Introduction
Down syndrome (DS) is the most frequent human
chromosomal disorder with a frequency of 1/400 conceptions and
1/1,000 births worldwide (1,2).
Since the initial discovery of Lejeune et al (3), it is known that the presence of full
or partial chromosome 21 (Hsa21) in three copies (trisomy 21) in
the cells of the affected subjects is responsible for the typical
features of DS, in particular intellectual disability (ID),
cardiovascular defects (4,5)
and craniofacial dysmorphism. Importantly, a highly restricted
'Down syndrome critical region' of 34 kb on distal 21q22.13 appears
to be specifically duplicated in all individuals with DS (6,7).
In addition, a recent study conducted on plasma and urine samples
of individuals with DS demonstrated a systematic deviation of
metabolites involved in central metabolic processes associated with
mitochondrial metabolism including the Krebs cycle, glycolysis and
oxidative phosphorylation in DS (8).
Despite intense efforts of the scientific community,
the pathogenic mechanism linking chromosome 21 and DS is remains
largely uncharacterized, and pharmacological therapies targeting
genes located on Hsa21 have not been developed so far. Therefore,
current research shifted to study the involvement of non-coding
RNAs (e.g., microRNAs) in the process (7). microRNAs (miRNAs/miR) are small
single stranded nucleic acids that regulate gene expression
post-transcriptionally via binding to different mRNA targets,
resulting in inhibition of mRNA translation (9,10).
Increasing evidence indicates that miRNAs serve important roles in
a large variety of biological processes including development,
differentiation, proliferation and apoptosis (11). It is well documented that
intracellularly produced miRNAs can be secreted in the
extracellular milieu, mostly associated with Ago proteins, bound to
lipoproteins or secreted in extracellular vesicles (EVs) (12-15). Due to their nature, the latter two
may provide an enriched and preserved source of miRNAs (16-19), therefore they may be better
associated with the disease state than total plasma analysis
(20,21), allowing a deeper understanding of
the ongoing pathological processes and complementing previous
studies.
Intriguingly, several circulating miRNAs have been
differentially retrieved from either placenta or plasma samples of
euploid and subjects with DS (22,23). However, only a few of them
(miR-99a, miR-125b, let-7c and miR-155) were mapped on Hsa21
(24). It should also be noted
that in other studies no Hsa21-derived miRNAs were identified to be
differentially expressed in association with DS (25,26). This variability may derive from
the type of sample analyzed and the extraction procedure used to
recover the nucleic acids.
The present study focused for the first time on a
subset of miRNAs carried by circulating biogenic nanoparticles,
namely EVs and high-density lipoproteins (HDLs). Biogenic
nanoparticles formulations, also referred to as
nanoparticle-enriched fraction (NEF), were obtained from the plasma
of young subjects with DS and their miRNA expression profiles
systematically compared with those of NEF from their non-trisomic
siblings. Interestingly, three novel candidate miRNAs emerged from
the present study (miR-16-5p, miR-144-3p and miR-99b-5p) that could
help unraveling molecular aspects underlying the complex DS
pathogenic phenotype.
Materials and methods
Subject enrolment
For all participants involved in the present study
(approved by the competent Ethics Committee of Sant'Orsola-Malpighi
Hospital in Bologna, Italy; approval no. 39/2013/U/Tess) written
informed consent was obtained from the parents in the case of
subjects with age below 18 years and from the subjects themselves
if aged over 18 years, according to the approved protocol.
Patient enrolment was performed at the Unit of
Neonatology of Sant'Orsola-Malpighi Hospital in Bologna, Italy from
February 2014 to March 2016. All methods were performed in
accordance with ethical principles for Medical Research involving
human subjects of the Helsinki Declaration.
The exclusion criteria of the enrolment to the study
were: Age <2 years old, distress at birth, strongly premature
(EG <35 weeks) and severe central nervous system (CNS) disease
at birth. The presence or absence of each clinical feature and
comorbidity was assessed based on the agreed clinical judgment of
at least two pediatricians with long-time experience in the
follow-up of children with DS, following a complete, systematic
visit of the subject and an interview with the parents.
Case selection
In this study, a dataset containing 30 participants:
15 young subjects with DS (DS; 7 females and 8 males, aged 4-20
years, mean 11.71±5.12 years) and the corresponding 15 healthy
siblings (C; 4 females and 11 males, aged 2-25 years, mean
14.24±6.42 years) was used. For each subject with DS, information
was collected on the age of the parent, birth weight, development
stage in the first months of life (sitting, babbling, walking and
sphincter control), the presence/absence of 37 typical Down
syndrome traits (dichotomous variables) and the evaluation of the
neurological impairment (mild, moderate and severe). In the absence
of the availability of cognitive test quantitative results, the
latter evaluation was based, following accurate visit of the
subject and an interview with the parents, on the agreed clinical
judgment of at least two pediatricians with long-time experience in
the follow-up of children with DS, aimed to assess if each subject
fell in the relative top, medium or lower class of cognitive and
global performance within the population.
Collection of blood samples
Blood samples (3 ml) were collected in EDTA-coated
blood collection tubes and processed within 2 h from blood draw
(27,28). All traceable identifiers were
removed prior to analysis to protect patient confidentiality, all
samples were analyzed anonymously (29). The sample was transferred in a new
tube and centrifuged at 1,250 × g for 10 min at room temperature
(centrifuge ALC 4235 A, rotor ALC T111). The plasma fraction was
isolated and centrifuged for a second time at 800 × g for 30 min at
room temperature (centrifuge ALC4214, rotor 6642) and the
supernatant was transferred to new tubes without touching the
pellet or the bottom of the tube, and divided in aliquots of 300
µl. All plasma samples were rapidly frozen and stored at
−80°C until needed for subsequent analysis.
The exclusion criteria of plasma samples from the
subsequent analysis were: Blood sample treatment after 2 h from the
draw, or evident contamination of plasma samples by residual
erythrocytes at the end of the treatment.
NEF separation from plasma and miRNA
isolation
A total of 300 µl of plasma were used to
obtain the NEF with Exosome Precipitation Solution (Serum/Plasma;
Macherey-Nagel GmbH, Düren, Germany), according to manufacturer's
protocol. Briefly the plasma was centrifuged at 10,000 × g for 15
min at 4°C to pellet intact cells, cellular debris and bigger
vesicles. Then the supernatant was mixed with 'Exosome
precipitation solution', incubated for 30 min on ice and finally
centrifuged at 600 × g for 5 min at 4°C. The supernatant was kept
for further analyses. The pellet was then resuspended in 300
µl RNAse free water and miRNAs were extracted using
'NucleoSpin miRNA Plasma' kit (Machery-Nagel GmbH), according to
the manufacturer's protocol.
For reverse transcription-quantitative polymerase
chain reaction (RT-qPCR) analyses, 3 µl of a 4.16 nM
solution of the synthetic miRNA cel-miR-39-3p from C.
elegans (custom synthesized by Integrated DNA Technologies,
Coralville, IO, USA) were added. miRNA fractions were eluted in 35
µl of nuclease-free water.
Western blotting
Proteins from NEF preparations obtained from C and
DS plasma and associated supernatants were extracted with reducing
SDS sample buffer (80 mM Tris, pH 6.8, 2% SDS, 7.5% glycerol, and
0.01% bromophenol blue) supplemented with 2% 2-mercaptoethanol for
5 min at 95°C and quantified by a Bradford's assay. A total of 50
µg of the preparations were loaded onto a 10%
acrylamide/bis-acrylamide gel, electrophoretically separated by
SDS-PAGE and transferred to polyvinylidene difluoride membranes
(30,31). Membranes were then blocked at 37°C
for 1 h in 5% non-fat milk, 10 mM Tris-HCl pH 7.5, 100 mM NaCl,
0.1% Tween-20 and probed with the following primary antibodies (all
at 1:500 dilution) at 4°C for 16 h: Mouse anti-Lamp1 (BD
Biosciences, San Jose, CA, USA; cat. no. 611042), mouse
anti-cluster of differentiation (CD)63 (EMD Millipore, Billerica,
MA, USA; clone RFAC4, cat. no. CBL553), rabbit anti-Adam10 (Acris
OriGene Technologies GmbH, Germany, clone 3C6, cat. no.
AP05830PU-N), mouse anti-TSG101 (Santa Cruz Biotechnology, Inc.,
Dallas, TX, USA; clone C-2, cat. no. sc-7964), mouse
anti-Flotillin2 (BD Biosciences, San Jose, CA, USA; cat. no.
610383), mouse anti-CD81 (Santa Cruz Biotechnology, Inc.; clone
B11, cat. no. sc-7637), mouse anti GM130 (BD Biosciences, San Jose,
CA, USA; clone 35/GM130, cat. no. 610822) (32), mouse anti-ApoAI (Thermo Fischer
Scientific, Inc.; clone 19H20L19, cat. no. 701239), mouse anti-Ago2
(Origene Technologies, Inc., Rockville, MD, USA; cat. no. TA352430)
and incubated in the presence of specific horseradish-peroxidase
conjugated immunoglobulin (Ig)G, Rabbit anti-mouse (cat. no.
A90-117P) or Goat anti-rabbit (cat. no. A120-101P) (Bethyl
Laboratories, Inc, USA, 1:3,000 dilution) at room temperature for 1
h (33,34). Immunoreactive bands were
identified using the Luminata classico detection system (EMD
Millipore). Images were acquired using a G:Box Chemi XT Imaging
system and quantified using Gene Tools software version 4.01
(Syngene, Frederick, MD, USA) (35).
Atomic force microscopy (AFM)
imaging
AFM imaging was conducted as described (36,37). Briefly, NEF preparations were
diluted 1:200 with deionized water. A total of 5-10 µl of
samples were then spotted onto freshly cleaved mica sheets (Grade
V-1, thickness 0.15 mm, size 10×10 mm). All mica substrates were
dried at room temperature and analyzed using a Nanosurf NaioAFM
(Nanosurf AG, Liestal, Switzerland), equipped with Tap190AI-G tips
(Budget Sensors; Innovative Solutions Bulgaria Ltd., Sofia,
Bulgaria). Images were captured in tapping mode; the scan size
ranged from 0.5-15 mm; the scan speed ranged from 0.6 to 1.5 sec ×
line.
miRNA expression by qPCR
Mature hsa-miR-16-5p, hsa-miR-99b-5p and
hsa-miR-144-3p were amplified by a two-step Taq-Man RT-PCR
analysis, using primers and probes obtained from Thermo Fisher
Scientific, Inc., (hsa-miR-144-3p, cat. no. 002676; hsa-miR-16-5p,
cat. no. 000391; hsa-miR-99b-5p, cat. no. 000436; cel-miR-39-3p,
cat. no. 000200). cDNA was synthesized from 5 µl of the each
RNA fraction in 15 µl reactions, using TaqMan MicroRNA
Reverse Transcription kit (Thermo Fisher Scientific, Inc.). The
reverse transcription reaction was performed by incubating the
samples at 16°C for 30 min, followed by incubation at 42°C for 30
min and 85°C for 5 min. The RT-qPCR reaction (20 µl)
contained 1.3 µl of reverse transcription product, 10
µl of Taq-Man 2X Universal PCR Master Mix and 1 µl of
the appropriate TaqMan MicroRNA Assay (20X) specific for the miRNA
targeted by the assay. The PCR mixtures were incubated at 95°C for
10 min, followed by 40 cycles at 95°C for 15 sec and 60°C for 60
sec. PCRs were performed in triplicate using the 7500 real time PCR
system (Applied Biosystems; Thermo Fisher Scientific, Inc.). The
expression of miRs was based on the 2−∆∆Cq method
(38), using cel-miR-39-3p as
reference.
Bioinformatic analysis
For functional enrichment analysis of gene ontology
terms and molecular signalling pathway, the following online
software packages were used: Database for Annotation, Visualization
and Integrated Discovery (DAVID) (39) and WebGestalt (40) and Kyoto Encyclopedia of Genes and
Genomes analysis (KEGG; www.genome.jp/kegg/). The human disease database
MalaCards (41) was used to
search the diseases and the genes associated with DS. The miRNA
targets were predicted by miRWALK (42). Considering that these procedures
may have a certain false positive rate, the target genes of miRNA
only considered the genes predicted by at least 7 procedures.
Statistical analysis
Descriptive statistics
The descriptive statistics for all quantitative
variables available in the dataset (minimum, maximum, first, second
and third quartile, mean ± standard deviation, number and
corresponding percentage of missing values) and the frequency
distributions of 37 typical DS pathologies (presence/absence of the
pathology-in absolute value and %-) have been computed.
The investigation on the linear association between
the quantitative variables collected on 15 individuals with DS has
been performed by calculating the Pearson correlation coefficient
ρ.
For the graphical representation of the significant
associations (correlation tests with P<0.05) between variables
under inspection, bubble charts were used that introduce in a
simple scatterplot a third dimension, namely the diameter of the
bubble. This dimension is proportional to the categorical variable
called 'number of comorbidities'. In detail, for each individual
with DS his/her number of comorbidities were summed, obtaining a
quantitative score; then the first (Q1) and third (Q3) quartile
were computed and the quantitative score were turned into the
following three categories: i) First quartile (Q1=17.5), each
subject with number of comorbidities <17.5 was classified as Low
comorbidities; ii) third quartile (Q3=21), each subject with number
of comorbidities >=17.5 and <21 was classified as Medium
comorbidities; and iii) each subject with number of comorbidities
>=21 was classified as High comorbidities.
Consequently, the scatterplots that highlight the
significative association between quantitative variables report the
dot circumference proportional to the number of assigned
comorbidities: Small dots when subjects with DS are classified as
'Low comorbidities' (green); medium size dots for 'Medium
comorbidities' (orange) and big dots when they are labeled as 'High
comorbidities' (red).
For each of the qualitative variables reported
(Table SI; considering also the
neurological severity), the median values of the miRNAs expression
levels in correspondence of subgroups of subjects defined by its
categories were computed. Then, the non-parametric Wilcoxon
signed-rank test (or Kruskal Wallis test when the subgroups are
>2) was computed in order to compare the medians obtained
(Table SII). These
non-parametric tests do not require any assumptions on the shape
distributions of the three miRNAs and permit analysis of the
possible association between clinical variables and miRNAs
expression. The unique significant associations were visualized by
means of a boxplot for each category (Low, Medium and High
comorbidities).
Heatmap generation
The data matrix with the investigated miRNAs was
visualized by means of a heatmap. Starting from the standardized
matrix (each miRNA has mean 0 and standard deviation 1), a heatmap
was generated to visualize the miRNA expression (low values are
represented with blue while high values with red). In this graph,
similar values were placed near each other according to the
clustering algorithm used in the analysis, thereby realizing one
dendrogram appended on the left y-axis.
k-medoids cluster analysis
Starting from the data matrix containing only miRNA
expression levels, we use the k-medoids cluster analysis
generating k=2 groups. For evaluating the distance between
subjects the Manhattan distance was used, which is based on
absolute value distance. This choice should provide more robust
results, whereas Euclidean distance would be influenced by unusual
values in the data matrix.
Inside each group, subjects exhibit a high degree of
similarity based on miRNA expression while subjects belonging to
different groups are as dissimilar as possible. The
k-medoids cluster analysis (known also with the acronym
PAM-Partitioning Around Medoids) searched for k
representative subjects (in the present study k=2) among all
the subjects in the dataset. These representative subjects are
called medoids (43) and the
algorithm assigns each patients of the dataset to nearest
medoid.
By means of silhouettes a graphical representation
of cluster analysis was provided where the entire clustering was
displayed by plotting the silhouette into a diagram highlighting
the cluster quality. Denoting Cluster 1 with C1
and Cluster 2 with C2, silhouette for the subject
i contained in C1 was:
where: a(i)=average dissimilarity of I to all subjects of
C1 and b(i)=average dissimilarity of I to all
subjects of C2.
From the ratio above, it is evident that each
silhouette belongs to the interval -1≤ s(i) ≤+1. This means
that when s(i) is close to 1 the within dissimilarities
a(i) is smaller than the between dissimilarities b(i)
and i is well classified. When s(i) is about 0, it is
not clear where i should be assigned (cluster
C1 or C2) and finally when
s(i) is close to -1, a(i) is larger than b(i)
and this means that i has been misclassified.
All statistical analyses were performed using R
version 3.4.4.
Results
NEF separation, characterization and
miRNA extraction
NEF samples were obtained with a commercial
polymer-based precipitation (PBP) kit as described in the Materials
and methods section. PBP is a simple, timesaving and cheap EV
concentration method (44)
characterized by low specificity. Formulations obtained by PBP
consist in a heterogeneous mix of nanosized particles that overlap
EVs in size and morphology, including HDLs.
NEF morphological properties were determined by AFM
(45). The preparation was
adsorbed on a freshly cleaved mica surface and imaged under ambient
conditions. Samples were composed by spherical nano-objects with a
size ranging from 10s to a few 100s of nm, which are visible
despite the presence of background material, very like the
polymeric matrix on which the precipitation kit is based (44,46) (Fig.
1A). The resulting fraction is mainly composed by small EVs and
HDLs, as can be deduced by the enrichment of specific EV markers
(47), including Lamp1, CD63,
Adam 10, TSG101, Ago2 and CD81, as compared with the supernatant
fractions by western blot analysis (Fig. 1B). On the other hand, NEF is
devoid of both GM130s and Flotillin, excluding the presence of
intracellular contaminants and implying that that large EVs are not
efficiently enriched by this protocol. On the other hand Apo-A1, a
biomarker characteristic of HDL lipoproteins (48), is detectable in supernatant
fractions and in a lesser extent also in NEFs (Fig. 1B). Overall, the above results
indicate that the final NEF is specifically enriched with nanosized
RNA-carriers, namely small EVs and HDLs.
 | Figure 1NEF characterization by AFM and
western blotting. (A) Morphological analysis by AFM of C and DS NEF
preparations. Images are representative of 3 independent
experiments. (B) NEF obtained from C and DS plasma and the
corresponding supernatants (SNC and SNDS)
were immunoblotted for the indicated extracellular vesicles and
high density lipoprotoeins markers (see text for details). Due to
antibody-unspecific signals, the original image of western blotting
membranes has been cropped. The uncropped, original image is
available upon request. DS, Down syndrome; C, healthy control; NEF,
nanoparticle-enriched fraction; AFM, atomic force microscopy; miR,
microRNA; SNC, SNDS, C and DS plasma corresponding
supernatants; M, marker; CD, cluster of differentiation. |
NEF miRNA extraction and expression
analysis
miRNAs were subsequently extracted from NEF using
commercial columns. First, Agilent miR microarray analysis aimed to
search for NEF miRNA content and was performed on samples derived
from 4 couples of siblings: 4 subjects with DS and their 4
correspondent healthy siblings. This allowed identification of the
most abundant miRNAs in the NEF recognized at least by 3 probes and
expressed in all the 8 subjects included in the analysis (Table SIII). Consequently, the
expression levels of the three most abundant miRNAs (hsa-miR-16-5p,
hsa-miR-144-3p and hsa-miR-99b-5p) were further assessed in a
larger cohort of DS and C by qPCR (15 couples of siblings;
n=30).
As presented in Fig.
2A-C, the expression levels of these three miRNAs were very
different among the couples of siblings considered and in general
the results demonstrated an increased average expression trend in
DS NEF with respect to C [miR-16-5p: Mean Relative Quantification
(RQ) of DS: 29.5±13.3, mean RQ of C: 11.9±3.3, Fig. 2D; miR-144-3p: Mean RQ of DS
66.6±18.7, mean RQ of C: 49.5±14.3, Fig. 2E; miR-99b-5p: Mean RQ of DS:
42.0±15.8, mean RQ of C: 27.7±10.6 Fig. 2F].
Since hsa-miR-16-5p, hsa-miR-144-3p and
hsa-miR-99b-5p have all been previously identified as
exosome-associated miRNAs and, in certain cases, associated with
distinct pathologies (49-52),
although never associated with the DS phenotype previously, it was
decided to statistically correlate the expression levels of the
three miRNAs with the available quantitative clinical data of the
participants (quantitative variables, Table SIV) in order to have insight into
their functional role in DS.
Correlation analysis of miRNA expression
with clinical features of subjects with DS
In Table I the
correlation matrix computed on the quantitative variables obtained
from the subjects with DS was reported (correlation test with
P<0.05). Bubble chart analysis (Fig. 3A-E) was subsequently performed
only on the significant associations, introducing a third
dimension, the diameter of the bubble. This dimension was
proportional to the categorical variable named 'number of
comorbidities', obtained as described in Material and methods
section and reported in Table SV
for each subject. An inverse significant linear association between
the clinical feature 'development babbling' expressed in months and
miR-16-5p and miR-144-3p expression levels was evident (Fig. 3A and B). From the bubble charts it
is clear that these two miRNAs demonstrated high expression levels
in subjects with DS with high number of comorbidities and beginning
to babble earlier.
 | Table ICorrelation matrix. |
Table I
Correlation matrix.
| Variables | Mother age | Father age | Birth weight
(g) | Sitting months | Babbling
months | Walking months | Sphincter control
months | miR-16-5p | miR-144-3p | miR-99b-5p |
|---|
| Mother age | 1,0000 | | | | | | | | | |
| Father age | 0,9197 | 1,0000 | | | | | | | | |
| Birth weight
(g) | −0,0982 | −0,1577 | 1,0000 | | | | | | | |
| Sitting months | 0,2095 | 0,2532 | −0,4586 | 1,0000 | | | | | | |
| Babbling
months | 0,1798 | −0,0132 | −0,1090 | −0,0157 | 1,0000 | | | | | |
| Walking months | 0,3488 | 0,1835 | 0,2421 | 0,2210 | 0,1501 | 1,0000 | | | | |
| Sphincter control
months | 0,2018 | 0,2531 | −0,3816 | 0,7177 | 0,2275 | 0,5274 | 1,0000 | | | |
| miR-16-5p | −0,1994 | −0,0205 | −0,0940 | −0,1439 | −0,7441 | −0,4159 | −0,2803 | 1,0000 | | |
| miR-144-3p | −0,1070 | 0,0451 | −0,2181 | 0,1122 | −0,8070 | −0,1911 | −0,0212 | 0,8383 | 1,0000 | |
| miR-99b-5p | −0,1954 | −0,0176 | −0,2089 | −0,1102 | −0,6024 | −0,1794 | −0,0208 | 0,8780 | 0,8516 | 1,0000 |
Concerning miR-99b-5p, it was observed that the
median value of the expression level was significantly increased
(Kruskal test: P=0.05) in individuals with DS presenting high
comorbidities (Fig. 4 and
Table SII) suggesting a
potentially more relevant role of miR-99b-5p upregulation in this
subgroup of subjects with DS. Instead, no significant differences
were identified for miR-144-3p and miR-16-5p (Table SII).
Fig. 3C highlights
a significant linear positive association between miR16-5p and
miR-144-3p expression. The same consideration can be extended to
miR16-5p versus miR-99b-5p and miR144-3p versus miR-99b-5p,
respectively (Fig. 3D and E).
Furthermore, individuals with DS with the highest expression levels
of miRNAs exhibited a high number of DS-associated typical clinical
features. It is interesting to outline that when the same
correlations were calculated by Pearson correlation analysis on the
healthy siblings (on the basis of the descriptive statistics
reported in Table SVI), we
obtained low values of ρ (miR-16-5p vs. miR-144-3p:
ρ= 0.1895; miR-16-5p vs. miR-99b-5p: ρ=
0.5123; miR-144-3p vs. miR-99b-5p: ρ=0.6820). This
well agrees with the finding that the three selected miRNAs were
upregulated in the DS cases in the present study, indicating a
possible role in DS-associated typical clinical anomalies, probably
mainly in subjects with 'high comorbidities'. As expected, by
repeating the same analysis on all the 30 participants (DS+C),
significant positive correlations were again obtained between
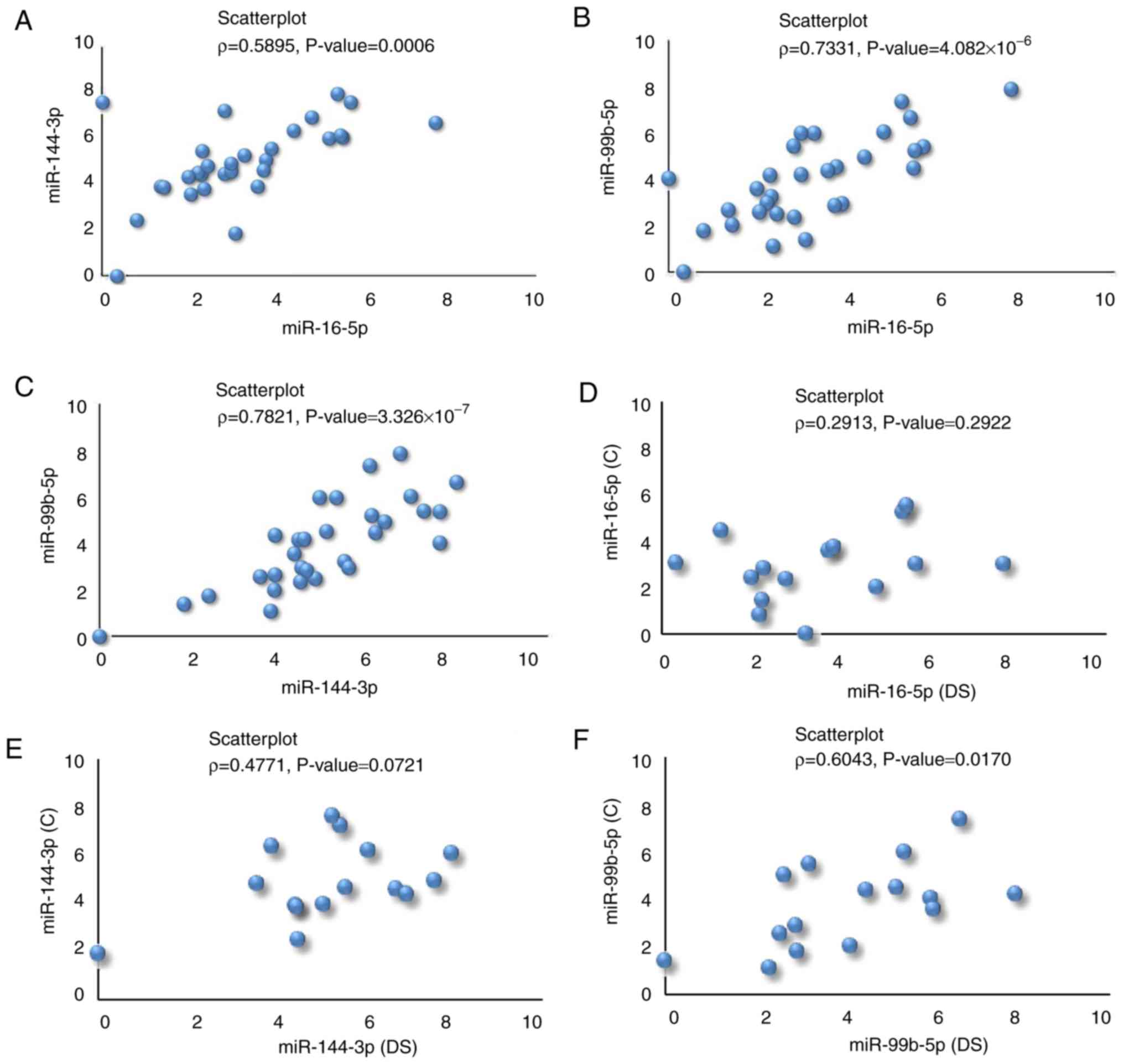
miRNAs (Fig. 5A-C).
ϱ was also computed on the expression level
of each miRNA comparing couples of siblings (Fig. 5D-F). The only significant positive
linear correlation was for miR-99b-5p (Fig. 5F). By computing ρ for
couples of unrelated subjects (C subjects and subjects with DS were
repeatedly randomized computing the correlation coefficients and
corresponding test) it was noted that there was no correlation for
miR-99b-5p (data and Figure provided upon request). This result
demonstrates the same expression trend of miR-99b-5p among
brothers, demonstrating the importance of the genetic background
when analyzing miRs.
miRNA expression was visualized using heatmaps
either on the entire sample (DS+C) (Fig. 6A), or separating C subjects and
subjects with DS (Fig. 6B and C,
respectively). The subjects with the highest miRNA expression (at
the top three positions of the graph) belonged to the DS group.
Furthermore, the C subjects at the top positions were siblings of
the DS with highest miRNA expression. Focusing the attention on
heatmaps in Fig. 6B and C, it was
evident that miRNA expression levels were different between DS and
C (in DS the color key is red-shifted compared with C); among the
subjects with DS there was only one case exhibiting very low miRNA
expression and notably this subject also presented low
comorbidities; the miRNAs less expressed were miR-144-3p and
miR-99b-5p in DS and C, respectively.
Using the Partitioning Around Medoids algorithm
(PAM) 2 clusters of subjects were identified (Fig. 7A): The first one, called
C1, contained subjects with low median and mean
expression values for each miRNA with a prevalence of C subjects
(55% of individuals in C1 were C which
corresponded to 73% of healthy siblings in the study, see Table SVII for major details). Cluster
2, called C2, contained a number of subjects with
DS with increased miRNA expression (60% of individuals in
C2 were DS which corresponded to 40% of subjects
with Down syndrome in the study). Furthermore, certain subjects
with DS in C2 were classified together with their
siblings (DS10 and C10, DS12 and C12) confirming similar miRNA
expression levels.
From the PAM algorithm it was also clear that miRNA
levels were able to generate clusters of subjects that were well
classified. In fact, the 'within dissimilarities' was smaller than
the 'between dissimilarities' and 73% of individuals were well
classified, as presented in Fig.
7B, where 22 subjects on 30 analyzed (on y-axis)
exhibited a width silhouette s(i) (on x-axis)
higher than 0.4. Therefore, this representation confirmed the
goodness of the classification induced by the miRNAs matrix (note
that s(i) reaches its maximum in correspondence of
1). Furthermore, in Fig. 7B each
subject with DS is colored using the classification based on number
of comorbidities: In C2 cluster, subjects with DS
with high comorbidities prevailed.
miRNAs gene target prediction and
enrichment study
The main putative targets of the 16 most abundant
miRNAs identified in the NEFs (Table
SIII) were subsequently predicted using the online software
miRWalk (6). The analysis in the
present study identified 2,515 targeted genes, including 25 of the
536 genes located on Hsa21. Functional annotation analysis of the
2,515 genes was performed next. In a GO analysis conducted by DAVID
(1,865 unique IDs identified), the candidate genes were analyzed in
all categories including biological process (Table II), molecular function (Table III) and cellular component
(Table IV). For the biological
process category, the first 4 enriched terms were referred to the
transcriptional process; the fifth biological term was 'nervous
system development' with 53 predicted genes included in this term
(P=0.0140). In the molecular function category, 1,010 genes were
included in 'protein binding' term and the other enriched terms
belonged essentially to 'transcription process' categories. In the
cellular component category, the 'nucleus' was the predominant term
with 620 associated-genes, the fourth term was 'neuronal cell body'
with 54 genes (P=0.0080). Therefore, >50 candidate miRNAs target
genes may have a role in 'nervous system development' or in the
'neuronal cell body' formation.
| Table IIGO Top 10 significant terms belonging
to the 'biological process' category. |
Table II
GO Top 10 significant terms belonging
to the 'biological process' category.
| GO term (biological
process) | Number of
genes | % | P-value | Benjamini corrected
P-value |
|---|
| Positive regulation
of transcription. DNA-templated | 102 | 5.5 |
1.40×10−11 |
6.80×10−08 |
| Positive regulation
of transcription from RNA polymerase II promoter | 163 | 8.7 |
2.80×10−11 |
7.10×10−08 |
| Negative regulation
of transcription from RNA polymerase II promoter | 126 | 6.8 |
2.50×10−10 |
4.20×10−07 |
| Transcription from
RNA polymerase II promoter | 96 | 5.1 |
1.40×10−09 |
1.70×10−06 |
| Nervous system
development | 53 | 2.8 |
1.40×10−05 |
1.40×10−02 |
| Protein
phosphorylation | 73 | 3.9 |
4.90×10−05 |
4.00×10−02 |
| Steroid hormone
mediated signaling pathway | 17 | 0.9 |
9.30×10−05 |
6.40×10−02 |
| Dentate gyrus
development | 9 | 0.5 |
1.00×10−04 |
6.00×10−02 |
| Stem cell
differentiation | 11 | 0.6 |
1.60×10−04 |
8.40×10−02 |
| Transcription.
DNA-templated | 241 | 12.9 |
1.9×10−4 |
9.1×10−2 |
| Table IIIGO Top 10 significant terms belonging
to 'molecular function' category. |
Table III
GO Top 10 significant terms belonging
to 'molecular function' category.
| GO term (molecular
function) | Number of
genes | % | P-value | Benjamini corrected
P-value |
|---|
| Protein
binding | 1,010 | 54.2 |
1.6×10−13 |
2.4×10−10 |
| Transcriptional
activator activity. RNA polymerase II core promoter proximal region
sequence-specific binding | 56 | 3.0 |
1.0×10−9 |
7.5×10−7 |
| Transcription
factor activity. sequence-specific DNA binding | 154 | 8.3 |
1.1×10−9 |
5.2×10−7 |
| Sequence-specific
DNA binding | 88 | 4.7 |
5.1×10−7 |
1.9×10−4 |
| RNA polymerase II
core promoter proximal region | 66 | 3.5 |
6.9×10−7 |
2.0×10−4 |
| sequence-specific
DNA binding | | | | |
| Protein
serine/threonine kinase activity | 66 | 3.5 |
5.2×10−6 |
1.3×10−3 |
| RNA polymerase II
regulatory region sequence-specific | 41 | 2.2 |
2.9×10−5 |
6.0×10−3 |
| DNA binding | | | | |
| GDP binding | 17 | 0.9 |
4.2×10−5 |
7.7×10−3 |
| Zinc ion
binding | 157 | 8.4 |
4.7×10−5 |
7.6×10−3 |
| Transcriptional
repressor activity. RNA polymerase II core promoter proximal region
sequence-specific binding | 26 | 1.4 |
6.6×10−5 |
9.5×10−3 |
| Table IVGO Top 10 significant terms belonging
to 'cellular component' category. |
Table IV
GO Top 10 significant terms belonging
to 'cellular component' category.
| GO term (cellular
component) | Number of
genes | % | P-value | Benjamini corrected
P-value |
|---|
| Nucleus | 620 | 33.2 |
1.1×10−7 |
8.2×10−5 |
| Nucleoplasm | 345 | 18.5 |
2.2×10−7 |
8.0×10−5 |
| Transcription
factor complex | 38 | 2.0 |
3.8×10−5 |
9.2×10−3 |
| Neuronal cell
body | 54 | 2.9 |
4.3×10−5 |
8.0×10−3 |
| Cytosol | 383 | 20.5 |
4.6×10−5 |
6.8×10−3 |
| Protein-DNA
complex | 11 | 0.6 |
5.7×10−5 |
7.1×10−3 |
| Perinuclear region
of cytoplasm | 89 | 4.8 |
1.5×10−4 |
1.6×10−2 |
| Golgi
apparatus | 116 | 6.2 |
2.1×10−4 |
1.9×10−2 |
| Golgi membrane | 84 | 4.5 |
3.1×10−4 |
2.5×10−2 |
| Cytoplasm | 567 | 30.4 |
3.6×10−4 |
2.6×10−2 |
To characterize the predominant pathways, putative
targets were searched by Kyoto Encyclopedia of Genes and Genomes
analysis, which revealed the term 'chronic myeloid leukemia'
(P<<0.01) with 23 target genes among the top 10 signal
pathways (Table V). A disease
association analysis of the predicted miRNA targets was also
performed using Webgestalt demonstrating that terms as 'leukemia
T-cell', 'leukemia, myeloid, acute', 'chromosome aberration',
'Precursor T-Cell Lymphoblastic Leukemia-Lymphoma', 'leukemia
myeloid' 'secondary leukemia' were among the top 10 statistically
significant (Table VI). In this
context, the predicted target genes of miR-16-5p, miR-99b-5p and
miR-144-3p were reported together with their expression values in
peripheral blood leucocytes from individuals with DS and controls
(Table SVIII). Expression values
were obtained from a publicly available transcriptome map resulted
from integration of microarray datasets through TRAM (Transcriptome
Mapper) software previously (53). A total of 12 predicted target
genes of miR-16-5p and 22 of miR-144-3p were identified to be
differentially expressed in individuals with DS respect to controls
(Ratio <0.8 and Ratio >1.2; in bold).
| Table VTop 10 significant terms belonging to
KEGG pathway. |
Table V
Top 10 significant terms belonging to
KEGG pathway.
| KEGG pathway | Number of
genes | % | P-value | Benjamini corrected
P-value |
|---|
| Pathways in
cancer | 77 | 4.1 |
1.8×10−8 |
5.0×10−6 |
| FoxO signaling
pathway | 35 | 1.9 |
3.6×10−7 |
5.0×10−5 |
| Chronic myeloid
leukemia | 23 | 1.2 |
1.5×10−6 |
1.4×10−4 |
| Glioma | 21 | 1.1 |
4.1×10−6 |
2.9×10−4 |
| Pancreatic
cancer | 20 | 1.1 |
1.6×10−5 |
9.1×10−4 |
| Prostate
cancer | 24 | 1.3 |
1.7×10−5 |
7.8×10−4 |
| Long-term
depression | 19 | 1.0 |
1.8×10−5 |
7.3×10−4 |
| AMPK signaling
pathway | 28 | 1.5 |
8.0×10−5 |
2.8×10−3 |
| cGMP-PKG signaling
pathway | 34 | 1.8 |
1.3×10−4 |
4.0×10−3 |
| Signaling pathways
regulating pluripotency of stem cells | 30 | 1.6 |
1.5×10−4 |
4.3×10−3 |
| Table VIDisease association of genes targeted
by miRNAs. |
Table VI
Disease association of genes targeted
by miRNAs.
| Disease association
(Term) | Number of
genes | FDR |
|---|
| Cell
transformation, neoplastic | 56 |
7.02×10−04 |
| Neoplastic
processes | 81 |
3.59×10−03 |
| Leukemia,
T-cell | 41 |
3.59×10−03 |
| Leukemia,
myeloid, acute | 45 |
3.59×10−03 |
| Neoplasms | 123 |
3.64×10−03 |
| Chromosome
aberrations | 64 |
4.38×10−03 |
| Costello
syndrome | 9 |
5.93×10−03 |
| Precursor T-cell
lymphoblastic leukemia-lymphoma | 22 |
7.53×10−03 |
| Leukemia,
myeloid | 50 |
9.16×10−03 |
| Secondary
leukemia | 20 |
1.25×10−02 |
Furthermore, the miRNA enrichment analysis conducted
by Webgestalt demonstrated that the major part (102 genes;
P<<0.01) of the predicted miRNA target genes were targeted by
miR-144-3p, which recognizes the binding site ATACTGT (Table VII).
| Table VIImiRNA enrichment analysis. Top 10
significant miRs targeting the gene list. |
Table VII
miRNA enrichment analysis. Top 10
significant miRs targeting the gene list.
| ID (putative
binding site and miR) | Number of
genes | FDR |
|---|
| AGCACTT, MIR-93,
MIR-302A, MIR-302B, MIR-302C, MIR-302D, MIR-372, | 134 | 0 |
| MIR-373, MIR-520E,
MIR-520A, MIR-526B, MIR-520B, MIR-520C, MIR-520D | | |
| ATACTGT,
MIR-144 | 102 | 0 |
| CAGTGTT, MIR-141,
MIR-200A | 124 | 0 |
| GCACTTT, MIR-17-5P,
MIR-20A, MIR-106A, MIR-106B, MIR-20B, MIR-519D | 190 | 0 |
| TGCTGCT, MIR-15A,
MIR-16, MIR-15B, MIR-195, MIR-424, MIR-497 | 199 | 0 |
| TGCACTT, MIR-519C,
MIR-519B, MIR-519A | 136 |
7.13×10−11 |
| AAGCACT,
MIR-520F | 85 |
1.33×10−10 |
| TTGCACT, MIR-130A,
MIR-301, MIR-130B | 119 |
8.25×10−9 |
| ATGCTGC, MIR-103,
MIR-107 | 75 |
2.31×10−8 |
| AGCACTT, MIR-93,
MIR-302A, MIR-302B, MIR-302C, MIR-302D, MIR-372, MIR-373, MIR-520E,
MIR-520A, MIR-526B, MIR-520B, MIR-520C, MIR-520D | 134 | 0 |
Among the predicted targets of miR-144-3p, two
DS-associated genes, DYRK1A, encoding for dual-specificity
tyrosine-(Y)-phosphorylation regulated kinase 1a and SIM1,
encoding for the transcriptional factor, have been respectively
reported at the 2nd and 26th positions in the Malacard gene list.
DYRK1A and SIM1 gene transcripts were predicted to be
targeted by miR-144-3p respectively by 8 and 6 prediction software
programs (miRWalk). TargetScan (release 7.1, June 2016) predicted 2
conserved putative binding sites for DYRK1A and 1 for
SIM1 gene transcripts in the 3′UTR portions.
Discussion
The present study compares the expression of miRNAs
carried by biogenic nanoparticles separated from plasma samples of
young individuals with DS and their healthy siblings. This is the
first study that tackles noncoding RNAs encased in EVs and HDLs in
association with DS pathogenesis, demonstrating specific
miRNA-signatures (19). So far,
only two studies have addressed an 'exosome approach' regarding
this syndrome, although focusing on proteins rather than on miRNAs
(54,55).
The adopted comprehensive approach of plasma NEF
analysis was very informative since it allowed identification novel
miRNAs probably involved in DS pathogenetic mechanisms. This
approach represents a first breakthrough in respect to previous
findings about miRNA and DS and sets the proof of concept for
further study. It will be interesting to indicate a sharp
stratification of EV and HDL miRNAs, provided the availability of
sufficient starting media to perform the very challenging
separation of the two nanoparticle populations.
Gene target prediction on the over-represented
miRNAs identified during the initial discovery phase by microarray
analysis and subsequent GO analysis of the predicted targets,
suggested the possible involvement of the identified miRNAs in
'nervous system development', 'neuronal cell body' and in the
pathogenesis of certain DS-associated diseases like leukemia. These
are interesting results since the nervous system is remarkably
affected during development and aging in individuals with DS.
Furthermore, it is well known that children with DS have a markedly
increased risk for a subtype of myeloid leukemia (ML) (56) classified by the World Health
Organization as ML-DS (Table
VIII).
| Table VIIIDown syndrome-associated diseases
according to Malacards. |
Table VIII
Down syndrome-associated diseases
according to Malacards.
| Related disease
ID | Score |
|---|
| Leukemia,
megakaryoblastic, with or without | 12.0 |
| Down syndrome,
somatic | |
| Acute
megakaryoblastic leukemia in Down syndrome | 12.0 |
| Acute
megakaryoblastic leukemia without | 11.9 |
| Down syndrome | |
| Ayme-gripp
syndrome | 11.1 |
| intellectual
disability | 10.4 |
| Leukemia | 10.4 |
| Macroglossia | 10.2 |
| Dementia | 10.1 |
| Myeloid
leukemia | 10.1 |
| Neuronitis | 10.1 |
Expression profiling of the three most abundant
miRNAs (miR-16-5p, miR-144-3p and miR-99b-5p) by RT-qPCR identified
a trend of dysregulation in individuals with DS compared with
C.
The expression of these miRNAs has not been
previously associated with DS and in general little data were
published regarding the expression of such miRNAs in human plasma.
miR-16-5p was identified in circulating exosomes of patients with
multiple myeloma (57) as well as
in circulating serum exosomes of patients with esophageal
adenocarcinoma (49). miR-99b-5p
is known as one of five most common miRNA in human plasma exosomes
by RNA deep sequencing (58).
Concerning function, miR-16-5p was previously
described in rat neurons as a negative regulator of dendritic
complexity and mediates brain derived neurotrophic factor
(BDNF)-induced dendritogenesis by regulating the translation of the
BDNF mRNA itself, supporting the hypothesis that miR-16-5p may be
important during neuronal development (59).
miR-144-3p has emerged as an important miRNA
implicated in certain human CNS pathologies (60,61). In a mouse model, miR-144-3p
targets a number of genes implicated in the control of neuronal
plasticity-associated signaling cascades, including PTEN, Spred1
and Notch1 (62).
Among the predicted targets of miR-144-3p, two
DS-associated genes, DYRK1A and the transcription factor
SIM1 were identified. While no experimental evidence
about miR-144-3p and both DS-genes expression regulation has
previously been demonstrated, it is known that DYRK1A expression
can be regulated by miRNAs (63,64).
DYRK1A kinase produced by Hsa21 serves a pivotal
role in the CNS and its overexpression in individuals with DS may
be implicated in cognitive impairments, therefore it has been
suggested as a putative therapeutic target for treating the
cognitive deficiencies observed in DS (58,65,66). Deregulation of DYRK1A levels are
also frequently observed in Alzheimer and Parkinson diseases
(67-69).
A recent study about the transcriptome map analysis
of DS vs. control human tissues (6) has demonstrated that in the
differential transcriptome map of brain, induced pluripotent stem
cells, blood and fibroblasts of DS and controls samples DYRK1A gene
has expression ratios of 1.51, 1.36, 1.30 and 1.37, respectively.
These ratios are consistent with a possible DYRK1A gene expression
alteration in trisomy 21 samples due to the presence of the Hsa21
in three copies in the cells of subjects with DS. Furthermore, it
is interesting to note that the SIM1 gene, which maps on human
chromosome 6 is homologous to SIM2 gene mapping on Hsa21. These two
genes are homologs to Drosophila single-minded (sim) gene
and have expression ratios in DS vs. control brain samples of 0.75
and 0.73 respectively. It was already proposed that the human SIM1
gene may be involved in the manifestation of brain development and
cognitive disability of DS (70).
Therefore, it would be interesting to
experimentally verify whether DYRK1A and SIM1 genes may be targeted
by miR-144-3p in order to shed light on their post-transcriptional
gene regulation. Notably, the amyloid precursor protein (APP) gene,
one of the most recognized genes to be overexpressed in DS
(6), was not identified among the
targets of the investigated miRNAs, on the basis of the selection
criteria. In future studies, it would be intriguing to study if APP
overexpression could be mediated by circulating miRNAs, not
enclosed in nanoparticles.
The correlation of the expression levels of the
three identified miRNAs with clinical features of the subjects with
DS by a non-parametric test like Kruskal Wallis allowed
identification of an inverse significant linear association between
the development of babbling (expressed in months) and miR-16-5p and
miR-144-3p in subjects with DS, which also demonstrates a high
number of comorbidities. Kruskal Wallis has been applied as the
present study used a small sample size where the quantitative
variables analyzed, in particular miRNAs, have extremely asymmetric
distributions. On the basis of the results, it can be hypothesized
that miR-16-5p and miR-144-3p may be involved in controlling the
timing of the babbling development: Indeed the role of miRNAs has
been suggested by previous studies about speech and language
impairment (71).
By means of two exploratory statistical methods,
namely heatmap and cluster analysis it was demonstrated that
individuals with DS expressing high levels of the three miRNAs
displayed also a high number of DS-associated typical clinical
features and allowed us to generate clusters of subjects well
classified on the basis of the three miRNA expression data. Cluster
analysis is not frequently applied to biological data, but together
with heatmaps, follows the visual mining philosophy. This is
an interesting strategy for understanding association and patterns
into the data, visualizing simple and understandable plots.
In conclusion, the results of the present study
demonstrate the importance of the comprehensive analysis of plasma
miRNA vehicles, enriching for different substructures, which may
lead to the discovery of unknown pathways. In the case of DS, this
approach could lead to the discovery of novel molecular entities
(miRNA, proteins, metabolites) that are involved in the pathology
but have never been identified earlier.
Supplementary Materials
Funding
The present study was partially supported by a
BIOMANE grant (University of Brescia, Brescia, Italy) to AR, AS,
GDP and PB. AR was also supported from Fondo per il Finanziamento
delle Attività Base di Ricerca (Ministero dell'Istruzione,
dell'Università e della Ricerca) and University of Brescia research
fund (ex 60%). This study was partially supported by Centro
Bresciano Down (CBD), Brescia, Italy to AR and DR.
Availability of data and materials
The analyzed datasets generated during the study
are available from the corresponding author on reasonable
request.
Authors' contributions
AR and DR conceived the project. MC, FA, AP, CL and
GC were involved in sample and medical record recruitment. AS, MV,
AR wrote the manuscript. AR, LP, SB performed NEF separation and
biophysical and biochemical characterization. AS, TF performed the
qPCR experiments; AS, EA performed the GO analysis. GDP, GA and PB
were involved in the study conception and design, analysis and
interpretation of data, and critical revision of the manuscript.
All authors read and approved the manuscript.
Ethics approval and consent to
participate
The present study was approved by the competent
Ethics Committee of Sant'Orsola-Malpighi Hospital in Bologna,
approval number: 39/2013/U/Tess.
Patient consent for publication
Informed consent was obtained from the subjects for
participation in this study.
Competing interests
No potential conflict of interest was reported by
the authors.
Acknowledgments
Not applicable.
References
|
1
|
Strippoli P, Pelleri MC, Caracausi M,
Vitale L, Piovesan A, Locatelli C, Mimmi MC, Berardi AC, Ricotta D,
Radeghieri A, et al: An integrated route to identifying new
pathogenesis-based therapeutic approaches for trisomy 21 (Down
Syndrome) following the thought of Jérôme Lejeune. Sci Postprint.
1:e000102013. View Article : Google Scholar
|
|
2
|
Delabar JM, Allinquant B, Bianchi D,
Blumenthal T, Dekker A, Edgin J, O'Bryan J, Dierssen M, Potier MC,
Wiseman F, et al: Changing paradigms in down syndrome: The first
international conference of the trisomy 21 research society. Mol
Syndromol. 7:251–261. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Lejeune J, Gauthier M and Turpin R: Human
chromosomes in tissue cultures. C R Hebd Seances Acad Sci.
248:602–603. 1959.In French. PubMed/NCBI
|
|
4
|
Chen YQ, Li T, Guo WY, Su FJ and Zhang YX:
Identification of altered pathways in down syndrome-associated
congenital heart defects using an individualized pathway aberrance
score. Genet Mol Res. 15:2016.
|
|
5
|
Pelleri MC, Gennari E, Locatelli C,
Piovesan A, Caracausi M, Antonaros F, Rocca A, Donati CM, Conti L,
Strippoli P, et al: Genotype-phenotype correlation for congenital
heart disease in down syndrome through analysis of partial trisomy
21 cases. Genomics. 109:391–400. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Pelleri MC, Cattani C, Vitale L, Antonaros
F, Strippoli P, Locatelli C, Cocchi G, Piovesan A and Caracausi M:
Integrated quantitative transcriptome maps of human trisomy 21
tissues and cells. Front Genet. 9:1252018. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Pelleri MC, Cicchini E, Locatelli C,
Vitale L, Caracausi M, Piovesan A, Rocca A, Poletti G, Seri M,
Strippoli P and Cocchi G: Systematic reanalysis of partial trisomy
21 cases with or without down syndrome suggests a small region on
21q22.13 as critical to the phenotype. Hum Mol Genet. 25:2525–2538.
2016.PubMed/NCBI
|
|
8
|
Caracausi M, Ghini V, Locatelli C, Mericio
M, Piovesan A, Antonaros F, Pelleri MC, Vitale L, Vacca RA, Bedetti
F, et al: Plasma and urinary metabolomic profiles of down syndrome
correlate with alteration of mitochondrial metabolism. Sci Rep.
8:29772018. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ambros V: The functions of animal
microRNAs. Nature. 431:350–355. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Bartel DP: MicroRNAs: Genomics,
biogenesis, mechanism, and function. Cell. 116:281–297. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Bartel DP: MicroRNAs: Target recognition
and regulatory functions. Cell. 136:215–233. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Turchinovich A, Weiz L, Langheinz A and
Burwinkel B: Characterization of extracellular circulating
microRNA. Nucleic Acids Res. 39:7223–7233. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Arroyo JD, Chevillet JR, Kroh EM, Ruf IK,
Pritchard CC, Gibson DF, Mitchell PS, Bennett CF,
Pogosova-Agadjanyan EL, Stirewalt DL, et al: Argonaute2 complexes
carry a population of circulating microRNAs independent of vesicles
in human plasma. Proc Natl Acad Sci USA. 108:5003–5008. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Vickers KC, Palmisano BT, Shoucri BM,
Shamburek RD and Remaley AT: MicroRNAs are transported in plasma
and delivered to recipient cells by high-density lipoproteins. Nat
Cell Biol. 13:423–433. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Valadi H, Ekström K, Bossios A, Sjöstrand
M, Lee JJ and Lötvall JO: Exosome-mediated transfer of mRNAs and
microRNAs is a novel mechanism of genetic exchange between cells.
Nat Cell Biol. 9:654–659. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Michell DL and Vickers KC: Lipoprotein
carriers of microRNAs. Biochim Biophys Acta. 1861:2069–2074. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Cheng L, Sharples RA, Scicluna BJ and Hill
AF: Exosomes provide a protective and enriched source of miRNA for
biomarker profiling compared to intracellular and cell-free blood.
J Extracell Vesicles. 3:2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Mateescu B, Kowal EJ, van Balkom BW,
Bartel S, Bhattacharyya SN, Buzás EI, Buck AH, de Candia P, Chow
FW, Das S, et al: Obstacles and opportunities in the functional
analysis of extracellular vesicle RNA-an ISEV position paper. J
Extracell Vesicles. 6:12860952017. View Article : Google Scholar
|
|
19
|
Li K, Rodosthenous RS, Kashanchi F,
Gingeras T, Gould SJ, Kuo LS, Kurre P, Lee H, Leonard JN, Liu H, et
al: Advances, challenges, and opportunities in extracellular RNA
biology: Insights from the NIH exRNA strategic workshop. JCI
Insight. 3:989422018. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Momen-Heravi F, Saha B, Kodys K, Catalano
D, Satishchandran A and Szabo G: Increased number of circulating
exosomes and their microRNA cargos are potential novel biomarkers
in alcoholic hepatitis. J Transl Med. 13:2612015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Berardocco M, Radeghieri A, Busatto S,
Gallorini M, Raggi C, Gissi C, D'Agnano I, Bergese P, Felsani A and
Berardi AC: RNA-seq reveals distinctive RNA profiles of small
extracellular vesicles from different human liver cancer cell
lines. Oncotarget. 8:82920–82939. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Svobodová I, Korabečná M, Calda P, Břešťák
M, Pazourková E, Pospíšilová Š, Krkavcová M, Novotná M and Hořínek
A: Differentially expressed miRNAs in trisomy 21 placentas. Prenat
Diagn. 36:775–784. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Xu Y, Li W, Liu X, Chen H, Tan K, Chen Y,
Tu Z and Dai Y: Identification of dysregulated microRNAs in
lymphocytes from children with Down syndrome. Gene. 530:278–286.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Alexandrov PN, Percy ME and Lukiw WJ:
Chromosome 21-encoded microRNAs (mRNAs): Impact on down's syndrome
and trisomy-21 linked disease. Cell Mol Neurobiol. 38:769–774.
2018. View Article : Google Scholar
|
|
25
|
Brás A, Rodrigues AS, Gomes B and Rueff J:
Down syndrome and microRNAs. Biomed Rep. 8:11–16. 2018.PubMed/NCBI
|
|
26
|
Lim JH, Kim DJ, Lee DE, Han JY, Chung JH,
Ahn HK, Lee SW, Lim DH, Lee YS, Park SY and Ryu HM: Genome-wide
microRNA expression profiling in placentas of fetuses with Down
syndrome. Placenta. 36:322–328. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Lacroix R, Judicone C, Mooberry M,
Boucekine M, Key NS and Dignat-George F: Standardization of
pre-analytical variables in plasma microparticle determination:
Results of the International Society on Thrombosis and Haemostasis
SSC Collaborative workshop. J Thromb Haemost. 11:1190–1193. 2013.
View Article : Google Scholar
|
|
28
|
Witwer KW, Buzás EI, Bemis LT, Bora A,
Lässer C, Lötvall J, Nolte-'t Hoen EN, Piper MG, Sivaraman S, Skog
J, et al: Standardization of sample collection, isolation and
analysis methods in extracellular vesicle research. J Extracell
Vesicles. 2:2013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Paolini L, Di Noto G, Maffina F,
Martellosio G, Radeghieri A, Luigi C and Ricotta D: Comparison of
Hevylite™ IgA and IgG assay with conventional techniques for the
diagnosis and follow-up of plasma cell dyscrasia. Ann Clin Biochem.
52:337–345. 2015. View Article : Google Scholar
|
|
30
|
Alvisi G, Roth DM, Camozzi D, Pari GS,
Loregian A, Ripalti A and Jans DA: The flexible loop of the human
cytomegalovirus DNA polymerase processivity factor ppUL44 is
required for effi-cient DNA binding and replication in cells. J
Virol. 83:9567–9576. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Alvisi G, Avanzi S, Musiani D, Camozzi D,
Leoni V, Ly-Huynh JD and Ripalti A: Nuclear import of HSV-1 DNA
polymerase proces-sivity factor UL42 is mediated by a C-terminally
located bipartite nuclear localization signal. Biochemistry.
47:13764–13777. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Montis C, Zendrini A, Valle F, Busatto S,
Paolini L, Radeghieri A, Salvatore A, Berti D and Bergese P: Size
distribution of extracellular vesicles by optical correlation
techniques. Colloids Surf B Biointerfaces. 158:331–338. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Paolini L, Radeghieri A, Civini S, Caimi L
and Ricotta D: The Epsilon Hinge-Ear region regulates membrane
localization of the AP-4 complex. Traffic. 12:1604–1619. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Alvisi G, Paolini L, Contarini A, Zambarda
C, Di Antonio V, Colosini A, Mercandelli N, Timmoneri M, Palù G,
Caimi L, et al: Intersectin goes nuclear: Secret life of an
endocytic protein. Biochem J. 475:1455–1472. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Paolini L, Orizio F, Busatto S, Radeghieri
A, Bresciani R, Bergese P and Monti E: Exosomes secreted by HeLa
cells shuttle on their surface the plasma membrane-associated
sialidase NEU3. Biochemistry. 56:6401–6408. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Radeghieri A, Savio G, Zendrini A, Di Noto
G, Salvi A, Bergese P and Piovani G: Cultured human amniocytes
express hTERT, which is distributed between nucleus and cytoplasm
and is secreted in extracellular vesicles. Biochem Biophys Res
Commun. 483:706–711. 2017. View Article : Google Scholar
|
|
37
|
Vescovi R, Monti M, Moratto D, Paolini L,
Consoli F, Benerini L, Melocchi L, Calza S, Chiudinelli M, Rossi G,
et al: Collapse of the plasmacytoid dendritic cell compartment in
advanced cutaneous melanomas by components of the tumor cell
secretome. Cancer Immunol Res. 7:12–28. 2019. View Article : Google Scholar
|
|
38
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
39
|
Huang da W, Sherman BT and Lempicki RA:
Systematic and integrative analysis of large gene lists using DAVID
bioinfor-matics resources. Nat Protoc. 4:44–57. 2009. View Article : Google Scholar
|
|
40
|
Wang J, Vasaikar S, Shi Z, Greer M and
Zhang B: WebGestalt 2017: A more comprehensive, powerful, flexible
and interactive gene set enrichment analysis toolkit. Nucleic Acids
Res. 45:W130–W137. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Rappaport N, Twik M, Plaschkes I, Nudel R,
Iny Stein T, Levitt J, Gershoni M, Morrey CP, Safran M and Lancet
D: MalaCards: An amalgamated human disease compendium with diverse
clinical and genetic annotation and structured search. Nucleic
Acids Res. 45:D877–D887. 2017. View Article : Google Scholar
|
|
42
|
Dweep H and Gretz N: miRWalk2.0: A
comprehensive atlas of microRNA-target interactions. Nat Methods.
12:6972015. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Kaufman L and Rousseeuw PJ: Clustering by
means of medoids Statistical Data Analysis Based on the L1 Norm.
North-Holland/Elsevier Amsterdam: pp. 405–416. 1987
|
|
44
|
Andreu Z, Rivas E, Sanguino-Pascual A,
Lamana A, Marazuela M, González-Alvaro I, Sánchez-Madrid F, de la
Fuente H and Yáñez-Mó M: Comparative analysis of EV isolation
procedures for miRNAs detection in serum samples. J Extracell
Vesicles. 5:316552016. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Paolini L, Zendrini A and Radeghieri A:
Biophysical properties of extracellular vesicles in diagnostics.
Biomark Med. 12:383–391. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Paolini L, Zendrini A, Di Noto G, Busatto
S, Lottini E, Radeghieri A, Dossi A, Caneschi A, Ricotta D and
Bergese P: Residual matrix from different separation techniques
impacts exosome biological activity. Sci Rep. 6:235502016.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Théry C, Witwer KW, Aikawa E, Alcaraz MJ,
Anderson JD, Andriantsitohaina R, Antoniou A, Arab T, Archer F,
Atkin-Smith GK, et al: Minimal information for studies of
extracellular vesicles 2018 (MISEV2018): A position statement of
the international society for extracellular vesicles and update of
the MISEV2014 guidelines. J Extracell Vesicles. 7:15357502018.
View Article : Google Scholar
|
|
48
|
Karimi N, Cvjetkovic A, Jang SC,
Crescitelli R, Hosseinpour Feizi MA, Nieuwland R, Lötvall J and
Lässer C: Detailed analysis of the plasma extracellular vesicle
proteome after separation from lipoproteins. Cell Mol Life Sci.
75:2873–2886. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Huang X, Yuan T, Tschannen M, Sun Z, Jacob
H, Du M, Liang M, Dittmar RL, Liu Y, Liang M, et al:
Characterization of human plasma-derived exosomal RNAs by deep
sequencing. BMC Genomics. 14:3192013. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Shurtleff MJ, Temoche-Diaz MM, Karfilis
KV, Ri S and Schekman R: Y-box protein 1 is required to sort
microRNAs into exosomes in cells and in a cell-free reaction.
Elife. 5:e192762016. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Pu C, Huang H, Wang Z, Zou W, Lv Y, Zhou
Z, Zhang Q, Qiao L, Wu F and Shao S: Extracellular
vesicle-associated mir-21 and mir-144 are markedly elevated in
serum of patients with hepato-cellular carcinoma. Front Physiol.
9:9302018. View Article : Google Scholar
|
|
52
|
Garcia-Contreras M, Shah SH, Tamayo A,
Robbins PD, Golberg RB, Mendez AJ and Ricordi C: Plasma-derived
exosome characterization reveals a distinct microRNA signature in
long duration Type 1 diabetes. Sci Rep. 7:59982017. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Pelleri MC, Piovesan A, Caracausi M,
Berardi AC, Vitale L and Strippoli P: Integrated differential
transcriptome maps of acute megakaryoblastic leukemia (AMKL) in
children with or without down syndrome (DS). BMC Med Genomics.
7:632014. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Hamlett ED, Goetzl EJ, Ledreux A,
Vasilevko V, Boger HA, LaRosa A, Clark D, Carroll SL,
Carmona-Iragui M, Fortea J, et al: Neuronal exosomes reveal
Alzheimer's disease biomarkers in Down syndrome. Alzheimers Dement.
13:541–549. 2017. View Article : Google Scholar
|
|
55
|
Gauthier SA, Perez-Gonzalez R, Sharma A,
Huang FK, Alldred MJ, Pawlik M, Kaur G, Ginsberg SD, Neubert TA and
Levy E: Enhanced exosome secretion in down syndrome brain-a
protective mechanism to alleviate neuronal endosomal abnormalities.
Acta Neuropathol Commun. 5:652017. View Article : Google Scholar
|
|
56
|
Zhang L, Pan L, Xiang B, Zhu H, Wu Y, Chen
M, Guan P, Zou X, Valencia CA, Dong B, et al: Potential role of
exosome-associated microRNA panels and in vivo environment to
predict drug resistance for patients with multiple myeloma.
Oncotarget. 7:30876–30891. 2016.PubMed/NCBI
|
|
57
|
Chiam K, Wang T, Watson DI, Mayne GC,
Irvine TS, Bright T, Smith L, White IA, Bowen JM, Keefe D, et al:
Circulating serum exosomal miRNAs as potential biomarkers for
esophageal adenocarcinoma. J Gastrointest Surg. 19:1208–1215. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Neumann F, Gourdain S, Albac C, Dekker AD,
Bui LC, Dairou J, Schmitz-Afonso I, Hue N, Rodrigues-Lima F,
Delabar JM, et al: DYRK1A inhibition and cognitive rescue in a Down
syndrome mouse model are induced by new fluoro-DANDY derivatives.
Sci Rep. 8:28592018. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Antoniou A, Khudayberdiev S, Idziak A,
Bicker S, Jacob R and Schratt G: The dynamic recruitment of TRBP to
neuronal membranes mediates dendritogenesis during development.
EMBO Rep. 19:e448532018. View Article : Google Scholar :
|
|
60
|
Cheng C, Li W, Zhang Z, Yoshimura S, Hao
Q, Zhang C and Wang Z: MicroRNA-144 is regulated by activator
protein-1 (AP-1) and decreases expression of Alzheimer
disease-related a disintegrin and metalloprotease 10 (ADAM10). J
Biol Chem. 288:13748–13761. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Katsuura S, Kuwano Y, Yamagishi N,
Kurokawa K, Kajita K, Akaike Y, Nishida K, Masuda K, Tanahashi T
and Rokutan K: MicroRNAs miR-144/144* and miR-16 in peripheral
blood are potential biomarkers for naturalistic stress in healthy
Japanese medical students. Neurosci Lett. 516:79–84. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Murphy CP, Li X, Maurer V, Oberhauser M,
Gstir R, Wearick-Silva LE, Viola TW, Schafferer S, Grassi-Oliveira
R, Whittle N, et al: MicroRNA-mediated rescue of fear extinction
memory by miR-144-3p in extinction-impaired mice. Biol Psychiatry.
81:979–989. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Zhang Y, Liao JM, Zeng SX and Lu H: p53
downregulates Down syndrome-associated DYRK1A through miR-1246.
EMBO Rep. 12:811–817. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
da Costa Martins PA, Salic K, Gladka MM,
Armand AS, Leptidis S, el Azzouzi H, Hansen A, Coenen-de Roo CJ,
Bierhuizen MF, van der Nagel R, et al: MicroRNA-199b targets the
nuclear kinase Dyrk1a in an auto-amplification loop promoting
calcineurin/NFAT signalling. Nat Cell Biol. 12:1220–1227. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Feki A and Hibaoui Y: DYRK1A protein, A
promising therapeutic target to improve cognitive deficits in down
syndrome. Brain Sci. 8:E1872018. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Najas S, Arranz J, Lochhead PA, Ashford
AL, Oxley D, Delabar JM, Cook SJ, Barallobre MJ and Arbonés ML:
DYRK1A-mediated cyclin D1 degradation in neural stem cells
contributes to the neurogenic cortical defects in down syndrome.
EBioMedicine. 2:120–134. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Arbones ML, Thomazeau A, Nakano-Kobayashi
A, Hagiwara M and Delabar JM: DYRK1A and cognition: A lifelong
relationship. Pharmacol Ther. 194:199–221. 2019. View Article : Google Scholar
|
|
68
|
Janel N, Sarazin M, Corlier F, Corne H, de
Souza LC, Hamelin L, Aka A, Lagarde J, Blehaut H, Hindié V, et al:
Plasma DYRK1A as a novel risk factor for Alzheimer's disease.
Transl Psychiatry. 4:e4252014. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Barallobre MJ, Perier C, Bove J, Laguna A,
Delabar JM, Vila M and Arbonés ML: DYRK1A promotes dopaminergic
neuron survival in the developing brain and in a mouse model of
Parkinson's disease. Cell Death Dis. 5:e12892014. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Chrast R, Scott HS, Chen H, Kudoh J,
Rossier C, Minoshima S, Wang Y, Shimizu N and Antonarakis SE:
Cloning of two human homologs of the Drosophila single-minded gene
SIM1 on chromosome 6q and SIM2 on 21q within the Down syndrome
chromosomal region. Genome Res. 7:615–624. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Fu L, Shi Z, Luo G, Tu W, Wang X, Fang Z
and Li X: Multiple microRNAs regulate human FOXP2 gene expression
by targeting sequences in its 3′ untranslated region. Mol Brain.
7:712014. View Article : Google Scholar
|