Introduction
Diabetes mellitus is estimated to affect
>366,000,000 individuals worldwide and is characterized by
chronic hyperglycemia (1).
Studies in humans and animal models have reported an association
between diabetes and neurological conditions that affect learning
and memory, including Alzheimer’s disease (AD) (2-4).
Diabetic encephalopathy is now recognized as a complication of
diabetes (5). Hyperglycemia has
been shown to significantly decrease cell viability and induce
apoptosis and loss of hippocampal neurons. The effect of high
glucose accumulation involves the intracellular accrual of reactive
oxygen species (ROS) (6,7). Therefore, it is necessary to develop
neuroprotective strategies to inhibit diabetic encephalopathy. One
avenue of investigation has focused on neurotrophic factors, which
are important for neuronal survival and regeneration and are
considered potential therapeutics for AD and other
neurodegenerative diseases (8).
Brain-derived neurotrophic factor (BDNF) is a
specific neurotrophic factor that is expressed in neurons and is
involved in the growth and differentiation of new neurons and
synapse development. BDNF binds to two receptors, namely
tropomyosin-related kinase B (TrkB) and low-affinity nerve growth
factor receptor, and is involved in the process of long-term
memory. BDNF provides trophic support to neurons and exerts a
neuroprotective effect against brain injury. In addition to its
well-established role in the survival, differentiation and
plasticity of neurons (9), BDNF
and its cognate receptor TrkB are implicated in the regulation of
energy and glucose homeostasis through their effects on the central
nervous system (10). Perturbed
BDNF signaling in the brain triggers hyperphagia and obesity in
mice, suggesting that BDNF acts as an anorexigenic signaling factor
(10). Studies have suggested
that BDNF regulates glucose metabolism by improving insulin
sensitivity and increasing pancreatic insulin production (11,12). In addition, in rodents with
impaired leptin signaling through diet-induced obesity and/or
deficient leptin signaling (db/db mice), systemic or central BDNF
administration has been shown to reduce food intake and body weight
(11) in a dose-dependent manner
(13).
Synaptic plasticity is defined as the ability of
synapses to reinforce and/or weaken their connections over time,
depending on their relative activity levels. Synaptic plasticity is
considered as one of the most important neurochemical processes
involved in learning and memory (14). In animals, diabetes can cause
changes in synaptic proteins, and hyperglycemia is one of the
factors contributing to these alterations (15). Synaptophysin (Syn) is a key
protein involved in the biogenesis and exo-endocytosis of synaptic
vesicles. Syn is considered to be a specific marker of synaptic
density and is closely associated with activity-dependent synapse
formation and synaptic plasticity (16,17). Syn is mainly degraded through the
ubiquitin-proteasome system (18), and evidence suggests that reduced
expression levels of Syn may contribute to hyperglycemia-induced
cognitive impairment in mice (19). Other factors have also been
implicated in synaptic plasticity and memory. Cyclic AMP response
element-binding protein (CREB) is a nuclear transcription factor
that is essential for the formation and retention of memory. The
activation of CREB occurs by phosphorylation at serine residue 133
and is required for neuronal growth and survival (20). CREB is necessary for the
maintenance of normal synapses in hippocampal neurons (21), and reduced levels of
phosphorylated CREB (p-CREB) have been observed in the postmortem
brains of patients with AD and experimental models of AD (22,23). Arc protein, which is transcribed
from the Arc/Arg3.1 gene, is another factor associated with the
progression of AD (24-26). The dysregulation of Arc in
cerebral neurons interferes with their normal activity and causes
synaptic damage and neuron loss, leading to the degradation of
specific neural circuit functions and a decrease in neuronal
network activity that may be involved in AD (24-26).
Previous studies have investigated the interaction
of the TrkB receptor with hyperglycemia and neuronal function
(27,28). BDNF upregulates TrkB and increases
the phosphorylation levels of TrkB and ERK in retinal neurons
exposed to hyperglycemic conditions (29). A previous study demonstrated that
the phosphorylation levels of the cell signaling molecule Akt
(protein kinase B) and transcription factor CREB are reduced in
diabetic rats compared with those in control animals (30), suggesting that these factors may
be involved in diabetes-induced cognitive dysfunction. Chen et
al (31) demonstrated that
the neuroprotective effects of BDNF, acting via the TrkB receptor,
were induced by activation of the phosphatidylinositol-3-kinase
(PI3K)-Akt pathway and the increased expression of Arc.
However, whether BDNF protects hippocampal neurons
from high glucose-induced apoptosis and/or synaptic plasticity
dysfunction remains to be fully elucidated. Therefore, the aim of
the present study was to evaluate whether long-term elevated
glucose, which mimics prolonged hyperglycemia, causes significant
changes in neuronal survival and synaptic plasticity, and whether
exogenous BDNF exerts neuroprotective effects.
Materials and methods
Primary culture of rat hippocampal
neurons
All animal experiments were performed in accordance
with the National Institutes of Health Guidelines for the Care and
Use of Laboratory Animals and approved by the Ethics Committee of
Animal Experiments of the Shanghai Sixth People’s Hospital
affiliated to Shanghai Jiao Tong University [Shanghai, China;
permit no. SYXK (Shanghai) 2011-0128].
Primary cultures of rat hippocampal neurons were
prepared from the hippocampi of 10 neonatal Sprague-Dawley rats
within 24 h of birth (Shanghai Laboratory Animal Co., Ltd.,
Shanghai, China), weighing between 4.5-6.5 g, as described
previously (32), with minor
modifications. The hippocampi were dissected from the rat brain
tissues and were placed on ice. Subsequently, the blood vessels and
meninges were thoroughly removed, and the hippocampi were washed
with phosphate-buffered saline (PBS). The tissues were then
transferred into Eppendorf tubes containing 1 ml 0.123% trypsin
(Gibco; Thermo Fisher Scientific, Inc., Waltham, MA, USA). The
hippocampi were cut into small pieces with sterile ophthalmic
scissors (Kun Sheng Medical Instrument Co., Ltd., Shanghai, China).
The hippocampal pieces were digested for 15 min at 37°C with
vortexing every 5 min. The digestion procedure was terminated by
the addition of 5 ml Dulbecco’s modified Eagle’s medium (DMEM;
Gibco; Thermo Fisher Scientific, Inc.) containing 20% fetal bovine
serum (FBS; Gibco; Thermo Fisher Scientific, Inc.). The cell
suspension was passed through a 200-mesh cell strainer and
separated by centrifugation at 300 × g for 5 min at room
temperature. The pellets were resuspended in 2 ml of DMEM
containing 20% FBS, at ~70 cells per ml. The neurons were seeded on
poly-D-lysine (0.1 mg/ml; Gibco; Thermo Fisher Scientific,
Inc.)-coated glass coverslips (Corning Incorporated, Corning, NY,
USA), 96-well plates and/or 6-well plates in 60-70 µl
medium. Following 6-12 h of incubation, the cells were cultured in
Neurobasal medium supplemented with B27 (1:50, Gibco; Thermo Fisher
Scientific, Inc.). Half of the medium was replaced with fresh
medium every 2 or 3 days.
Immunofluorescence staining
The primary hippocampal neurons were fixed with 4%
paraformaldehyde (China National Medicines Corporation, Ltd.,
Beijing, China) for 1 h at room temperature and incubated in PBS
containing 0.5% Triton for 20 min at room temperature. Non-specific
antibody binding was blocked by incubation at room temperature for
30 min with normal goat serum (Gibco; Thermo Fisher Scientific,
Inc.). Each coverslip was incubated with 20 µl rabbit
anti-NeuN primary antibody (1:200; cat. no. ab177487, Abcam,
Cambridge, MA, USA) or rabbit anti-synaptophysin primary antibody
(1:200; cat. no. ab32127; Abcam) at 4°C overnight. The coverslips
were subsequently washed three times in PBS and incubated with
donkey anti-rabbit secondary antibody (1:500; cat. no. A0453, Alex
fluor 555; Beyotime Institute of Biotechnology, Shanghai, China) or
goat anti-rabbit secondary antibody (1:500; cat. no. A0423; Alex
fluor 488; Beyotime Institute of Biotechnology) for 30 min at room
temperature. The coverslips were finally incubated with 20
µl of 4′,6-diamidino-2-phenylindole (DAPI; Roche
Diagnostics, Basel, Switzerland) for 5 min at room temperature in
the dark. The cells were observed and images were captured using a
Volocity Demo imaging system (PerkinElmer, Inc., Waltham, MA,
USA).
High glucose exposure and experimental
grouping
The hippocampal neurons in primary culture for 3
days were divided into four experimental groups, including the
control group (CON), high glucose group (HG), high glucose + BDNF
group (HG + BDNF) and high glucose + BDNF + wortmanin group (HG +
BDNF + wort). The hippocampal neurons were seeded in 96-well plates
at a density of 5,000-10,000 cells in each well and maintained at
37°C in a humidified incubator supplemented with 5% CO2.
Each of the four groups was exposed to different intervention
measures. The control group was exposed to normal medium containing
25 mM glucose. The primary hippocampal neurons were exposed to 75
mM glucose (China National Medicines Corporation, Ltd.) for 72 h
(33), which has no effect on
normal metabolism. To establish the HG + BDNF group, the primary
hippocampal neurons were incubated for 24 h with 50 ng/ml BDNF
(Sigma; Merck KGaA, Darmstadt, Germany) prior to stimulation with
75 mM glucose for 72 h. Primary hippocampal neurons in the HG +
BDNF + wort group were pretreated with 0.5 µM of wortmannin
(Selleck Chemicals, Houston, TX, USA) to suppress PI3K for 2 h, and
further treatments were the same as those for the HG + BDNF
group.
Assessment of apoptosis by flow
cytometry
The apoptotic rate was measured using an Annexin
V-fluorescein isothiocyanate (FITC)/propidium iodide (PI) apoptosis
detection kit (Gibco; Thermo Fisher Scientific, Inc.). Flow
cytometric data were acquired on FACSCalibur flow cytometer (BD
Biosciences, San Jose, CA, USA) and analysed with the use of FlowJo
v10 software (FlowJo, LLC, Ashland, OR, USA). Following 72 h of
incubation, the primary hippocampal neurons were washed twice with
ice-cold PBS and stained with 190 µl Annexin V-FITC (Gibco;
Thermo Fisher Scientific, Inc.) and 10 µl PI (Roche
Diagnostics) according to the manufacturer’s protocol. Following 30
min of incubation at 37°C, the stained neurons were analyzed by
flow cytometry, and the rate of cellular apoptosis was determined.
Annexin V was set as the horizontal axis and PI as the vertical
axis. Apoptotic or necrotic cells were indicated in the upper right
quadrant of the flow-cytogram, whereas early apoptotic cells were
indicated in the lower right quadrant.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR) analysis
Total RNA was isolated from the primary hippocampal
neurons using TRIzol (Invitrogen; Thermo Fisher Scientific, Inc.).
cDNA synthesis was performed at 37°C for 15 min followed by 85°C
for 5 sec using the PrimeScript™ RT reagent kit (Takara
Biotechnology Co., Ltd., Dalian, China). Specific mRNA
quantification was performed by real-time PCR using
SYBR® Premix Ex Taq™ II (Tli RNase H Plus; Takara
Biotechnology Co., Ltd.) in a FTC3000HT real-time PCR system
(Funglyn Biotech, Inc., Toronto, ON, Canada). The gene-specific
primers used are presented in Table
I. Each PCR mixture contained 1 µl cDNA, 0.8 µl
each primer (10 µmol/l), 7 µl ddH2O, 0.4
µl ROX and 10 µl SYBR Green Premix (Takara Bio, Inc.,
Shiga, Japan). All reactions involved initial denaturation at 95°C
for 30 sec followed by 40 cycles of 95°C for 5 sec and 60°C for 34
sec. The 2−ΔΔCq method (34) was used to determine the relative
mRNA expression of the target genes. Glyceraldehyde 3-phosphate
dehydrogenase (GAPDH) was used as an internal control.
 | Table IPrimer sequences for reverse
transcription-quantitative polymerase chain reaction. |
Table I
Primer sequences for reverse
transcription-quantitative polymerase chain reaction.
| Gene | Primer sequence
(5′-3′) | Product length
(bp) | Temperature
(°C) |
|---|
| GAPDH | F:
CAGGGCTGCCTTCTCTTGTG | 111 | 60.70 |
| R:
AACTTGCCGTGGGTAGAGTC | | 60.54 |
| Arc | F:
TATGTGGACGCTGAGGAGGA | 77 | 60.77 |
| R:
CGCAGAAAGCGCTTGAACTT | | 60.75 |
| CREB | F:
AGGGCCTGCAGACATTAACC | 88 | 60.03 |
| R:
TGTCCATCAGTGGTCTGTGC | | 60.04 |
| Syn | F:
TCGTGTTCAAGGAGACAGGC | 78 | 60.80 |
| R:
CAGGTGCTGGTTGCTTTTCC | | 60.82 |
Western blot analysis
The cells were lysed in radioimmunoprecipitation
assay buffer (Beyotime Institute of Biotechnology) containing
phenylmethylsulfonyl fluoride (final concentration 1 mM; Ameresco,
Inc., Framingham, MA, USA) and centrifuged at 10,000 × g for 5 min
at 4°C. The protein concentration was determined using a
Bicinchoninic Acid Protein Assay kit (Beyotime Institute of
Biotechnology). Equal quantities of protein (60 µg) were
separated by 10% sodium dodecyl sulfate-polyacrylamide gel
electrophoresis and transferred onto polyvinylidene difluoride
membranes (EMD Millipore, Burlington, MA, USA). The membranes were
subsequently incubated in blocking buffer for 1 h, 5% bovine serum
albumin (Gibco; Thermo Fisher Scientific, Inc.) in Tris-buffered
saline with 0.1% Tween-20, followed by overnight incubation at 4°C
with rabbit anti-CREB (1:1,000; cat. no. ab32515; Abcam), rabbit
anti-Arc/Arg3.1 (1:1,000; cat. no. ab183183; Abcam), rabbit
anti-TrkB (1:1,000; cat. no. 4603; Cell Signaling Technology,
Inc.), rabbit anti-pAkt (1:1,000; cat. no. cst-4060s; Cell
Signaling Technology, Inc.), rabbit anti-Akt (1:1,000; cat. no.
cst-4691s; Cell Signaling Technology, Inc.), rabbit anti-Syn
(1:5,000; cat. no. ab32127; Abcam) or mouse anti-GAPDH (1:1,000;
cat. no. sc-293335; Santa Cruz Biotechnology, Inc., Dallas, TX,
USA) antibodies. The membranes were subsequently incubated for 1 h
at room temperature with goat anti-rabbit (1:5,000; cat. no.
sc-2030) or anti-mouse (1:5,000; cat. no. sc-516180) IgG conjugated
to horseradish peroxidase (Santa Cruz Biotechnology, Inc.). The
detection of specific bands was achieved with enhanced
chemiluminescence reagent (Pierce; Thermo Fisher Scientific, Inc.),
and the immunoreactive bands were visualized on an ImageQuant
LAS4000 mini apparatus (GE Healthcare, Chicago, IL, USA).
Semi-quantification was performed using Image-Pro Plus v6.0
software (www.mediacy.com).
Statistical analysis
Statistical analyses were performed using Prism 5
software (GraphPad Software, Inc., La Jolla, CA, USA). Data are
presented as the mean ± standard deviation of three or four
independent experiments. Statistical significance was determined by
two-way analysis of variance followed by the Newman-Keuls post-hoc
test. P<0.05 was considered to indicate a statistically
significant difference.
Results
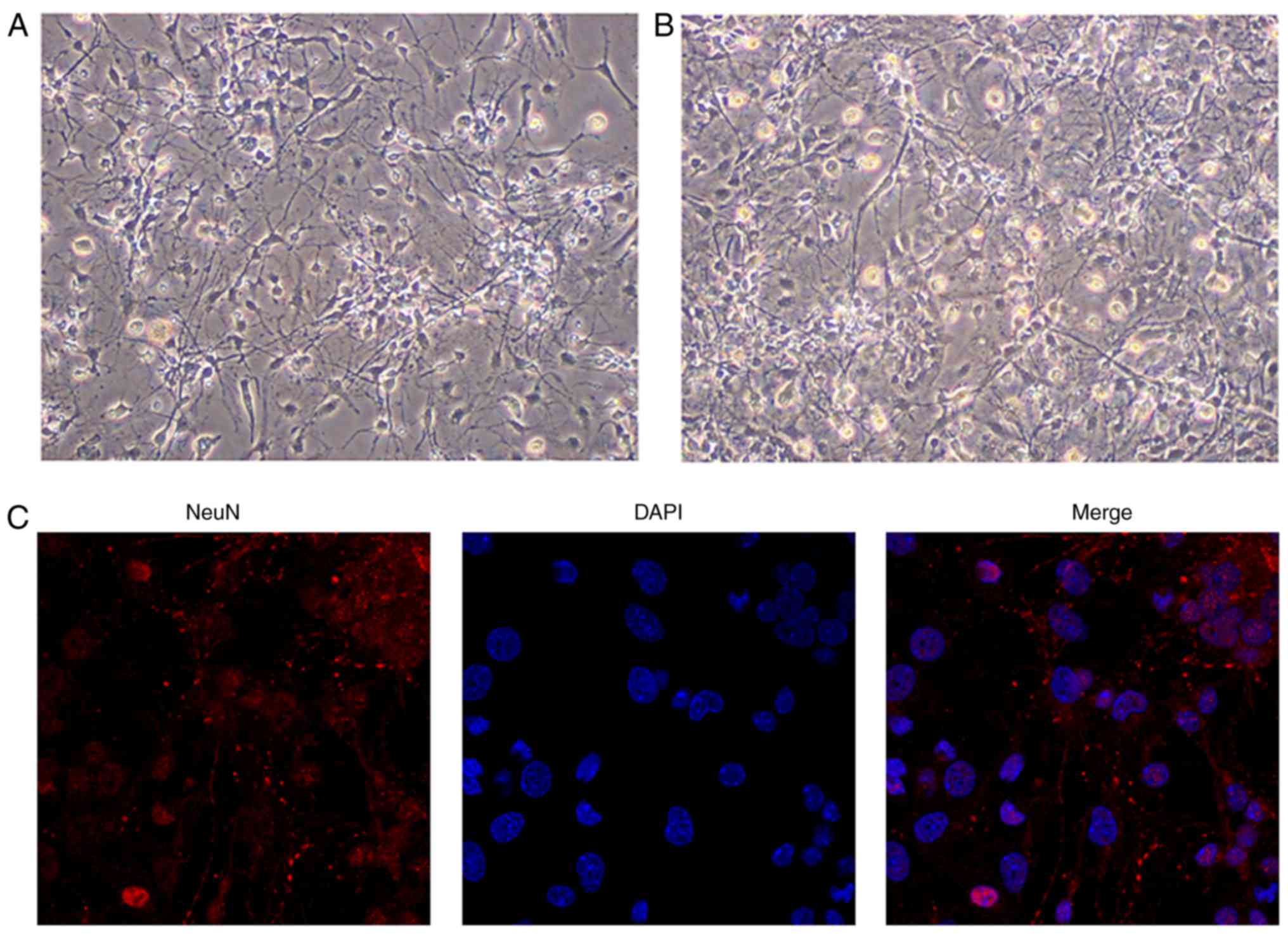
Identification of hippocampal
neurons
Previous studies have used hippocampal neurons from
fetal tissues obtained from 18 days of pregnancy (35) or newborn rats (36,37). Chen et al (38) demonstrated no difference in the
neuronal survival rates between hippocampal neurons from fetal rats
and those from corresponding newborn rats. In the present study,
hippo-campal neurons from newborn rats were selected for culture
in vitro. On days 3 and 5 of culture, the neurites were
observed to interconnect with each other to form a loose network of
cells (Fig. 1A and B), which is a
typical function of cultured hippocampal neurons. Nuclear staining
of the neurons was achieved using DAPI, and neurite growth was
demonstrated by immunofluorescence staining of NeuN (red staining,
Fig. 1C). The purity of the
neurons, calculated as the ratio of the number of positive cells
(identified by nuclear staining) to the total number of cells, was
estimated to be ~95% (Fig.
1C).
BDNF inhibits the high glucose-induced
apoptosis of hippocampal neurons, and wortmannin reverses this
effect
The apoptotic rate was significantly higher in
hippocampal neurons treated with high glucose than in neurons
exposed to normal glucose (36.32±1.80, vs. 2.68±0.60%, P<0.001;
Fig. 2A and B). BDNF suppressed
the apoptotic rate of neurons exposed to high glucose (11.75±1.10,
vs. 36.32±1.80%, P<0.001; Fig. 2A
and B). However, this effect of BDNF was attenuated by
wortmannin, an inhibitor of PI3K (24.72±1.06, vs. 11.75±1.10%,
P<0.01; Fig. 2A and B). These
data indicated that high glucose induced the apoptosis of
hippocampal neurons cultured in vitro, which was suppressed
by BDNF via PI3K signaling.
 | Figure 2Effect of BDNF on HG-induced neuronal
apoptosis. (A) Neuronal apoptosis was assayed by flow cytometry
(Annexin V-FITC/PI staining). CON: 25 mM glucose; HG: 75 mM glucose
for 72 h; HG + BDNF: 50 ng/ml BDNF for 24 h followed by 75 mM
glucose for 72 h; HG + BDNF + wort: 0.5 µM wort pretreatment
for 2 h to suppress PI3K, followed by ng/ml BDNF for 24 h and then
75 mM glucose for 72 h. (B) Data are presented as the mean ±
standard deviation of three independent triplicate experiments.
***P<0.001, vs. CON group; ###P<0.001,
vs. HG group; &&P<0.01, vs. HG + BDNF
group. FITC, fluorescein isothiocyanate; PI, propidium iodide; CON,
control; BDNF, brain-derived neurotrophic factor; HG, high glucose;
wort, wortmannin. |
High glucose suppresses the expression
levels of synaptic plasticity-related proteins, and BDNF reverses
these effects
To examine the mechanism underlying the protective
effect of BDNF on hippocampal neurons under hyperglycemic
conditions, RT-qPCR and western blot experiments were performed to
assess the expression levels of the synaptic plasticity-related
proteins, CREB, Arc and Syn. The RT-qPCR experiments revealed that
the mRNA expression levels of Syn, Arc and CREB were significantly
reduced on exposure to high glucose (all P<0.001; Fig. 3A-C). BDNF significantly inhibited
the effects of high glucose on the mRNA expression levels of Syn,
Arc and CREB (all P<0.01; Fig.
3A-C). In addition, prior administration of wortmannin
significantly attenuated the ability of BDNF to reverse the effects
of high glucose on the mRNA expression levels of Syn (P<0.001),
Arc (P<0.05) and CREB (P<0.01; Fig. 3A-C). When the protein levels of
Syn, Arc and CREB were assessed by western blotting (Fig. 4A–D), the results were consistent
with those of the RT-qPCR experiments. Taken together, these data
indicated that high glucose may lead to an imbalance in the
synaptic plasticity of hippocampal neurons, and that this effect is
suppressed by BDNF via PI3K signaling.
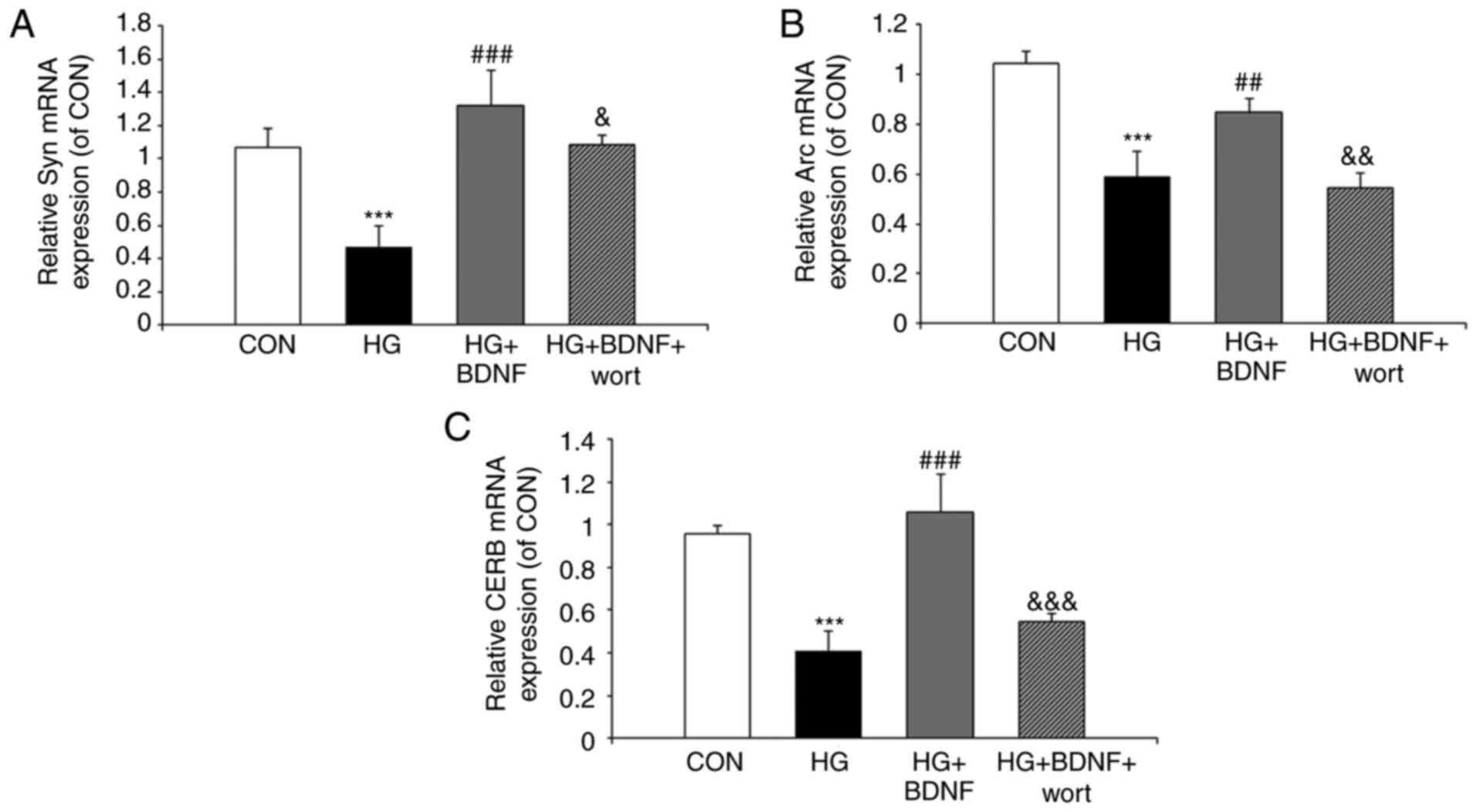 | Figure 3Effects of BDNF on the mRNA
expression levels of synaptic plasticity-related proteins in
primary hippocampal neurons under high glucose conditions. The mRNA
expression of (A) Syn, (B) Arc and (C) CREB (C) in primary
hippocampal neurons was monitored by reverse
transcription-quantitative polymerase chain reaction analysis.
GAPDH was used as an internal control. Data are presented as the
mean ± standard deviation of three independent experiments.
***P<0.001, vs. CON group; ##P<0.01 and
###P<0.001, vs. HG group; &P<0.05,
&&P<0.01 and
&&&P<0.001, vs. HG + BDNF group. Syn,
synapto-physin; CREB, cyclic AMP response element-binding protein;
GAPDH, glyceraldehyde 3-phosphate dehydrogenase; CON, control;
BDNF, brain-derived neurotrophic factor; HG, high glucose; wort,
wortmannin. |
 | Figure 4Effects of BDNF on the protein
expression levels of synaptic plasticity-related proteins in
primary hippocampal neurons under high glucose conditions. (A)
Western blot analysis was used to measure the protein expression
levels of (B) Syn, (C) Arc and (D) CREB in primary hippocampal
neurons. GAPDH was used as an internal control. Data are presented
as the mean ± standard deviation of three independent experiments.
***P<0.001, vs. CON group; #P<0.05 and
###P<0.001, vs. HG group; &P<0.05
and &&P<0.01, vs. HG + BDNF group.
Syn, synaptophysin; CREB, cyclic AMP response element-binding
protein; GAPDH, glyceraldehyde 3-phosphate dehydrogenase; CON,
control; BDNF, brain-derived neurotrophic factor; HG, high glucose;
wort, wortmannin. |
BDNF regulates the level and distribution
of Syn in primary hippocampal neurons under high glucose
conditions
The level and distribution of Syn can be used as an
indirect measure of synaptic density (19). Therefore, the distribution of Syn
in primary hippocampal neurons was assessed using
immunofluorescence techniques. In the CON group, Syn protein was
expressed in the neurites of hippocampal cells (Fig. 5A–C). However, the expression level
of Syn was markedly reduced in hippocampal neurons in the HG group,
with loss of expression in the neurites (Fig. 5D–F). BDNF normalized the levels
and distribution of Syn protein in the neurons, and this effect of
BDNF was prevented by wortmannin (Fig. 5G-L). These findings indicated that
high glucose reduced synaptic density in primary hippocampal
neurons, and that BDNF was able to reverse this effect via PI3K
signaling.
 | Figure 5Effects of BDNF on the expression and
distribution of Syn in primary hippocampal neurons under high
glucose conditions. The expression and distribution of Syn were
determined using immunofluorescence techniques. Syn is stained
green, nuclei are stained blue with DAPI. (A) Syn, (B) DAPI and (C)
Merge staining in the CON group; (D) Syn, (E) DAPI and (F) Merge
staining in the HG group; (G) Syn, (H) DAPI and (I) Merge staining
in the HG + BDNF group; (J) Syn, (K) DAPI and (L) Merge staining in
the HG + BDNF + wort group. Magnification, x630. CON, control;
BDNF, brain-derived neurotrophic factor; HG, high glucose; wort,
wortmannin; DAPI, 4′,6-diamidino-2-phenylindole. |
BDNF upregulates protein expression
levels of TrkB, Akt and p-Akt in primary hippocampal neurons under
high glucose conditions
The expression levels of the downstream signaling
proteins of BDNF were assessed to further investigate the molecular
mechanisms underlying the neuroprotective effect of BDNF on
hippocampal neurons under high glucose conditions (Fig. 6A). The protein expression levels
of TrkB in the HG group were similar to those in the CON group,
whereas combined treatment of HG + BDNF resulted in increased
protein expression levels of TrkB compared with those in the HG
group (P<0.001; Fig. 6B). The
expression levels of TrkB were significantly increased in the HG +
BDNF + wort group compared with those in the HG group (P<0.001;
Fig. 6B), but did not differ
significantly compared with those in the HG + BDNF group. The
expression levels of p-Akt, Akt, and p-Akt/Akt were significantly
decreased in the HG + BDNF + wort group compared with those in the
HG + BDNF group (P<0.01, P<0.05 and P<0.01, respectively;
Fig. 6C–E), whereas the HG + BDNF
group exhibited significantly higher protein expression levels of
p-Akt, AKT and p-Akt/Akt compared with those in the HG group of
cells (P<0.01, P<0.05 and P<0.01, respectively; Fig. 6C–E). The expression levels of
p-Akt and p-Akt/Akt were significantly decreased in the HG group
compared with those in the CON group (both P<0.01; Fig. 6C and E), Taken together, these
data suggested that high glucose may lead to abnormal plasticity in
hippocampal neuronal synapses, which is reversed by BDNF via its
cognate receptor TrkB and the downstream signaling protein Akt.
 | Figure 6Effects of BDNF on protein expression
levels of TrkB, Akt and p-Akt in primary hippocampal neurons under
high glucose conditions. (A) Expression levels of TrkB, Akt and
p-Akt were determined by western blot analysis, with data for (B)
TrkB, (C) p-Akt, (D) Akt and (E) p-Akt/Akt presented as the mean ±
standard deviation of three independent experiments.
**P<0.01, vs. CON group; #P<0.05,
##P<0.01 and ###P<0.001, vs. HG group;
&P<0.05, &&P<0.01, vs. HG +
BDNF group. CON, control; BDNF, brain-derived neurotrophic factor;
HG, high glucose; wort, wortmannin; TrkB, tropomyosin-related
kinase B; p-, phosphorylated; GAPDH, glyceraldehyde 3-phosphate
dehydrogenase. |
Discussion
In the present study, an in vitro model of
rat hippocampal neurons was established from newborn rats, and
neurite growth was evaluated by immunostaining for NeuN (39). Furthermore, hyperglycemic
conditions were established for neuronal growth and it was
demonstrated that BDNF protected against neuronal cell apoptosis
induced by high glucose. In addition, BDNF increased the expression
levels of synaptic plasticity-related proteins under high glucose
conditions. Notably, the various effects of BDNF in rat hippocampal
neurons treated with high glucose were dependent on PI3K-Akt
signaling.
In the present study, BDNF protected neuronal cells
from high glucose-induced apoptosis as demonstrated by flow
cytometry. The neuronal apoptotic rate was decreased markedly
(11.49, vs. 38.86%) when the neurons were exposed to 75 mM glucose
for 72 h and administered with 50 ng/ml BDNF. A previous study
demonstrated that the addition of BDNF prior to anoxia resulted in
neuroprotective effects (40).
The protective effect of BDNF was decreased following prolonged
anoxia and irreversible neuronal injury. Therefore, in the present
study, the hippocampal neurons were pretreated with BDNF for 24 h
prior to their culture in the presence of high glucose, and a BDNF
concentration of 50 ng/ml was selected based on the previous
studies (41,42). Consistent with the findings in the
present study, Bathina et al (43) reported that RIN5F cells exhibited
reduced viability following treatment with streptozotocin, which
was reverted by BDNF.
The present study also demonstrated that the mRNA
and protein expression levels of Syn, Arc and CREB were lower in
hippocampal neurons exposed to high glucose than in neurons of the
control group. Notably, the immunofluorescence experiments revealed
a decrease in the protein expression of Syn in the neurites
following treatment with high glucose, and this abnormal
distribution of Syn was consistent with a reduction in synaptic
density. These findings suggested that high glucose may lead to
abnormal plasticity in hippocampal neuronal synapses via
alterations in the levels, and thus functions, of proteins that are
closely associated with synaptic plasticity. Consistent with these
observations, Zhao et al (18) demonstrated that Syn was
downregulated in primary neuronal cultures subjected to high
glucose and hypoxia. Furthermore, a previous study demonstrated
that the combination of hyperglycemia and hypoxia in mice resulted
in cognitive impairment and was associated with significantly
reduced protein levels of Syn in the hippocampus (19). It was suggested that the effects
of high glucose and hypoxia on the protein levels of Syn may result
from the enhanced degradation of Syn involving the E3 ubiquitin
ligase, siah family (19).
Another report found abnormal levels of certain synaptic proteins
(synaptosomal-associated protein-25, synaptotagmin-1 and vesicular
glutamate transporter-1) following long-term exposure of
hippocampal neurons to hyperglycemia, suggesting that the
trafficking of proteins to the synapse may be impaired (15).
In the present study, BDNF caused an increase in the
mRNA and protein expression levels of Syn, Arc and CREB in
hippocampal neurons treated with high glucose. BDNF also normalized
the distribution of Syn in these cells. These observations
suggested that the protective effect of BDNF on hippocampal neurons
was achieved, at least in part, through enhancement of synaptic
plasticity. Leal et al (44) demonstrated that BDNF can regulate
hippocampal synaptic plasticity. In addition, a previous study
found that rats fed on a high-fat, high-glucose diet to induce
experimental diabetes exhibited impaired spatial learning,
decreased hippocampal dendritic spine density and reduced long-term
potentiation, and these changes were associated with a reduction in
hippocampal BDNF levels (45).
Arc has been demonstrated to exert a neuroprotective effect via
decreased AMPA receptor current and glutamate receptor 2
internalization (46), therefore,
the upregulation of Arc levels by BDNF may contribute to the
neuroprotective effects of BDNF.
BDNF binds to TrkB and recruits proteins that
activate several signal transduction cascades, including the
sequential activation of insulin receptor substrate-1, PI3K and Akt
(47). The BDNF signaling
pathways activate CREB and CREB-binding protein, regulating the
genes involved in neural plasticity (47). The PI3K-Akt signaling pathway is
involved in synaptic plasticity, memory consolidation and synaptic
morphogenesis (48,49). In terms of the role of this
pathway in diabetes, asiaticoside, a glycosylated triterpene from
Centella asiatica, has been shown to attenuate
diabetes-induced cognitive impairment and upregulate the expression
of synaptic proteins via PI3K/Akt signaling (50). However, no previous reports have
examined the role of the PI3K-Akt pathway in mediating the effects
of BDNF in diabetic encephalopathy. In the present study, BDNF was
demonstrated to activate PI3K-Akt signaling under high glucose
conditions, as the levels of p-Akt and Akt were increased. In
addition, BDNF enhanced the mRNA and protein expression levels of
Arc, Syn and CREB, all of which can influence synaptic plasticity
through the PI3K-Akt pathway as the effects of BDNF were inhibited
by wortmannin. These findings indicated that BDNF-TrkB activates
Akt under hyper-glycemic conditions to reverse the abnormalities in
synaptic plasticity and inhibit apoptosis. Taken together, these
data indicated that BDNF protects hippocampal neurons partially
through the upregulation of CREB and Arc, which is mediated through
the PI3K-Akt signaling pathway.
In the present study, the expression of TrkB was
increased following treatment with BDNF under high glucose
conditions. Although it has been reported that the expression of
TrkB is regulated by the cyclic AMP/CREB pathway in neurons
(51), the administration of PI3K
inhibitor did not decrease the expression level of TrkB, despite a
reduction in CREB and p-Akt/Akt levels. This suggests that the
regulation of the expression of TrkB by BDNF occurs upstream of
Akt.
Of note, BDNF has also been demonstrated to protect
retinal neurons from hyperglycemia via the TrkB/ERK/MAPK pathway
and attenuate diabetic hyperglycemia via an insulin-independent
mechanism in rats (13,29). The regulation of long-term
synaptic plasticity and memory formation by Arc are dependent on
its phosphorylation by ERK protein, suggesting that MAPK kinases
are important in the memory process (52). The findings of the present study
indicate the possibility of potential interplay between the
ERK/MAPK and PI3K-Akt pathways in the regulation of neuronal
plasticity by BDNF.
In conclusion, the present study demonstrated that
BDNF can activate the PI3K-Akt signaling pathway and induce the
expressions of synaptic plasticity-related proteins in hippocampal
neurons cultured under high glucose conditions. This improves
synaptic plasticity in the hippocampal neurons and protects them
from high glucose-induced apoptosis. These findings provide a
theoretical basis for subsequent investigations on the mechanism of
BDNF-mediated hippocampal neuroprotection. In addition, the present
study provides novel insights into therapeutically targeting BDNF
and PI3K-Akt signaling for the prevention of diabetic
encephalopathy.
Acknowledgments
Not applicable.
Funding
The present study was funded by the Shanghai Sixth
People’s Hospital Group Science Foundation, the Shanghai Science
and Technology Commission Foundation Research Project (grant no.
13JC1401504) and the Chinese National Natural Science Foundation
(grant no. 81300933).
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors’ contributions
YZ, YM and YTZ performed the experiments, were
involved in data collection and drafted the manuscript. TH and QL
performed the statistical analyses and were involved in study
design. WL assisted in drafting the manuscript. All authors read
and approved the final manuscript.
Ethics approval and consent to
participate
All animal experiments were performed in accordance
with the National Institutes of Health Guidelines for the Care and
Use of Laboratory Animals and approved by the Ethics Committee of
Animal Experiments of The Shanghai Sixth People’s Hospital
affiliated to Shanghai Jiao Tong University [permit no. SYXK
(Shanghai) 2011-0128].
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Alam U, Asghar O, Azmi S and Malik RA:
General aspects of diabetes mellitus. Handb Clin Neurol.
126:211–222. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Shekhar S, Wang S, Mims PN,
Gonzalez-Fernandez E, Zhang C, He X, Liu CY, Lv W, Wang Y, Huang J
and Fan F: Impaired cerebral Autoregulation-A common neurovascular
pathway in diabetes may play a critical role in diabetes-related
Alzheimer’s disease. Curr Res Diabetes Obes J. 2:pii:
5555872017.
|
|
3
|
Ninomiya T: Diabetes mellitus and
dementia. Curr Diab Rep. 14:4872014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Bitel CL, Kasinathan C, Kaswala RH, Klein
WL and Frederikse PH: Amyloid-β and tau pathology of Alzheimer’s
disease induced by diabetes in a rabbit animal model. J Alzheimers
Dis. 32:291–305. 2012. View Article : Google Scholar
|
|
5
|
Sima AA: Encephalopathies: The emerging
diabetic complications. Acta Diabetol. 47:279–293. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Chen S, Liu AR, An FM, Yao WB and Gao XD:
Amelioration of neurodegenerative changes in cellular and rat
models of diabetes-related Alzheimer’s disease by exendin-4. Age
(Dordr). 34:1211–1224. 2012. View Article : Google Scholar
|
|
7
|
Liu D, Zhang H, Gu W and Zhang M: Effects
of exposure to high glucose on primary cultured hippocampal
neurons: Involvement of intracellular ROS accumulation. Neurol Sci.
35:831–837. 2014. View Article : Google Scholar
|
|
8
|
Allen SJ, Watson JJ, Shoemark DK, Barua NU
and Patel NK: GDNF, NGF and BDNF as therapeutic options for
neurodegeneration. Pharmacol Ther. 138:155–175. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Noble EE, Billington CJ, Kotz CM and Wang
C: The lighter side of BDNF. Am J Physiol Regul Integr Comp
Physiol. 300:R1053–R1069. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Jo YH and Chua SC Jr: The brain-liver
connection between BDNF and glucose control. Diabetes.
62:1367–1368. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Nakagawa T, Tsuchida A, Itakura Y,
Nonomura T, Ono M, Hirota F, Inoue T, Nakayama C, Taiji M and
Noguchi H: Brain-derived neurotrophic factor regulates glucose
metabolism by modulating energy balance in diabetic mice. Diabetes.
49:436–444. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Nonomura T, Tsuchida A, Ono-Kishino M,
Nakagawa T, Taiji M and Noguchi H: Brain-derived neurotrophic
factor regulates energy expenditure through the central nervous
system in obese diabetic mice. Int J Exp Diabetes Res. 2:201–209.
2001. View Article : Google Scholar
|
|
13
|
Meek TH, Wisse BE, Thaler JP, Guyenet SJ,
Matsen ME, Fischer JD, Taborsky GJ Jr, Schwartz MW and Morton GJ:
BDNF action in the brain attenuates diabetic hyperglycemia via
insulin-independent inhibition of hepatic glucose production.
Diabetes. 62:1512–1518. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Sheng M, Sabatini BL and Südhof TC:
Synapses and Alzheimer’s disease. Cold Spring Harb Perspect Biol.
4:pii: a0057772012. View Article : Google Scholar
|
|
15
|
Gaspar JM, Castilho A, Baptista FI,
Liberal J and Ambrosio AF: Long-term exposure to high glucose
induces changes in the content and distribution of some exocytotic
proteins in cultured hippocampal neurons. Neuroscience.
171:981–992. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Masliah E, Terry RD, Alford M and DeTeresa
R: Quantitative immunohistochemistry of synaptophysin in human
neocortex: An alternative method to estimate density of presynaptic
terminals in paraffin sections. J Histochem Cytochem. 38:837–844.
1990. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Tarsa L and Goda Y: Synaptophysin
regulates activity-dependent synapse formation in cultured
hippocampal neurons. Proc Natl Acad Sci USA. 99:1012–1016. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zhao Y, Li Q, Jin A, Cui M and Liu X: E3
ubiquitin ligase Siah-1 downregulates synaptophysin expression
under high glucose and hypoxia. Am J Transl Res. 7:15–27.
2015.PubMed/NCBI
|
|
19
|
Li Q, Zhu XL, Jin AP, Liu XY and Zhao YX:
Inhibition of synaptophysin ubiquitination may improve the
intelligent drop due to high glucose and hypoxia. Int J Clin Exp
Med. 7:5021–5030. 2014.
|
|
20
|
Lonze BE and Ginty DD: Function and
regulation of CREB family transcription factors in the nervous
system. Neuron. 35:605–623. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Middei S, Houeland G, Cavallucci V,
Ammassari-Teule M, D’Amelio M and Marie H: CREB is necessary for
synaptic maintenance and learning-induced changes of the AMPA
receptor GluA1 subunit. Hippocampus. 23:488–499. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Pugazhenthi S, Wang M, Pham S, Sze CI and
Eckman CB: Downregulation of CREB expression in Alzheimer’s brain
and in Aβ-treated rat hippocampal neurons. Mol Neurodegener.
6:602011. View Article : Google Scholar
|
|
23
|
Zhang N, Wen Q, Ren L, Liang W, Xia Y,
Zhang X, Zhao D, Sun D, Hu Y, Hao H, et al: Neuroprotective effect
of arctigenin via upregulation of P-CREB in mouse primary neurons
and human SH-SY5Y neuroblastoma cells. Int J Mol Sci.
14:18657–18669. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Lacor PN, Buniel MC, Chang L, Fernandez
SJ, Gong Y, Viola KL, Lambert MP, Velasco PT, Bigio EH, Finch CE,
et al: Synaptic targeting by Alzheimer’s-related amyloid beta
oligomers. J Neurosci. 24:10191–10200. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wegenast-Braun BM, Fulgencio Maisch A,
Eicke D, Radde R, Herzig MC, Staufenbiel M, Jucker M and Calhoun
ME: Independent effects of intra- and extracellular Abeta on
learning-related gene expression. Am J Pathol. 175:271–282. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Dickey CA, Gordon MN, Mason JE, Wilson NJ,
Diamond DM, Guzowski JF and Morgan D: Amyloid suppresses induction
of genes critical for memory consolidation in APP + PS1 transgenic
mice. J Neurochem. 88:434–442. 2004. View Article : Google Scholar
|
|
27
|
Ennis K, Dotterman H, Stein A and Rao R:
Hyperglycemia accentuates and ketonemia attenuates
hypoglycemia-induced neuronal injury in the developing rat brain.
Pediatr Res. 77:84–90. 2015. View Article : Google Scholar
|
|
28
|
Liao GY, Li Y and Xu B: Ablation of TrkB
expression in RGS92 cells leads to hyperphagic obesity. Mol Metab.
2:491–497. 2013. View Article : Google Scholar :
|
|
29
|
Liu Y, Tao L, Fu X, Zhao Y and Xu X: BDNF
protects retinal neurons from hyperglycemia through the
TrkB/ERK/MAPK pathway. Mol Med Rep. 7:1773–1778. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Xiang Q, Zhang J, Li CY, Wang Y, Zeng MJ,
Cai ZX, Tian RB, Jia W and Li XH: Insulin resistance-induced
hyperglycemia decreased the activation of Akt/CREB in hippocampus
neurons: Molecular evidence for mechanism of diabetes-induced
cognitive dysfunction. Neuropeptides. 54:9–15. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Chen TJ, Wang DC and Chen SS: Amyloid-beta
interrupts the PI3K-Akt-mTOR signaling pathway that could be
involved in brain-derived neurotrophic factor-induced Arc
expression in rat cortical neurons. J Neurosci Res. 87:2297–2307.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Kaech S and Banker G: Culturing
hippocampal neurons. Nat Protoc. 1:2406–2415. 2006. View Article : Google Scholar
|
|
33
|
Chen Y, Cao CP, Li CR, Wang W, Zhang D,
Han LL, Zhang XQ, Kim A, Kim S and Liu GL: Ghrelin modulates
insulin sensitivity and tau phosphorylation in high glucose-induced
hippocampal neurons. Biol Pharm Bull. 33:1165–1169. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
35
|
Fath T, Ke YD, Gunning P, Götz J and
Ittner LM: Primary support cultures of hippocampal and substantia
nigra neurons. Nat Protoc. 4:78–85. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Chen YJ, Huang XB, Li ZX, Yin LL, Chen WQ
and Li L: Tenuigenin protects cultured hippocampal neurons against
methylglyoxal-induced neurotoxicity. Eur J Pharmacol. 645:1–8.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Shao JL, Wan XH, Chen Y, Bi C, Chen HM,
Zhong Y, Heng XH and Qian JQ: H2S protects hippocampal neurons from
anoxia-reoxygenation through cAMP-mediated PI3K/Akt/p70S6K
cell-survival signaling pathways. J Mol Neurosci. 43:453–460. 2011.
View Article : Google Scholar
|
|
38
|
Chen WS, Yueh CY, Huang YA and Hwang E: An
inverted method for culturing dissociated mouse hippocampal
neurons. Neurosci Res. 70:118–123. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Boldrini M, Santiago AN, Hen R, Dwork AJ,
Rosoklija GB, Tamir H, Arango V and John Mann J: Hippocampal
granule neuron number and dentate gyrus volume in
antidepressant-treated and untreated major depression.
Neuropsychopharmacology. 38:1068–1077. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Luo XL, Mao M, Zhou H, Sun XM and Li SF:
Neuroprotective effect of BDNF on hypoxia for embryonic rat
cortical neurons in vitro. Sichuan Da Xue Xue Bao Yi Xue Ban.
37:373–377. 2006.In Chinese. PubMed/NCBI
|
|
41
|
Rankin SL, Guy CS, Rahimtula M and Mearow
KM: Neurotrophin-induced upregulation of p75NTR via a protein
kinase C-delta-dependent mechanism. Brain Res. 1217:10–24. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Smith ED, Prieto GA, Tong L,
Sears-Kraxberger I, Rice JD, Steward O and Cotman CW: Rapamycin and
interleukin-1β impair brain-derived neurotrophic factor-dependent
neuron survival by modulating autophagy. J Biol Chem.
289:20615–20629. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Bathina S, Srinivas N and Das UN:
Streptozotocin produces oxidative stress, inflammation and
decreases BDNF concentrations to induce apoptosis of RIN5F cells
and type 2 diabetes mellitus in Wistar rats. Biochem Biophys Res
Commun. 486:406–413. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Leal G, Afonso PM, Salazar IL and Duarte
CB: Regulation of hippocampal synaptic plasticity by BDNF. Brain
Res. 1621:82–101. 2015. View Article : Google Scholar
|
|
45
|
Stranahan AM, Norman ED, Lee K, Cutler RG,
Telljohann RS, Egan JM and Mattson MP: Diet-induced insulin
resistance impairs hippocampal synaptic plasticity and cognition in
middle-aged rats. Hippocampus. 18:1085–1088. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Cohan CH, Stradecki-Cohan HM,
Morris-Blanco KC, Khoury N, Koronowski KB, Youbi M, Wright CB and
Perez-Pinzon MA: Protein kinase C epsilon delays latency until
anoxic depolarization through arc expression and GluR2
internalization. J Cereb Blood Flow Metab. 37:3774–3788. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Bathina S and Das UN: Brain-derived
neurotrophic factor and its clinical implications. Arch Med Sci.
11:1164–1178. 2015. View Article : Google Scholar
|
|
48
|
Garelick MG and Kennedy BK: TOR on the
brain. Exp Gerontol. 46:155–163. 2011. View Article : Google Scholar
|
|
49
|
Kennedy MB, Beale HC, Carlisle HJ and
Washburn LR: Integration of biochemical signalling in spines. Nat
Rev Neurosci. 6:423–434. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Yin Z, Yu H, Chen S, Ma C, Ma X, Xu L, Ma
Z, Qu R and Ma S: Asiaticoside attenuates diabetes-induced
cognition deficits by regulating PI3K/Akt/NF-kB pathway. Behav
Brain Res. 292:288–299. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Deogracias R, Espliguero G, Iglesias T and
Rodriguez-Peña A: Expression of the neurotrophin receptor trkB is
regulated by the cAMP/CREB pathway in neurons. Mol Cell Neurosci.
26:470–480. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Nikolaienko O, Eriksen MS, Patil S, Bito H
and Bramham CR: Stimulus-evoked ERK-dependent phosphorylation of
activity-regulated cytoskeleton-associated protein (Arc) regulates
its neuronal subcellular localization. Neuroscience. 360:68–80.
2017. View Article : Google Scholar : PubMed/NCBI
|