JY, Wei
Y, Abbruzzese JL, Hortobagyi GN and Hung MC: Epidermal growth
factor receptor cooperates with signal transducer and activator of
transcription 3 to induce epithelial-mesenchymal transition in
cancer cells via up-regulation of TWIST gene expression. Cancer
Res. 67:9066–9076. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Pozharskaya V, Torres-Gonzalez E, Rojas M,
Gal A, Amin M, Dollard S, Roman J, Stecenko AA and Mora AL: Twist:
A regulator of epithelial-mesenchymal transition in lung fibrosis.
PLoS One. 4:e75592009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Frid MG, Brunetti JA, Burke DL, Carpenter
TC, Davie NJ, Reeves JT, Roedersheimer MT, van Rooijen N and
Stenmark KR: Hypoxia-induced pulmonary vascular remodeling requires
recruitment of circulating mesenchymal precursors of a
monocyte/macrophage lineage. Am J Pathol. 168:659–669. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2−ΔΔC T method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
21
|
Spencer VA, Sun JM, Li L and Davie JR:
Chromatin immunoprecipitation: A tool for studying histone
acetylation and transcription factor binding. Methods. 31:67–75.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wohrley JD, Frid MG, Moiseeva EP, Orton
EC, Belknap JK and Stenmark KR: Hypoxia selectively induces
proliferation in a specific subpopulation of smooth muscle cells in
the bovine neonatal pulmonary arterial media. J Clin Invest.
96:273–281. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Arciniegas E, Frid MG, Douglas IS and
Stenmark KR: Perspectives on endothelial-to-mesenchymal transition:
Potential contribution to vascular remodeling in chronic pulmonary
hypertension. Am J Physiol Lung Cell Mol Physiol. 293:L1–L8. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Stenmark KR, Davie N, Frid M,
Gerasimovskaya E and Das M: Role of the adventitia in pulmonary
vascular remodeling. Physiology. 21:134–145. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Arciniegas E, Sutton AB, Allen TD and
Schor AM: Transforming growth factor beta 1 promotes the
differentiation of endothelial cells into smooth muscle-like cells
in vitro. J Cell Sci. 103:521–529. 1992.PubMed/NCBI
|
|
26
|
Zhu P, Huang L, Ge X, Yan F, Wu R and Ao
Q: Transdifferentiation of pulmonary arteriolar endothelial cells
into smooth muscle-like cells regulated by myocardin involved in
hypoxia-induced pulmonary vascular remodelling. Int J Exp Pathol.
87:463–474. 2006. View Article : Google Scholar
|
|
27
|
Jones R and Reid L: Vascular Remodeling in
Clinical and Experimental Pulmonary Hypertensions. Portland Press;
London: 1995
|
|
28
|
Yu AY, Shimoda LA, Iyer NV, Huso DL, Sun
X, McWilliams R, Beaty T, Sham JS, Wiener CM, Sylvester JT and
Semenza GL: Impaired physiological responses to chronic hypoxia in
mice partially deficient for hypoxia-inducible factor 1alpha. J
Clin Invest. 103:691–696. 1999. View
Article : Google Scholar : PubMed/NCBI
|
|
29
|
Higgins DF, Kimura K, Bernhardt WM,
Shrimanker N, Akai Y, Hohenstein B, Saito Y, Johnson RS, Kretzler
M, Cohen CD, et al: Hypoxia promotes fibrogenesis in vivo via HIF-1
stimulation of epithelial-to-mesenchymal transition. J Clin Invest.
117:3810–3820. 2007.PubMed/NCBI
|
|
30
|
Ansieau S, Bastid J, Doreau A, Morel AP,
Bouchet BP, Thomas C, Fauvet F, Puisieux I, Doglioni C, Piccinin S,
et al: Induction of EMT by twist proteins as a collateral effect of
tumor-promoting inactivation of premature senescence. Cancer Cell.
14:79–89. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Yang J, Mani SA, Donaher JL, Ramaswamy S,
Itzykson RA, Come C, Savagner P, Gitelman I, Richardson A and
Weinberg RA: Twist, a master regulator of morphogenesis, plays an
essential role in tumor metastasis. Cell. 117:927–939. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Sun S, Ning X, Zhang Y, Lu Y, Nie Y, Han
S, Liu L, Du R, Xia L, He L and Fan D: Hypoxia-inducible
factor-1alpha induces Twist expression in tubular epithelial cells
subjected to hypoxia, leading to epithelial-to-mesenchymal
transition. Kidney Int. 75:1278–1287. 2009. View Article : Google Scholar : PubMed/NCBI
|