Introduction
Keloid disease (KD) is a complex fibroproliferative
disorder which appears as a pathological response to cutaneous
wound healing (1). Keloids are
defined as persistent existing scars that spread beyond the
boundaries of the original wound (2). Keloid scars not only affect the
appearance of the skin, but also cause itching and pain, and may
lead to necrosis, hemorrhaging and suppuration (1). Histologically, keloids are
characterized by the excessive accumulation of extracellular matrix
(ECM) components, such as type I collagen (3). Asians are susceptible to keloids,
with an incidence rate of 4-16% (4). KD is difficult to treat, since
keloids are prone to recurrence following surgical resection and
continue to proliferate for decades. Multiple therapies have been
established for KD; however, none of these are fully effective
(2,3,5,6).
Keloids are considered to be benign tumors of the dermis (7). The development of novel treatment
strategies for KD is thus required.
Although the etiopathogenesis of KD remains
incompletely understood, keloid fibroblasts (KFs) are deemed as the
main inductive cells for keloid formation. KFs exhibit a marked
infiltration in lesion tissue and display an increased rate of
proliferation; the aberrant behavior of KFs is considered to
contribute to disease progression (8). KFs are implicated as mediators of
elevated growth factor, chemokine or cytokine production, as well
as of the excessive accumulation of ECM components, such as type
I/III collagen and fibronectin (9,10).
The suppression of the malignant proliferation of KFs and the
blocking of their abnormal secretion of growth factors and ECM
components is crucial for KD treatment.
The Janus kinase (JAK)/signal transducer and
activator of transcription (STAT) signaling pathway is an important
pathway that is responsible for cell signal transduction from the
surface into the nucleus. STAT3 is a key member belonging to the
JAK/STAT signaling pathway, which plays a leading role in the
modulation of diverse processes, including cell proliferation,
migration, cytokine or chemokine production and inflammation
(11). It has been reported that
the expression and phosphorylation of STAT3 is abnormally increased
in keloid tissues, suggesting a role of STAT3 in the pathogenesis
of keloids (12). STAT3 is in
turn subject to multiple regulations by various cytokines, such as
interleukin (IL)-6, IL-10 and interferon (IFN)-α/γ (13), which were largely produced during
the progression of KD. The RNA interference of STAT3 has also been
reported to be associated with the inhibition of the proliferation
of keloids (12). As STAT3 is not
essential for the functioning of normal cells (14), it is rational to consider it as a
valuable target for the treatment of diseases, such as KD. However,
due to the lack of specific inhibitors, strategies have to be
developed to target the STAT3signaling pathway. AG490, as a
tyrosine kinase inhibitor for JAK2, can decrease the
phosphorylation of STAT3 by selectively inactivating JAK2 (15). The therapeutic potential of AG490
has been illustrated in brain hemorrhage (16), liver injury and fibrosis (17). It thus worthwhile to determine
whether it has a special role in the treatment of KD.
The present study hereby investigated the mechanisms
through which the inhibition of the JAK2/STAT3 pathway contributes
to the regulation of the aberrant behavior of KFs in vitro.
The effects of AG490 on the cellular and molecular behavior of
human KFs (HKFs), human normal fibroblasts (HNFs) and hypertrophic
scar fibroblasts (HSFs) were further examined.
Materials and methods
Reagents and antibodies
AG490 was purchased from MedChem Express Inc.
Stocking solution of AG490 was prepared as 100 mmol/l using
dimethylsulfoxide (DMSO) (Sigma-Aldrich; Merck KGaA) and stored at
−20°C. AG490 stock solution was further diluted in cell culture
medium to various concentrations (0, 12.5, 25, 50, 75 and 100
µmol/l) according to previous reports (15,18-21). The final concentration of DMSO was
not >0.1%. The recombinant human TGF-β was purchased from
PeproTech Inc. TGF-β powder was dissolved by 10 mmol/l citric acid
to prepare a liquid at 1.0 mg/ml (pH 3.0, containing 0.1% BSA).
Long-term storage was achieved by refrigeration −20°C. According to
the experiment requirements, TGF-β was diluted in cell culture
medium to a final concentration of 5 µg/ml. TGF-β was used
to treat the cells 2 h prior to the application of AG490.
Primary antibodies were purchased from Abcam,
including antibodies against STAT3 (ab119352), phosphorylated
(p-)STAT3 (Tyr705) (ab76315), CTGF (ab6992) and β-actin (ab179467).
Horseradish peroxidase (HRP)-conjugated secondary antibodies
produced in rabbit (SAB1301585) and mouse (SAB1411905) were
purchased from Sigma-Aldrich; Merck KGaA. The primers used in this
study were synthesized by Takara Biotechnology Co., Ltd (Dalian,
China). Other chemical reagents without special indication were
obtained from Sigma.
Cell culture
The cell lines, including HNFs, HKFs and HSFs were
kind gifts from Dr Qian Tan (Department of Burns and Plastic
Surgery, Nanjing University Medical School Affiliated Nanjing Drum
Tower Hospital) (22,23). Cells were routinely cultivated in
DMEM (HyClone; GE Healthcare Life Sciences) supplemented with 10%
fetal bovine serum (FBS; Invitrogen; Thermo Fisher Scientific,
Inc.), 100 µg/ml streptomycin and 100 U/ml penicillin
(Invitrogen; Thermo Fisher Scientific, Inc.) (37°C, 5%
CO2).
Cell proliferation assay (CCK-8
assay)
Cells were plated overnight in a 96-well plate
(5×104/well, 100 µl) until 80% confluence was
achieved. The cells were then treated with 0.1% DMSO (vehicle
control) or AG490 at various concentrations (0, 12.5, 25, 50, 75
and 100 µmol/l) for 24, 48 or 72 h. The culture supernatant
was discarded and the cells were washed with DMEM once. Following 2
h of incubation at 37°C with 10 µl CCK-8 (Sigma-Aldrich;
Merck KGaA), the OD absorbance was recorded at 450 nm using a
microplate reader (Tecan Group Ltd.). Cell viability was measured
using the following formula: Cell viability
%=[(ODtest-ODblank)/(ODcontrol-ODblank)]
×100%.
Cell cycle assay
During the exponential growth phase, HKFs were
digested by 0.25% trypsin and suspended in DMEM containing no FBS.
Cells were plated in a 6-well plate (2×106/well, 2 ml)
followed by 24 h of serum starvation. Subsequently, the cells were
treated with the vehicle control (0.1% DMSO) or various
concentrations (12.5, 25, 50, 75 and 100 µmol/l) of AG490
for 24 h. Following treatment, cells were harvested and washed with
cold PBS, followed by fixation in 70% ethanol at 4°C overnight.
Fixed cells were then treated with RNase (100 µg/ml, 50
µl) at 37°C in the dark for 30 min and stained with
propidium iodide (PI, 200 µl from 50 µg/ml stock
solution) at room temperature for 5 min. Cell cycle assay was
performed using a flow cytometer (BD Biosciences). Data were
analyzed using FlowJo7.6 software (Tree Star, Inc.).
Annexin-V/PI binding apoptosis assay
The serum-starved cells were prepared and cultured
as described above. Following treatment with AG490 at the indicated
concentrations for 24 h, cells were washed with cold PBS 3 times,
and then incubated with Annexin V-FITC and PI at room temperature
in the dark for 15 min. Samples were analyzed by flow cytometry (BD
Biosciences) within 1 h. Data were analyzed using FlowJo7.6
software (Tree Star, Inc.).
Western blot analysis
Following proper treatment, cells were collected and
lysed in RIPA buffer (WEIAO BioTech Co. Ltd.) with 1 mmol/l PMSF.
The same amount (30 µg) of proteins was loaded on 10%
SDS/PAGE gels and transferred to PVDF membranes (0.45 µM).
After blocking with 5% skim milk in the TBST bufer (50 mmol/l
Tris-HCl, 150 mmol/l NaCl, 0.1% Tween-20, pH 7.4) at room
temperature for 1 h, the membranes were incubated with primary
antibodies (1:1,000) overnight at 4°C. The membranes were then
washed with TBST buffer and incubated with horseradish peroxidase
(HRP)-conjugated secondary antibodies (1:20,000) for 1 h at room
temperature. The chemiluminescence detection kit (Thermo Fisher
Scientific, Inc.) was used to detect the protein signals with an
X-ray film (Fuji Film). The quantification of the western blots was
performed using ImageJ 1.52u software. The expression levels of
each protein were normalized to those of corresponding β-actin,
respectively.
RNA isolation, reverse transcription and
quantitative PCR
Differently treated cells were washed with PBS and
collected respectively. Total RNA was isolated using TRIzol Reagent
(Invitrogen; Thermo Fisher Scientific, Inc.). cDNA was prepared
using the ReverTra Ace qPCR RT kit (Toyobo) and amplified by
real-time PCR on StepOne Plus (Thermo Fisher Scientific, Inc.). The
primers sets and sequences are presented in Table I. Reactions were run in duplicate
as follows: 95°C for 5 min, followed by 40 cycles of 15 sec at 95°C
and 1 min at 60°C. The ending cycle was maintained at 60°C for 5
min. Gene expression levels were determined by relative
quantification according to the ΔΔCq method as previously described
(24).
 | Table IPrimer sequences used in RT-qPCR. |
Table I
Primer sequences used in RT-qPCR.
| Primer | Sequence
(5′→3′) |
|---|
| STAT3 forward |
CACATGCCACTTTGGTGTTTCA |
| STAT3 reverse |
GGGCAATCTCCATTGGCTTC |
| Cyclin D1
forward |
TCGGTGTCCTACTTCAAATGTGT |
| Cyclin D1
reverse |
GAAGCGGTCCAGGTAGTTCA |
| CTGF forward |
CTTGCGAAGCTGACCTGGAA |
| CTGF reverse |
AAAGCTCAAACTTGATAGGCTTGGA |
| MMP-2 forward |
ACCTACACCAAGAACTTCCGTCTG |
| MMP-2 reverse |
CTCGTATACCGCATCAATCT |
| TIMP-2 forward |
TCTGGCATCAGGCACCTGGATTGAG |
| TIMP-2 reverse |
CTATCCTAACCCCCATATCACT |
| VEGF forward |
GCTCTACTTCCCCAAATCACT |
| VEGF reverse |
CCCAAAAGCAGGTCACTCACT |
| β-actin
forward |
TGGCACCCAGCACAATGAA |
| β-actin
reverse |
CTAAGTCATAGTCCGCCTAGAAGCA |
STAT3 decoy and scramble
oligodeoxynucleotides (ODNs)
STAT3 decoy and scrambled control ODNs were designed
as previously described (25) and
were synthesized by Sangon Biotechnology Co., Ltd. The STAT3 decoy
ODN sequence (SODNs) was 5′-CAT TTC CCG TAA ATC-3′ and
3′-GTAAAGGGCATTTAG-5′. The scrambled ODN sequence (MODN) was 5′-CAT
TTC CCT TAA ATC-3′ and 3′-GTA AAG GGA ATT TAG-5′. The ODNs and the
Lipofectamine™ 2000 (Invitrogen; Thermo Fisher Scientific, Inc.)
were respectively diluted in equal volumes of serum-free medium
before being mixed at a ratio of 2:5 (µg:µl). The
mixture was incubated for 20 min at room temperature and then
transfected into the cells.
Cell invasion assay
Cell invasion assay in vitro was performed
using a 24-well Transwell chamber (Corning, Inc.) with an
8-µm pore size polycarbonate filter coated with ECMatrix gel
(Chemicon; Thermo Fisher Scientific, Inc.) to form a continuous
thin layer. HKFs transfected with ODNs (1.0×106/sample)
were harvested in serum-free DMEM containing 0.1% BSA and added to
the upper chamber. The lower chamber contained 500 µl DMEM
and 5% FBS. Cells were incubated for 72 h (37°C, 5% CO2)
followed by complete removal from the upper surface of the filter
using cotton swabs. The filters were fixed in 95% ethanol and
stained with crystal violet (Sigma-Aldrich; Merck KGaA) at room
temperature for 20 min. Cells migrating across the Matrigel and
reaching the lower surface of the filter were counted under a light
microscope [Nikon Imaging (China) Sales Co., Ltd.]. Samples were
acquired in triplicate and data were measured as the average cell
number in 10 fields.
Electrophoretic mobility shift assay
(EMSA)
After the cells were transfected with ODNs for 72 h
as described above, the nuclear protein was prepared as reported
previously (7). EMSA was
performed using the double-stranded synthetic oligo-nucleotides
mimicking the STAT3 binding sites present within the promoters of
the c-fos gene as follows: Sense, 5′-AGC TTC ATT TCC CGT AAA TCC
CTA-3′ and antisense, 5′-TAG GGA TTT ACG GGA AAT GAA GCT-3′. The
synthetic probes were 5′-end labeled using γ-32P-dATP
and T4 polynucleotide kinase. Nuclear proteins (10 µg) from
each sample were incubated with γ-32P-labeled
oligonucleotide probe (0.1 µg/µl, 1 µl) in 20
µl of binding buffer containing 10 mM HEPES (pH 7.8), 50
mmol/l KCl, 1 mmol/l EDTA, 5 mmol/l MgCl2, 10% glycerol,
5 mmol/l DTT, 1 mg/ml bovine serum albumin, and 1 mmol/l
Na3VO4. Following a 15-min incubation at room
temperature, the samples were separated on a 6% non-denaturing
polyacrylamide gel. For competition analyses, nuclear protein was
incubated with cold probe (unlabeled oligonucleotide) for 15 min at
room temperature prior to the addition of the labeled
oligonucleotides. Gels were dried and subjected to standard
autoradiographic procedures at -70°C. The quantification of STAT3
activation levels was performed using ImageJ 1.52u software.
Statistical analysis
Data are presented as the means ± SD obtained from
at least 3 independent tests. Data were analyzed by one-way ANOVA
followed by Tukey's multiple comparisons test (GraphPad Prism 7,
GraphPad Prism, Inc.). P<0.05 was considered to indicate a
statistically significant difference. The association between mRNA
expression levels was examined by a simple linear regression model.
Linear regression analysis was performed using SPSS®
Statistical software (IBM, Inc.).
Results
AG490 inhibits the proliferation and
induces the G1 cell cycle arrest of HKFs
The effects of AG490 (chemical structure presented
in Fig. 1A), the JAK2/STAT3
pathway inhibitor, were examined in HNFs and HKFs. Increasing
concentrations (0, 12.5, 25, 50, 75 and 100 µmol/l) of AG490
were respectively prepared to ensure that the final concentration
of DMSO in working assays was not >0.1%. Pre-experimental tests
were performed to confirm that DMSO had no deleterious effects on
cell proliferation at concentrations <0.2% (Fig. S1A). As was expected, the
expression levels of both STAT3 and p-STAT3 were decreased by the
application of AG490 in a dose-dependent manner (Fig. 1). In addition, the HKFs expressed
significantly higher mRNA levels of STAT3 than the HNFs, as
detected by RT-qPCR (Fig. S1B),
which is in accordance with the findings of previous findings
(12). Compared with the control
group, AG490 inhibited the proliferation of HNFs and HKFs at 24, 48
and 72 h in a time- and concentration-dependent manner (P<0.05,
Fig. 2A). At all testing time
points, the inhibition was more evident in the HKFs than the HNFs
(P<0.05). To elucidate the mechanisms through which AG490
suppressed HKF proliferation, the cell cycle distribution was
subsequently detected by flow cytometry. Following 24 h of
pre-treatment with AG490 at concentrations up to 100 µmol/l,
the cell proportion markedly declined from approximately 50 to 25%
at the G2/M phase, while increased it from 20 to 42% at the G0/G1
phase in response to AG490 (Fig. 2B
and C). By contrast, the cell numbers in the S phase remained
unaltered upon the application of AG490. The results indicate that
AG490 mainly induced cell cycle arrest at G1 phase, which is
different from most clinical DNA damage drugs that induced G2/M
arrest. A diagram of the cell cycle for each phase based on flow
cytometry (Fig. 2C) also revealed
that the effective concentration of AG490 to reduce HKF
proliferation was at least 50 µmol/l.
 | Figure 1AG490 decreases the expression of
both STAT3 and activated STAT3. HNFs and HKFs were treated with 0,
12.5, 25, 50, 75, 100 µmol/l AG490 for 24 h, respectively.
(A) Chemical structure of AG490 is shown. (B) Relative expression
of STAT3 and phosphorylated STAT3 (p-STAT3) proteins in HNFs and
HKFs detected by western blot analysis. β-actin was used as a
loading control. (C and D) The ratio of total STAT3 and p-STAT3 to
β-actin in HNFs (C) and HKFs (D) was examined by densitometry,
respectively. (E and F) The ratio of p-STAT3 to total STATs in HNFs
(E) and HKFs (F) was calculated and shown, respectively.
Statistical analysis was performed using one-way ANOVA followed by
post-hoc Tukey's multiple comparisons test. *P<0.05,
**P<0.01 and ***P<0.005 vs. the control; n=3.
STAT3, signal transducer and activator of transcription 3; HNFs,
human normal fibroblasts; HKFs, human keloid fibroblasts. |
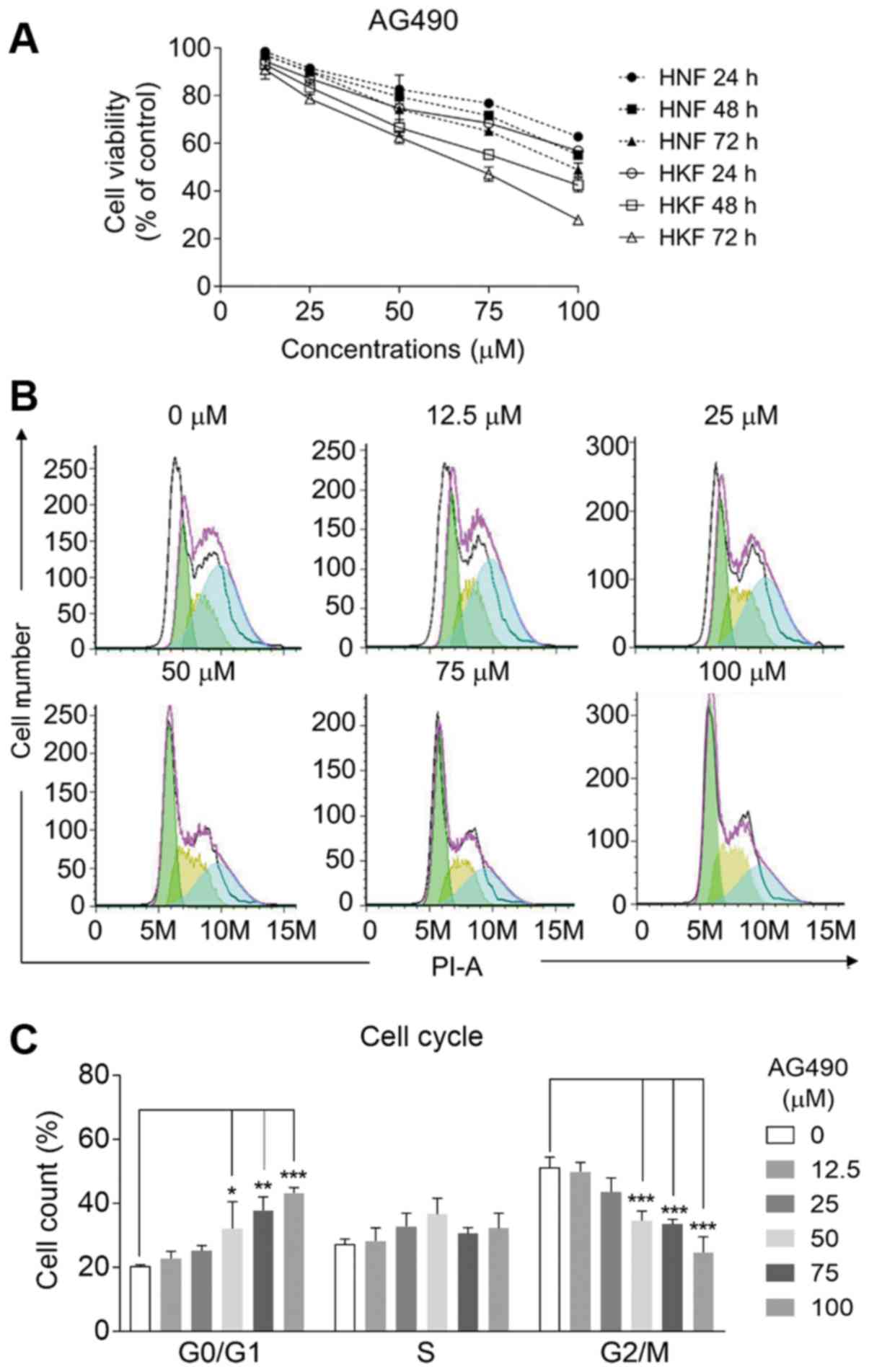 | Figure 2AG490 inhibits the proliferation of
fibroblasts and induces G1 phase arrest of HKFs. (A) Proliferation
rates of HNFs and HKFs was detected by CCK-8 assay. HNFs and HKFs
were treated with 0, 12.5, 25, 50, 75, 100 µmol/l AG490 for
24, 48 and 72 h, respectively. Compared with the control group,
AG490 inhibited the proliferation of HNFs and HKFs in both time-
and concentration-dependent manner. (B) The cell numbers in the
G0/G1, S, and G2/M phases were analyzed by flow cytometry following
treatment with AG490 for 24 h. (C) The total number in different
cell cycle phase was measured, respectively. Values represent the
means ± SD, n=3/each group. *P<0.05,
**P<0.01 and ***P<0.005 vs. the
control. HNFs, human normal fibroblasts; HKFs, human keloid
fibroblasts. |
AG490 induces the apoptosis of HKFs
To demonstrate whether the cytotoxicity of AG490 to
HKFs was associated with the induction of apoptosis, HKFs were
stained with Annexin V-FITC/PI following exposure to AG490 for 24
h. Cells were harvested and analyzed by flow cytometry. The results
were plotted in two-dimensional dot plots, which could be divided
in 4 regions (Q1, Q2, Q3 and Q4; Fig.
3). According to a previous report, Annexin V-FITC-positive and
PI-negative cells (Annexin V-FITC+/PI-, Q3)
are generally considered apoptotic cells in the early stage, while
Annexin V/PI-double-positive cells (Annexin
V-FITC+/PI+, Q2) are considered apoptotic
cells in the late stage (26). Q1
(Annexin V-FITC-/PI+) and Q4 (Annexin
V-FITC-/PI-) indicate necrotic cells and
viable cells, respectively. Consistent with the findings of cell
proliferation assay, a significant enhancement of HKF apoptosis was
observed when AG490 was used at concentrations of 50, 75 and 100
µmol/l (Fig. 3A and B).
Specifically, the proportions of HKFs at the early stages of
apoptosis were markedly promoted in response to AG490, while those
in the late stages of apoptosis were moderately enhanced (Fig. 3C).
 | Figure 3AG490 markedly enhances the apoptosis
of HKFs. HKFs were treated with 0, 12.5, 25, 50, 75, 100
µmol/l AG490 for 24 h, respectively. (A) Cell apoptosis was
detected by Annexin V/PI double staining assay. The data were
generated by flow cytometry are plotted in two-dimensional dot
plots, which can be divided into 4 regions (Q1, Q2, Q3 and Q4). (B)
The proportion of total apoptotic (Annexin V-positive) cells
increased concentration-dependently with AG490 treatment. (C) The
proportion of apoptotic cells at the early phase (Q3, Annexin
V-positive and PI-negative) and late phase (Q2, Annexin V and PI
double-positive). Values represent the means ± SD, n=3;
*P<0.05, **P<0.01 and
***P<0.005 vs. control. HKFs, human keloid
fibroblasts. |
AG490 decreases the mRNA levels of CTGF
and cyclin D1 in HKFs
To gain further insight into the mechanisms through
which AG490 suppresses the proliferation and induces the G1 phase
arrest of HKFs, the expression levels of cyclin D1, the G1/S cell
cycle checkpoint controller and CTGF, the primary regulator of
fibroblast proliferation and differentiation, were assessed in HKFs
following 24 h of treatment (Fig.
4). Compared with the control group, the relative mRNA
expression levels of STAT3, cyclin D1 and CTGF in the HKFs
decreased significantly in response to increasing concentrations of
AG490 (P<0.05, Fig. 4A-C).
Moreover, there was a strong association between the mRNA
expression levels of STAT3 and cyclin D1 (F=43.647, P<0.005;
Fig. 4D and E) and CTGF
(F=42.138, P<0.005; Fig. 4F and
G) as shown by a linear regression analysis. These results
indicated that the downregulation of cyclin D1 and CTGF were
associated with the AG490-induced decrease in STAT3 expression.
 | Figure 4AG490 decreases the mRNA expression
levels of STAT3, cyclin D1 and CTGF in HKFs. HKFs were treated with
AG490 at concentrations of 0, 12.5, 25, 50, 75, and 100
µmol/l for 24 h. Cells were then collected and total RNA was
isolated, respectively. (A-C) The mRNA expression levels for (A)
STAT3, (B) cyclin D1 and (C) CTFG were quantified by RT-qPCR.
Values represent the means ± SD, n=3; **P<0.01 and
***P<0.005 vs. control. (D-G) Simple linear
regression analysis revealed that cyclin D1 and CTGF mRNA
expression were highly associated with STAT3 mRNA levels in HKFs
that were treated with AG490 at various concentrations. To
efficiently deduce the linear regression model, the
SPSS® Statistics software platform was used. In the
regression model, STAT3 was used as a predictor (independent
variable), while cyclin D1 and CTGF were used as the dependent
variables. (D and F) The scatter plots represented relative mRNA
expression levels of cyclin D1 (D) and CTGF (F) against that of
STAT3 in AG490-treated HKF cells. The F-values in one-way ANOVA
test were 43.647 for cyclin D1 and 42.138 for CTGF. The P-values
were both <0.001. The coefficients of determination
(R2) were 0.732 for cyclin D1 and 0.725 for CTGF. The
residuals were calculated and used for subsequent analysis. (E and
G) The predicted probability (P-P) plots of standardized residuals
were established for normality and homoscedasticity analysis. The
plotted points (residuals) of (E) cyclin D1 and (G) CTGF were both
evenly distributed and followed the straight lines, suggesting the
regression model was valid. Observed Cum Prob, observed cumulative
probability. Expected Cum Prob, expected cumulative probability.
STAT3, signal transducer and activator of transcription 3; CTGF,
connective tissue growth factor; HKFs, human keloid
fibroblasts. |
Suppressive effects of AG490 on the
excessive proliferation of fibroblasts induced by TGF-β
It has been well established that TGF-β plays a
critical role in the pathogenesis of keloids by promoting the
proliferation, collagen formation and differentiation of dermal
fibroblasts (27). In the present
study, to evaluate the effects of AG490 on TGF-β-stimulated
fibroblasts, HNFs, HSFs and HKFs were maintained in the absence or
presence of 5 µg/ml TGF-β1 and 50 µmol/l AG490 for 24
h before assessing cell viability. The results revealed that the
TGF-β1-induced excessive proliferation was markedly inhibited by
the application of AG490 by 16.7% (HNFs), 21.2% (HSFs) and 36.2%
(HKFs) (P<0.05; Fig. 5A). The
HKFs and HSFs exhibited higher levels of the constitutive protein
expression of total STAT3 and CTGF than the HNFs (Fig. 5B and D). Following treatment with
AG490, the expression levels of both STAT3 and phosphorylation
(p-STAT3) were decreased in the HNFs, HSFs and HKFs that were
stimulated with TGF-β1 (Fig.
5B-D). These results were consistent with the above-mentioned
findings that AG490 dose-dependently reduced both the expression
and phosphorylation of STAT-3 in the HNFs and HKFs (Fig. 1). In addition, the inhibitory
effects of AG490 were observed on the TGF-β1-induced production of
CTGF in fibroblasts (Fig. 5B and
C). The relative mRNA expression of CTGF in HNFs, HSFs and HKFs
was decreased by approximately 45, 39 and 51%, respectively
(P<0.05; Fig. 5E).
 | Figure 5AG490 significantly inhibits the
effects of TGF-β on HNFs, HSFs and HKFs. HNFs, HSFs and HKFs were
incubated with TGF-β (5 µg/ml) for 2 h, followed by
treatment with AG490 (50 µmol/l) or not. HNFs. The control
HSFs and HKFs were incubated with medium in the absence of either
TGF-β or AG490. (A) AG490 inhibited TGF-β-induced excessive
proliferation of HNFs, HSFs and HKFs. After 24 h of treatment,
cells were collected and the proliferation rates were detected by
CCK-8 assay. (B) AG490 reduced the expression of phosphorylated
STAT3 (p-STAT3) and CTGF proteins that were promoted by TGF-β in
HNFs, HSFs and HKFs. The expression of p-STAT3 and CTGF proteins in
the different groups were detected by western blot analysis.
β-actin was used as a loading control. (C) The ratio of
p-STAT3/STAT3 and p-STAT3/β-actin was measured, respectively. (D)
The ratio of STAT3/β-actin and CTGF/β-actin was measured,
respectively. (E) The mRNA expression levels for total STAT3 and
CTFG were quantified by RT-qPCR. Values represent the means ± SD,
n=3; *P<0.05, **P<0.01 and
***P<0.005 vs. control. HNFs, human normal
fibroblasts; HKFs, human keloid fibroblasts; HNFs, human normal
fibroblasts. |
STAT3 ODNs inhibit HKF invasion
The delivery of the decoy ODNs into the cell has
been reported as a promising strategy with which to hinder the
transcription of disease related genes (28,29). The ODNs usually consist of
cis-element sequences that serve as the decoy for the target
transcription factor and then reduce its transcriptional activity.
In the present study, to assess the effects of STAT3 decoy ODNs on
KFs, SODNs were transfected into HKFs in vitro. The results
of EMSA demonstrated that SODNs markedly decreased the DNA-binding
activity of STAT3, MONDs or control medium did not affect the
activation of STAT3 (Fig. 6A).
Consistent with the results obtained above, the proliferation of
HKFs was also inhibited by the application of SODNs (Fig. S1C). Moreover, there was a 2-fold
decrease in the number of migrated HKFs in the presence of SODNs
(Fig. 6B-E), indicating that
SODNs remarkably inhibited HKFs invasion and progression. The
expression levels of ECM components, including tissue inhibitor of
metalloproteinase-2 (TIMP-2), matrix metalloproteinase-2 (MMP-2)
and vascular endothelial growth factor (VEGF), were detected by
RT-qPCR. Transfection with SODNs also attenuated the mRNA
expression of MMP-2 and VEGF by 57 and 46%, respectively, and
increased the expression of TIMP-2 >2-fold (Fig. 6F). These results demonstrated that
decoy STAT3 can block the constitutive activation of STAT3, thereby
inhibiting the invasion and progression of HKFs by upregulating the
expression of TIMP-2, and downregulating that of MMP-2 and
VEGF.
Discussion
The JAK/STAT signal transduction pathway plays a
leading role in the regulation of diverse processes, including cell
proliferation, migration, cytokine or chemokine production and
inflammation (30). During the
process of cutaneous wound healing, various chemokines, cytokines
(such as TNF, IL-1 and IL-6) and growth factors are secreted by
inflammatory cells in a well-orchestrated manner (31). The JAK/STAT signaling pathway is
activated in response to most of these mediators. The enhanced
expression and phosphorylation of STAT3 has been observed in keloid
tissues and KFs cultured in vitro, suggesting an important
role of STAT3 the pathogenesis of keloids (12,32). The inhibition of STAT3 expression
by siRNA or inhibitor (cucurbitacin I) has been shown to lead to
corresponding decrease in collagen expression, and in the
proliferation and migration of KFs (12). STAT3 can be used as a therapeutic
target for keloids.
AG490, as a potent inhibitor of the JAK2/STAT3
pathway, inhibits the proliferation of HKFs effectively. It has
been reported that AG490 can inhibit epidermal growth factor
receptor (EGFR) in cell-free assays (33). It has previously been demonstrated
that KFs exhibit a higher expression and phosphorylation levels of
molecules associated with tyrosine phosphorylation signaling,
including EGFR, compared with normal dermal fibroblasts (34). As a tyrosine kinase blocker, AG490
may significantly suppress KF growth by exerting combined
inhibitory effects on EGFR and STAT3. The reduction of EGFR and
STAT3 activation can also suppress the transcription of the
downstream genes. In the present study, decreasing the expression
of cyclin D1 resulted in cell cycle arrest in the G1 phase and in
the promotion of cell apoptosis. The downregulation of CTGF
inhibited the proliferation and differentiation of HKFs, as well as
the fibrotic effects of fibroblasts. Another possible mechanism for
the AG490 inhibitory effect was that the production of collagen
type I and type II may be decreased through the inhibition of the
activation of STAT3 in HKFs, as suggested by a previous study
(12). However, it remains
unclear as to whether collagen production is reduced and which type
of collagen is affected by AG490 treatment, as collagen production
was not examined in the present study.
CTGF is involved in ECM remodeling, whose
overexpression is closely related to the occurrence and development
of proliferative or fibrotic diseases (35). In fibroblasts, CTGF expression can
be induced by both TGF-β and downstream signals of TGF-β (36). It has been well established that
the TGF-β/Smad signaling pathway plays a critical role in the
pathogenesis of keloids by promoting collagen synthesis, ECM
deposition and sustained fibrosis (37,38). However, blocking the TGF-β/Smad
pathway can only prevent the initiation or progression of fibrosis;
it cannot effectively reverse the fibrosis that has already
occurred (39). The treatment of
keloids may be more optimal by inhibiting the abnormal increase in
the expression of CTGF, which has been proven to be a central
mediator of tissue remodeling and fibrosis and its inhibition
reverses the process of fibrosis in liver and lung tissues
(40). The results of the present
study demonstrated that blocking STAT3 activation decreased
TGF-β-induced CTGF overexpression, suggesting that AG490 may
reverse the profibrotic effects of TGF-β by regulating CTGF in KFs.
The present study demonstrated that blocking the STAT3 pathway by
molecular targeted drugs or gene therapy can inhibit the abnormal
proliferation and invasion of HKFs and promote apoptosis, thereby
treating keloids.
In conclusion, the present study demonstrated that
STAT3 can be used as a therapeutic target for keloids. By
inhibiting the STAT3 pathway using AG490 or decoy ODNs, the
proliferation and abnormal behavior of HKFs were significantly
suppressed. The TGF-β1-stimulated activation of STAT3 and the
production of CTGF were also decreased by AG490 in fibroblasts.
These results suggested that AG490 plays a protective role in
keloids mainly by inducing G1 phase arrest, promoting apoptosis and
suppressing inflammatory responsiveness. AG490, as a selective
inhibitor of the JAK2/STAT3 pathway, may be a therapeutic candidate
for inhibiting the development of keloids.
Supplementary Data
Funding
The present study was supported by the Natural
Science Foundation of Jiangsu Province (grant no. BK20171117).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
JB and WF designed the study. YZ and YS performed
the experiments and drafted the manuscript. WH and LM participated
in data analysis. YT, DL and CX were involved in the discussion and
interpretation of the results. All authors read and approved the
final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
Not applicable.
References
|
1
|
Shih B, Garside E, McGrouther DA and Bayat
A: Molecular dissection of abnormal wound healing processes
resulting in keloid disease. Wound Repair Regen. 18:139–153. 2010.
View Article : Google Scholar
|
|
2
|
Cosman B, Crikelair GF, Ju DMC, Gaulin JC
and Lattes R: The surgical treatment of keloids. Plast Reconstr
Surg. 27:335–358. 1961. View Article : Google Scholar
|
|
3
|
Viera MH, Vivas AC and Berman B: Treatment
of keloids and scars. Ethn Dermatology Princ Pract:. 159–172. 2013.
View Article : Google Scholar
|
|
4
|
Tuan TL and Nichter LS: The molecular
basis of keloid and hypertrophic scar formation. Mol Med Today.
4:19–24. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ledon JA, Savas J, Franca K, Chacon A and
Nouri K: Intralesional treatment for keloids and hypertrophic
scars: A review. Dermatol Surg. 39:1745–1757. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Manca G, Pandolfi P, Gregorelli C, Cadossi
M and de Terlizzi F: Treatment of keloids and hypertrophic scars
with bleomycin and electroporation. Plast Reconstr Surg.
132:621e–630e. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Vincent AS, Phan TT, Mukhopadhyay A, Lim
HY, Halliwell B and Wong KP: Human skin keloid fibroblasts display
bioener-getics of cancer cells. J Invest Dermatol. 128:702–709.
2008. View Article : Google Scholar
|
|
8
|
Naitoh M, Hosokawa N, Kubota H, Tanaka T,
Shirane H, Sawada M, Nishimura Y and Nagata K: Upregulation of
HSP47 and collagen type III in the dermal fibrotic disease, keloid.
Biochem Biophys Res Commun. 280:1316–1322. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Uitto J, Perejda AJ, Abergel RP, Chu ML
and Ramirez F: Altered steady-state ratio of type I/III procollagen
mRNAs correlates with selectively increased type I procollagen
biosynthesis in cultured keloid fibroblasts. Proc Natl Acad Sci
USA. 82:5935–5939. 1985. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Babu M, Diegelmann R and Oliver N: Keloid
fibroblasts exhibit an altered response to TGF-beta. J Invest
Dermatol. 99:650–655. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Banerjee K and Resat H: Constitutive
activation of STAT3 in breast cancer cells: A review. Int J Cancer.
138:2570–2578. 2016. View Article : Google Scholar :
|
|
12
|
Lim CP, Phan TT, Lim IJ and Cao X: Stat3
contributes to keloid pathogenesis via promoting collagen
production, cell proliferation and migration. Oncogene.
25:5416–5425. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Abroun S, Saki N, Ahmadvand M, Asghari F,
Salari F and Rahim F: STATs: An old story, yet mesmerizing. Cell J.
17:395–411. 2015.PubMed/NCBI
|
|
14
|
Schlessinger K and Levy DE: Malignant
transformation but not normal cell growth depends on signal
transducer and activator of transcription 3. Cancer Res.
65:5828–5834. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Meydan N, Grunberger T, Dadi H, Shahar M,
Arpaia E, Lapidot Z, Leeder JS, Freedman M, Cohen A, Gazit A, et
al: Inhibition of acute lymphoblastic leukaemia by a Jak-2
inhibitor. Nature. 379:645–648. 1996. View
Article : Google Scholar : PubMed/NCBI
|
|
16
|
An JY, Pang HG, Huang TQ, Song JN, Li DD,
Zhao YL and Ma XD: AG490 ameliorates early brain injury via
inhibition of JAK2/STAT3-mediated regulation of HMGB1 in
subarachnoid hemorrhage. Exp Ther Med. 15:1330–1338.
2018.PubMed/NCBI
|
|
17
|
Xu MY, Hu JJ, Shen J, Wang ML, Zhang QQ,
Qu Y and Lu LG: Stat3 signaling activation crosslinking of TGF-β1
in hepatic stel-late cell exacerbates liver injury and fibrosis.
Biochim Biophys Acta. 1842:2237–2245. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Nielsen M, Kaltoft K, Nordahl M, Röpke C,
Geisler C, Mustelin T, Dobson P, Svejgaard A and Odum N:
Constitutive activation of a slowly migrating isoform of Stat3 in
mycosis fungoides: Tyrphostin AG490 inhibits Stat3 activation and
growth of mycosis fungoides tumor cell lines. Proc Natl Acad Sci
USA. 94:6764–6769. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Burdelya L, Catlett-Falcone R, Levitzki A,
Cheng F, Mora LB, Sotomayor E, Coppola D, Sun J, Sebti S, Dalton
WS, et al: Combination therapy with AG-490 and interleukin 12
achieves greater antitumor effects than either agent alone. Mol
Cancer Ther. 1:893–899. 2002.PubMed/NCBI
|
|
20
|
Samanta AK, Lin H, Sun T, Kantarjian H and
Arlinghaus RB: Janus kinase 2: A critical target in chronic
myelogenous leukemia. Cancer Res. 66:6468–6472. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Abe M, Funakoshi-Tago M, Tago K,
Kamishimoto J, Aizu-Yokota E, Sonoda Y and Kasahara T: The
polycythemia vera-associated Jak2 V617F mutant induces
tumorigenesis in nude mice. Int Immunopharmacol. 9:870–877. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ma J, Yan X, Lin Y and Tan Q: Hepatocyte
growth factor secreted from human adipose-derived stem cells
inhibits fibrosis in hypertrophic scar fibroblasts. Curr Mol Med.
Jan 5–2020.Epub ahead of print. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wu X, Bian D, Dou Y, Gong Z, Tan Q, Xia Y
and Dai Y: Asiaticoside hinders the invasive growth of keloid
fibroblasts through inhibition of the GDF-9/MAPK/Smad pathway. J
Biochem Mol Toxicol. 31:2017. View Article : Google Scholar
|
|
24
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
25
|
Dunkel Y, Ong A, Notani D, Mittal Y, Lam
M, Mi X and Ghosh P: STAT3 protein up-regulates Gα-interacting
vesicle-associated protein (GIV)/Girdin expression, and GIV
enhances STAT3 activation in a positive feedback loop during wound
healing and tumor invasion/metastasis. J Biol Chem.
287:41667–41683. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wlodkowic D, Telford W, Skommer J and
Darzynkiewicz Z: Apoptosis and beyond: Cytometry in studies of
programmed cell death. Methods Cell Biol. 103:55–98. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Pakyari M, Farrokhi A, Maharlooei MK and
Ghahary A: Critical role of transforming growth factor beta in
different phases of wound healing. Adv Wound Care (New Rochelle).
2:215–224. 2013. View Article : Google Scholar
|
|
28
|
Hanagata N: CpG oligodeoxynucleotide
nanomedicines for the prophylaxis or treatment of cancers,
infectious diseases, and allergies. Int J Nanomedicine. 12:515–531.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Tomita N, Ogihara T and Morishita R:
Transcription factors as molecular targets: Molecular mechanisms of
decoy ODN and their design. Curr Drug Targets. 4:603–608. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Rawlings JS, Rosler KM and Harrison DA:
The JAK/STAT signaling pathway. J Cell Sci. 117:1281–1283. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Efron PA and Moldawer LL: Cytokines and
wound healing: The role of cytokine and anticytokine therapy in the
repair response. J Burn Care Rehabil. 25:149–160. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Ghazizadeh M, Tosa M, Shimizu H, Hyakusoku
H and Kawanami O: Functional implications of the IL-6 signaling
pathway in keloid pathogenesis. J Invest Dermatol. 127:98–105.
2007. View Article : Google Scholar
|
|
33
|
Gazit A, Osherov N, Posner I, Yaish P,
Poradosu E, Gilon C and Levitzki A: Tyrphostins. 2. Heterocyclic
and alpha-substituted benzylidenemalononitrile tyrphostins as
potent inhibitors of EGF receptor and ErbB2/neu tyrosine kinases. J
Med Chem. 34:1896–1907. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Chin GS, Liu W, Steinbrech D, Hsu M,
Levinson H and Longaker MT: Cellular signaling by tyrosine
phosphorylation in keloid and normal human dermal fibroblasts.
Plast Reconstr Surg. 106:1532–1540. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Lipson KE, Wong C, Teng Y and Spong S:
CTGF is a central mediator of tissue remodeling and fibrosis and
its inhibition can reverse the process of fibrosis. Fibrogenesis
Tissue Repair. 5(Suppl 1): S242012. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Jurzak M, Adamczyk K, Antończak P,
Garncarczyk A, Kuśmierz D and Latocha M: Evaluation of genistein
ability to modulate CTGF mRNA/protein expression, genes expression
of TGFβ isoforms and expression of selected genes regulating cell
cycle in keloid fibroblasts in vitro. Acta Pol Pharm. 71:972–986.
2014.
|
|
37
|
Biernacka A, Dobaczewski M and
Frangogiannis NG: TGF-β signaling in fibrosis. Growth Factors.
29:196–202. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Walton KL, Johnson KE and Harrison CA:
Targeting TGF-β mediated SMAD signaling for the prevention of
fibrosis. Front Pharmacol. 8:4612017. View Article : Google Scholar
|
|
39
|
Song R, Li G and Li S: Aspidin PB, a novel
natural anti-fibrotic compound, inhibited fibrogenesis in
TGF-β1-stimulated keloid fibroblasts via PI-3K/Akt and Smad
signaling pathways. Chem Biol Interact. 238:66–73. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Bickelhaupt S, Erbel C, Timke C, Wirkner
U, Dadrich M, Flechsig P, Tietz A, Pföhler J, Gross W, Peschke P,
et al: Effects of CTGF blockade on attenuation and reversal of
radiation-induced pulmonary fibrosis. J Natl Cancer Inst. 109:2017.
View Article : Google Scholar : PubMed/NCBI
|















