Introduction
Individuals with diabetes often suffer from coronary
artery diseases that contribute to their morbidity or mortality
(1). The high incidence rate and
unfavorable prognosis for patients with diabetes who develop
coronary artery disease are associated with the diabetes-mediated
impairment of angiogenesis (1,2).
Inhibited angiogenesis contributes to the impaired coronary
collateral vessel formation and cardiac repair in patients with
diabetes (3).
Accumulating evidence suggests that bone
marrow-derived endothelial progenitor cells (EPCs) contribute to
vascular repair. Preclinical and clinical studies have confirmed
that EPCs improve heart function and delay cardiac remodeling after
ischemic damage, by reducing fibrosis and increasing angiogenesis;
in addition, autologous EPC treatment prevents immune rejection
(4,5). However, diabetic EPCs are
insufficient in function and survival (6), thus limiting the self-repair
capability of diabetic myocardial infarction (DMI) (7) and hindering autologous cell therapy
(8). Effective methods of
restoring diabetic EPCs to improve their angiogenic capability may
be required to maximize the benefits of autologous stem cell
therapy.
Hedgehog proteins are well-known morphogens that
have critical roles in several tissues during embryonic and
postnatal development and in adult life. The hedgehog protein
family comprises Desert hedgehog, Indian hedgehog and Sonic
hedgehog (Shh) (9). The Shh
signal has been studied and characterized extensively during
embryogenesis, tissue regeneration and repair after severe injury.
This pathway occurs when the Shh protein interacts with its
receptor patched 1, removing the inhibition of another Shh pathway
receptor, the smoothened frizzled class receptor (Smo), and
activating the transcription factor GLI family zinc finger (Gli),
which induces the expression of downstream target genes, including
Gli1 itself, resulting in various cellular activities (10-14). Our laboratory has recently
demonstrated that the Shh pathway activity is remarkably reduced in
diabetic EPCs, and that activation of the Shh pathway with the Smo
receptor agonist SAG improves the function of diabetic EPCs
(15). However, the Shh pathway
in diabetic EPC survival or apoptosis remains poorly understood. In
addition, the efficacy of EPC therapy for ischemic diseases is
compromised by the poor survival of these cells under ischemic
conditions. Hence, the present study examined the effect of the Shh
pathway on diabetic EPC apoptosis under hypoxic conditions.
In specific, the present study investigated the
effect of Shh upregulation on diabetic EPC apoptosis when
transplanted into the ischemic hearts of diabetic mice, the EPC
repair capability and cardiac function of diabetic mice with
myocardial infarction (MI). In addition, in vitro
experiments were performed to investigate the effects and molecular
mechanism of Shh on diabetic EPC apoptosis under basal or hypoxic
conditions.
Materials and methods
Type-1 diabetic mouse model
This study was conducted in accordance with the
recommendations of the Institutional Animal Ethics Committee of
Guangzhou Medical University (Guangzhou, China). The protocol was
approved by the Institutional Animal Ethics Committee of Guangzhou
Medical University.
The experiments were performed on healthy adult male
C57/B6 mice (weight, 20±2 g; age, 6-8 weeks) obtained from
Guangdong Medical Laboratory Animal Center (Foshan, China). The
animals were housed in a pathogen-free environment, in a 12-h
light/dark cycle, and given access to food and water ad
libitum. The animals were given 2 weeks to acclimate prior to
the experiments.
The MI model was established as previously described
(13). Mice were injected
intraperitoneally with streptozotocin (STZ; 45 mg/kg;
Sigma-Aldrich; Merck KGaA) dissolved in sterile citrate buffer
(0.05 mol/l sodium citrate, pH 4.5). STZ or citrate buffer (vehicle
control) was administered daily for 5 consecutive days during the
first week of the study. Blood samples were collected from the vena
caudalis. Whole blood glucose levels were measured using the
glucose analyzer One Touch Ultra Mini Blood Glucose Monitoring
System (Johnson & Johnson), and mice with a blood glucose level
≥16.7 mmol/l were considered diabetic. A total of 83 mice were
established as type-1 diabetic models, and 22 mice were used as
vehicle controls.
Bone marrow (BM) EPC culture and drug
treatment
Human umbilical vein endothelial cells (HUVECs;
American Type Culture Collection) were used as a positive control
in the characterization of the EPCs, and the HUVECs were supplied
by another team from our lab. EPCs were generated from BM
mononuclear cells (MNCs), as previously described (15). In brief, the BM was flushed out
from tibias and femurs, and BM-derived MNCs were isolated by
density gradient centrifugation using Histopaque 1083
(Sigma-Aldrich; Merck KGaA) and resuspended. BM-MNCs isolated from
mice were counted and plated (1×106 cells/well) on
fibronectin-coated 24-well plates (BD Biosciences) and then grown
in endothelial cell basal medium-2 (EBM-2) supplemented with 5%
fetal calf serum (FCS) containing EPC growth cytokine cocktail
[hydrocortisone, human basic fibroblast growth factor-B, vascular
endothelial growth factor (VEGF), R3-insullin-like growth factor-1,
ascorbic acid, human epidermal growth factor and gentamicin
sulfate-amphotericin; all from Lonza Group Ltd.]. After 3 days in
culture, non-adherent cells were removed, and fresh endothelial
cell growth medium-2 (EGM-2; EBM-2 plus 5% FCS and growth factor
cocktail) was added every two days. All cells used in molecular
assays were performed after day 7. Shh protein (0.5 µg/ml;
Merck KGaA), p53 agonist Tenovin-1 (10 µM; Selleck
Chemicals) or antioxidant Tempol (0.5 mM; Merck KGaA) were
administered for 24 h. BMI1 proto-oncogene (Bmi1)-targeting small
interfering (si)RNA or Adv-Shh were applied for 48 h. Fifty-eight
mice were used for establishing the BM-derived EPCs.
Dil-conjugated acetylated low density
lipoprotein (Dil-acLDL) and lectin double staining
After 7 day in culture, attached EPCs were labeled
with Dil-acLDL (10 µg/ml; Invitrogen; Thermo Fisher
Scientific, Inc.) and FITC-labeled UEA-1 lectin (10 µg/ml;
Sigma-Aldrich; Merck KGaA) for 1 h. After washing with PBS three
times, cells were observed under an inverted fluorescent microscope
(Nikon Corporation). Cells demonstrating double-positive
fluorescence of Dil-acLDL and lectin were identified as
differentiating EPCs. EPCs from one mouse were used for
fluorescence staining.
Immunofluorescence staining of CD31 and
CD34
After 7 days in culture, attached EPCs were fixed by
4% paraformaldehyde for 10 min at room temperature (RT). After
washing with PBS three times, cells were stained with anti-CD31
antibody (cat. no. 550274; 1:100; BD Biosciences) or anti-CD34
antibody (cat. no. ab81289; 1:100; Abcam) at 4°C overnight. Next,
the cells were incubated with Dylight 488-conjugated goat anti-rat
secondary antibody (cat. no. A23220; 1:500; Abbkine Scientific,
Co., Ltd.) or Dylight 594-conjugated goat anti-rabbit secondary
antibody (cat. no. A23420; 1:500; Abbkine Scientific, Co., Ltd.)
for 1 h at RT. Nuclei were counterstained with DAPI (10
µg/ml; Beyotime Institute of Biotechnology), and cells were
examined with a fluorescent microscope (Nikon Corporation).
Hypoxic treatment
The EPCs were divided into control and hypoxia
groups. In the hypoxia groups, the culture medium was changed into
EBM-2, deprived of 5% FCS and EPC growth cytokine cocktail. Then,
the culture dishes were put into a hypoxia chamber
(Billups-Rothenberg, Inc.), flushed with a gas mixture of 1%
O2, 5% CO2 and 94% N2, and
maintained at 37°C for 6, 12 and 24 h. Oxygen concentration was
measured with an oxygen analyzer (Billups-Rothenberg, Inc.).
Hypoxic culture for 12 h was selected as the optimum analysis
timepoint (Fig. S1). EPCs from
12 mice were used for hypoxic treatment.
Cell migration and tube formation
assays
For migration, EPCs (3.75×104 cells) were
placed into the upper compartment of Boyden chambers (Chemicon;
Thermo Fisher Scientific, Inc.). VEGF (50 ng/ml; R&D Systems,
Inc.) and stromal cell-derived factor-1 (100 ng/ml; R&D
Systems, Inc.) were added in the lower compartment. The medium in
the upper and lower compartments was serum-free EBM-2. After 16 h,
the EPCs which migrated across the membrane were counted under an
inverted light microscope (Nikon Corporation), quantified and
averaged by examining 5 random microscopic fields (magnification,
×100). For tube formation assay, 48-well plates were coated with
growth factor-reduced Matrigel (150 µl; Corning, Inc.).
After 7 days in culture, BM-derived EPCs (1×105 cells)
were plated in 400 µl EGM-2 medium and incubated at 37°C
with 5% CO2 for 16 h. Tube formation was counted in 4
random fields at ×40 magnification, with a Nikon 2000 anatomical
lens (15). EPCs from eight mice
were used for analysis of migration and tube formation.
Transfection with Bmi1-siRNA
siRNA against the mouse Bmi1 gene (GGA CAT TGC CTA
CAT TTA T; Guangzhou RiboBio Co., Ltd.) was transiently transfected
into cells using Hiperfect transfection reagent (Qiagen GmbH),
according to the method previously published (16). Briefly, the siRNA (60 nM) was
diluted in serum-free EBM-2, then Hiperfect transfection reagent
was added to the diluted siRNA, mixed by vortexing, and allowed to
form complexes for 5-10 min at RT. The complexes were then added
drop-wise onto the cells for 6 h, after which the media were
replaced with normal growth media. Non-targeting siRNA was used as
negative control (Guangzhou RiboBio Co., Ltd.). At 48 h
post-transfection, the cells were used for subsequent
experiments.
Shh gene transfer of EPCs
To upregulate the Shh expression in EPCs,
recombinant adenovirus encoding mouse Shh (cat. no. 20160722003) or
the control adenovirus (cat. no. CV0001) was used (Shandong Vigene
Biosciences). Ex vivo gene transfer studies were conducted
as described previously (17),
with minor modifications. Briefly, replication incompetent
adenoviral vectors, driven by a cytomegalovirus promoter, were used
to deliver Shh (Adv-Shh) or control adenovirus (Adv-Null) to
isolated EPCs in EGM-2 medium [multiplicity of infection (MOI),
250] for 12 h. EPCs were used 48 h after the initial infection.
EPCs from 50 mice were used for gene transfer.
MI model and EPC transplantation
MI was performed as previously described (13). Adult male C57/B6 mice (12 weeks of
diabetes) were anaesthetized by an intraperitoneal injection of
sodium pentobarbital (50 mg/kg) and artificially ventilated with a
respirator. To provide analgesia, buprenorphine (0.1 mg/kg) was
injected subcutaneously immediately prior to the operation, and
subsequently administered every 8 h for the next 48 h. After
administration of the anesthetic, 7-10 min were allowed for it to
take effect. The depth of general anesthesia was assessed by
pinching the toe, tail or ear of the animal. Any reaction from the
mouse indicated that the anesthesia was too light and that
additional anesthetic agent should be given. Mice were subjected to
either ligation of the left anterior descending coronary artery or
to sham operation with an 8-0 suture needle. Immediately after
coronary artery ligation, 2×105 EPCs were injected at
three sites per mouse heart along the anterior and posterior left
ventricle (LV) wall of infarct zone. PBS was used as a vehicle
control, because the EPCs were suspended in PBS in this experiment.
Forty mice were used for MI models, and EPCs from eight mice were
used for transplantation.
CM-Dil staining for EPC retention
EPCs (1×106) were stained in 1 ml M199
medium (Invitrogen; Thermo Fisher Scientific, Inc.) containing 4
mg/ml CM-Dil dye (chloromethyl-Dil; Invitrogen; Thermo Fisher
Scientific, Inc.) for 15 min at 37°C, then the stained EPCs
(2×105) were injected in the ischemic myocardium. After
3 days of MI, the fresh myocardium was collected and tissue
OCT-freeze medium was injected into the LV. After freezing the
tissue, the myocardium was cut into serial frozen sections from the
ligation point to the apex of myocardium, every 100 µm. The
frozen sections were observed using a fluorescent microscope (Nikon
Corporation; magnification, ×40). The average fluorescence of the
serial frozen sections represented the result of one sample. EPCs
from three mice were used for CM-Dil staining, and 15 mice were
used for the EPC retention assay.
Terminal deoxynucleotidyl
transferase-mediated dUTP nick end-labeling (TUNEL) staining in
myocardium in vivo and in vitro
At 3 days post-MI, cell survival in the myocardium
was determined by TUNEL staining on 5 µm thick frozen
sections, or in vitro in EPCs, as per the manufacturer's
instructions (Cell death detection assay; Roche Diagnostics). DAPI
(10 µg/ml; Beyotime Institute of Biotechnology) staining was
used to count the total number of nuclei. In vivo, apoptotic
cells were observed in the peri-infarct and infarct myocardium
under z100 magnification, with a Nikon TI fluorescence microscope
(Nikon Corporation). In vitro, five cell fields were
captured randomly per sample using confocal fluorescence microscopy
(Nikon A1; Nikon Corporation). Twenty mice were used for in
vivo detection, and EPCs from 12 mice were used for in
vitro detection.
Dihydroethidium (DHE) dye
Superoxide production was detected using DHE
staining (Sigma-Aldrich; Merck KGaA). EPCs were fixed with 4%
paraformaldehyde at 4°C overnight, and incubated with 10 µM
DHE for 45 min at 37°C in a humidified chamber in the dark. The
average fluorescence intensity of the nuclei was then analyzed
using Image Pro-Plus software 6.0 (Media Cybernetics, Inc.).
Immunofluorescence staining of capillary
vessels
On day 14 after induction of MI, the hearts were
harvested and embedded into frozen OTC. Immunofluorescence staining
of frozen sections was performed as described previously (18). Tissue sections were permeabilized
and stained with anti-CD31 antibody (1:50; BD Biosciences) to label
the capillaries, followed by incubation with the respective
secondary antibody. Nuclei were counterstained with DAPI (10
µg/ml; Beyotime Institute of Biotechnology), and sections
were examined with a fluorescent microscope (Nikon TI;
magnification, ×100). The number of capillaries (CD31-positive,
green) was assessed in the border zone of the infarcted myocardium.
Twenty mice were used for capillary vessel analysis.
Assessment of percentage myocardial
infarct and fibrosis (Masson's trichrome staining)
On day 14 after induction of MI, the hearts were
perfusion-fixed with 10% buffered formalin, horizontally sectioned
at 0.5 mm thickness between the point of ligation and the apex, and
then embedded in paraffin. The percentage myocardial infarct was
obtained on Masson's trichrome-stained tissue sections using Image
Pro-Plus software (Media Cybernetics, Inc.). The fibrosis and total
LV area were measured and expressed as a final percentage
myocardial infarct (19). Twenty
mice were used for Masson's trichrome staining.
Echocardiography
Transthoracic two-dimensional M-mode echocardiogram
was performed using Vevo 2100 (VisualSonics) equipped with 30 MHz
transducer. Echocardiographic studies were performed before
(baseline) and at 14 days post-MI on mice anesthetized with a
mixture of 1.5% isoflurane and oxygen (1 l/min). M-mode tracings
were used to measure LV wall thickness, LV end-diastolic dimension
(LVIDd) and LV end-systolic dimension (LVIDs). The mean value of
three measurements was determined for each sample. Percent
fractional shortening (FS) and ejection fraction (EF) were
automatically calculated by the software (13). Twenty mice were used for
echocardiography.
Western blot analysis
Western blot analysis was performed as previously
described (20). EPCs were lysed
in lysis buffer (Cell Signaling Technology, Inc.) with protease
inhibitor cocktail (Merck KGaA) for 30 min on ice. Then the lysed
liquid was collected with a cell scraper. After centrifugation for
10 min at 12,000 × g at 4°C, the supernatant was used for the
detection of protein concentration with a bicinchoninic acid
protein assay kit (Pierce; Thermo Fisher Scientific, Inc.). A total
of 30 µg of protein was loaded onto 12% SDS-PAGE and
transferred to polyvinylidene fluoride membranes (EMD Millipore).
After blocking in 5% skimmed milk for 1 h at RT, membranes were
incubated with primary antibodies against Gli1 (cat. no. ab151796;
1:1,000; Abcam), Bmi1 (cat. no. 5856; 1:1,000; Cell Signaling
Technology, Inc.), p53 (cat. no. 2524; 1:1,000; Cell Signaling
Technology, Inc.), cleaved caspase-3 (cat. no. 9664; 1:1,000; Cell
Signaling Technology, Inc.) and eNOS (cat. no. 32027; 1:1,000; Cell
Signaling Technology, Inc.) overnight at 4°C. After washing with
TBST (TBS/1% Tween-20), membranes were incubated with horseradish
peroxidase-conjugated secondary antibodies anti-mouse or
anti-rabbit (cat. no. BS13278; 1:5,000; Bioworld Technology, Inc.)
at RT for 1 h. GAPDH (cat. no. BS60630; 1:5,000; Bioworld
Technology, Inc.) was used as a loading control. Bands were
visualized using a chemiluminescence kit (Cell Signaling
Technology, Inc.) and X-ray films (Kodak). The band densities were
quantified using the Image-J software (version 1.48u; National
Institutes of Health). EPCs from 31 mice were used for western blot
analysis.
Statistical analysis
All data were analyzed with the statistical software
GraphPad Prism 5.0 (GraphPad Software, Inc.), and all values were
expressed as means ± SEM. Differences between two groups were
analyzed using Student's unpaired t-test, and differences between
three or more groups were evaluated via one-way ANOVA with
Bonferroni correction. P<0.05 was considered to indicate a
statistically significant difference.
Results
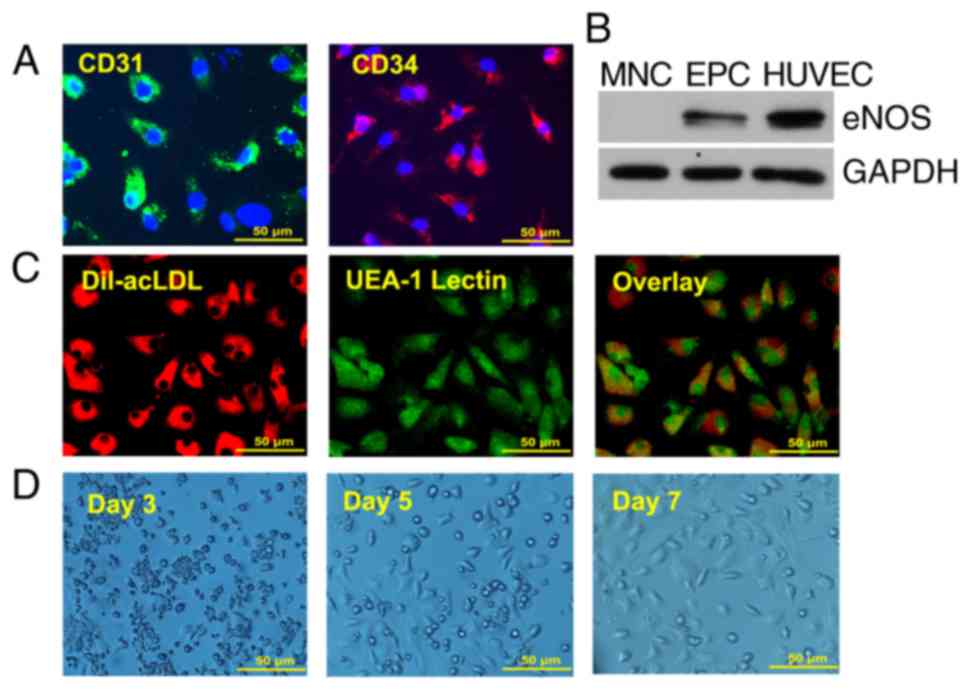
Characterization of EPCs
Although EPCs are heterogeneous in nature, they
possess stem cell characteristics and can be differentiated into
endothelial cells. To characterize BM-derived EPCs, the expression
of the stem cell marker CD34 and the endothelial cell marker CD31
was examined by immunofluorescence (Fig. 1A). Western blot analysis indicated
that, similar to HUVECs, cultured EPCs expressed the endothelial
marker eNOS, whereas no eNOS expression was evident in the MNCs
(Fig. 1B). These data suggested
that the 7 day-cultured EPCs maintained stem cell-characteristics
and contained higher populations of endothelial cells than the
freshly isolated MNCs. The 7 day-cultured EPCs were also confirmed
by Dil-ac-LDL and FITC-lectin double staining, a previously defined
characteristic of EPCs (Fig. 1C).
Fig. 1D displays the growth of
EPCs for 7 days. Spindle cells and colonies occurred from day 3,
several cobblestone cells appeared from day 5, and the density of
cobblestone cells increased on day 7.
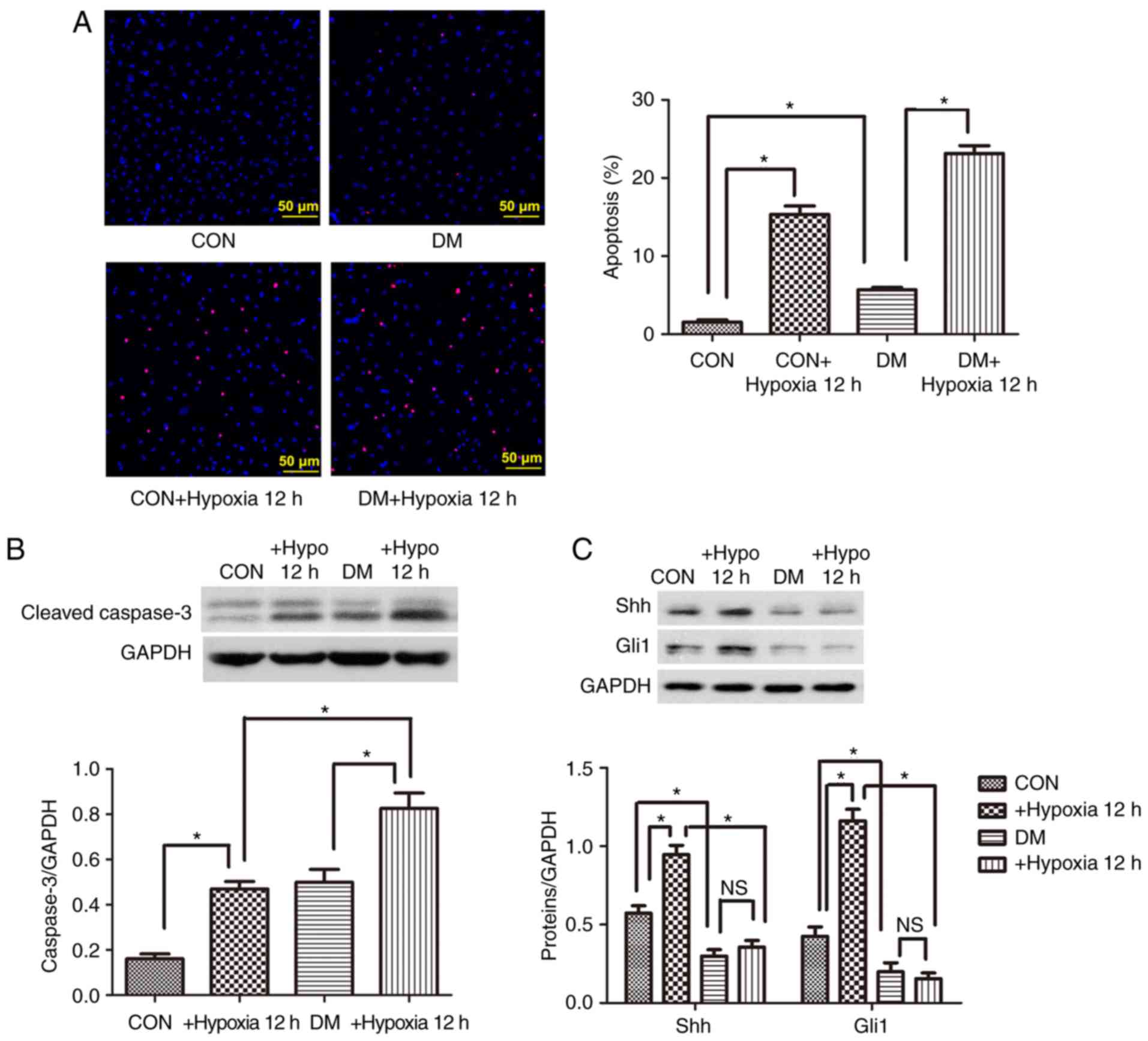
Cell apoptosis is increased and Shh
pathway is impaired in diabetic EPCs under basal or hypoxic
conditions
Control and diabetic EPCs were cultured for 12 h
under hypoxic conditions, and then cell apoptosis was evaluated.
The percentage of apoptotic cells (positive for TUNEL staining) was
signifi-cantly higher in the diabetic group compared with the
control group under basal and hypoxic conditions (Fig. 2A). Similarly, the protein
expression levels of the proapoptotic cleaved caspase-3 were
significantly upregulated in the diabetic EPCs under basal or
hypoxic conditions (Fig. 2B). By
contrast, the expression levels of the Shh pathway proteins Shh and
Gli1 were significantly decreased in the diabetic EPCs (Fig. 2C). When administered with the
antioxidant Tempol (0.5 mM), the diabetic EPCs displayed reduced
reactive oxygen species (ROS) and upregulated protein levels of Shh
and Gli1 (Fig. S2), implying
that oxidative stress may be involved in the impaired Shh pathway.
After hypoxia treatment, the protein expression levels of Shh and
Gli1 were significantly upregulated in the control EPCs, but were
not changed in the diabetic EPCs (Fig. 2C). The present data suggested a
possible link between Shh pathway downregulation and increased
diabetic EPC apoptosis under basal or hypoxic conditions.
Activation of the Shh pathway in diabetic
EPCs decreases apoptosis under basal or hypoxic conditions
Diabetic EPCs were stimulated with Shh protein, and
cellular apoptosis was evaluated to further determine the
relationship between the Shh pathway and diabetic EPC apoptosis.
After treatment with the Shh protein (0.5 µg/ml), Gli1
protein expression levels were significantly increased, suggesting
the rescue of the impaired Shh pathway (Fig. 3A). The protein expression levels
of the proapoptotic protein cleaved caspase-3 were significantly
reduced (Fig. 3A), and the ratio
of TUNEL-positive cells was significantly decreased in the diabetic
EPCs (Fig. 3B). This protection
from apoptosis was also observed under hypoxic conditions. Western
blot and TUNEL assays demonstrated that the Shh protein (0.5
µg/ml) significantly reduced the cleaved caspase-3 protein
expression levels and the ratio of TUNEL-positive cells in the
diabetic EPCs under hypoxic conditions (Fig. 3). These results suggested that
activation of the Shh pathway protected diabetic EPCs from cell
death.
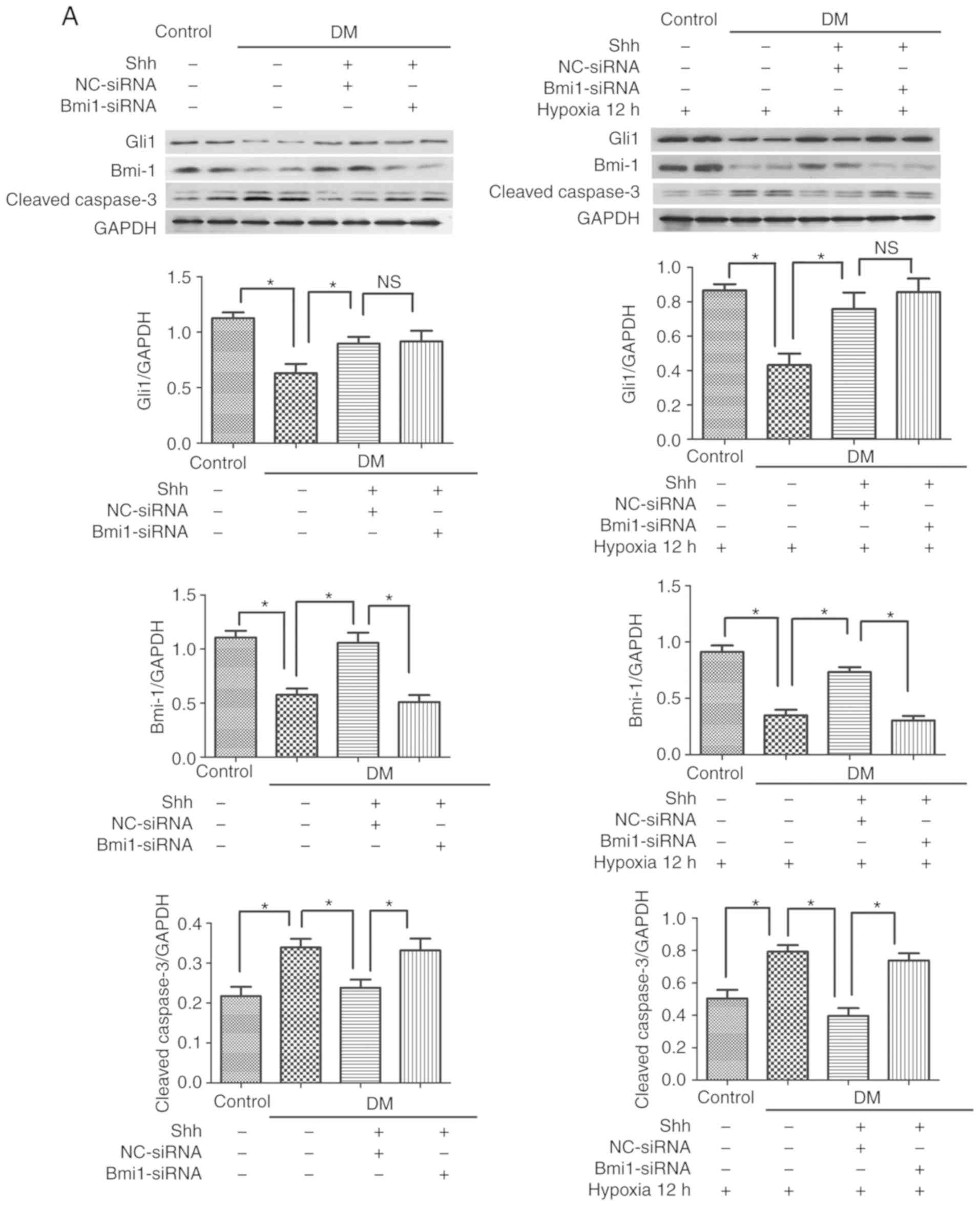 | Figure 3Bmi1 mediates the antiapoptotic
effect of the Shh pathway in diabetic EPCs under basal or hypoxic
conditions. The 7 day-cultured EPCs were transfected with
Bmi1-siRNA or NC-siRNA for 24 h, and then stimulated with the Shh
protein (0.5 µg/ml) for another 24 h. After that, the cells
were incubated under hypoxic conditions for 12 h. (A) Western blot
analysis of Gli1, Bmi1 and cleaved caspase-3 protein expression
levels. (B) Representative images and quantitative analysis of
TUNEL-positive cells (red). Nuclei were counterstained with DAPI
(blue). Three independent experiments were performed. Values are
presented as means ± standard error of the mean.
*P≤0.05, with comparisons indicated by brackets. Bmi1,
BMI1 proto-oncogene; Shh, sonic hedgehog; EPCs, endothelial
progenitor cells; si, small interfering; NC, negative control;
Gli1, GLI family zinc finger 1; DM, diabetes mellitus; NS, not
significant. |
The antiapoptotic effect of Shh in
diabetic EPCs is attenu- ated by Bmi1 silencing
The mechanism by which the Shh pathway may regulate
apoptosis in diabetic EPC remains unknown. Bmi1 is a polycomb group
protein that enters into the nucleus to repress gene transcription
and regulate the function and viability of various types of cells
(21). A potential link exists
between Shh signaling and Bmi1in the proliferation of cerebellar
granule cell progenitors, glioma cells, and medulloblastoma brain
tumor-initiating cells (22-24); however, the antiapoptotic effect
of the Shh/Bmi1 pathway is poorly understood. In the present study,
it was demonstrated that, similar to the Shh pathway, Bmi1 protein
expression levels were significantly reduced in the diabetic EPCs
under basal or hypoxic conditions, while stimulation with the Shh
protein (0.5 µg/ml) significantly rescued the protein
expression levels of Bmi1 (Fig.
3A). When Bmi1-siRNA was delivered in the Shh
protein-stimulated diabetic EPCs, the ratio of TUNEL-positive EPCs
and the protein expression levels of cleaved caspase-3 showed a
reverse trend. Bmi1-siRNA was continuously applied in the diabetic
EPCs under hypoxic conditions; Bmi1 silencing also reversed the
declining numbers of TUNEL-positive EPCs and the reduced protein
expression levels of cleaved caspase-3 mediated by the Shh protein
stimulation (Fig. 3).
Next, the effect of the Shh/Bmi1 pathway was
investigated on normal EPCs. When stimulated with the Shh protein
(0.5 µg/ml), the protein expression levels of Gli1 and Bmi1
were significantly upregulated under basal and hypoxic conditions,
whereas the cleaved caspase-3 levels and the ratio of
TUNEL-positive EPCs were reduced under hypoxic conditions, but
hardly changed under basal conditions (Fig. 4). Following Bmi1 silencing, the
protein expression levels of Bmi1 were downregulated, but the
levels of Gli1 were unchanged (Fig.
4A). In addition, the cleaved caspase-3 levels and the ratio of
TUNEL-positive EPCs increased under basal conditions, but hardly
changed under hypoxic conditions (Fig. 4). When Shh protein plus Bmi1-siRNA
were administered, the protein expression levels of Bmi1 were
significantly reduced, but the levels of Gli1 were unchanged, and
the cleaved caspase-3 levels and the ratio of TUNEL-positive EPCs
increased under basal and hypoxic conditions, compared with the Shh
stimulation alone group (Fig. 4).
These data indicated that the antiapoptotic effect of the Shh/Bmi1
pathway was evident under basal/hypoxic conditions and under
diabetic/normal conditions.
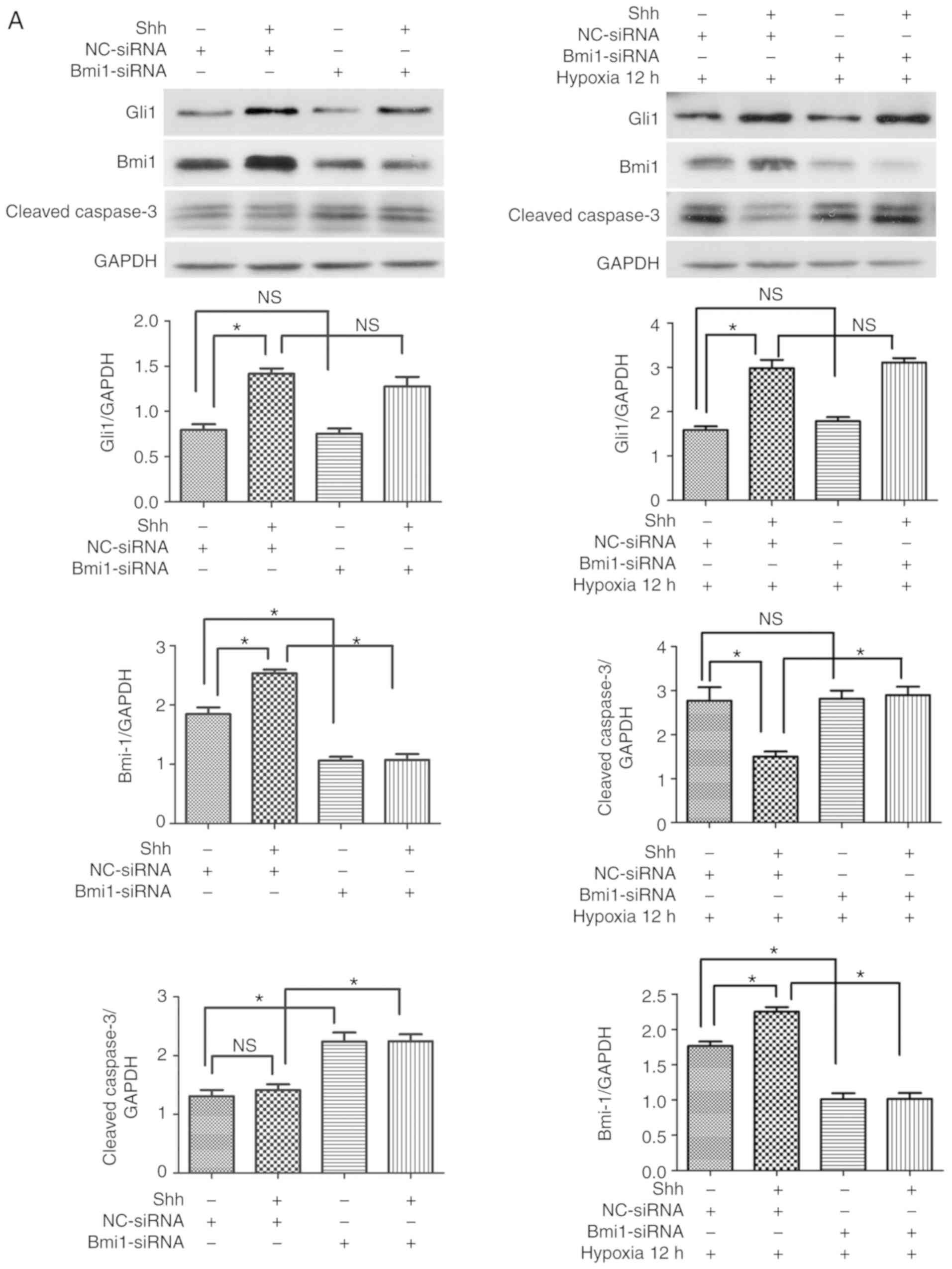 | Figure 4Bmi1 mediates the antiapoptotic
effect of the Shh pathway in normal EPCs under hypoxic conditions.
The 7 day-cultured normal EPCs were transfected with Bmi1-siRNA or
NC-siRNA for 24 h, and then stimulated with the Shh protein (0.5
µg/ml) for another 24 h. After that, the cells were
incubated under hypoxic conditions for 12 h. (A) Western blot
analysis of Gli1, Bmi1 and cleaved caspase-3 protein expression
levels. (B) Representative images and quantitative analysis of
TUNEL-positive cells (red). Nuclei were counterstained with DAPI
(blue). Three independent experiments were performed. Values are
presented as means ± standard error of the mean.
*P≤0.05, with comparisons indicated by brackets. Bmi1,
BMI1 proto-oncogene; Shh, sonic hedgehog; EPCs, endothelial
progenitor cells; si, small interfering; NC, negative control;
Gli1, GLI family zinc finger 1; DM, diabetes mellitus; NS, not
significant. |
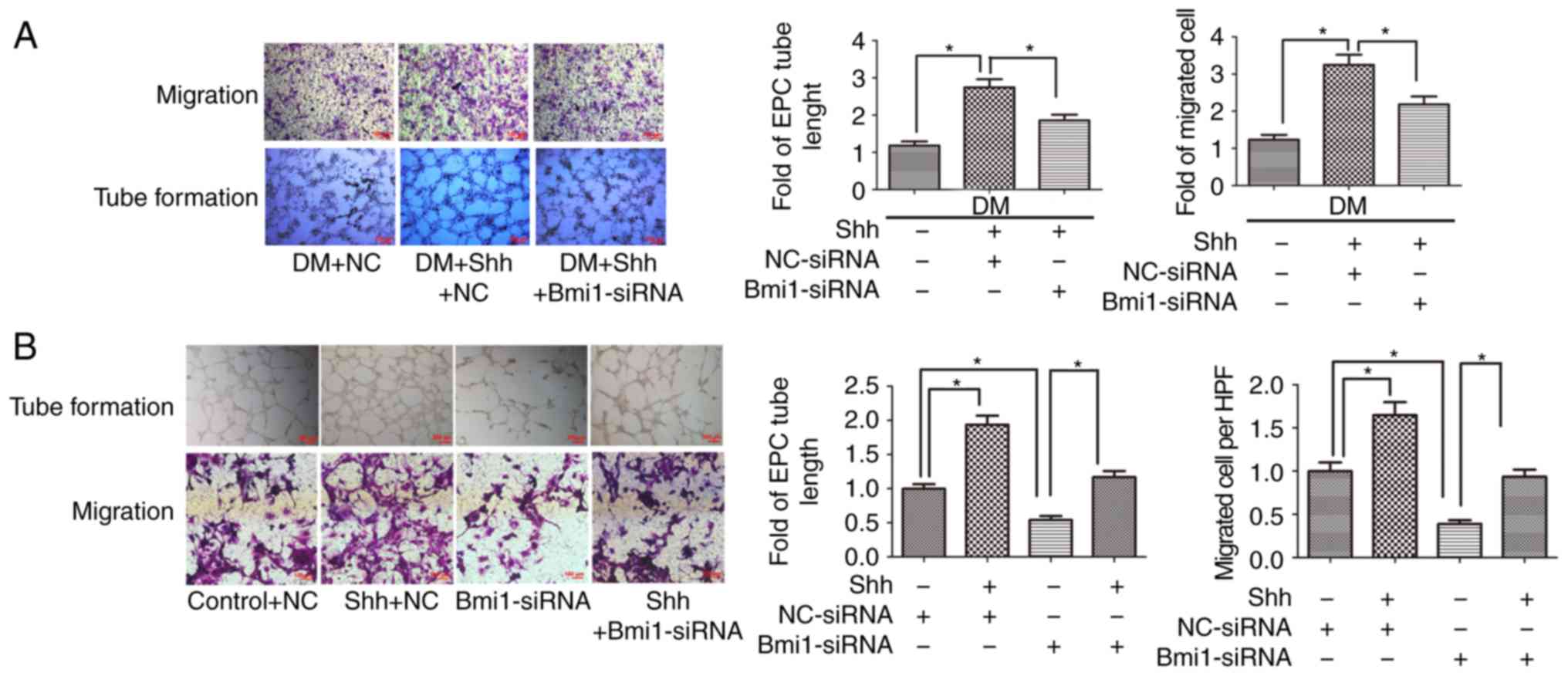
The protective effect of Shh in diabetic
EPC function is attenuated by Bmi1 silencing
It was previously revealed that the Shh pathway is
closely related to the malfunction of diabetic EPCs (15); however, the involvement of
downstream Bmi1 is unknown. In the present study, EPC function was
assessed through tube formation and cell migration assays. The
results demonstrated that the stimulation of tube formation induced
by the Shh protein (0.5 µg/ml) was significantly hindered by
Bmi1 silencing in the diabetic EPCs. Cell migration was also
substantially diminished following Bmi1 silencing (Fig. 5A).
The association between Shh and Bmi1 was also
analyzed in normal EPCs. The results demonstrated that tube
formation and cell migration were significantly enhanced after
stimulation with the Shh protein (0.5 µg/ml). When
Bmi1-siRNA was added in the Shh protein group, the EPCs had
decreased tube formation and cell migration compared with the Shh
protein alone group (Fig. 5B). In
addition, treatment with Bmi1-siRNA in the control group also
significantly reduced tube formation and cell migration (Fig. 5B). These data indicated that the
Shh/Bmi1 pathway affected EPC function under diabetic and normal
conditions.
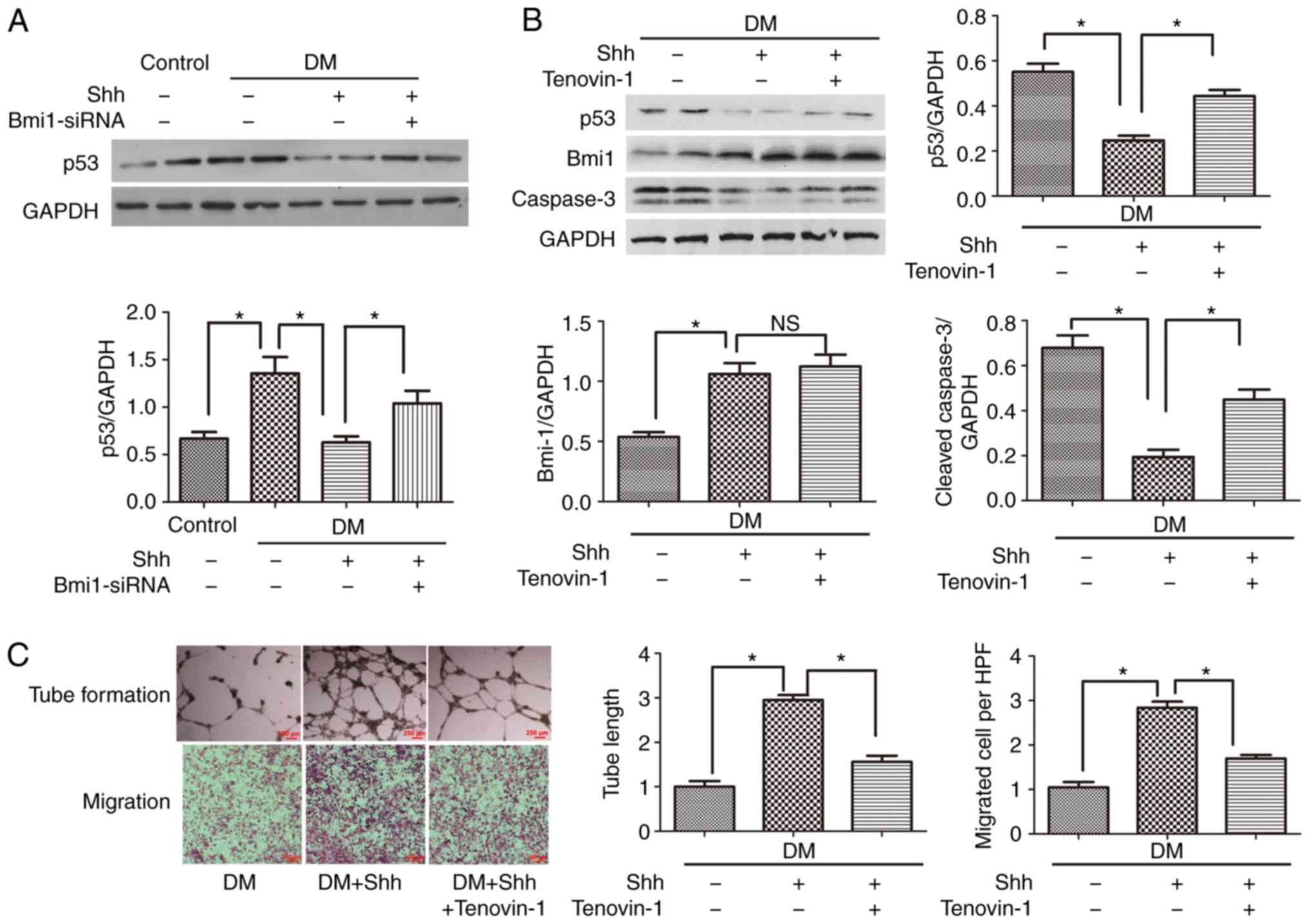
P53 mediates the positive role of the
Shh/Bmi1 axis in diabetic EPCs
The present study focused on the downstream factor
that mediates the Shh/Bmi1 axis in diabetic EPCs. p53, an important
protein related to proliferation and apoptosis, was investigated.
The results demonstrated that p53 was significantly activated in
diabetic EPCs compared with normal EPCs (Fig. 6A); this effect was opposite to the
reduced expression of Shh and Bmi1 in diabetic EPCs (Fig. 3). Treatment with the Shh protein
significantly reduced the protein expression levels of p53 in
diabetic EPCs (Fig. 6A).
When the p53 agonist Tenovin-1 (10 µM) was
added, the Shh protein-stimulated EPCs presented increased protein
expression levels of cleaved caspase-3 (Fig. 6B) and reduced numbers of tube
formation and cell migration (Fig.
6C), compared with the Shh protein group alone. By contrast,
Tenovin-1 barely affected Bmi1 protein expression (Fig. 6B), whereas the treatment with
Bmi1-siRNA significantly reversed the Shh protein-induced p53
reduction (Fig. 6A). These
results indicated that p53 was a downstream factor in the Shh/Bmi1
pathway.
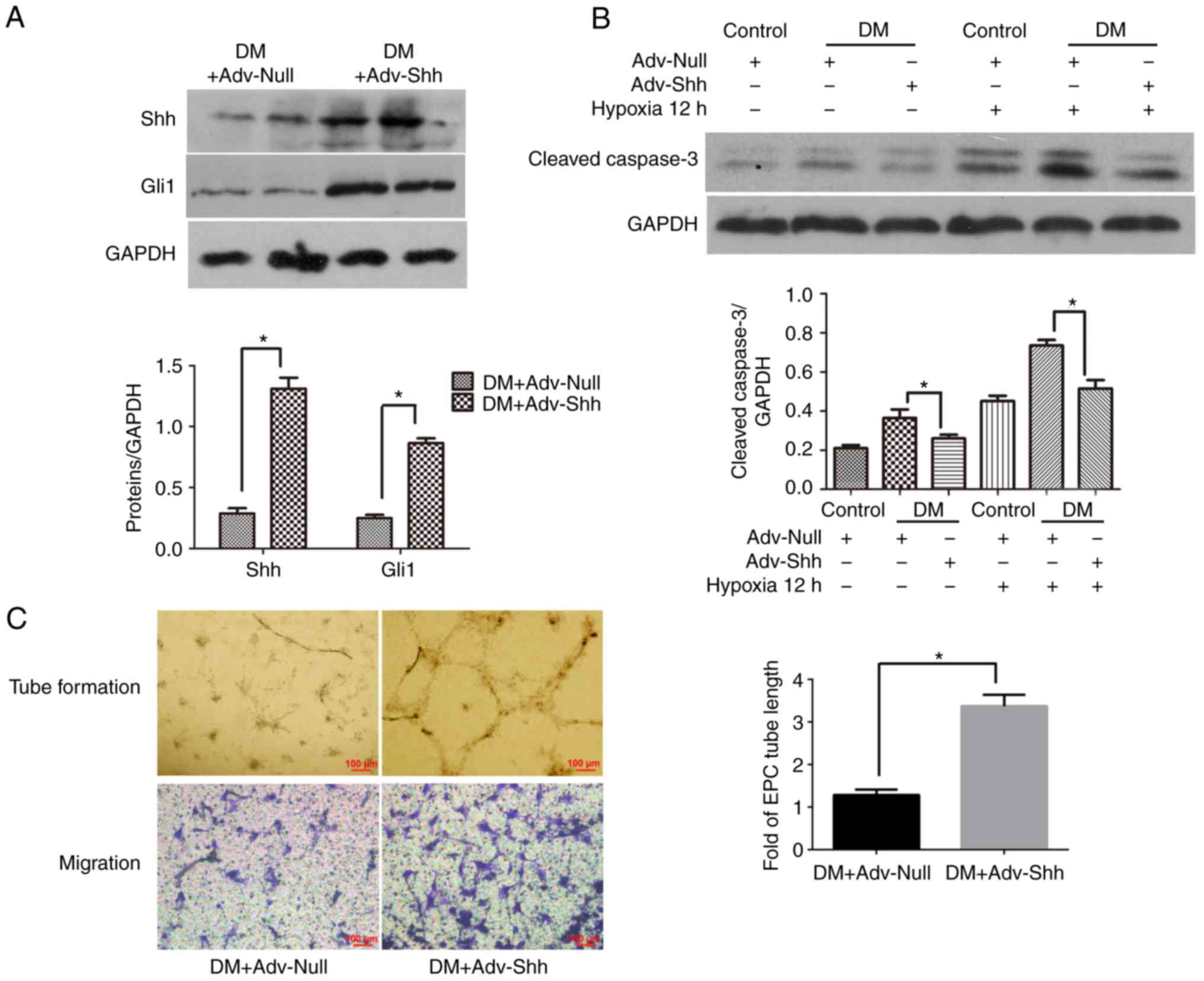
Shh overexpression by adenoviral
infection decreases apop- tosis and improves the function of
diabetic EPCs
To evaluate whether modifying the Shh pathway in
diabetic EPCs could improve the beneficial effects of autologous
stem cell therapy, the cells were infected with recombinant
adenovirus overex-pressing Shh or control adenovirus, and then
implanted into the infarcted hearts of diabetic mice. For the
infection, an MOI of 250 was used, according to a a previous study
(17). To confirm the transfer
efficacy, the protein expression levels of Shh and Gli1 were
examined in cell lysates by western blot analysis. As shown in
Fig. 7A, the protein expression
levels of Shh and Gli1 were significantly higher in the
Adv-Shh-infected cells compared with cells transfected with control
adenovirus. These results indicated the effective gene modification
of diabetic EPCs with adenovirus ex vivo, resulting in the
activation of the cellular Shh pathway in diabetic EPCs.
To investigate the effect of Shh overexpression on
the status of EPCs, diabetic EPCs were infected with Shh or Null
adenovirus for 2 days. First, the expression of the proapoptotic
cleaved caspase-3 was investigated in the Shh-modified diabetic
EPCs. The results demonstrated that Shh overexpression
significantly reduced the cleaved caspase-3 expression levels in
the diabetic EPCs under normal or hypoxic conditions (Fig. 7B). Second, the function of the
Shh-modified diabetic EPCs was evaluated. As shown in Fig. 7C, Shh overexpression significantly
increased the tube length and cell migration in the diabetic EPCs.
These results confirmed the enhanced capabilities of the diabetic
EPCs following Shh gene modification.
Injection of diabetic EPCShh
preserves cardiac function and alleviates cardiac remodeling in DMI
mice
To determine whether this endogenous impairment of
the Shh pathway in diabetic EPCs may be associated with DMI and
whether Shh gene therapy could improve the in vivo
angiogenic capability of diabetic EPCs and enhance the cardiac
function in DMI, Shh gene therapy was performed ex vivo on
diabetic EPCs (DM-EPCShh) prior to transplantation.
Control adenovirus was transfected to diabetic EPCs
(DM-EPCNull) or normal EPCs (CON-EPCNull),
and a vehicle PBS injection group served as control.
After 14 days of MI, injection of 2×105
CON-EPCNull robustly improved the EF and FS values and
decreased systolic and diastolic ventricular dilation (Fig. 8A). By contrast, injection of equal
numbers of DM-EPCNull showed no significant functional
changes (Fig. 8A). When injected
with equal numbers of DM-EPCShh, the diabetic heart with
MI exhibited functional benefit over DM-EPCNull. The
improvements between CON-EPCNull and
DM-EPCShh groups had no significant difference (Fig. 8A).
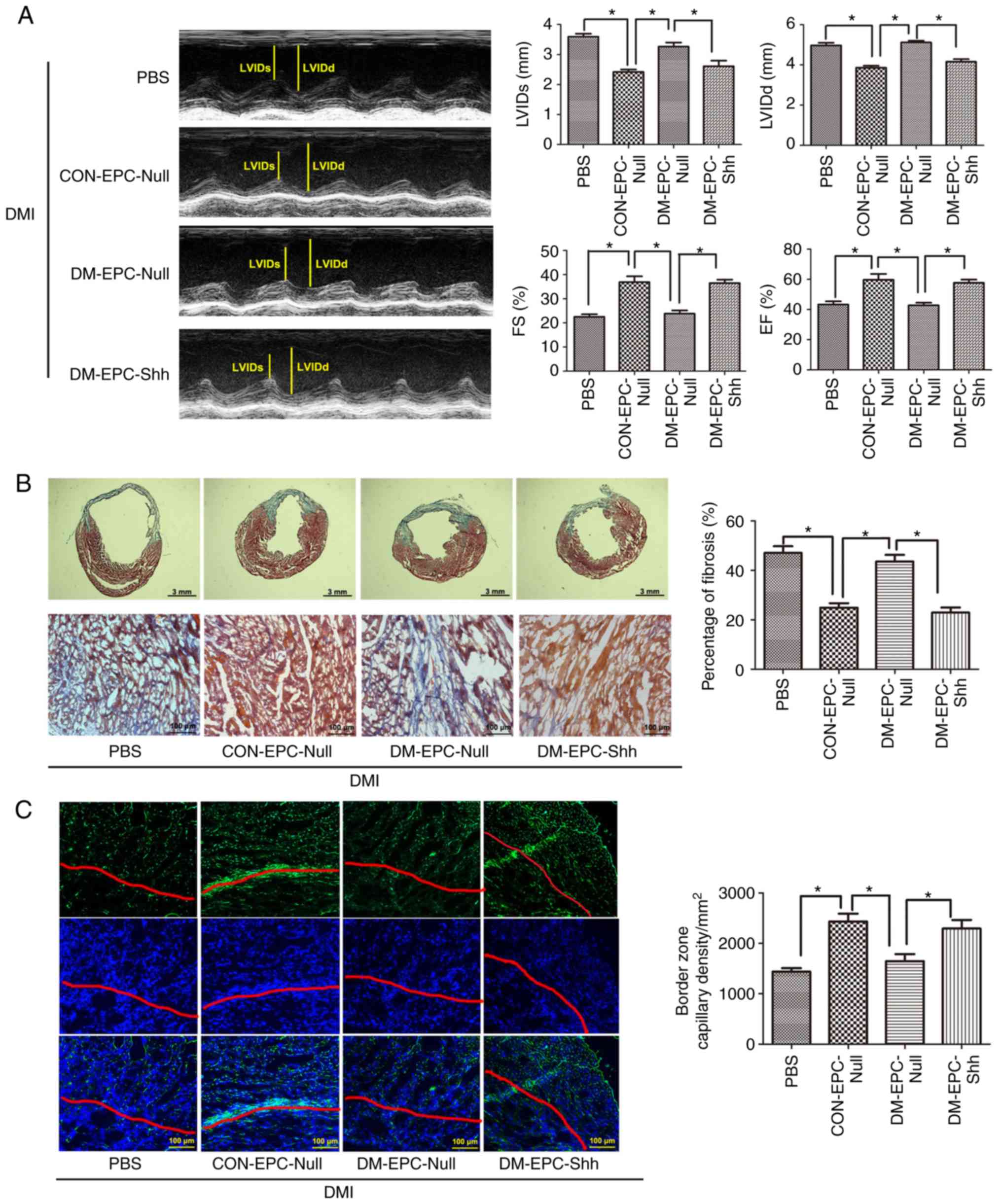 | Figure 8Injection of diabetic
EPCShh preserves cardiac function and alleviates cardiac
remodeling in DMI mice. EPCs (2×105 per heart) were
injected immediately after coronary artery ligation.
Echocardiography, Masson's trichrome staining, and CD31 detection
were implemented after 2 weeks. (A) Representative M-mode images
and quantitative analysis of LVIDs, LVIDd, FS and EF alues (n=5).
(B) Representative images of Masson's trichrome staining (the whole
frozen section and the border zone of myocardial infarction) and
quantitative analysis of fibrosis ratio (n=5). (C) Representative
images of capillary vessels (CD31-positive cells; green). Nuclei
were counterstained with DAPI (blue). The zone under the red line
is the infarct area, while the upper zone is the border area.
Quantitative analysis is also shown of the capillary density in the
border zone of the infarcted myocardium (n=5). Values are presented
as means ± standard error of the mean. *P≤0.05, with
comparisons indicated by brackets. DMI, diabetic myocardial
infarction; EPCs, endothelial progenitor cells; LVIDd, LV
end-diastolic dimension; LVIDs, LV end-systolic dimension; FS,
fractional shortening; EF, ejection fraction; CON-EPC-Null, control
EPCs modified by control adenovirus; DM-EPC-Null, diabetic EPCs
modified by control adenovirus; DM-EPC-Shh, diabetic EPCs modified
by Shh-overexpressing adenovirus. |
At the end of the experiment, 2 weeks after MI, the
hearts from the CON-EPCNull-treated mice exhibited
significantly reduced infarct dimensions and border zone
interstitial fibrosis compared with those from the PBS group
(Fig. 8B). By contrast, equal
numbers of DM-EPCNull induced no significant changes
(Fig. 8 B). The diabetic heart
with MI showed less infarct size and fibrosis following treatment
with DM-EPCShh compared with treatment with
DM-EPCNull (Fig. 8B).
The improvements between CON-EPCNull and
DM-EPCShh groups had no significant difference (Fig. 8A).
Using CD31 staining, the capillary density was
evaluated within the infarct border zone of all the hearts. The
density of capillaries within the border zone was significantly
higher in the hearts of the mice treated with
CON-EPCNull compared with those of the control groups
(Fig. 8C). Equal numbers of
DM-EPCNull induced no therapeutic change (Fig. 8C). The diabetic heart with MI
showed more capillary vessels in the border zone following
treatment with DM-EPCShh compared with treatment with
DM-EPCNull (Fig. 8C).
Ina ddition, the improvements between CON-EPCNull and
DM-EPCShh groups were not significantly different
(Fig. 8C).
The present results suggested that the impaired Shh
pathway in diabetic EPCs resulted in poor repair in diabetic
myocardial ischemia (CON-EPCNull vs.
DM-EPCNull; CON-EPCNull vs.
DM-EPCShh). However, activation of the Shh pathway via
gene modification greatly accelerated EPC-mediated capillary
development, thereby reducing myocardial fibrosis and enhancing the
cardiac function of diabetic MI (DM-EPCShh vs.
DM-EPCNull).
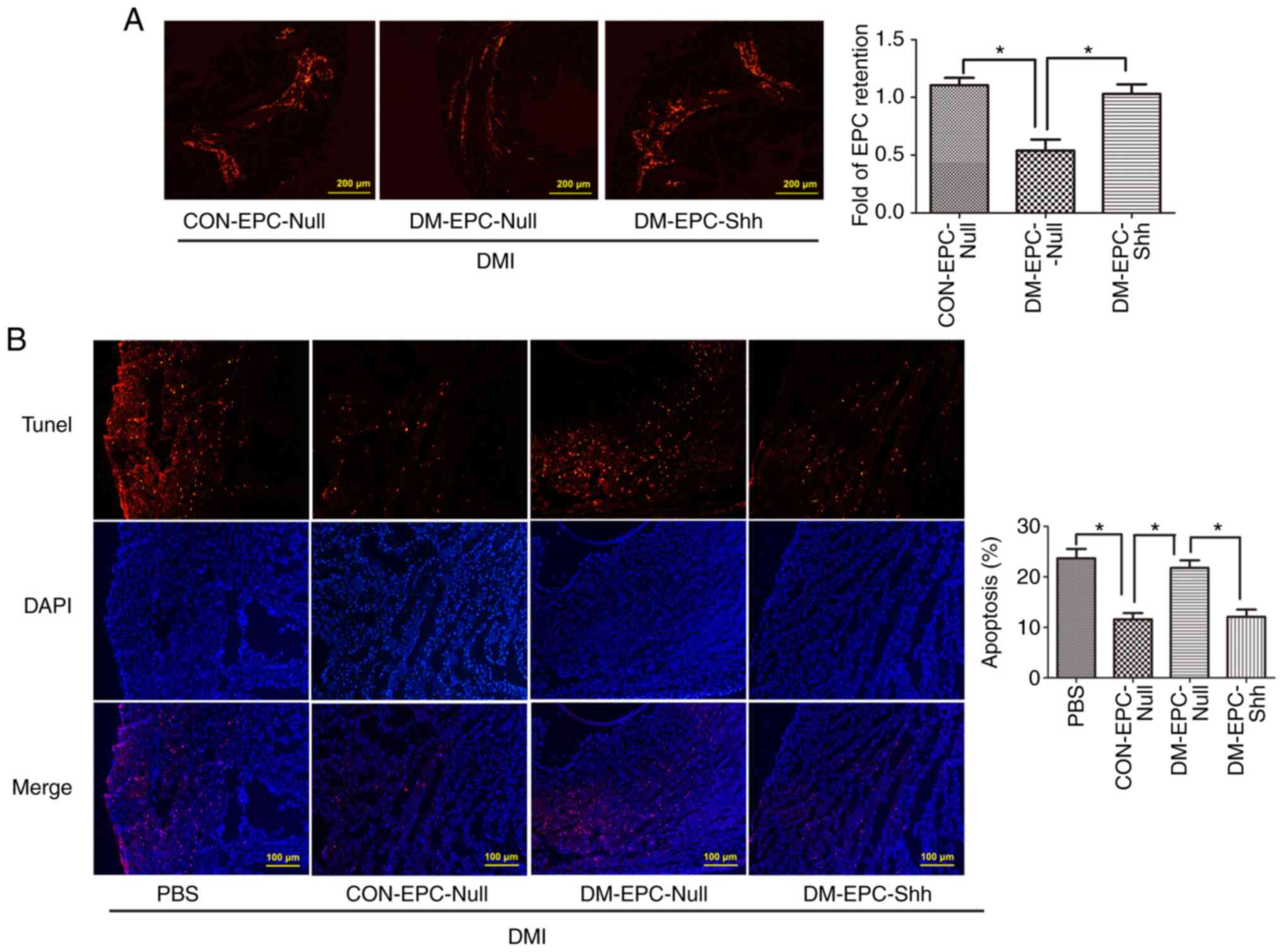
Injection of diabetic EPCShh
exhibits enhanced EPC retention and cell apoptosis in DMI mice
Next, the retention of EPCs after their
transplantation in the ischemic myocardium (3 days post-MI) was
investigated, by assessing the number of CM-Dil (red)-labeled EPCs
in the myocardium. As shown in Fig.
9A, the retention of labeled EPCs in the myocardium was higher
in the mice receiving CON-EPCNull compared with those
receiving DM-EPCNull, and the delivery with
DM-EPCShh significantly compensated for the deficiency
of DM-EPCNull retention. In addition,
DM-EPCShh greatly reduced the apoptosis of cells in the
injured myocardium, similar to the CON-EPCNull
administration (Fig. 9B). By
contrast, DM-EPCNull induced no change in myocardial
apoptosis (Fig. 9B). These data
suggested that Shh-modified diabetic EPCs exhibited higher
retention capability in the myocardial tissue, which may be an
indispensable property for improved repair function.
Discussion
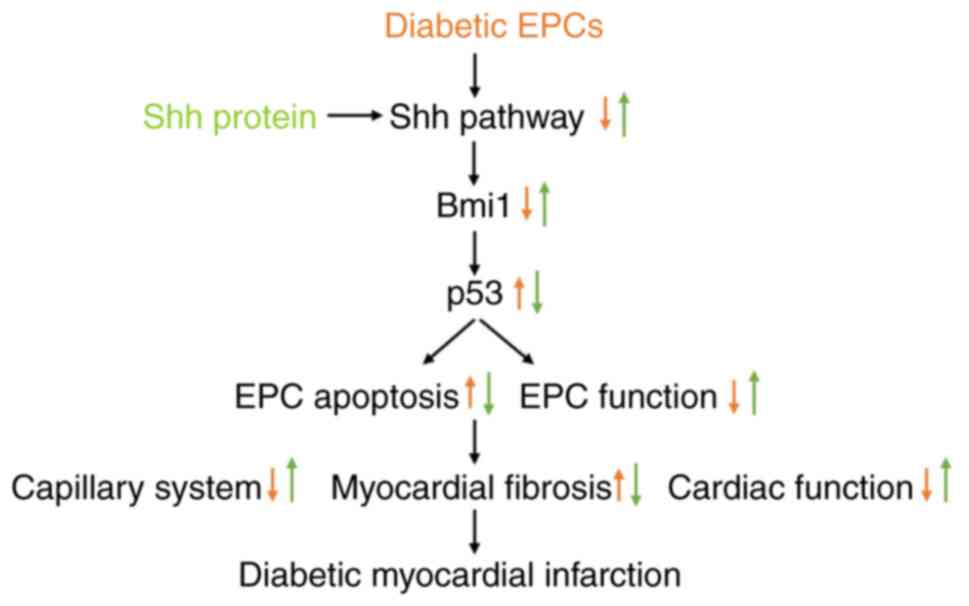
The predominant findings in the present study were
as follows (Fig. 10): i) the Shh
pathway alleviated the apoptosis of diabetic EPCs under baseline or
hypoxic conditions; ii) Bmi1, found in the present study for the
first time to be related to EPCs, mediated the benefits of the Shh
pathway by inhibiting p53; and iii) Shh gene therapy of diabetic
EPCs rescued the myocardial repair of diabetic mice through
enhanced angiogenesis and reduced apoptosis.
Patients with type 1 or type 2 diabetes mellitus
have decreased numbers and impaired angiogenic function of EPCs,
which have a causative role in the development and progression of
virtually all diabetic cardiovascular complications, such as
diabetic cardiomyopathy and diabetes combined with MI (25,26). Despite evidence indicating that
local or systematic administration of EPCs can significantly
improve angiogenesis in injured myocardium, MI in diabetic
background remains an important clinical problem leading to poor
prognosis and mortality, due to the impaired function and survival
of autologous EPCs (27).
The Shh pathway enhances the angiogenic function of
EPCs under normal and diabetic conditions (15,28,29). This effect was also confirmed in
the present study, which demonstrated that delivery of the Shh gene
significantly improved the migration and tube formation of diabetic
EPCs. Furthermore, the present results demonstrated that
stimulation with the Shh protein significantly alleviated the
apoptosis of diabetic EPCs under normal and hypoxic conditions. The
evidence of resistance to hypoxic conditions may be useful in
coping with the inefficiency of clinical EPC injection to injured
myocardium.
The present study also found that cleaved-caspase-3
levels and TUNEL-positive cells were increased by hypoxia in
control conditions, despite the Shh pathway being activated. This
effect seems to be contradictory to the protective role of Shh
pathway. Actually, this stimulation of the Shh pathway is a
stress-induced reaction, which is insufficient to counteract the
damage induced by hypoxia. Previous studies have reported that Shh
is stimulated under stress conditions, such as hypoxia and
ischemia, when stress-induced injury occurs (30,13). Inhibition of this stress-induced
Shh expression exacerbates the damage, implying that the
upregulated Shh pathway induced by stress has a protective role in
alleviating cell damage (13).
Furthermore, delivery of exogenous Shh protein reduces the damage,
which in turn reveals the protective role of Shh pathway (30-33).
In the present study, the Shh pathway was
demonstrated to be stimulated in the control EPCs following
hypoxia, which is consistent with previous reports (34-37). This effect can be explained by
previous literature: Hypoxia-inducible factor-1 (HIF-1), a
transcriptional regulator of the Shh gene, is upregulated and then
activates the Shh pathway under cell hypoxia and stress (38). However, the present results found
that the Shh pathway was not activated in the diabetic EPCs; the
unresponsiveness may be related to the impaired Shh pathway itself
and/or potential impaired upstream factors (such as HIF-1 or nitric
oxide) under diabetic conditions (13,39,40).
The Shh pathway improved the function and apoptosis
of EPCs in diabetic EPCs, but its downstream signaling remains
elusive. Bmi1 is important for the self-renewal of stem cells. Bmi1
modulates mitochondrial function (41), ROS generation, and DNA
double-strand break repair (42),
and regulates the function and apoptosis of various types of cells,
such as stem cells and cancer cells (43,44). Therefore, the present study
examined whether Shh pathway activation regulates Bmi1 in diabetic
EPCs. The present data indicated that Bmi1 was significantly
reduced in diabetic EPCs and that stimulation with the Shh protein
efficiently reversed this reduction. In addition, Bmi1 silencing
reversed the protection of the Shh pathway in diabetic EPC function
and apoptosis. Bmi1 silencing also affected the function and
apoptosis of normal EPCs. The present study is the first to uncover
the relationship of Shh and Bmi1 in diabetic EPCs; impaired
Shh/Bmi1 axis mediated the damaged status in diabetic EPCs, and
maintained the status of normal EPCs. The present study is also the
first to elucidate the function of Bmi1 in EPCs and its role as a
potential target in EPC therapy.
The most known downstream factor of Bmi1 is p53,
which is downregulated by Bmi1 through the negative regulation of
the INK4A/ARF/p19/p14 pathway (45,46). In EPCs, p53 is associated with
diabetic EPC apoptosis, and the delivery of p53-silenced EPCs
significantly improves ischemia in diabetic peripheral vascular
disease (47). The present study
revealed that the Shh pathway activation reduced p53 expression,
and this effect was reversed by Bmi1 silencing. Accumulating p53 by
Tenovin-1 reversed the benefits induced by Shh stimulation in the
function and apoptosis of diabetic EPCs. This effect was similar to
that observed following of Bmi1-siRNA. These results suggested that
p53 downregulation mediated the protection of Shh/Bmi1 axis
activation in diabetic EPCs.
Diabetes-induced oxidative stress is well documented
in the pathophysiology of diabetic complications (48). Suppression of the Shh pathway
resulting from oxidative stress, which has been reported in BM
stromal cells in vitro (49,50), was confirmed in the diabetic EPCs
in the present study. The results suggested that the diabetic EPCs
had a higher ROS level (DHE dye) compared with control EPCs, and
delivery of the antioxidant Tempol significantly reduced the ROS
level and restored the impaired Shh pathway in the diabetic EPCs.
(Fig. S2) These results
indicated that oxidative stress may be involved in the impaired Shh
pathway of diabetic EPCs.
The present study demonstrated that the Shh
gene-modified diabetic EPCs exhibited significantly improved
cardiac function compared with the untreated diabetic EPCs. In
addition, treatment with DM-EPCshh robustly increased
the capillary development within the infarct border zone,
accompanied by reduced infarct sizes and interstitial fibrosis. The
indicated mechanism, i.e. autocrine secretion of the angiogenic
factor Shh by engineered EPCs, may be involved; however, other
hypotheses cannot be excluded. Modified EPCs themselves may respond
better to angiogenic or noxious stimuli (such as oxidative stress
at reperfusion) from the microenvironment, which might also
contribute to their improved function.
In addition to the long-term effects of Shh-modified
DM-EPCs, the present results also demonstrated improved cell
retention of these cells and myocardial apoptosis, which may be
involved in the protection of DM-EPCShh in diabetic MI.
It was assumed that the autocrine Shh signaling may increase the
retention numbers of DM-EPCshh and a paracrine loop with
the myocardial cells may alleviate their apoptosis on day 3 after
cardiac ischemia. Further analysis of the secretome of Shh-modified
EPCs and further experiments, such as investigations of cell-cell
interactions, are needed to investigate these mechanisms in depth.
Indeed, normal EPCs can repair the ischemic myocardium through
angiogenesis and paracrine signaling, thus restoring blood supply
and salvaging cells, and eventually improving cardiac function
(19,51); these effects are consistent with
the effects of CON-EPCNull presented in the current
study. In addition, the effectiveness of repair is related to the
numbers of EPCs. For example, 2×106 EPCs are more
effective than 1×106 EPCs in wound healing (17). In the present study,
2×105 EPCs were used, as described in previous reports
(19,52). The status of EPCs is also related
to repair effectiveness. Diseases, such as hypertension and
diabetes, significantly damage the function and numbers of EPCs and
influence their repair potential (53), which is consistent with the
present results of the DM-EPCNull.
In the present study, the relationship between the
impaired Shh pathway of diabetic EPCs and diabetic myocardium was
elucidated. Treatment with diabetic EPCs did not significantly
change the status of diabetic myocardium with ischemia. This was
greatly improved by diabetic EPCs harboring a Shh gene
modification, to a level similar to normal EPCs. These findings
indicated that the impaired Shh pathway undermined the behavior of
diabetic EPCs in injured diabetic myocardium, leading to
deteriorated myocardial repair, suggesting poor prognosis and
eventually high mortalities in diabetic MI. Amending this pathway
in diabetic EPCs may remarkably improve the potential of autologous
EPC therapy in diabetic MI.
In conclusion, the enhanced Shh pathway in diabetic
EPCs improved their angiogenic function and survival through Bmi1
upregulation and p53 downregulation, and consequently rescued the
inefficient repair in diabetic myocardium under ischemia. For the
development of improved autologous cell therapies for patients with
diabetes, alterations in the progenitor cells through gene therapy
prior to transplantation may be necessary to compensate for their
defective viability and function.
Supplementary Materials
Funding
This study was supported by the National Natural
Science Foundation of China (grant no. 81302767 to Dr Qing Xiao,
and grant no. 81573433 to Dr Jian-Dong Luo), the Science and
Technology Planning Project of Guangdong Province (grant nos.
2014A020212361 and 2017A020215194 to Dr Qing Xiao), the Medical
Scientific Research Foundation of Guangdong Province (grant no.
A2015444 to Dr Qing Xiao, and grant no. B2018054 to Dr Xiao-Ling
Zhang), the Natural Science Foundation of Guangzhou (grant no.
2016A030310271 to Dr Yuan Qin), the Scientific and Technological
Planning Program of Guangzhou (grant no. 2017071010458 to Dr
Ying-Hua Liu), and the Municipal Education Bureau Program of
Guangzhou (grant no. 1201610286 to Dr Ying-Hua Liu).
Availability of data and materials
The datasets used or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
QX contributed to conception and design, provision
of study material, collection and/or assembly of data, data
analysis and interpretation, manuscript writing, and final approval
of the manuscript. XYZ, RCJ, XHC, XZ, KFC and SYC contributed to
collection and/or assembly of data, and final approval of the
manuscript. XLZ, YQ and YHL contributed to provision of study
material and final approval of the manuscript. JDL contributed to
conception and design, financial support, administrative support,
provision of study material, data analysis and interpretation,
manuscript writing, and final approval of manuscript.
Ethics approval and consent to
participate
All experimental protocols involving animals were
approved by the Animal Care and Welfare Committee of Guangzhou
Medical University.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interest
Abbreviations:
|
EPCs
|
endothelial progenitor cells
|
|
STZ
|
streptozotocin
|
|
MI
|
myocardial infarction
|
|
DM
|
diabetes mellitus
|
|
DMI
|
diabetic myocardial infarction
|
|
Shh
|
sonic hedgehog
|
|
EF
|
ejection fraction
|
|
FS
|
fractional shortening
|
|
LV
|
left ventricle
|
|
LVIDd
|
LV end-diastolic dimension
|
|
LVIDs
|
LV end-systolic dimension
|
Acknowledgments
The authors thank Professor Gui-Ping Zhang
(Department of Pharmacology, Guangzhou Medical University) for his
valuable advice regarding methods in this manuscript.
References
|
1
|
Patel NB and Balady GJ: Diagnostic and
prognostic testing to evaluate coronary artery disease in patients
with diabetes mellitus. Rev Endocr Metab Disord. 11:11–20. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Cheng R and Ma J: Angiogenesis in diabetes
and obesity. Rev Endocr Metab Disord. 16:67–75. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Howangyin KY and Silvestre JS: Diabetes
mellitus and ischemic diseases: Molecular mechanisms of vascular
repair dysfunction. Arterioscler Thromb Vasc Biol. 34:1126–1135.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Co M, Tay E, Lee CH, Poh KK, Low A, Lim J,
Lim IH, Lim YT and Tan HC: Use of endothelial progenitor cell
capture stent (Genous Bio-Engineered R Stent) during primary
percutaneous coronary intervention in acute myocardial infarction:
Intermediate- to long-term clinical follow-up. Am Heart J.
155:128–132. 2008. View Article : Google Scholar
|
|
5
|
Zhu J, Song J, Yu L, Zheng H, Zhou B, Weng
S and Fu G: Safety and efficacy of autologous thymosin β4
pre-treated endothelial progenitor cell transplantation in patients
with acute ST segment elevation myocardial infarction: A pilot
study. Cytotherapy. 18:1037–1042. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Georgescu A, Alexandru N, Constantinescu
A, Titorencu I and Popov D: The promise of EPC-based therapies on
vascular dysfunction in diabetes. Eur J Pharmacol. 669:1–6. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ling L, Shen Y, Wang K, Jiang C, Fang C,
Ferro A, Kang L and Xu B: Worse clinical outcomes in acute
myocardial infarction patients with type 2 diabetes mellitus:
Relevance to impaired endothelial progenitor cells mobilization.
PLoS One. 7:pp. e507392012, View Article : Google Scholar : PubMed/NCBI
|
|
8
|
António N, Fernandes R, Ribeiro CF and
Providência LA: Challenges in vascular repair by endothelial
progenitor cells in diabetic patients. Cardiovasc Hematol Disord
Drug Targets. 10:161–166. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Briscoe J and Thérond PP: The mechanisms
of Hedgehog signalling and its roles in development and disease.
Nat Rev Mol Cell Biol. 14:416–429. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Álvarez-Buylla A and Ihrie RA: Sonic
hedgehog signaling in the postnatal brain. Semin Cell Dev Biol.
33:105–111. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lopez-Rios J: The many lives of SHH in
limb development and evolution. Semin Cell Dev Biol. 49:116–124.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Rimkus TK, Carpenter RL, Qasem S, Chan M
and Lo HW: Targeting the sonic hedgehog signaling pathway: Review
of smoothened and gli inhibitors. Cancers (Basel). 8. pp. E222016,
View Article : Google Scholar
|
|
13
|
Xiao Q, Hou N, Wang YP, He LS, He YH,
Zhang GP, Yi Q, Liu SM, Chen MS and Luo JD: Impaired sonic hedgehog
pathway contributes to cardiac dysfunction in type 1 diabetic mice
with myocardial infarction. Cardiovasc Res. 95:507–516. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Xiao Q, Yang YA, Zhao XY, He LS, Qin Y, He
YH, Zhang GP and Luo JD: Oxidative stress contributes to the
impaired sonic hedgehog pathway in type 1 diabetic mice with
myocardial infarction. Exp Ther Med. 10:1750–1758. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Qin Y, He YH, Hou N, Zhang GS, Cai Y,
Zhang GP, Xiao Q, He LS, Li SJ, Yi Q and Luo JD: Sonic hedgehog
improves ischemia-induced neovascularization by enhancing
endothelial progenitor cell function in type 1 diabetes. Mol Cell
Endocrinol. 423:30–39. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Liu Y, Gao M, Ma MM, Tang YB, Zhou JG,
Wang GL, Du YH and Guan YY: Endophilin A2 protects H2O2-induced
apoptosis by blockade of Bax translocation in rat basilar artery
smooth muscle cells. J Mol Cell Cardiol. 92:122–133. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Marrotte EJ, Chen DD, Hakim JS and Chen
AF: Manganese superoxide dismutase expression in endothelial
progenitor cells accelerates wound healing in diabetic mice. J Clin
Invest. 120:4207–4219. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Krishnamurthy P, Thal M, Verma S, Hoxha E,
Lambers E, Ramirez V, Qin G, Losordo D and Kishore R: IL-10
deficiency impairs bone marrow-derived endothelial progenitor cell
(EPC) survival and function in ischemic myocardium. Circ Res.
109:1280–1289. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Sun YY, Bai WW, Wang B, Lu XT, Xing YF,
Cheng W, Liu XQ and Zhao YX: Period 2 is essential to maintain
early endothelial progenitor cell function in vitro and
angiogenesis after myocardial infarction in mice. J Cell Mol Med.
18:907–918. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhang H, He Y, Zhang G, Li X, Yan S, Hou
N, Xiao Q, Huang Y, Luo M, Zhang G, et al: HDAC2 is required by the
physiological concentration of glucocorticoid to inhibit
inflammation in cardiac fibroblasts. Can J Physiol Pharmacol.
95:1030–1038. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Cao L, Bombard J, Cintron K, Sheedy J,
Weetall ML and Davis TW: BMI1 as a novel target for drug discovery
in cancer. J Cell Biochem. 112:2729–2741. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Shahi MH, Farheen S, Mariyath MP and
Castresana JS: Potential role of Shh-Gli1-BMI1 signaling pathway
nexus in glioma chemoresistance. Tumour Biol. 37:15107–15114. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wang X, Venugopal C, Manoranjan B,
McFarlane N, O'Farrell E, Nolte S, Gunnarsson T, Hollenberg R,
Kwiecien J, Northcott P, et al: Sonic hedgehog regulates Bmi1 in
human medulloblastoma brain tumor-initiating cells. Oncogene.
31:187–199. 2012. View Article : Google Scholar
|
|
24
|
Subkhankulova T, Zhang X, Leung C and
Marino S: Bmi1 directly represses p21Waf1/Cip1 in Shh-induced
proliferation of cerebellar granule cell progenitors. Mol Cell
Neurosci. 45:151–162. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Savvatis K, Westermann D, Schultheiss HP
and Tschöpe C: Kinins in cardiac inflammation and regeneration:
Insights from ischemic and diabetic cardiomyopathy. Neuropeptides.
44:119–125. 2010. View Article : Google Scholar
|
|
26
|
Aragona CO, Imbalzano E, Mamone F, Cairo
V, Lo Gullo A, D'Ascola A, Sardo MA, Scuruchi M, Basile G, Saitta A
and Mandraffino G: Endothelial progenitor cells for diagnosis and
prognosis in cardiovascular disease. Stem Cells Int.
2016:80437922016. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
António N, Fernandes R, Soares A, Soares
F, Lopes A, Carvalheiro T, Paiva A, Pêgo GM, Providência LA,
Gonçalves L and Ribeiro CF: Reduced levels of circulating
endothelial progenitor cells in acute myocardial infarction
patients with diabetes or pre-diabetes: Accompanying the glycemic
continuum. Cardiovasc Diabetol. 13:1012014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Fu JR, Liu WL, Zhou JF, Sun HY, Xu HZ, Luo
L, Zhang H and Zhou YF: Sonic hedgehog protein promotes bone
marrow-derived endothelial progenitor cell proliferation, migration
and VEGF production via PI 3-kinase/Akt signaling pathways. Acta
Pharmacol Sin. 27:685–693. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kanaya K, Ii M, Okazaki T, Nakamura T,
Horii-Komatsu M, Alev C, Akimaru H, Kawamoto A, Akashi H, Tanaka H,
et al: Sonic Hedgehog signaling regulates vascular differentiation
and function in human CD34 positive cells: Vasculogenic CD34(+)
cells with Sonic Hedgehog. Stem Cell Res. 14:165–176. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kusano KF, Pola RT, Murayama T, Curry C,
Kawamoto A, Iwakura A, Shintani S, Ii M, Asai J, Tkebuchava T, et
al: Sonic hedgehog myocardial gene therapy: Tissue repair through
transient reconstitution of embryonic signaling. Nat Med.
11:1197–1204. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
31
|
Ahmed RP, Haider KH, Shujia J, Afzal MR
and Ashraf M: Sonic Hedgehog gene delivery to the rodent heart
promotes angiogenesis via iNOS/netrin-1/PKC pathway. PLoS One.
5:pp. e85762010, View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Xiao Q, Yang Y, Qin Y, He YH, Chen KX, Zhu
JW, Zhang GP and Luo JD: AMP-activated protein kinase-dependent
autophagy mediated the protective effect of sonic hedgehog pathway
on oxygen glucose deprivation-induced injury of cardiomyocytes.
Biochem Biophys Res Commun. 457:419–425. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Roncalli J, Renault MA, Tongers J, Misener
S, Thorne T, Kamide C, Jujo K, Tanaka T, Ii M, Klyachko E and
Losordo DW: Sonic hedgehog-induced functional recovery after
myocardial infarction is enhanced by AMD3100-mediated
progenitor-cell mobilization. J Am Coll Cardiol. 57:2444–2452.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Liu Z, Tu K, Wang Y, Yao B, Li Q, Wang L,
Dou C, Liu Q and Zheng X: Hypoxia accelerates aggressiveness of
hepatocellular carcinoma cells involving oxidative stress,
epithelial-mesenchymal transition and non-canonical hedgehog
signaling. Cell Physiol Biochem. 44:1856–1868. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Hung YH, Chang SH, Huang CT, Yin JH, Hwang
CS, Yang LY and Yang DI: Inhibitor of differentiation-1 and
hypoxia-inducible factor-1 mediate sonic hedgehog induction by
amyloid beta-peptide in rat cortical neurons. Mol Neurobiol.
53:793–809. 2016. View Article : Google Scholar
|
|
36
|
Spivak-Kroizman TR, Hostetter G, Posner R,
Aziz M, Hu C, Demeure MJ, Von Hoff D, Hingorani SR, Palculict TB,
Izzo J, et al: Hypoxia triggers hedgehog-mediated tumor-stromal
interactions in pancreatic cancer. Cancer Res. 73:3235–3247. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Onishi H, Kai M, Odate S, Iwasaki H,
Morifuji Y, Ogino T, Morisaki T, Nakashima Y and Katano M: Hypoxia
activates the hedgehog signaling pathway in a ligand-independent
manner by upregulation of Smo transcription in pancreatic cancer.
Cancer Sci. 102:1144–1150. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Bijlsma MF, Groot AP, Oduro JP, Franken
RJ, Schoenmakers SH, Peppelenbosch MP and Spek CA: Hypoxia induces
a hedgehog response mediated by HIF-1alpha. J Cell Mol Med.
13:2053–2060. 2009. View Article : Google Scholar
|
|
39
|
Marfella R, D'Amico M, Di Filippo C,
Piegari E, Nappo F, Esposito K, Berrino L, Rossi F and Giugliano D:
Myocardial infarction in diabetic rats: Role of hyperglycaemia on
infarct size and early expression of hypoxia-inducible factor 1.
Diabetologia. 45:1172–1181. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Ceriello A, Quagliaro L, D'Amico M, Di
Filippo C, Marfella R, Nappo F, Berrino L, Rossi F and Giugliano D:
Acute hyperglycemia induces nitrotyrosine formation and apoptosis
in perfused heart from rat. Diabetes. 51:1076–1082. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Banerjee Mustafi S, Aznar N, Dwivedi SK,
Chakraborty PK, Basak R, Mukherjee P, Ghosh P and Bhattacharya R:
Mitochondrial BMI1 maintains bioenergetic homeostasis in cells.
FASEB J. 30:4042–4055. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Lin X, Ojo D, Wei F, Wong N, Gu Y and Tang
D: A novel aspect of tumorigenesis—BMI1 functions in regulating dna
damage response. Biomolecules. 5:3396–3415. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Allegra E, Trapasso S, Pisani D and Puzzo
L: The role of BMI1 as a biomarker of cancer stem cells in head and
neck cancer: A review. Oncology. 86:199–205. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Ren H, Du P, Ge Z, Jin Y, Ding D, Liu X
and Zou Q: TWIST1 and BMI1 in cancer metastasis and
chemoresistance. J Cancer. 7:1074–1080. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Yao D, Wang Y, Xue L, Wang H, Zhang J and
Zhang X: Different expression pattern and significance of
p14ARF-Mdm2-p53 pathway and Bmi-1 exist between gastric cardia and
distal gastric adenocarcinoma. Hum Pathol. 44:844–851. 2013.
View Article : Google Scholar
|
|
46
|
Xu XH, Liu Y, Li DJ, Hu J, Su J, Huang Q,
Lu MQ, Yi F, Bao D and Fu YZ: Effect of shRNA-mediated gene
silencing of Bmi-1 expression on chemosensitivity of CD44+
nasopharyngeal carcinoma cancer stem-like cells. Technol Cancer Res
Treat. 15:NP27–NP39. 2016. View Article : Google Scholar
|
|
47
|
Kundu N, Domingues CC, Chou C, Ahmadi N,
Houston S, Jerry DJ and Sen S: Use of p53-silenced endothelial
progenitor cells to treat ischemia in diabetic peripheral vascular
disease. J Am Heart Assoc. 6:pp. e0051462017, View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Brownlee M: Biochemistry and molecular
cell biology of diabetic complications. Nature. 414:813–820. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Feldman EL: Oxidative stress and diabetic
neuropathy: A new understanding of an old problem. J Clin Invest.
111:431–433. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Kim WK, Meliton V, Bourquard N, Hahn TJ
and Parhami F: Hedgehog signaling and osteogenic differentiation in
multipotent bone marrow stromal cells are inhibited by oxidative
stress. J Cell Biochem. 111:1199–1209. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Larrick JW and Mendelsohn A: Applied
healthspan engineering. Rejuvenation Res. 13:265–280. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Chen X, Gu M, Zhao X, Zheng X, Qin Y and
You X: Deterioration of cardiac function after acute myocardial
infarction is prevented by transplantation of modified endothelial
progenitor cells over-expressing endothelial NO synthases. Cell
Physiol Biochem. 31:355–365. 2013. View Article : Google Scholar
|
|
53
|
Bianconi V, Sahebkar A, Kovanen P,
Bagaglia F, Ricciuti B, Calabrò P, Patti G and Pirro M: Endothelial
and cardiac progenitor cells for cardiovascular repair: A
controversial paradigm in cell therapy. Pharmacol Ther.
181:156–168. 2017. View Article : Google Scholar : PubMed/NCBI
|