Introduction
In the last 25 years, the rate of preterm births has
increased by approximately one-third globally (1). The majority of preterm infants are
born during the saccular stage of lung development (26 to 36
gestational weeks), while the lung structure is immature, with
deficient surfactant proteins (SPs), delayed absorption of
intrapulmonary fluid and inefficient gas exchange (2). Accumulating evidence in previous
years has demonstrated a consistently high risk of respiratory
morbidity in infants born at <37 weeks of gestation (3). Lung development is a complex process
with a series of orchestrated events. Premature birth interrupts
normal lung development, which results in a series of respiratory
diseases (4). Neonatal
respiratory distress syndrome (RDS), a condition of pulmonary
insufficiency that in its natural course commences at or shortly
following birth and increases in severity over the first 48 h of
life, is a common disease in preterm babies (5). RDS occurs in ~45% of early or
moderate-preterm infants (23–33 weeks of pregnancy) (5). The main cause of RDS is a deficiency
of SPs along with structural immaturity (4,5).
Thus, improving the understanding of lung development may hold
promise for improving the prevention and treatment of respiratory
diseases in preterm infants.
MicroRNAs (miRNAs/miRs) are a class of small
non-coding RNAs that are ~22 nucleotides long, and that inhibit the
translation of target mRNAs, serving an important function in cell
proliferation, differentiation and organ development (6). A number of studies have documented
that >100 miRNAs undergo substantial changes in expression
during lung development (6,7).
However, little is known about if or how they regulate lung
development. A previous study reported that miR-431 expression is
higher in preterm infants with RDS compared with those without RDS
(8). Additionally, miR-431 was
revealed to be downregulated in the lung tissues of Sprague-Dawley
rats at 3 time points, embryonic day 19, embryonic day 21 and
postnatal day 3, by reverse transcription quantitative PCR
(RT-qPCR) and fluorescence in situ hybridization (9).
Alveolar epithelial type II (AECII) cells are
specialized epithelial cells for maintaining lung function and
homeostasis, and are able to regulate the metabolism of alveolar
surfactants, express innate immune molecules, and regenerate and
restore alveoli in response to an injury (10,11). Tumor AECII cell lines fail to
fully recapitulate alveolar epithelial cell phenotypes in
vivo. However, the A549 cell line is a classic human type II
alveolar epithelial cell line, which contains lamellar bodies
producing surfactant and containing a phospholipid content similar
to that of AECII cells in situ (12). Therefore, A549 cells have the
majority of the functions of type II cells. In a previous study,
SMAD family member 4 (SMAD4) was identified as an important target
of miR-431 in A549 cells; the overexpression of miR-431 was able to
accelerate the proliferation of A549 cells, inhibit apoptosis and
downregulate the expression of SPs (13,14). Thus, it was hypothesized that
miR-431 is critical for the expression of SPs and it serves a
regulatory function similar to the bone morphogenetic protein 4
(BMP4)/activin/transforming growth factor β (TGF-β) signaling
pathway.
As a member of the SMAD family, SMAD4 is a common
mediator of the TGF-β signaling pathway. One previous study
reported that, in the canonical signaling pathway, activated SMAD2
and SMAD3 form a complex with SMAD4, a component shared by all
TGF-β family members, and this complex translocates to the nucleus
to regulate the transcription of target genes (15). Notably, increasing evidence
suggests that SMAD4-dependent TGF-β signaling serves an important
function in the differentiation of endodermal cells and inhibits
the epithelial to mesenchymal transition process, which is
essential for lung branching morphogenesis (16,17). BMP4 belongs to the TGF-β
superfamily and is a multifunctional peptide with important
functions in normal lung development (18).
In the present study, the functions of miR-431 and
its target gene SMAD4 were investigated to follow on from previous
studies. The present study aimed to determine the potential
mechanisms of miR-431 in the regulation of lung development. The
results of the present study may provide crucial molecular and
cellular evidence of the potential of miR-431 as a marker of fetal
lung development and as a therapeutic target for lung developmental
diseases.
Materials and methods
Cell culture
The human non-squamous cell lung carcinoma line
(A549 cell line), obtained from the American Type Culture
Collection (Manassas, VA, USA), were seeded and grown in Dulbecco’s
modified Eagle’s medium (DMEM; Gibco; Thermo Fisher Scientific,
Inc., Waltham, MA, USA) supplemented with 10% fetal bovine serum
(FBS; Gibco; Thermo Fisher Scientific, Inc.) and (100 U/ml) 1%
penicillin and 100 μg/ml streptomycin; Gibco; Thermo Fisher
Scientific, Inc.) at the condition of 37°C in a humidified
incubator containing 5% CO2.
Generating stable cell lines with
lentivirus
The lentivirus of the overexpression of SMAD4
(LV-SMAD4+), the lentivirus of the knockdown of SMAD4
(LV-SMAD4−) and their corresponding controls
(LV-SMAD4+-NC and LV-SMAD4−-NC) were obtained
from Shanghai GenePharma Co., Ltd. (Shanghai, China).
LV-SMAD4+ and LV-SMAD4+-NC were used with
adenovirus-based vectors containing the green fluorescent protein
(GFP) gene and puromycin-resistant marker (cat. no.
AAV5-EF-1a-GFP&Puro; Shanghai GenePharma Co., Ltd.), while
LV-SMAD4− and LV-SMAD4−-NC were used with
different adenovirus-based vectors containing the GFP gene and
puromycin-resistant marker (cat. no. AAV3-H1-GFP&Puro; Shanghai
GenePharma Co., Ltd.). In detail, LV-SMAD4+ synthesized
and cloned the SMAD4 gene (gene ID: NM_005359.5) into the vector
AAV5-EF-1a-GFP&Puro, and LV-SMAD4+-NC used the empty
vector AAV5-EF-1a-GFP&Puro. Meanwhile, LV-SMAD4−
synthesized and cloned the SMAD4-homo-1506
(5′-GGTGTTCCATTGCTTACTTTG-3′) into the vector AAV3-H1-GFP&Puro,
and LV-SMAD4−-NC synthesized and cloned a universal
sequence (5′-TTCTCCGAACGTGTCACGT-3′) into the vector
AAV3-H1-GFP&Puro. In additional, the LV-SMAD4−
knockdown of endogenous SMAD4 was performed. To minimize the
off-target effects of RNA interference, three potential sequences
targeting the human SMAD4 gene were constructed and the shRNA
sequence (5′-GGTGTTCCATTGCTTACTTTG-3′) was selected for the
subsequent experiment. The selected SMAD4 shRNA sequence and a
scrambled sequence (5′-TTCTCCGAACGTGTCACGT-3′) with no homology to
any known human genes were synthesized and ligated into the
AAV3-H1-GFP&Puro adenovirus-based vector. A549 cell lines were
cultured in the 96-well plate at a concentration of
5x104 cells/well, mixed and incubated at the condition
of 37°C in a humidified incubator containing 5% CO2 for
24 h. Then, 10 μl lentiviral stock was diluted 1:10 in DMEM medium
containing 10% FBS, and polybrene was added at a concentration of 5
μg/ml. Next, the culture medium was aspirated in a 96-well plate,
and 100 μl of each of the aforementioned diluted virus solution was
added to each well, a blank control group was set up, and incubated
with Polybrene (Shanghai GenePharma Co., Ltd.) to a final
concentration 6 μg/ml for 24 h at 37°C in a humidified incubator
containing 5% CO2. Finally, the diluted virus solution
was aspirated from the 96-well plate and 100 μl DMEM containing 10%
FBS was added to each well and cultured for 72 or 96 h, and the
lentivirus infection efficiency was observed under an optical
microscope (x100 magnification; DM2500; Leica Microsystems GmbH,
Wetzlar, Germany) and a fluorescent microscope (BX51; Olympus
Corporation, Tokyo, Japan). Meanwhile, a concentration gradient of
puromycin was established (0, 0.1, 0.2, 0.4, 0.6, 0.8, 1.0, 1.2,
1.5, 2.0, 2.5 and 3.0 μg/ml; Sigma-Aldrich; Merck KGaA, Darmstadt,
Germany) to screen for the optimal puromycin concentration that
completely killed the cells. Finally, the stable cell lines were
verified by RT-qPCR and western blotting.
miR-431 mimic, miR-431 inhibitor and cell
transfection
miR-431 mimic, miR-431 inhibitor and the
corresponding control (mimic-NC and inhibitor-NC) were synthesized
by Guangzhou RiboBio Co., Ltd. Stable cell lines
(LV-SMAD4+ and LV-SMAD4−) were transfected
with miR-431 mimics, an miR-431 inhibitor and the corresponding
negative control with Lipofectamine 3000 (Invitrogen; Thermo Fisher
Scientific), according to the manufacturer’s protocol. Mixture A
contained 125 μl DMEM and 5 μl Lipofectamine 3000. Mixture B
contained 125 μl DMEM and 10 μl mimic or inhibitor or the
corresponding control. The two mixtures were left to stand at room
temperature for 10 min initially, and then mixed and left for a
further 10 min. The complex was then added to each well of the
6-well plate and incubated for 24 h at 37°C. Finally, the
Lipofectamine 3000 mixture was aspirated, and DMEM supplemented
with 10% FBS was added and cultured for 48 h. In brief, cells
(LV-SMAD4+ and LV-SMAD4−) were plated in
6-well plates. When cells reached a confluence of ~50% in complete
growth medium, cells were transfected with 50 nM mimics or 100 nM
inhibitors and incubated for 48 or 72 h for RNA or protein
extraction. The miR-431 sequence used was accession no.
MIMAT0001625, 5′-UGUCUUGCAGGCCGUCAUGCA-3′.
RNA extraction, cDNA synthesis and
RT-qPCR
Total RNA was isolated from cells (A549 cells
treated as above) by TRIzol reagent (Invitrogen; Thermo Fisher
Scientific, Inc.). The RNA concentration were detected using a
NanoDrop2000 Spectrophotometer (Thermo Fisher Scientific, Inc.).
The conversion of miR-431 into cDNA was performed with the
HiScript®II Q Select RT SuperMix for qPCR (cat. no.
R232-01; Vazyme, Piscataway, NJ, USA) according to the
manufacturer’s protocol [including 4 μl 5x HiScript II Select qRT
SuperMix, 1 μl miR-431-primer (50 ng/μl) and 1 μg RNA]. The RT
primer of miR-431 was designed and synthesized by Generay Biotech
Co., Ltd. and is listed in Table
I. Reverse transcription of 1 μg RNA isolated from A549 cells
into cDNA was performed using the HiScript®II Q Select
RT SuperMix for qPCR (cat. no. R222-01; Vazyme) according to the
manufacturer’s protocol [including 4 μl 5x HiScript II Select qRT
SuperMix and 1 μg RNA]. The RT reaction was conducted at 37°C for
15 min and 85°C for 2 min. Next, the RT-qPCR reaction was performed
with AceQ® qPCR (cat. no. Q131-01; Vazyme) according to
the manufacturer’s protocol (including 1 μl cDNA, 5 μl
SYBR® Green Master Mix, 0.2 μl reverse and forward
primers and 3.6 μl diethypyrocarbonate water) on an ABI 7500
thermal cycler (Applied Biosystems; Thermo Fisher Scientific,
Inc.). The thermocycling conditions included an initial step at
95°C for 5 min, 40 cycles at 90°C for 15 sec and at 60°C for 15
sec, 72°C for 1 min and final extension at 72°C for 10 min. U6 was
used as the internal control of miR-431. GAPDH was used as the
internal control of all other genes (SMAD4, BMP4 and SPs). All
primer sequences used were designed and synthesized by Generay
Biotech Co., Ltd. and are listed in Table I. The relative mRNA expression was
measured using the 2−ΔΔCq method (19).
 | Table IPrimers for reverse
transcription-quantitative PCR. |
Table I
Primers for reverse
transcription-quantitative PCR.
| Gene name | Primers | Ta (°C) |
|---|
| miR-431 reverse
transcription primer |
5′-GTCGTATCCAGTGCAGGGTCCGAGGTATTC | 60 |
|
GCACTGGATACGACTGCATG-3′ | |
| miR-431 | F:
5′-ACGCGTGTCTTGCAGGCCGT-3′ | |
| R:
5′-ATCCAGTGCAGGGTCCGAGG-3′ | 60 |
| SP-A | F:
5′-TGTGTGCGAAGTGAAGGACG-3′ | |
| R:
5′-CTTTGAGACCATCTCTCCCGT-3′ | 60 |
| SP-B | F:
5′-AGGACACGATGAGGAAGT-3′ | |
| R:
5′-AGTCTGGTTCTGGAAGTAGT-3′ | 60 |
| SP-C | F:
5′-CACCTGAAACGCCTTCTTATCG-3′ | |
| R:
5′-TTTCTGGCTCATGTGGAGACC-3′ | 60 |
| SMAD4 | F:
5′-CTCATGTGATCTATGCCCGTC-3′ | |
| R:
5′-AGGTGATACAACTCGTTCGTAGT-3′ | 60 |
| BMP4 | F:
5′-GACCACCTCAACTCAACCAACCA-3′ | |
| R:
5′-GCACCCACATCCCTCTACTACCAT-3′ | 60 |
| U6 | F:
5′-CTCGCTTCGGCAGCACA-3′ | |
| R:
5′-AACGCTTCACGAATTTGCGT-3′ | 60 |
| GAPDH | F:
5′-AGAAGGCTGGGGCTCATTTG-3′ | |
| R:
5′-AGGGGCCATCCACAGTCTTC-3′ | 60 |
Western blotting
Cells (A549 cells treated as above) were harvested
and lysed in RIPA buffer (Beyotime Institute of Biotechnology)
containing a protease inhibitor and phosphatase inhibitor cocktail
(Beyotime Institute of Biotechnology). Following incubation at 4°C
for 10 min, the total lysates were centrifuged at 1,400 x g for 15
min at 4°C, and then the supernatant was extracted. Protein
concentration was determined with bicinchoninic acid protein assay
kit (cat. no. G2026; Servicebio Inc.), according to the
bicinchoninic acid protein quantification method (sourced from
Beyotime Institute of Biotechnology). Subsequently, 2 μl 5xSDS-PAGE
(Beyotime Institute of Biotechnology) loading buffer was added to 8
μl supernatant for 10 min at 100°C. Approximately 40 μg protein per
well was separated by 10% SDS-PAGE electrophoresis (Bio-Rad
Laboratories, Inc.), transferred to polyvinylidene fluoride
membranes (EMD Millipore), blocked with 5% skimmed milk powder in
tris-buffered saline with 0.05% Tween-20 (TBST) buffer for 1 h at
room temperature, and then immunoblotted with primary antibodies
overnight at 4°C. Subsequent to washing four times with TBST, the
membranes were further incubated with the horseradish
peroxidase-conjugated anti-rabbit secondary antibody (1:2,000; cat.
no. 7074; Cell Signaling Technology, Inc.) for 2 h at room
temperature. Finally, immunodetection was performed using an
Enhanced Chemiluminescence system (cat. no. G2014; Servicebio Inc.)
and analyzed using ImageJ software (version 2.1; National
Institutes Health), following a further four washes with TBST. All
antibodies used for western blotting were as follows: Anti-Smad4
(1:1,000; cat. no. 46535; Cell Signaling Technology, Inc.),
anti-BMP4 (1:10,000; cat. no. ab124715; Abcam, Cambridge, MA, USA),
anti-SP-A (1:1,000; cat. no. sc-80621; Santa Cruz Biotechnology,
Inc.), anti-SP-B (1:1,000; cat. no. sc-133143; Santa Cruz
Biotechnology, Inc.), Anti-proSP-C (1:500; cat. no. 2464523; EMD
Millipore) and anti-β-actin (1:5,000; cat. no. 4967; Cell Signaling
Technology, Inc.).
Statistical analysis
All data were presented as the mean ± standard
deviation. The data were analyzed using SPSS 17.0 (SPSS, Inc.) and
GraphPad Prism 5.0 (GraphPad Software, Inc.) statistical packages,
and evaluated using a Student’s t-test between the two groups.
P<0.05 was considered to indicate a statistically significant
difference.
Results
Effect of SMAD4 on the expression of SPs
and the BMP4/activin/TGF-β signaling pathway
The optimal puromycin concentration was determined
to be 1.2 μg/ml, and the efficiency of lentivirus infection was
observed by microscope after 72 and 96 h (Fig. 1). To confirm the transfection
efficiency of LV-SMAD4+ and LV-SMAD4− in
A549, SMAD4 expression levels were determined by RT-qPCR and
western blotting. LV-SMAD4+ transfection significantly
increased the expression levels of SMAD4 in cells compared with
LV-SMAD4+-NC (P<0.0005), and LV-SMAD4−
significantly decreased the expression of SMAD4 compared with
LV-SMAD4−-NC (P<0.05; Figs. 2A and 3A and B). Furthermore, to investigate
whether SMAD4 serves a function in SP expression via the
BMP4/activin/TGF-β signaling pathway, the expression levels of SPs
(SP-A, SP-B and SP-C) and BMP4 were determined by RT-qPCR. It was
revealed that SP-A, SP-B, SP-C and BMP4 exhibited increased
expression levels in LV-SMAD4− cells compared with
LV-SMAD4−-NC (P<0.001). On the contrary, the
expression levels of the SPs and BMP4 were significantly decreased
in LV-SMAD4+ cells compared with LV-SMAD4+-NC
cells (P<0.0005; Fig. 2). To
confirm this effect at the protein level, cell lines with stable
lentivirus transfection were harvested for western blotting.
Similar results were observed via western blotting, although the
differences were not as significant as those observed by RT-qPCR
(Fig. 3). In addition, miR-431
expression levels were also significantly increased when SMAD4 was
overexpressed compared with LV-SMAD4+-NC cells
(P<0.0001; Fig. 2).
 | Figure 2Validation of the expression of
SMAD4, BMP4, SPs and miR-431 in four types of stable cell lines
with lentivirus (LV-SMAD4+, LV-SMAD4+-NC,
LV-SMAD4− and LV-SMAD4−-NC) determined using
reverse transcription-quantitative PCR. (A) SMAD4 mRNA expression
levels in cell lines with lentivirus. (B) BMP4 mRNA expression
levels in cell lines with lentivirus. (C) SP-A mRNA expression
levels in cell lines with lentivirus. (D) SP-B mRNA expression
levels in cell lines with lentivirus. (E) SP-C mRNA expression
levels in cell lines with lentivirus. (F) miR-431 mRNA expression
levels in cell lines with lentivirus. **P<0.001,
***P<0.0005 and ****P<0.0001 with
comparisons shown by lines. LV, lentivirus; SMAD4, SMAD family
member 4; NC, negative control; BMP4, bone morphogenetic protein 4;
SP, surfactant proteins; miR, microRNA. |
 | Figure 3Western blotting was used to detect
the protein expression levels of SMAD4, BMP4 and SPs in four types
stable cell lines with lentivirus (LV-SMAD4+,
LV-SMAD4+-NC, LV-SMAD4− and
LV-SMAD4−-NC). (A) Western blotting results for SMAD4,
BMP4 and SPs (SP-A, SP-B and SP-C), with β-actin used as an
internal control. (B) Relative expression levels of SMAD4 protein.
The data confirmed the transfection efficiency of four types of
stable cell lines with lentivirus (LV-SMAD4+,
LV-SMAD4+-NC, LV-SMAD4− and
LV-SMAD4−-NC). (C) Relative expression levels of BMP4
protein. (D) Relative expression levels of SP-A protein. (E)
Relative expression levels of SP-B protein. (F) Relative expression
levels of SP-C protein. *P<0.05,
**P<0.001 and ***P<0.0005 with
comparisons shown by lines. LV, lentivirus; SMAD4, SMAD family
member 4; NC, negative control; BMP4, bone morphogenetic protein 4;
SP, surfactant proteins. |
miR-431 inhibits the expression of SPs by
inhibiting the BMP4/activin/TGF-β signaling pathway
A previous study demonstrated that miR-431 may
negatively regulate lung development by targeting SMAD4 (13). In the present study, the mechanism
of the miR-431-mediated inhibition of the expression of SPs via
BMP4/activin/TGF-β signaling in A549 cells treated with SMAD4 was
investigated. A549 cells were treated with miR-431 mimic in
combination with LV-SMAD4−, which resulted in the
significantly increased expression levels of BMP4 (P<0.0001) and
significantly reduced expression levels of the SPs (P<0.05) at
the mRNA level compared with cells transfected with miR-431
mimic-NC. On the contrary, cells treated with the miR-431 inhibitor
in combination with LV-SMAD4− exhibited significantly
reduced expression levels of BMP4 (P<0.001) and significantly
increased expression levels of SPs (P<0.05) at the mRNA level
compared with cells transfected with miR-431 inhibitor-NC (Fig. 4). The changes in protein
expression levels of the SPs were consistent with the changes at
mRNA level, while the relative expression levels of BMP4 at the
protein level was different to that observed at the mRNA level
(Fig. 5). Overexpression of
miR-431 combined with LV-SMAD4+ resulted in the
significantly reduced expression levels of SPs (P<0.05), and the
expression levels of BMP4 were significantly increased at the mRNA
and protein levels (P<0.05) compared with the negative control.
Cells with the inhibition of miR-431 combined with
LV-SMAD4+ exhibited significantly increased expression
levels of SPs (P<0.05) and reduced expression levels of BMP4
(P<0.05) at the mRNA and protein levels compared with the
inhibitor-NC group. In addition, the expression of SMAD4 was
determined subsequent to transfecting miR-431 into
LV-SMAD4+ cells, and it was revealed that miR-431
significantly downregulated the expression of SMAD4 compared with
the mimic-NC group (P<0.001; Figs.
6 and 7).
 | Figure 4Relative expression levels of
miR-431, BMP4, SPs and SMAD4 in stable cell lines with
LV-SMAD4− transfected with miR-431 mimic, inhibitor and
the corresponding control (mimic-NC and inhibitor-NC) by reverse
transcription-quantitative PCR. (A) miR-431 mRNA expression levels
were determined in LV-SMAD4− cells to confirm the
transfection efficiency of the mimic, inhibitor and the
corresponding controls. (B) BMP4 mRNA expression levels in
LV-SMAD4− cells transfected with miR-431 mimic,
inhibitor and the corresponding control. (C) SP-A mRNA expression
levels. (D) SP-B mRNA expression levels. (E) SP-C mRNA expression
levels. (F) SMAD4 mRNA expression levels. *P<0.05,
**P<0.001, ***P<0.0005 and
****P<0.0001 with comparisons shown by lines. miR,
microRNA; LV, lentivirus; SMAD4, SMAD family member 4; NC, negative
control; BMP4, bone morphogenetic protein 4; SP, surfactant
proteins. |
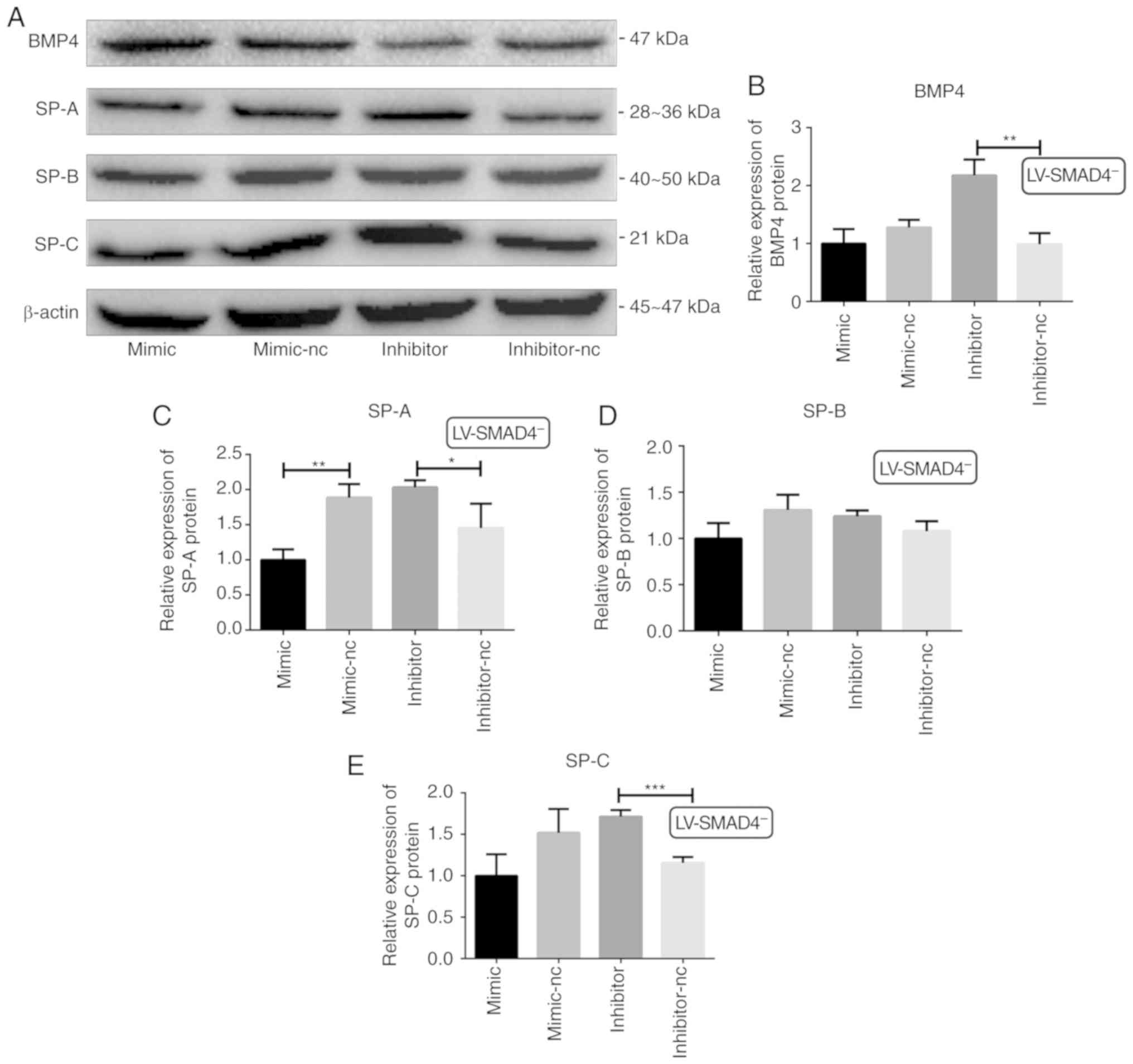 | Figure 5Validation of the expression of BMP4
and SPs (SP-A, SP-B and SP-C) protein levels in stable cell lines
with LV-SMAD4− transfected with an miR-431 mimic,
inhibitor and the corresponding control (mimic-NC and inhibitor-NC)
via western blotting. (A) Western blotting results for BMP4 and SPs
(SP-A, SP-B and SP-C), with β-actin used as the internal control.
(B) Relative expression levels of BMP4 protein. (C) Relative
expression levels of SP-A protein. (D) Relative expression levels
of SP-B protein. (E) Relative expression levels of SP-C protein.
*P<0.05, **P<0.001 and
***P<0.0005 with comparisons shown by lines. LV,
lentivirus; SMAD4, SMAD family member 4; NC, negative control;
BMP4, bone morphogenetic protein 4; SP, surfactant proteins. |
 | Figure 6Relative expression of miR-431, BMP4,
SPs and SMAD4 in stable cell lines with LV-SMAD4+
transfected with miR-431 mimic, inhibitor and the corresponding
control (mimic-NC and inhibitor-NC) determined using reverse
transcription-quantitative PCR. (A) miR-431 mRNA expression levels
in LV-SMAD4+ cells were determined to confirm the
transfection efficiency of mimic, inhibitor and the corresponding
controls. (B) BMP4 mRNA expression levels. (C) SP-A mRNA expression
levels. (D) SP-B mRNA expression levels. (E) SP-C mRNA expression
levels. (F) SMAD4 mRNA expression levels. *P<0.05,
**P<0.001 and ***P<0.0005 with
comparisons shown by lines. LV, lentivirus; SMAD4, SMAD family
member 4; NC, negative control; BMP4, bone morphogenetic protein 4;
SP, surfactant proteins; miR, microRNA. |
 | Figure 7Determination of BMP4, SMAD4 and SPs
protein levels in stable cell lines with LV-SMAD4+
transfected with miR-431 mimic, inhibitor and the corresponding
control (mimic-NC and inhibitor-NC) though western blotting. (A)
Western blotting results for SMAD4, BMP4 and SPs (SP-A, SP-B and
SP-C) expression, with β-actin used as the internal control. (B)
Relative expression levels of BMP4 protein. (C) Relative expression
levels of SP-A protein. (D) Relative expression levels of SP-B
protein. (E) Relative expression levels of SP-C protein. (F)
Relative expression levels of SMAD4 protein. *P<0.05,
**P<0.001, ***P<0.0005 and
****P<0.0001 with comparisons shown by lines. LV,
lentivirus; SMAD4, SMAD family member 4; NC, negative control;
BMP4, bone morphogenetic protein 4; SP, surfactant proteins. |
Discussion
Lung development is divided into five stages, the
embryonic, pseudoglandular, canalicular, saccular and alveolar
stages, and it extends from the embryonic period through the fetal
period up to birth and subsequent development (20). Based on the morphological
maturity, and the synthesis and secretion of SPs, the main function
of the lung is effective gas and blood exchange (7,20).
Evidence has demonstrated that a number of miRNAs (21), including miR-302/367 (22) and the miR-17-92 cluster (23), and multiple signaling pathways,
including Hippo (24,25), WNT (26,27) and vascular endothelial growth
factor (28,29), participate in the regulation of
lung specification and maturation (7). In previous studies, the levels of
miR-431 were demonstrated to decline during rat lung development
and demonstrated a higher expression in preterm infants with RDS
compared with those without RDS (8,9,14,30). SMAD4 was identified as a target
gene of miR-431 via TargetScan and luciferase reporter assays, and
SPs were differentially expressed in A549 cells overexpressing
miR-431 or with miR-431 knocked down (13). In the present study, SMAD4 was
revealed to negatively regulate the expression of BMP4 and SPs in
A549 cells when overexpressed. As a member of the SMAD family,
SMAD4 is mainly expressed in the epithelial cytoplasm, and is
associated with other SMADs to negatively regulate lung
morphogenesis (31). In addition,
a previous study demonstrated that newborn mice with long-term
exposure to hyperoxia exhibited the upregulation of SMAD4 and
dampening of the BMP4 signaling pathway, which resulted in AECII
functional defects, obstruction of normal alveolarization and
formation of bronchopulmonary dysplasia (32). The results of the present study
are consistent with those reports. In addition, in the present
study, the expression level of miR-431 was lower in the SMAD4
knockdown group, indicating that miR-431 negatively regulates the
expression of SPs.
The molecular mechanism of miR-431-mediated
regulation of the expression of SPs was investigated in the present
study by overexpressing or knocking down miR-431, combined with the
overexpression or knockdown of SMAD4. The expression of SPs was
reduced in the SMAD4 knockdown group via overexpressing miR-431,
and increased in the SMAD4 overexpression group via inhibiting
miR-431. However, the expression of BMP4 was inconsistent at the
protein and mRNA levels in the LV-SMAD4− stable cell
line with overexpression or knockdown of miR-431. This
inconsistency may be due to the following reasons: i) Transcription
of mRNA and protein levels are not necessarily parallel; ii)
occasionally, the rate and degree of degradation of mRNA may have
impacted the results; iii) post-translational modification of the
protein may impact the rate of degradation. Meanwhile, this
inconsistency may result in the differences in the expression of
SPs at the protein level being not as significant as those observed
by RT-qPCR in the LV-SMAD4− stable cell line with
overexpression or knockdown of miR-431. Overall, the results
indicated that miR-431 negatively regulates the expression of SPs
through the inhibition of the BMP4/activin/TGF-β signaling
pathway.
BMPs are multifunctional growth factors belonging to
the TGF-β superfamily, and they function through serine/threonine
receptor kinases, composed of type I and II subtypes (33,34). The type I receptor kinases may be
transphosphorylated by the type II receptor kinases in the TGF-β
system, and interact with SMADs, including SMAD1, SMAD5, SMAD8 and
SMAD4, to translocate to the nucleus and participate in the
regulation of transcription together with other transcription
factors (33). The
BMP4/activin/TGF-β signaling pathway functions in lung development,
from mesodermal differentiation to the saccular and alveolarization
stages of late lung development (33,34). One study has demonstrated that
homozygous BMP4 mutant embryos die between embryonic stage 6.5 and
9.5, and have little or no mesodermal differentiation (35). Addition of BMP4 to the medium
surrounding lungs grown in organ culture stimulates cell
proliferation and branching morphogenesis (36). Additionally, BMP4/activin/TGF-β
signaling is active during late lung development, indicating that
the BMP4/activin/TGF-β signaling pathway serves a notable function
in septal and vascular development, and maintains homeostasis of
the epithelial layer of the large conducting airways in the mature
lung (34). The present results
suggest that the BMP4/activin/TGF-β signaling pathway also
regulates the expression of SPs. miR-431 may inhibit the
BMP4/activin/TGF-β signaling pathway by targeting SMAD4.
Pulmonary surfactant (PS) is stored in the lamellar
bodies of AECII cells (37). PS
is a complex of lipids, proteins and carbohydrates, containing at
least 4 types of proteins, including SP-A, SP-B, SP-C and SP-D.
SP-C is the predominant PS-associated protein, followed by SP-B,
SP-A and SP-D in terms of concentration, SP-D is the only type that
does not have lipid-containing structures, while the other 3 types
(SP-A, SP-B and SP-C) have lipid-containing structures (38,39). The quantity of PS is one of the
most important indicators of the grade of pulmonary maturity and is
closely associated with fetal development (40). The majority of preterm births
occur during the late canalicular or saccular stage (weeks 26–36),
when distal epithelial progenitors start to give rise to
bipotential alveolar epithelial progenitors, and AECII cells are
just starting to produce PS (2).
PS reduces the surface tension within the alveoli and enhance
alveolar expansion, allowing for gas exchange. PS deficiency
increases the incidence of alveolar atelectasis, which further
reduces the carbon dioxide and oxygen exchange between the
pulmonary capillaries and alveoli and ventilation perfusion ratio
(V/Q) mismatching ensues (41,42). PS deficiency exacerbates
hypoxemia, hypercarbia and acidosis (41). This is a key mechanism through
which PS deficiency causes RDS in preterm infants, resulting in
tachypnea, retractions of alveoli, nasal flaring, diminished breath
sounds, inspiratory crackles, cyanosis and pallor (42). RDS remains a common disease in
preterm infants, although prenatal steroids, respiratory support
and PS replacement for neonates have reduced the occurrence
(1). Thus, a complete
understanding of the molecular mechanisms of lung development,
particularly in the synthesis and secretion of PS, is critical for
improving clinical practice.
In conclusion, the present study revealed that
miR-431 negatively regulates the expression of SPs via inhibition
of the BMP4/activin/TGF-β signaling pathway by targeting SMAD4 in
A549 cells in vitro. These results indicate that the
suppression of miR-431 may promote the expression of SPs. However,
the present study was performed in vitro with only one cell
line and does not clarify the connection with other signaling
pathways in lung developmental. Therefore, in vivo studies
are required to verify whether miR-431 is able to regulate the
expression of SPs via inhibition of the BMP4/activin/TGF-β
signaling pathway by targeting SMAD4. Thus, the specific inhibitors
and agonists of key signaling molecules will be used to further
investigate the mechanisms of miR-431-mediated lung development
in vitro. A tissue-specific miR-431 knockout mouse model
will be built using the CRISPR/Cas9 system to further investigate
how miR-431 influences in vivo lung development, morphology
and function. Moving forward, the challenge will be to further
delineate the mechanisms of miR-431, and to develop therapeutic
strategies to selectively inhibit miR-431 or SMAD4 and enhance the
BMP4/activin/TGF-β signaling pathway to effectively prevent or
treat lung developmental diseases, particularly RDS.
Acknowledgements
Not applicable.
Funding
The present study was supported by the National
Natural Science Foundation of China (grant no. 81601321) and the
Jiangsu Science and Education Talents Program (grant no.
QNRC2016092).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors’ contributions
YY, XYZ and XGZ conceived and designed the
experiments of the current study. YQS, ZDB and JJP performed the
experiments. YQS, ZDB, XNM and RC analyzed the data. YQS, YY and
XYZ drafted the manuscript. ZDB revised the manuscript. All authors
read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Condò V, Cipriani S, Colnaghi M, Bellù R,
Zanini R, Bulfoni C, Parazzini F and Mosca F: Neonatal respiratory
distress syndrome: Are risk factors the same in preterm and term
infants? J Matern Fetal Neonatal Med. 30:1267–1272. 2017.
View Article : Google Scholar
|
|
2
|
Volckaert T and De Langhe SP: Wnt and FGF
mediated epithelial-mesenchymal crosstalk during lung development.
Dev Dyn. 244:342–366. 2015. View Article : Google Scholar
|
|
3
|
Colin AA, McEvoy C and Castile RG:
Respiratory morbidity and lung function in preterm infants of 32 to
36 weeks’ gestational age. Pediatrics. 126:115–128. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Mahoney AD and Jain L: Respiratory
disorders in moderately preterm, late preterm, and early term
infants. Clin Perinatol. 40:665–678. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Sweet DG, Carnielli V, Greisen G, Hallman
M, Ozek E, Plavka R, Saugstad OD, Simeoni U, Speer CP, Vento M, et
al: European consensus guidelines on the management of neonatal
respiratory distress syndrome in preterm infants-2013 update.
Neonatology. 103:353–368. 2013. View Article : Google Scholar
|
|
6
|
Ameis D, Khoshgoo N, Iwasiow BM, Snarr P
and Keijzer R: MicroRNAs in lung development and disease. Paediatr
Respir Rev. 22:38–43. 2017.PubMed/NCBI
|
|
7
|
Herriges M and Morrisey EE: Lung
development: Orchestrating the generation and regeneration of a
complex organ. Development. 141:502–513. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kan Q, Ding S, Yang Y and Zhou X:
Expression profile of plasma microRNAs in premature infants with
respiratory distress syndrome. Mol Med Rep. 12:2858–2864. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Sun ZY, Shen YQ, Chen XQ, Zhou XY, Cheng
R, Bao ZD and Yang Y: Expression and potential regulation of
miRNA-431 during lung development of Sprague-Dawley rats. Mol Med
Rep. 19:4980–4988. 2019.PubMed/NCBI
|
|
10
|
Wu J, Wang Y, Liu G, Jia Y, Yang J, Shi J,
Dong J, Wei J and Liu X: Characterization of air-liquid interface
culture of A549 alveolar epithelial cells. Braz J Med Biol Res.
51:e69502017. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Grek CL, Newton DA, Qiu Y, Wen X,
Spyropoulos DD and Baatz JE: Characterization of alveolar
epithelial cells cultured in semipermeable hollow fibers. Exp Lung
Res. 35:155–174. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ryndak MB, Singh KK, Peng Z and Laal S:
Transcriptional profile of Mycobacterium tuberculosis replicating
in type II alveolar epithelial cells. PLoS One. 10:e01237452015.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Li S, Sun Z, Chen T, Pan J, Shen Y, Chen
X, Zhou X, Cheng R and Yang Y: The role of miR-431-5p in regulating
pulmonary surfactant expression in vitro. Cell Mol Biol Lett.
24:252019. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Shen YQ, Yang Y, Sun ZY, Li SJ, Shen JX
and Zhou XY: Continuous expression of miR-431 during lung
development in Sprague-Dawley rats. Zhongguo Dang Dai Er Ke Za Zhi.
21:287–293. 2019.(In Chinese). PubMed/NCBI
|
|
15
|
Shi Y and Massagué J: Mechanisms of
TGF-beta signaling from cell membrane to the nucleus. Cell.
113:685–700. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zeng Y, Zhu J, Shen D, Qin H, Lei Z, Li W,
Huang JA and Liu Z: Repression of Smad4 by miR-205 moderates
TGF-β-induced epithelial-mesenchymal transition in A549 cell lines.
Int J Oncol. 49:700–708. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Xing Y, Li C, Hu L, Tiozzo C, Li M, Chai
Y, Bellusci S, Anderson S and Minoo P: Mechanisms of TGFbeta
inhibition of LUNG endodermal morphogenesis: The role of TbetaRII,
Smads, Nkx2.1 and Pten. Dev Biol. 320:340–350. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Geng Y, Dong Y, Yu M, Zhang L, Yan X, Sun
J, Qiao L, Geng H, Nakajima M, Furuichi T, et al: Follistatin-like
1 (Fstl1) is a bone morphogenetic protein (BMP) 4 signaling
antagonist in controlling mouse lung development. Proc Natl Acad
Sci USA. 108:7058–7063. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
20
|
Mullassery D and Smith NP: Lung
development. Semin Pediatr Surg. 24:152–155. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Johar D, Siragam V, Mahood TH and Keijzer
R: New insights into lung development and diseases: The role of
microRNAs. Biochem Cell Biol. 93:139–148. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Tian Y, Zhang Y, Hurd L, Hannenhalli S,
Liu F, Lu MM and Morrisey EE: Regulation of lung endoderm
progenitor cell behavior by miR302/367. Development. 138:1235–1245.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Carraro G, El-Hashash A, Guidolin D,
Tiozzo C, Turcatel G, Young BM, De Langhe SP, Bellusci S, Shi W,
Parnigotto PP and Warburton D: miR-17 family of microRNAs controls
FGF10-mediated embryonic lung epithelial branching morphogenesis
through MAPK14 and STAT3 regulation of E-Cadherin distribution. Dev
Biol. 333:238–250. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Lin C, Yao E and Chuang PT: A conserved
MST1/2-YAP axis mediates Hippo signaling during lung growth. Dev
Biol. 403:101–113. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Mahoney JE, Mori M, Szymaniak AD, Varelas
X and Cardoso WV: The hippo pathway effector Yap controls
patterning and differentiation of airway epithelial progenitors.
Dev Cell. 30:137–150. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zhang M, Shi J, Huang Y and Lai L:
Expression of canonical WNT/β-CATENIN signaling components in the
developing human lung. BMC Dev Biol. 12:212012. View Article : Google Scholar
|
|
27
|
Li C, Xiao J, Hormi K, Borok Z and Minoo
P: Wnt5a participates in distal lung morphogenesis. Dev Biol.
248:68–81. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Woik N and Kroll J: Regulation of lung
development and regeneration by the vascular system. Cell Mol Life
Sci. 72:2709–2718. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Liu X, Lin Y, Tian B, Miao J, Xi C and Liu
C: Maternal protein restriction alters VEGF signaling and decreases
pulmonary alveolar in fetal rats. Int J Clin Exp Pathol.
7:3101–3111. 2014.PubMed/NCBI
|
|
30
|
Yang Y, Kai G, Pu XD, Qing K, Guo XR and
Zhou XY: Expression profile of microRNAs in fetal lung development
of Sprague-Dawley rats. Int J Mol Med. 29:393–402. 2012.
|
|
31
|
Zhao J, Lee M, Smith S and Warburton D:
Abrogation of Smad3 and Smad2 or of Smad4 gene expression
positively regulates murine embryonic lung branching morphogenesis
in culture. Dev Biol. 194:182–195. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Alejandre-Alcázar MA, Kwapiszewska G,
Reiss I, Amarie OV, Marsh LM, Sevilla-Pérez J, Wygrecka M, Eul B,
Köbrich S, Hesse M, et al: Hyperoxia modulates TGF-beta/BMP
signaling in a mouse model of bronchopulmonary dysplasia. Am J
Physiol Lung Cell Mol Physiol. 292:L537–L549. 2007. View Article : Google Scholar
|
|
33
|
Chen D, Zhao M, Harris SE and Mi Z: Signal
transduction and biological functions of bone morphogenetic
proteins. Front Biosci. 9:349–358. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Alejandre-Alcázar MA, Shalamanov PD,
Amarie OV, Sevilla-Pérez J, Seeger W, Eickelberg O and Morty RE:
Temporal and spatial regulation of bone morphogenetic protein
signaling in late lung development. Dev Dyn. 236:2825–2835. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Winnier G, Blessing M, Labosky PA and
Hogan BL: Bone morphogenetic protein-4 is required for mesoderm
formation and patterning in the mouse. Genes Dev. 9:2105–2116.
1995. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Bragg AD, Moses HL and Serra R: Signaling
to the epithelium is not sufficient to mediate all of the effects
of transforming growth factor beta and bone morphogenetic protein 4
on murine embryonic lung development. Mech Dev. 109:13–26. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Sanders RL, Hassett RJ and Vatter AE:
Isolation of lung lamellar bodies and their conversion to tubular
myelin figures in vitro. Anat Rec. 198:485–501. 1980. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Crouch E and Wright JR: Surfactant
proteins a and d and pulmonary host defense. Annu Rev Physiol.
63:521–554. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Weaver TE and Conkright JJ: Function of
surfactant proteins B and C. Annu Rev Physiol. 63:555–578. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Parmigiani S, Solari E and Bevilacqua G:
Current concepts on the pulmonary surfactant in infants. J Matern
Fetal Neonatal Med. 18:369–380. 2005. View Article : Google Scholar
|
|
41
|
Rubarth LB and Quinn J: Respiratory
development and respiratory distress syndrome. Neonatal Netw.
34:231–238. 2015. View Article : Google Scholar
|
|
42
|
Reuter S, Moser C and Baack M: Respiratory
distress in the newborn. Pediatr Rev. 35:417–429. 2014. View Article : Google Scholar : PubMed/NCBI
|















