Introduction
Rheumatic heart disease (RHD) is a preventable heart
disease caused by Streptococcus pyogenes or group A
streptococcus (GAS) infection (1). RHD is a leading cause of mortality
and disability in young patients, and it remains a serious global
public health concern (2). The
number of individuals with RHD worldwide exceeds one quarter of the
number of individuals with cancer, and the number of associated
deaths caused by RHD annually is as high as 250,000 (3). However, studies on RHD continue to
examine its pathogenesis, which remains unclear. Precise
intervention targets for the prevention or treatment of RHD have
not yet been identified, at least to the best of our knowledge. The
majority of studies on RHD have focused on the association between
its pathogenesis and signalling pathways (4-6).
Research on intervention targets that attenuate valvular damage due
to RHD is lacking.
Sphingosine 1-phosphate receptor 1 (S1PR1) is a G
protein-coupled receptor belonging to the S1PR family. S1PR1
mediates lymphocyte migration, and it is associated with multiple
immune (7) and heart diseases
(8). S1PR1 primarily plays a
role in protecting the heart in patients with heart diseases
(9-11), and a high S1PR1 expression
generally protects the heart during the pathogenesis of heart
disease (9,12). Garris et al (13) found that the downregulation of
S1PR1 expression increased the levels of phosphorylated (p-) signal
transducer and activator of transcription 3 (p-STAT3). A recent
study also demonstrated that the S1PR1/STAT3 signalling pathway was
involved in the development of valvular damage due to RHD in a rat
model, in which S1PR1 expression was downregulated, and the levels
of p-STAT3 and T helper 17 (Th17)-related cytokines were increased
(14).
STAT3 is a cellular signal transcription factor that
is involved in the regulation of a number of cellular activities
(15). STAT3 may regulate the
differentiation of CD4+ T cells into Th17 cells
(16). Th17 cells and related
cytokines mediate inflammatory and autoimmune responses (17-19). Bas et al (20) demonstrated that the ratio of
Th17/Treg cells and the levels of IL-17A were significantly
increased in patients with RHD compared with those in subjects in
the control group. Similar results were observed in animal models
of RHD established in Lewis rats (21). A close association between S1PR1
and STAT3 has been identified, and a number of studies have
discussed the role of S1PR1 in the regulation of STAT3 in various
diseases (22-25). Therefore, it was hypothesised
that the STAT3 pathway is activated during the development of RHD
and that it induces CD4+ T cell differentiation into
Th17 cells, and Th17 cell-related inflammatory factors participate
in the development of RHD. However, researchers have not yet
clearly determined whether this pathway modulates or prevents RHD
following intervention. A more important goal is the development of
appropriate interventions targeting this pathway to modulate or
prevent the occurrence of RHD.
Therefore, the present study interfered with the
expression of S1PR1 and STAT3 by overexpressing S1PR1 and
inhibiting STAT3 to determine whether these treatments would
attenuate RHD-induced valvular damage.
Materials and methods
The present study aimed to determine whether
interfering with the expression of S1PR1 or STAT3 would attenuate
RHD-induced valvular damage. A rat model of RHD was established as
described in previous studies in order to achieve this goal
(14,26).
Antigen preparation
Brain heart infusion fluid medium (Guangdong Huankai
Microbial Sci. & Tech. Co., Ltd.) was used to culture GAS
[American Type Culture Collection (ATCC)19615], and the temperature
during the cultivation process was maintained at a constant value
of 37°C. After 24 h, GAS was washed with normal saline (NS) and
transferred onto 10% neutral formalin for 12 h for inactivation. NS
was used to wash and resuspend the inactivated GAS, and the
concentration was simultaneously adjusted to 4.0×1011
colony forming units (CFU)/ml (26). The antigen suspension was
obtained by emulsifying the suspension via sonication (Sonics &
Materials, Inc.).
In vivo gene therapy
Recombinant adeno-associated virus (AAV; serotype 9)
vectors carrying the rat S1PR1 overexpression sequence (S1PR1
overexpression; Hanbio Biotechnology Co., Ltd.) driven by the cTNT
promoter were used to overexpress S1PR1. As the main role of AAV
was to carry gene sequences as a vector, it did not affect the
experiment itself. Therefore, the AAV control group was injected
only with the AAV vector without any sequence to assess whether the
AAV vectors altered the results of the experiment. The S1PR1
overexpression sequence is listed in Table I. A rat STAT3 small interfering
RNA (siRNA) sequence (5′-GGCTGATCATTTATATAAA-3′; STAT3-siRNA;
Hanbio Biotechnology Co., Ltd.) driven by the cTNT promoter in a
recombinant AAV vector was used to directly silence STAT3
expression. An AAV control was also used as a negative control to
determine whether the AAV vector exerted an effect in the rats.
 | Table ISequencing result (S1PR1
overexpression sequence). |
Table I
Sequencing result (S1PR1
overexpression sequence).
| Sequence name | Sequencing
result |
|---|
| S1PR1
overexpression sequence |
ATGGTGTCCTCCACCAGCATCCCAGTGGTTAAGGCTCTCCGCAGCCAAGTCTCCGACTATGGCAACTATGATATCATAGTCCGGCATTACAACTACACAGGCAAGCTGAACATCGGAGTGGAGAAGGACCATGGCATTAAACTGACTTCAGTGGTGTTCATTCTCATCTGCTGCTTGATCATCCTAGAGAATATATTTGTCTTGCTAACTATTTGGAAAACCAAGAAGTTCCACCGGCCCATGTACTATTTCATAGGCAACCTAGCCCTCTCGGA
CCTGTTAGCAGGAGTGGCTTACACAGCTAACCTGCTGTTGTCTGGGGCCACCACCTACAAGCTCACACCTGCCCAGTGGTTTCTGCGGGAAGGAAGTATGTTTGTGGCTCTGTCTGCCTCAGTCTTCAGCCTCCTTGCTATCGCCATTGAGCGCTACATCACCATGCTGAAGATGAAACTACACAACGGCAGCAACAGCTCGCGCTCCTTTCTGCTGATCAGTGCCTGCTGGGTCATCTCCCTCATCCTGGGTGGGCTGCCCATCATGGGCTGGAACTGCATCAGCTCGCTGTCCAGCTGCTCCACCGTGCTCCCGCTCTACCACAAGCACTATATTCTCTTCTGCACCACCGTCTTCACCCTGCTCCTGCTTTCCATCGTCATCCTCTACTGCAGGATCTACTCCTTGGTGAGGACTCGAAGCCGCCGCCTGACCTTCCGCAAGAACATCTCCAAGGCCAGCCGCAGTTCCGAGAAGTCTCTGGCCTTGCTGAAGACAGTGATCATTGTCCTGAGTGTCTTCATTGCCTGCTGGGCCCCTCTCTTCATCCTACTACTTTTAGATGTGGGGTGCAAGGCGAAGACCTGTGACATCCTGTACAAAGCAGAGTACTTCCTGGTTCTGGCTGTGCTGAACTCAGGTACCAACCCCATCATCTACACTCTGACCAATAAGGAGATGCGCCGGGCCTTCATCAGGATCATATCTTGTTGCAAATGCCCCAACGGAGACTCCGCTGGCAAATTCAAGAGGCCCATCATCCCGGGCATGGAATTTAGCCGCAGCAAATCAGACAACTCCTCCCACCCCCAGAAGGATGATGGGGACAATCCAGAGACCATTATGTCTTCTGGAAACGTCAATTCTTCT
TCT |
Immunization of rats
A total of 30 Lewis rats (150-180 g) were purchased
from Beijing Vital River Animal Technology Co., Ltd.. All rats were
female and weighed 150-180 g at 8 weeks of age. The rats were
randomly divided into 5 groups (n=6 each) for the two parts of the
present study. Part I of the study examined the effects of S1PR1
overexpression, and part II examined the effects of STAT3
inhibition. The pathogen-free animal laboratory at the Animal
Experiment Centre of Guangxi Medical University provided a
satisfactory environment for the rats: The temperature was
constantly set to 23°C, and the fluctuation did not exceed 2°C; the
day/night cycle was 12 h; the movement of the rats in the cage was
completely unrestricted; sufficient drinking water and standard rat
feed were also provided. All the rats were allowed to adapt to the
environment for 5 days prior to the commencement of the
experiments. All animal experimental procedures were performed
according to the Guidelines for the Ethical Review of Laboratory
Animal Welfare of China (GB/T 35892-2018) for the care and use of
laboratory animals and were approved by the Medical Ethics
Committee of the First Affiliated Hospital of Guangxi Medical
University (Approval no. 2019-KY-E-069).
The rats were divided randomly into 5 groups as
follows: The control group, AAV control group, RHD group, S1PR1
overexpression group and the STAT3-siRNA group. Each group included
6 rats. The RHD group was the established RHD model. A footpad
injection of complete Freund's adjuvant (CFA) (each millilitre
contained 1 mg heat-inactivated dry Mycobacterium
tuberculosis (H37Ra, ATCC 25177), 0.85 ml paraffin oil and 0.15
ml mannitol mono-oleic acid; cat. no. F5881; Sigma-Aldrich, Merck
KGaA) was essential for establishing the rat model of RHD. All rats
were maintained on soft bedding to protect their hind feet. In
total, 9 weeks were required to establish the rat model of RHD. One
hind footpad of each rat was injected initially with 100 µl
of a solution of inactivated GAS (4.0×1011 CFU/ml) and
CFA mixed at a ratio of 1:1 (v/v). After 1 week, a subcutaneous
injection of 500 µl of inactivated GAS (4.0×1011
CFU/ml) and CFA mixed at a ratio of 1:1 (v/v) were administered
into the abdomen of the rats once weekly at the same interval for 4
weeks. Over the last 4 weeks, a subcutaneous abdominal injection
was administered once weekly at the same interval with an
adjustment of the injection solution to 500 µl of
inactivated GAS (4.0×1011 CFU/ml). Rats in the S1PR1
overexpression group were injected with 2.5×1011 viral
genome particles once through the tail vein (AAV-S1PR1
overexpression, diluted in 200 µl of NS) at the beginning of
the experiment. After 3 weeks, the rats received exactly the same
treatment as those in the RHD group. The rats in the AAV control
group received an injection of 2.5×1011 viral genome
particles once through the tail vein (AAV control, diluted with 200
µl of NS) at the beginning of the experiment. After 3 weeks,
these rats were injected according to the same protocol as that for
the RHD group. The rats in the control group were injected using
the same protocol as that for the RHD group from the beginning of
the experiment, although the injection solution included the same
volume of NS. The rats in the STAT3-siRNA group were injected using
the same protocol as that for the S1PR1 overexpression group,
except that the solution for the tail vein injection was changed
from 2.5×1011 viral genome particles (AAV-S1PR1
overexpression, diluted with 200 µl of NS) to
2.5×1011 viral genome particles (AAV-STAT3-siRNA,
diluted with 200 µl of NS).
Animal sacrifice
Following the administration of all treatments, 1 ml
of blood was collected from the tail vein of the rats in each group
without anaesthesia, and an intraperitoneal injection of sodium
pentobarbital (150 mg/kg) was then administered to euthanise the
rats. Animal death was determined when >5 min had elapsed
without breathing or a heartbeat. The humane endpoint in the
present study was defined as animals losing >15% of their body
weight with a decreased ability to consume food and water. None of
the rats reached this humane endpoint before the end of the
experimental period.
Sample preparation
Valvular samples were collected from each rat. All
samples were rapidly frozen in liquid nitrogen and stored at −80°C
for use in subsequent experiments. No animals died during the
modelling process. The following five experimental methods [reverse
transcription-quantitative PCR (RT-qPCR)], western blotting (WB),
immunohistochemistry, histochemistry and enzyme-linked
immunosorbent assay (ELISA)] were performed on the previously
mentioned experimental groups.
RT-qPCR
Total RNA was extracted from each sample.
TRIzol® reagent (Invitrogen; Thermo Fisher Scientific,
Inc.) was used to complete this step according to the
manufacturer's protocol. The RNA concentration was measured using a
NanoDrop 2000 spectrophotometer (NanoDrop Technologies; Thermo
Fisher Scientific, Inc.) for quantitative reverse transcription.
RNA was reverse transcribed into cDNA; 0.5 µg of total RNA
from each sample was reverse transcribed into cDNAs. The
PrimeScript RT reagent kit (cat. no. RR036A; Takara Bio, Inc.) was
used for reverse transcription. The entire reverse transcription
process was performed in accordance with the manufacturer's
instructions. RT-qPCR was performed using TB Green Premix Ex Taq II
(cat. no. RR820Q; Takara Bio, Inc.), a StepOne system (cat. no.
4376357; Applied Biosystems; Thermo Fisher Scientific, Inc.), and
the internal reference gene, β-actin. The entire process was
performed in accordance with the manufacturer's instructions. The
thermocycling conditions were as follows: 95°C for 30 min, followed
by 40 cycles at 95°C for 5 sec and 60°C for 30 sec. The sequences
of the primers are listed in Table
II. The final results are expressed as the fold change between
the expression level of each mRNA and the internal reference using
the 2−ΔΔCq method (27). All samples were measured three
times.
| Table IISequences of primers used in reverse
transcriptionquantitative PCR. |
Table II
Sequences of primers used in reverse
transcriptionquantitative PCR.
| Gene | Primer sequence
(5′-3′) |
|---|
| STAT3 | F:
TTTGAGACAGAGGTGTACCACCAAG |
| R:
ACCACAGGATTGATGCCCAAG |
| S1PR1 | F:
GCTTCATCACTCACTACCCTAGCA |
| R:
TTCTCCCTTCCCTCCCTCTC |
| Col3a1 | F:
ACTTCTGGTCCTCCTGGTCTGC |
| R:
CGCCTGGCTCACCCTTTTCAC |
| FSP1 | F:
TGGGGAGAAGGACAGACGAAGC |
| R:
TGGCAATGCAGGACAGGAAGAC |
| β-actin | F:
GGAGATTACTGCCCTGGCTCCTA |
| R:
GACTCATCGTACTCCTGCTTGCTG |
WB
Total protein was extracted from each sample using
RIPA lysis buffer (Sangon Biotech Co., Ltd.), according to the
manufacturer's instructions. The protein concentration was measured
using a bicinchoninic acid (BCA) protein assay kit (Sangon Biotech
Co., Ltd.). The same amounts of protein (30 µg) from each
sample were separated on 10% SDS-PAGE gels. The separation
conditions were 80 V for 30 min and 120 V for 60 min using a
blotting system (Bio-Rad Laboratories, Inc.), according to the
manufacturer's instructions. The separated proteins were
electrotransferred to 0.22-µm polyvinylidene fluoride (PVDF)
membranes (EMD Millipore), and the transfer conditions were a
constant voltage of 80 V for 80 min. The membranes were blocked for
1 h at room temperature in a 3% bovine serum albumin (BSA) blocking
solution (Sangon Biotech Co., Ltd.) and the membranes were then
incubated overnight at 4°C with the following antibodies:
Anti-S1PR1 (1:1,000; cat. no. 55133-1-AP; ProteinTech Group, Inc.),
anti-STAT3 (1:1,000; cat. no. ab68153; Abcam), anti-p-STAT3
(1:1,000; cat. no. 9145; Cell Signaling Technology, Inc.) and
anti-β-tubulin (1:3,000; cat. no. 10068-1-AP; ProteinTech Group,
Inc.). The membranes were subsequently incubated with an
HRP-conjugated secondary antibody (1:10,000; cat. no. ab6721;
Abcam) in the dark for 1 h at room temperature. Protein bands were
scanned using a chemiluminescence imaging system (Alpha FluorChem
FC3; Alpha, Inc.). The levels of the proteins were normalized to
β-tubulin and quantified using ImageJ software (1.51j, National
Institute of Health). All samples were measured three times.
Histochemistry
The valvular tissues were fixed in 4%
paraformaldehyde for 24 h at 4°C prior to decalcification and
embedding in paraffin blocks. All blocks were serially sectioned at
a thickness of 5 µm for Hematoxylin and eosin (H&E; cat.
no. G1120; Beijing Solarbio Science & Technology Co., Ltd.) and
Sirius Red staining (cat. no. S8060-5; Beijing Solarbio Science
& Technology Co., Ltd.). H&E staining was performed at room
temperature, and the sections were stained with Hematoxylin for
4-10 min followed by eosin for 0.5-2 min. A BX43 light microscope
(Olympus Corporation) was used to capture the images of H&E
staining. Sirius Red staining was also performed at room
temperature for 1 h. A BX43 confocal microscope (magnification,
×400; Olympus Corporation) was used to capture the images of Sirius
Red staining.
Immunohistochemistry
Immunohistochemistry was performed using the method
described in a previous study (26) to analyse the valvular tissues
stained with antibodies against IL-6 (1:65; cat. no. ab9324;
Abcam), IL-17 (1:90; cat. no. ab214588; Abcam), S1PR1 (1:80; cat.
no. ab77076; Abcam), STAT3 (1:75; cat. no. ab68153; Abcam), p-STAT3
(1:70; cat. no. ab76315; Abcam) and retinoic acid-related orphan
receptor γT (RORγt; 1:75; cat. no. 13205-1-AP; ProteinTech Group,
Inc.), which is the key transcription factor driving the
differentiation of IL-17-producing Th17 cells (28). Briefly, formalin-fixed valvular
tissues were embedded in paraffin. All blocks were sectioned at a
thickness of 5 µm. Following deparaffinization and
rehydration, a 5% BSA (Beijing Solarbio Science & Technology
Co., Ltd.) solution was used to block the sections at room
temperature for 1 h. Following the deactivation of endogenous
peroxidases with hydrogen peroxide, the sections were incubated
with the primary antibodies described above for 12 h at 4°C. A
horseradish peroxidase (HRP)-conjugated anti-rabbit (1:10; cat. no.
PV-6001; OriGene Technologies, Inc.) or anti-mouse secondary
antibody (1:10; cat. no. PV-6002; OriGene Technologies, Inc.) were
incubated with the sections for 30 min at room temperature. After
enhanced colour development using diaminobenzidine (DAB), the
immunostained tissues were examined under a BX43 light microscope
(Olympus Corporation), and positive expression was detected as
brownish yellow staining. Quantitative assessment was performed
using the methods described by in the study by Friedrichs et
al (29). The
immunohistochemical score (IHS) is equal to the staining intensity
(SI) multiplied by the percentage of positive cells (PP). The SI
was scored as follows: 0 points, negative; 1 point, weak; 2 points,
moderate; and 3 points, strong. The PP was scored as follows: 0
points, negative; 1 point, 10% positive cells; 2 points, 11-50%
positive cells; 3 points, 51-80% positive cells; and 4 points,
>80% positive cells. The IHS was calculated to describe the
results of the quantitative evaluation.
ELISA
ELISA kits (cat. nos. E04640r and E07451r; Cusabio)
were used to measure the serum levels of IL-6 and IL-17. The entire
process was performed according to the manufacturer's instructions.
All samples were measured three times. After the preparation of all
reagents, working standards, samples (serum) and assay plates, 100
µl of standard solutions and samples were added to each
well, covered with an adhesive strip, and incubated at 37°C for 2
h. The liquid of each well was removed, and 100 µl of biotin
antibody (1X) was added to each well. The assay plate was incubated
at 37°C for 1 h with a new adhesive strip covering. Washing buffer
(provided in the ELISA kit) was used to wash each well three times
following the removal of the liquid from each well. HRP-avidin (1X,
100 µl) was added to each well, covered with a new adhesive
strip, and incubated at 37°C for 1 h. Each well was washed five
times, 90 µl TMB substrate were added to each well, and the
assay plate was placed in the dark at 37°C for 15-30 min. A stop
solution (50 µl) was added to each well to stop the
reaction. The sample concentrations were calculated based on the
absorbance of each well.
Statistical analysis
For data other than IHS, the results are presented
as the means ± standard deviations of at least three independent
experiments. SPSS software 17.0 (SPSS, Inc.) was used for
statistical analyses. One-way ANOVA was used to compare differences
between the 5 groups with Tukey's test as the post hoc multiple
comparisons test. For the data of IHS, the Kruskal-Wallis test was
used, and Dunn's test was used as the post hoc test. The results
are expressed as the median and interquartile range. P<0.05 was
considered to indicate a statistically significant difference.
Results
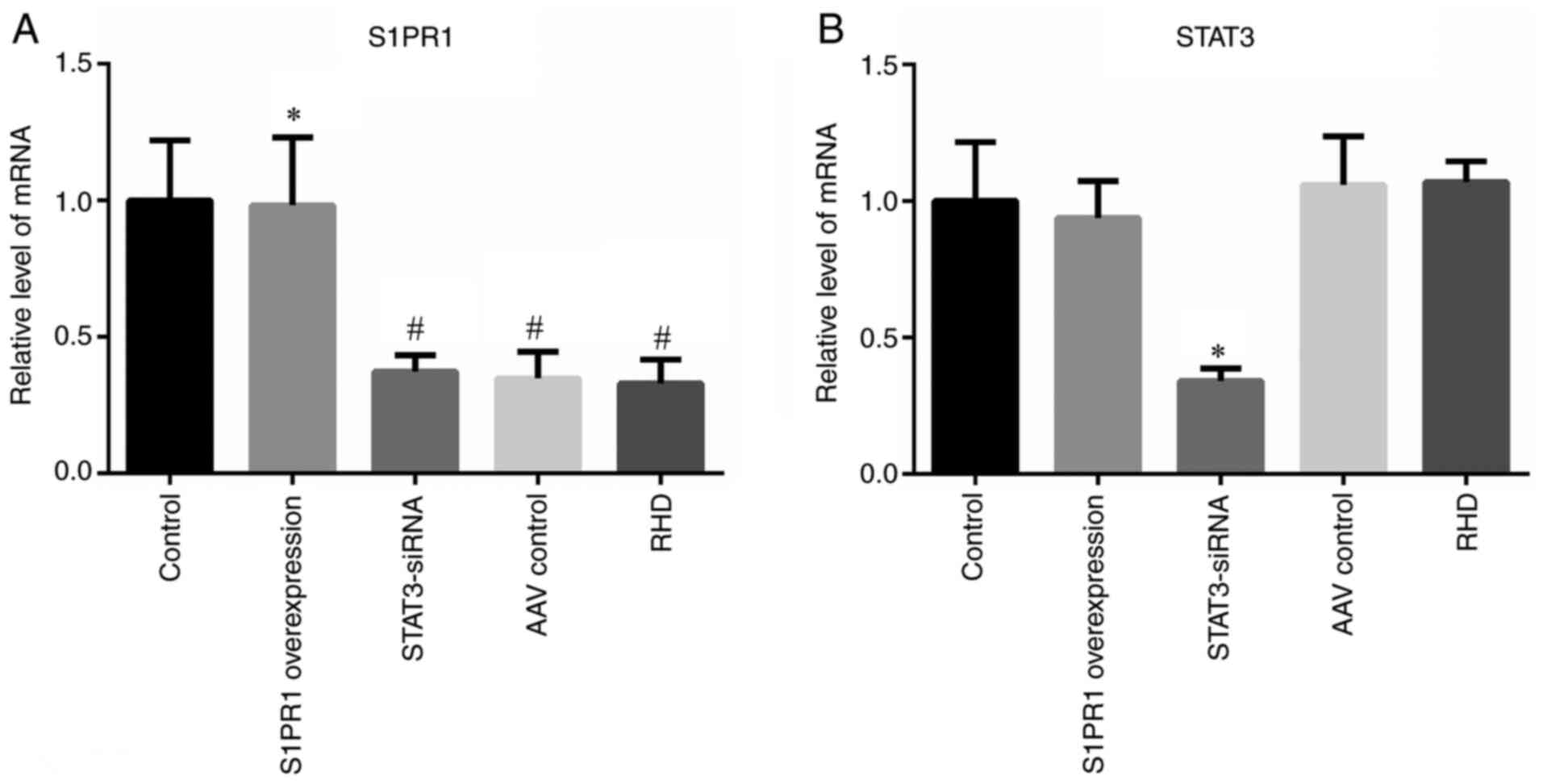
Overexpression of S1PR1
In vivo gene therapy increases S1PR1
expression
The results of RT-qPCR, WB and immunohistochemistry
revealed a significantly lower expression of S1PR1 in the AAV
control and RHD groups than in the control group (P<0.05); these
findings were consistent with those of previous studies (14,26). The expression in the S1PR1
overexpression group was similar to that in the control group, and
it was significantly higher than that in the RHD group (P<0.05;
Figs. 1A, 2A and B, and 3A-C). These results indicated that the
S1PR1 overexpression sequence used for overexpression in the
present study successfully increased the expression of S1PR1.
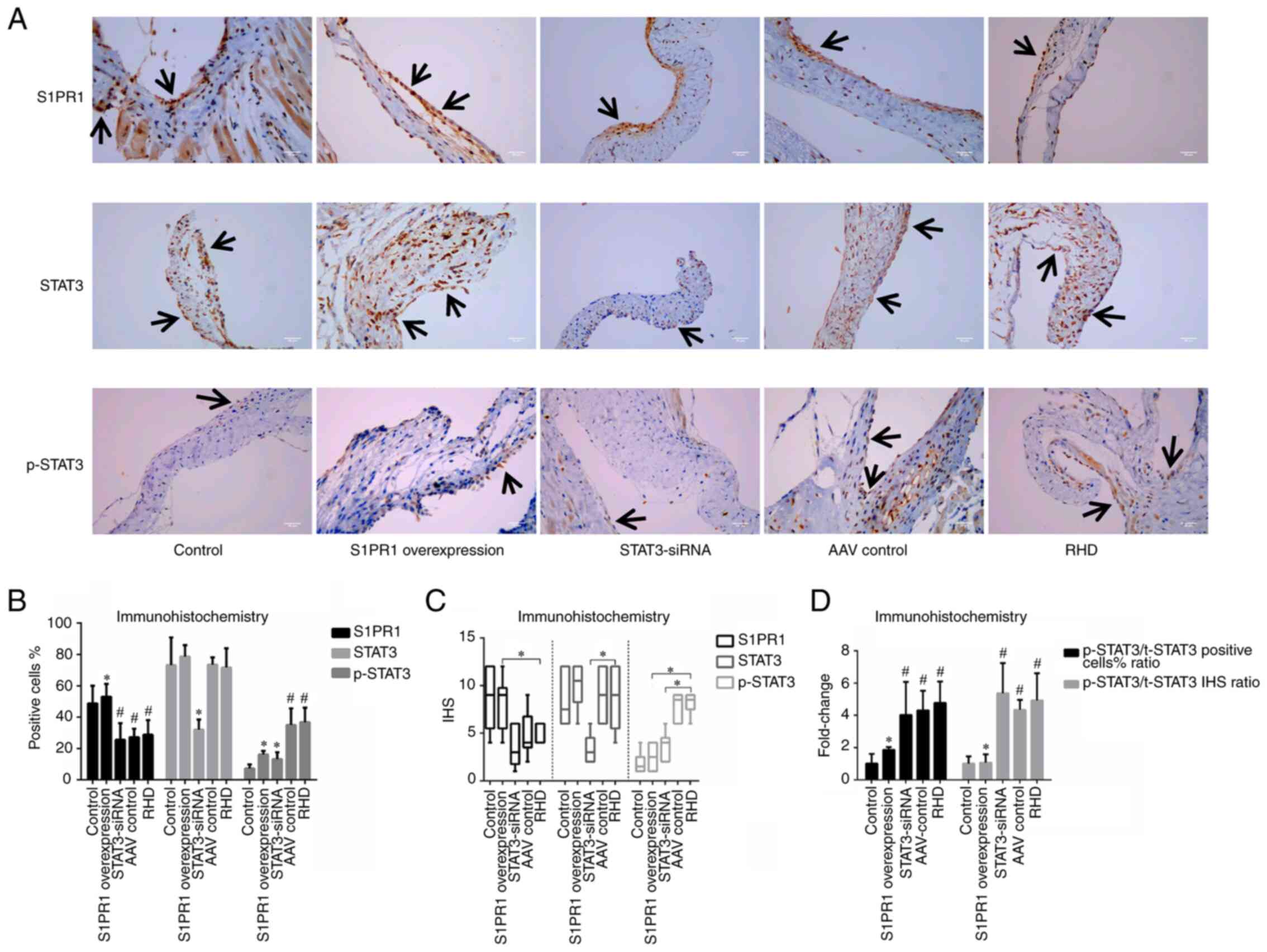 | Figure 3Immunohistochemical staining for
S1PR1, STAT3 and p-STAT3. (A) Immunohistochemical staining for
S1PR1, STAT3 and p-STAT3 in valvular tissues; magnification, ×400;
scale bar, 50 µm. Arrows indicate positively stained cells.
(B) Percentage of positive cells. (C) The IHS. (D) Ratio of
p-STAT3/t-STAT3. These results revealed that S1PR1 was expressed at
higher levels in the control group and S1PR1 overexpression group
than the other 3 groups, and the levels of p-STAT3 in the S1PR1
overexpression group were lower than those in the RHD group, and
the silencing of STAT3 by STAT3-siRNA decreased the level of the
STAT3 protein and the total amount of p-STAT3 in valvular tissue.
Data are presented as the mean ± standard deviation and the median
and interquartile range; #P<0.05 compared with the
control group. *P<0.05 compared with the RHD group.
S1PR1, sphingosine-1-phosphate receptor 1; STAT3, signal transducer
and activator of transcription 3; RHD, rheumatic heart disease;
IHS, immunohistochemical score; t-STAT3, total-signal transducer
and activator of transcription 3; p-STAT3, phosphorylated signal
transducer and activator of transcription 3. |
The level of phosphorylated STAT3 is
reduced with S1PR1 overexpression
The results of WB, RT-qPCR and immunohistochemistry
did not reveal significant differences in STAT3 expression between
the control, S1PR1 overexpression, AAV control and RHD groups
(Figs. 1B, 2A and C, and 3A-C). Significantly higher levels of
p-STAT3 were detected in the AAV control and RHD groups than in the
control group (P<0.05). Significantly lower levels of p-STAT3
were detected in the S1PR1 overexpression group than in the RHD
group (P<0.05; Figs. 2A and
D, and 3A-C). The ratio of
p-STAT3/total (t-)STAT3 also exhibited a similar trend (P<0.05;
Figs. 2E and 3D). Therefore, the level of
phosphorylated STAT3 was reduced.
The expression of Th17 cell-related
factors in the S1PR1 overexpression group is significantly lower
than that in the RHD group
Immunohistochemistry and ELISA were then performed
to determine the levels of Th17 cell-related factors. The results
revealed significantly higher levels of RORγt, IL-6 and IL-17 in
the AAV control and the RHD groups than in the control group
(P<0.05). The S1PR1 overexpression group exhibited significantly
lower levels of RORγt, IL-6 and IL-17 than the RHD group
(P<0.05; Fig. 4). Thus, S1PR1
overexpression reduced the levels of Th17 cell-related factors in
valvular tissue and serum.
Elevated S1PR1 expression attenuates
RHD-induced valvular damage
H&E and Sirius Red staining revealed
inflammation and fibrosis in the valvular tissue of the AAV control
and RHD groups. The control group exhibited a normal valvular
structure; however, the valves in the S1PR1 overexpression group
exhibited reduced inflammation (Fig.
5A) and fibrosis (Fig. 5B)
compared with those in the RHD group. Type 1 collagen (COL1) fibres
are the main type of collagen in non-fibrotic valves (30). The ratio of type 3 collagen
(COL3) and COL1 is used to reflect the degree of fibrosis in valve
tissue. During the process of fibrosis of valvular tissue, the
proportion of COL3 increases, indicating more severe fibrosis
(30). Therefore, the present
study calculated the COL3/COL1 (COL3/1) ratio to quantify the
degree of fibrosis in valve tissue. The Sirius Red staining images
revealed that COL1 fibres were closely packed yellow and red fibres
with obvious birefringence, and COL3 fibres were loosely arranged
green fibres with weak birefringence (Fig. 5B). The ratio of COL3/COL1 in the
S1PR1 overexpression group was significantly lower than that in the
RHD group (P<0.05; Fig. 5C).
The expression of COL3a1 and fibroblast-specific protein 1 (FSP1)
was also detected by RT-qPCR to examine the degree of valve
fibrosis at the mRNA level. COL3a1 and FSP1 were expressed at
significantly lower levels in the S1PR1 overexpression group than
the RHD group (P<0.05; Fig. 5D
and E). These results demonstrated that S1PR1 overexpression
attenuated RHD-induced valvular damage.
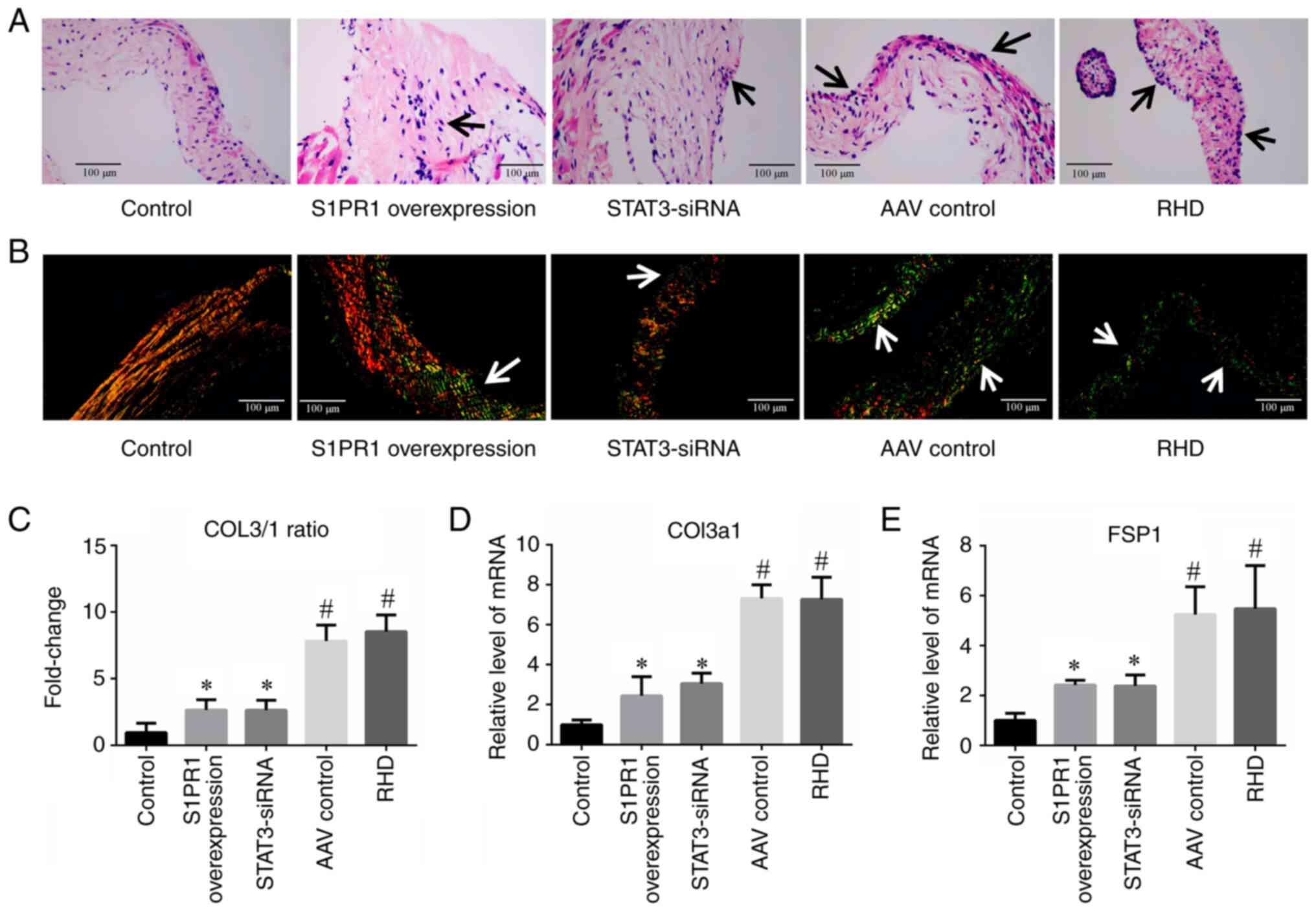 | Figure 5H&E and Sirius Red staining of
valvular tissues and RT-qPCR analysis of fibrosis-related factors.
(A) H&E staining revealed an inflammatory response in the heart
valves of the rats in the AAV control and RHD groups. In the S1PR1
overexpression and STAT3-siRNA groups, the inflammatory response
was significantly reduced compared with that in the RHD group;
magnification, ×400; scale bar, 100 µm. Arrows indicate the
inflammatory response. (B) Images of Sirius red staining of the
valves. The valves in the AAV control group and RHD group exhibited
marked fibrosis. In the S1PR1 overexpression and STAT3-siRNA
groups, the degree of fibrosis was markedly reduced compared with
that in the RHD group; magnification, ×400; scale bar, 100
µm. Arrows indicate COL3 expression. (C) A significantly
higher COL3/1 ratio was observed in the RHD group than in the
control group, and the COL3/1 ratio in the S1PR1 overexpression and
STAT3-siRNA groups was significantly lower than that in the RHD
group. (D and E) RT-qPCR analysis of COL3a1 and FSP1 expression.
These results revealed that the degree of valvular damage was at
significantly higher levels in the RHD group than the control
group. S1PR1 overexpression or STAT3-silencing reduced the level of
valvular damage. Data are presented as the mean ± standard
deviation. #P<0.05 compared with the control group;
*P<0.05 compared with the RHD group. H&E,
Hematoxylin and eosin staining; RT-qPCR, reverse
transcription-quantitative PCR; COL3, collagen fibre type 3; COL1,
collagen fibre type 1; Col3a1, collagen type III α1 chain; FSP1,
fibroblast-specific protein 1; RHD, rheumatic heart disease. |
Inhibition of STAT3
STAT3-siRNA pre-treatment decreases
STAT3 expression and reduces the total amount of p-STAT3
The silencing of STAT3 using STAT3-siRNA decreased
the expression of STAT3 mRNA (P<0.05; Fig. 1B). A significantly higher protein
level of p-STAT3 was observed in the RHD and AAV control groups
than in the control group (P<0.05). The silencing of STAT3 by
STAT3-siRNA decreased the protein levels of STAT3 and p-STAT3 in
valvular tissues (P<0.05; Figs.
2A, C and D, and 3A-C). The
ratio of p-STAT3/t-STAT3 was significantly higher in the
STAT3-siRNA, AAV control and RHD groups than in the control group
(P<0.05); however, a significant difference was not observed
between the STAT3-siRNA, AAV control and RHD groups (P<0.05;
Figs. 2E and 3D).
Expression of Th17-related
transcription factors and cytokines is reduced in the STAT3-siRNA
group compared with the RHD group
Immunohistochemistry and ELISA revealed
significantly higher levels of RORγt, IL-6 and IL-17 in the RHD and
AAV-control groups than in the control group (P<0.05). The
silencing of STAT3 by STAT3-siRNA decreased the levels of RORγt,
IL-6 and IL-17 in the serum and valvular tissues of the rats
(P<0.05; Fig. 4).
STAT3-siRNA pre-treatment attenuates
RHD-induced valvular damage
H&E staining revealed an inflammatory response
in the heart valves of rats in the AAV control and RHD groups. The
inflammatory response in the STAT3-siRNA group was reduced compared
with that in the RHD group (Fig.
5A). All changes were observed under a microscope. The Sirius
Red staining images revealed significantly more severe fibrosis in
the RHD group than in the control group. The COL3/1 ratio was also
significantly higher in the RHD group than in the control group.
The degree of fibrosis in the STAT3-siRNA group was lower than the
RHD group. The COL3/1 ratio was significantly lower in the
STAT3-siRNA group than the RHD group (P<0.05; Fig. 5B and C). The findings for the
COL3a1 and FSP1 expression levels were consistent with the results
of the histological examination (P<0.05; Fig. 5D and E). These results
demonstrated that the inflammatory response and fibrosis of the
valvular tissue were reduced following the silencing of STAT3
compared with the RHD group.
Discussion
RHD has a long history, and a number of patients
have succumbed to this disease. RHD caused 319,400 deaths in 2015
(31), 314,600 deaths in 2016
(32) and 285,500 deaths in 2017
(33); however, the pathogenesis
of this disease remains unknown. Recent research has primarily
focused on the signalling pathways related to the pathogenesis of
RHD. The efforts of numerous researchers have elucidated some of
the signalling pathways related to this disease. Recently,
researchers have discovered that the S1PR1/STAT3 signalling pathway
is involved in RHD-induced valvular damage in a rat model (14). However, the mechanisms through
which interventions targeting the expression of S1PR1 and STAT3
affect RHD-induced valvular damage remain unknown. Notably, the
most useful method which can be used to intervene with the
expression of S1PR1 and STAT3, and effectively attenuate
RHD-induced valvular damage is not yet clear.
S1PR1 has been extensively studied, and it is an
important factor in heart diseases, including RHD (14,26), myocardial infarction (9) and cardiac remodelling (10). S1PR1 primarily protects the heart
in these diseases (9-11), and a high S1PR1 expression
generally protects the heart during the pathogenesis of heart
disease (9,12). However, a previous study reported
that a high expression of S1PR1 exacerbated heart damage (34), and the role of the downregulation
of S1PR1 expression in mediating the pathogenesis of other
diseases, such as multiple sclerosis, has also been reported
(35). The low expression of
S1PR1 is not only present in RHD. For example, studies have
reported that in tumours, the low expression or lack of S1PR1
aggravates the growth of tumours, and the high expression of S1PR1
can enhance the antitumour ability of the body (36).
Studies on hypertension have also reported that the
expression of S1PR1 is downregulated, and increasing the expression
of S1PR1 is helpful for reducing hypertension (36). It may be related to RHD being an
autoimmune disease. The low expression of S1PR1 has also been
observed in autoimmune diseases (multiple sclerosis) and Crohn's
disease (37). S1PR1 may also be
regulated by microRNAs (miRNAs/miRs) to downregulate its expression
(38), such as miR-155-5p
(26). Therefore, it can be
concluded that the expression of S1PR1 in different heart diseases
is not static. Although S1PR1 expression varies among heart
diseases, S1PR1 expression is generally upregulated and it exerts a
cardioprotective effect. The present study found that RHD-induced
valvular damage was reduced with S1PR1 overexpression, which may
also be related to the cardioprotective effects of S1PR1. A close
association was identified between S1PR1 and STAT3, and a number of
studies have described the regulatory effect of S1PR1 on STAT3 in
various diseases (22-25). The present study demonstrated
that the expression of S1PR1 and STAT3 was closely related to the
pathogenesis of valvular damage in a rat model of RHD. Previous
studies on the process of valvular damage in RHD have found that
the downregulation of S1PR1 and the increased STAT3 phosphorylation
are involved in this process (14,26). This phenomenon of a downregulated
S1PR1 expression and an increased STAT3 phosphorylation has been
reported in previous studies. For example, Garris et al
(13) found that S1PR1
deficiency increased the level of p-STAT3 and promoted Th17 cell
differentiation in a mouse autoimmune encephalitis model with an
S1PR1 gene mutation. The present study demonstrated that the RHD
group also exhibited a decreased S1PR1 expression and an increased
STAT3 phosphorylation compared with that in the control group.
However, an increase in S1PR1 expression has been reported to
increase the phosphorylation of STAT3 (23,39); however, these studies did not
examine RHD. Combined with the uncertainty of the expression of
S1PR1 in the different heart diseases mentioned above, it can be
concluded that the expression of S1PR1 and its role in regulating
the phosphorylation of STAT3 in different diseases and different
physiological or pathological processes may not be static. However,
the opposite trend of S1PR1 and p-STAT3 also indicates the
possibility that p-STAT3 is indirectly regulated by S1PR1. Two
previous studies on the roles of S1PR1 and STAT3 in RHD (14,26) and the experimental results
presented in the present study demonstrated that the downregulation
of S1PR1 and the upregulation of phosphorylation of STAT3 during
RHD-induced valvular damage and induces the differentiation of Th17
cells. Therefore, the mechanism of the S1PR1 and STAT3 during the
process of RHD-induced valvular damage may be very similar to the
mechanism described by Garris et al (13), as autoimmune encephalitis and RHD
are autoimmune diseases. However, the similarity of the mechanisms
is purely a speculation. S1PR1 is downregulated in this pathway,
and whether the overexpression of S1PR1 would attenuate RHD-induced
valvular damage was not known.
STAT3 is a key pathogenic factor in many
inflammatory conditions. STAT3 mediates immune myocarditis due to
IL-6-induced liver complement component C3 production and Th17 cell
differentiation (40), and the
differentiation of Th17 cells plays an important role in the
occurrence and development of myocarditis (41). Tissue signalling cytokines, such
as IL-17 and IL-22, may affect the heart via a pathway that
involves STAT3 (42). Th17 cells
and related inflammatory factors (such as IL-17) play important
roles in the process of inflammation and the autoimmune response
(17-19). The levels of Th17 cell-related
factors are increased in the peripheral blood and serum of patients
with RHD (20), and the level of
Th17-related cytokines in the mitral valve is significantly
increased (21). Therefore, Th17
cells likely promote the development of RHD. Previous researchers
have reported high levels of p-STAT3 in individuals with rheumatoid
arthritis (43), and the present
study considered STAT3 a key component of this signalling pathway.
The present study wished to determine whether the suppression of
STAT3 expression would attenuate RHD-induced valvular damage.
Based on studies on the association between the
expression of S1PR1, STAT3 and RHD-induced valvular damage, it was
hypothesised that the level of p-STAT3 was increased during the
process of valvular damage, which promoted the differentiation of
CD4+ T cells into Th17 cells and the Th17 cell-related
cytokines to participate in the process of RHD-induced valvular
damage. Therefore, experiments were performed to overexpress S1PR1
and inhibit STAT3. The results obtained from in the first part of
the present study demonstrated that the originally downregulated
expression of S1PR1 was increased with S1PR1 overexpression, the
level of p-STAT3 was decreased, the levels of Th17 cell-related
cytokines in the valvular tissue and serum were also decreased, and
eventually, the level of RHD-induced inflammation and fibrosis of
the valve was attenuated. In previous studies, it was also
demonstrated that the overexpression of S1PR1 caused the expression
of p-STAT3 to decrease, and there was also no significant
difference in the amount of STAT3 between groups (14,26). This may have occurred as the
intervention of S1PR1 affected the activation of STAT3, and the
activation of STAT3 was mainly manifested in the amount of p-STAT3;
thus, the amount of STAT3 had no effect, as a number of previous
studies on STAT3 have observed the same phenomenon (44-46).
The results obtained from the second part of the
present study demonstrated decreased levels of total STAT3 and
p-STAT3, the decreased expression of Th17-related transcription
factors and cytokines, and the attenuation of the level of
RHD-induced inflammation and fibrosis of the valve following STAT3
silencing. There is a close association between STAT3, IL-6 and
IL-17, it has been suggested that the three factors are mutually
reinforcing (47). For example,
STAT3 can promote the production of the pro-inflammatory cytokine,
IL-6, and forms a positive feedback loop to regulate IL-6 level
(48). The pharmacological
inhibition of STAT3 with JAK or STAT3 inhibitors, or the inhibition
of STAT3 genetically with dominant negative STAT3 and short hairpin
STAT3 has been shown to reduce the level of IL-6 (49). The activation of STAT3 promotes
the differentiation of Th17 cells. IL-6 and IL-17 are Th17-related
cytokines; thus, the inhibition of STAT3 expression will affect the
expression of IL-6 and IL-17 (50,51). These results demonstrate that the
expression of S1PR1 and STAT3 is involved in the regulation of Th17
cell-related cytokine levels during RHD-induced valvular damage,
and strategies designed to interfere with the expression of S1PR1
and STAT3 may modulate the expression of Th17 cell-related
cytokines, and may subsequently attenuate RHD-induced valvular
damage.
Studies investigating the signalling pathways
related to the pathogenesis of RHD are limited. Significant
research progress was achieved in only six signalling pathways: The
RhoA/Rho-dependent kinase (RhoA/ROCK) signalling pathway,
mitogen-activated protein kinase (MAPK) signalling pathway, protein
kinase B/S6 kinase (AKT/S6K) signalling pathway, TGF-β1/Smad
signalling pathway, Wnt signalling pathway and S1PR1/STAT3
signalling pathway (52-57). Only three potential intervention
targets in these signalling pathways were identified: The
modulation of the expression of interferon (IFN)-γ and tumour
necrosis factor (TNF)-α to regulate extracellular matrix
remodelling and reduce RHD-induced heart damage, altering the
activity of the AKT/S6K signalling pathway to inhibit
TGF-β1-induced fibroblasts, and targeting the S1PR1/STAT3
signalling pathway to reduce RHD-induced valvular damage. Further
studies are required to determine whether these intervention
targets effectively prevent and treat RHD. International research
on the pathogenesis of RHD is lacking, and the pathogenesis of RHD
remains unclear. The threat to the lives and health of patients
with RHD is substantial, and damage to the health and quality of
life of patients is devastating. RHD is a severe condition, and
studies investigating its pathogenesis are worthwhile. S1PR1 and
STAT3 may prove to be two potential intervention targets for RHD.
The findings of the present study may enhance the current
understanding of the signalling pathways related to the
pathogenesis of RHD, thus contributing to the further understanding
of the pathogenesis of RHD. The findings presented herein may also
aid in the development of effective and inexpensive methods for
controlling RHD in the future.
The present study has some limitations. The present
study was performed using a rat model, and further studies using
human samples are required. S1PR1 expression is downregulated
during RHD-induced valvular damage, and greater technical
requirements and further experiments are required to examine the
effects of a complete inhibition of S1PR1 expression. The effects
of upregulating STAT3 expression on RHD are unknown. Cell-based
experiments may provide cell-level evidence to support the findings
of the present study; however, such experiments were not performed
herein. In addition, the present study did not detect the protein
levels of FSP1 and COL3a1. The absence of data evaluating cardiac
function in the RHD model following the overexpression of
S1PR1/knockdown of STAT3 is also a potential limitation of the
present study. The effect of other STAT3 inhibitors in the RHD
model may be a good direction for further studies. The specific
regulatory mechanisms between S1PR1 and STAT3, and whether there
are other regulatory mechanisms between these two proteins warrants
further investigation in the future.
The status of RHD remains severe, and although
primary and secondary prevention strategies have been clearly
identified, their global implementation is not ideal (58). The pathogenesis of RHD has long
been studied (2); however, this
is still not fully understood. By summarizing the results of
previous studies and the inflammatory mechanisms of RHD
pathogenesis, it was hypothesised that strategies targeting the
expression of S1PR1 and STAT3 may modulate the process of
RHD-induced valvular damage. In a previous study by the authors, it
was found that S1PR1 and STAT3 may be involved in RHD (14); however, the specific roles of
S1PR1 and STAT3 remain unclear. The animal experiments in the
present study revealed a role for the expression of S1PR1 and STAT3
in regulating the levels of Th17 cell-related cytokines during
RHD-induced valvular damage, and interfering with the expression of
S1PR1 and STAT3 may alter the expression of Th17 cell-related
cytokines and attenuate RHD-induced valvular damage. The present
study provides some insight into the pathogenesis of RHD, and
provides some references for discovering intervention targets for
RHD. However, the present study found that only intervention with
the expression of S1PR1 and STAT3 reduced RHD-induced valve damage.
The specific regulatory mechanisms between S1PR1 and STAT3, whether
there are other regulatory mechanisms between these two proteins,
and whether this strategy is effective in in vitro
experiments and in human samples needs to be further studied in the
future.
Availability of data and materials
All data generated or analysed during this study are
included in this published article.
Authors' contributions
ZZ and FH conceived and designed the study. SX and
AC participated in the experimental design. SX, AC, YW and CL
performed the experiments. SX, AC and HW analysed the data. SX and
FH confirm the authenticity of all the raw data. SX wrote the
manuscript, and all authors reviewed, and have read and approved
the final manuscript.
Ethics approval and consent to
participate
Protocols involving animals were approved by The
Medical Ethics Committee of the First Affiliated Hospital of
Guangxi Medical University (Nanning, China; approval no.
2019-KY-E-069).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgements
Not applicable.
Abbreviations:
|
RHD
|
rheumatic heart disease
|
|
S1PR1
|
sphingosine 1-phosphate receptor 1
|
|
STAT3
|
signal transducer and activator of
transcription 3
|
|
Th17
|
T helper 17
|
|
GAS
|
group A streptococcus;
|
|
RT-qPCR
|
reverse transcription-quantitative
PCR
|
|
Col3a1
|
collagen type III α1 chain
|
|
FSP1
|
fibroblast-specific protein 1
|
|
WB
|
western blotting
|
|
p-
|
phosphorylated
|
|
RORγt
|
retinoic acid-related orphan receptor
γT
|
|
ELISAs
|
enzyme-linked immunosorbent
assays;
|
|
IL
|
interleukin
|
|
H&E
|
Hematoxylin and eosin
|
|
NS
|
normal saline;
|
|
AAV
|
adeno-associated virus
|
|
t-
|
total
|
References
|
1
|
Watkins DA, Johnson CO, Colquhoun SM,
Karthikeyan G, Beaton A, Bukhman G, Forouzanfar MH, Longenecker CT,
Mayosi BM, Mensah GA, et al: Global, regional, and national burden
of rheumatic heart disease, 1990-2015. N Engl J Med. 377:713–722.
2017. View Article : Google Scholar
|
|
2
|
Leal MT, Passos LS, Guarçoni FV, Aguiar
JM, Silva RB, Paula TM, Santos RF, Nassif MC, Gomes NF, Tan TC and
Nunes MC: Rheumatic heart disease in the modern era: Recent
developments and current challenges. Rev Soc Bras Med Trop.
52:e201800412019. View Article : Google Scholar
|
|
3
|
Mirabel M, Narayanan K, Jouven X and
Marijon E: Cardiology patient page. Prevention of acute rheumatic
fever and rheumatic heart disease. Circulation. 130:e35–e37. 2014.
View Article : Google Scholar
|
|
4
|
Zhao Z, He D, Ling F, Chu T, Huang D, Wu H
and Ge J: CD4+ T cells and TGFβ1/MAPK signal pathway
involved in the valvular hyperblastosis and fibrosis in patients
with rheumatic heart disease. Exp Mol Pathol. 114:1044022020.
View Article : Google Scholar
|
|
5
|
Messias-Reason IJ, Schafranski MD,
Kremsner PG and Kun JF: Ficolin 2 (FCN2) functional polymorphisms
and the risk of rheumatic fever and rheumatic heart disease. Clin
Exp Immunol. 157:395–399. 2009. View Article : Google Scholar
|
|
6
|
Li M, Xu S, Geng Y, Sun L, Wang R, Yan Y,
Wang H, Li Y, Yi Q, Zhang Y, et al: The protective effects of
L-carnitine on myocardial ischaemia-reperfusion injury in patients
with rheumatic valvular heart disease undergoing CPB surgery are
associated with the suppression of NF-κB pathway and the activation
of Nrf2 pathway. Clin Exp Pharmacol Physiol. 46:1001–1012. 2019.
View Article : Google Scholar
|
|
7
|
Aoki M, Aoki H, Ramanathan R, Hait NC and
Takabe K: Sphingosine-1-phosphate signaling in immune cells and
inflammation: Roles and therapeutic potential. Mediators Inflamm.
2016:86068782016.
|
|
8
|
Jozefczuk E, Guzik TJ and Siedlinski M:
Significance of sphingosine-1-phosphate in cardiovascular
physiology and pathology. Pharmacol Res. 156:1047932020. View Article : Google Scholar
|
|
9
|
Deng S, Zhou X, Ge Z, Song Y, Wang H, Liu
X and Zhang D: Exosomes from adipose-derived mesenchymal stem cells
ameliorate cardiac damage after myocardial infarction by activating
S1P/SK1/S1PR1 signaling and promoting macrophage M2 polarization.
Int J Biochem Cell Biol. 114:1055642019. View Article : Google Scholar
|
|
10
|
Liu X, Wu J, Zhu C, Liu J, Chen X, Zhuang
T, Kuang Y, Wang Y, Hu H, Yu P, et al: Endothelial S1pr1 regulates
pressure overload-induced cardiac remodelling through AKT-eNOS
pathway. J Cell Mol Med. 24:2013–2026. 2020. View Article : Google Scholar
|
|
11
|
Liu Y, Zhi Y, Song H, Zong M, Yi J, Mao G,
Chen L and Huang G: S1PR1 promotes proliferation and inhibits
apoptosis of esophageal squamous cell carcinoma through activating
STAT3 pathway. J Exp Clin Cancer Res. 38:3692019. View Article : Google Scholar
|
|
12
|
Chen YZ, Wang F, Wang HJ and Liu HB:
Sphingosine 1 phosphate receptor-1 (S1PR1) signaling protects
cardiac function by inhibiting cardiomyocyte autophagy. J Geriatr
Cardiol. 15:334–345. 2018.
|
|
13
|
Garris CS, Wu L, Acharya S, Arac A, Blaho
VA, Huang Y, Moon BS, Axtell RC, Ho PP, Steinberg GK, et al:
Defective sphingosine 1-phosphate receptor 1 (S1P1) phosphorylation
exacerbates TH17-mediated autoimmune neuroinflammation. Nat
Immunol. 14:1166–1172. 2013. View Article : Google Scholar
|
|
14
|
Wu XD, Zeng ZY, Gong DP, Wen JL and Huang
F: Potential involvement of S1PR1/STAT3 signaling pathway in
cardiac valve damage due to rheumatic heart disease. Biotech
Histochem. 94:398–403. 2019. View Article : Google Scholar
|
|
15
|
Hu YS, Han X and Liu XH: STAT3: A
potential drug target for tumor and inflammation. Curr Top Med
Chem. 19:1305–1317. 2019. View Article : Google Scholar
|
|
16
|
Liu X, Hu H, Fan H, Zuo D, Shou Z, Liao Y,
Nan Z and Tang Q: The role of STAT3 and AhR in the differentiation
of CD4+ T cells into Th17 and Treg cells. Medicine
(Baltimore). 96:e66152017. View Article : Google Scholar
|
|
17
|
Gaffen SL, Jain R, Garg AV and Cua DJ: The
IL-23-IL-17 immune axis: From mechanisms to therapeutic testing.
Nat Rev Immunol. 14:585–600. 2014. View Article : Google Scholar
|
|
18
|
Whibley N, Tritto E, Traggiai E, Kolbinger
F, Moulin P, Brees D, Coleman BM, Mamo AJ, Garg AV, Jaycox JR, et
al: Antibody blockade of IL-17 family cytokines in immunity to
acute murine oral mucosal candidiasis. J Leukoc Biol. 99:1153–1164.
2016. View Article : Google Scholar
|
|
19
|
Zhang Y, Shao Z, Zhang X, Jia X, Xia Y,
Zhang Y, Xin N, Guo M, Chen J, Zheng S, et al: TIPE2 play a
negative role in TLR4-mediated autoimmune T helper 17 cell
responses in patients with myasthenia gravis. J Neuroimmune
Pharmacol. 10:635–644. 2015. View Article : Google Scholar
|
|
20
|
Bas HD, Baser K, Yavuz E, Bolayir HA,
Yaman B, Unlu S, Cengel A, Bagriacik EU and Yalcin R: A shift in
the balance of regulatory T and T helper 17 cells in rheumatic
heart disease. J Investig Med. 62:78–83. 2014. View Article : Google Scholar
|
|
21
|
Wen Y, Zeng Z, Gui C, Li L and Li W:
Changes in the expression of Th17 cell-associated cytokines in the
development of rheumatic heart disease. Cardiovasc Pathol.
24:382–387. 2015. View Article : Google Scholar
|
|
22
|
Lankadasari MB, Aparna JS, Mohammed S,
James S, Aoki K, Binu VS, Nair S and Harikumar KB: Targeting
S1PR1/STAT3 loop abrogates desmoplasia and chemosensitizes
pancreatic cancer to gemcitabine. Theranostics. 8:3824–3840. 2018.
View Article : Google Scholar
|
|
23
|
Lin Q, Ren L, Jian M, Xu P, Li J, Zheng P,
Feng Q, Yang L, Ji M, Wei Y and Xu J: The mechanism of the
premetastatic niche facilitating colorectal cancer liver metastasis
generated from myeloid-derived suppressor cells induced by the
S1P1STAT3 signaling pathway. Cell Death Dis. 10:6932019. View Article : Google Scholar
|
|
24
|
Lee H, Deng J, Kujawski M, Yang C, Liu Y,
Herrmann A, Kortylewski M, Horne D, Somlo G, Forman S, et al:
STAT3-induced S1PR1 expression is crucial for persistent STAT3
activation in tumors. Nat Med. 16:1421–1428. 2010. View Article : Google Scholar
|
|
25
|
Deng J, Liu Y, Lee H, Herrmann A, Zhang W,
Zhang C, Shen S, Priceman SJ, Kujawski M, Pal SK, et al:
S1PR1-STAT3 signaling is crucial for myeloid cell colonization at
future metastatic sites. Cancer Cell. 21:642–654. 2012. View Article : Google Scholar
|
|
26
|
Chen A, Wen J, Lu C, Lin B, Xian S, Huang
F, Wu Y and Zeng Z: Inhibition of miR-155-5p attenuates the
valvular damage induced by rheumatic heart disease. Int J Mol Med.
45:429–440. 2020.
|
|
27
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
28
|
Guendisch U, Weiss J, Ecoeur F, Riker JC,
Kaupmann K, Kallen J, Hintermann S, Orain D, Dawson J, Billich A
and Guntermann C: Pharmacological inhibition of RORγt suppresses
the Th17 pathway and alleviates arthritis in vivo. PLoS One.
12:e01883912017. View Article : Google Scholar
|
|
29
|
Friedrichs K, Gluba S, Eidtmann H and
Jonat W: Overexpression of p53 and prognosis in breast cancer.
Cancer. 72:3641–3647. 1993. View Article : Google Scholar
|
|
30
|
Purushothaman KR, Purushothaman M,
Turnbull IC, Adams DH, Anyanwu A, Krishnan P, Kini A, Sharma SK,
O'Connor WN and Moreno PR: Association of altered collagen content
and lysyl oxidase expression in degenerative mitral valve disease.
Cardiovasc Pathol. 29:11–18. 2017. View Article : Google Scholar
|
|
31
|
GBD 2015 Mortality and Causes of Death
Collaborators: global, regional, and national life expectancy,
all-cause mortality, and cause-specific mortality for 249 causes of
death, 1980-2015: A systematic analysis for the global burden of
disease study 2015. Lancet. 388:1459–1544. 2016. View Article : Google Scholar
|
|
32
|
GBD 2016 Causes of Death Collaborators:
global, regional, and national age-sex specific mortality for 264
causes of death, 1980-2016: A systematic analysis for the global
burden of disease study 2016. Lancet. 390:1151–1210. 2017.
View Article : Google Scholar
|
|
33
|
GBD 2017 Causes of Death Collaborators:
global, regional, and national age-sex-specific mortality for 282
causes of death in 195 countries and territories, 1980-2017: A
systematic analysis for the global burden of disease study 2017.
Lancet. 392:1736–1788. 2018. View Article : Google Scholar
|
|
34
|
Zhang F, Xia Y, Yan W, Zhang H, Zhou F,
Zhao S, Wang W, Zhu D, Xin C, Lee Y, et al: Sphingosine 1-phosphate
signaling contributes to cardiac inflammation, dysfunction, and
remodeling following myocardial infarction. Am J Physiol Heart Circ
Physiol. 310:H250–H261. 2016. View Article : Google Scholar
|
|
35
|
Edmonds Y, Milstien S and Spiegel S:
Development of small-molecule inhibitors of sphingosine-1-phosphate
signaling. Pharmacol Ther. 132:352–360. 2011. View Article : Google Scholar
|
|
36
|
Cartier A, Leigh T, Liu CH and Hla T:
Endothelial sphingosine 1-phosphate receptors promote vascular
normalization and antitumor therapy. Proc Natl Acad Sci USA.
117:3157–3166. 2020. View Article : Google Scholar
|
|
37
|
Abarca-Zabalía J, García MI, Lozano Ros A,
Marín-Jiménez I, Martínez-Ginés ML, López-Cauce B, Martín-Barbero
ML, Salvador-Martín S, Sanjurjo-Saez M, García-Domínguez JM and
López Fernández LA: Differential expression of SMAD genes and S1PR1
on circulating CD4+ T cells in multiple sclerosis and Crohn's
disease. Int J Mol Sci. 21:6762020. View Article : Google Scholar
|
|
38
|
Zhang C, Shen J, Kong S, Zhang M, Zhang Q,
Zhou J, Zhen X, Kang N, Jiang Y, Ding L, et al: MicroRNA-181a
promotes follicular granulosa cell apoptosis via
sphingosine-1-phosphate receptor 1 expression downregulation†. Biol
Reprod. 101:975–985. 2019. View Article : Google Scholar
|
|
39
|
Silva VR, Micheletti TO, Katashima CK,
Lenhare L, Morari J, Moura-Assis A, de Lima-Júnior JC, Camargo JA,
Passos GR, Gaspar RS, et al: Exercise activates the hypothalamic
S1PR1-STAT3 axis through the central action of interleukin 6 in
mice. J Cell Physiol. 233:9426–9436. 2018. View Article : Google Scholar
|
|
40
|
Camporeale A, Marino F, Papageorgiou A,
Carai P, Fornero S, Fletcher S, Page BD, Gunning P, Forni M,
Chiarle R, et al: STAT3 activity is necessary and sufficient for
the development of immune-mediated myocarditis in mice and promotes
progression to dilated cardiomyopathy. EMBO Mol Med. 5:572–590.
2013. View Article : Google Scholar
|
|
41
|
Yuan J, Yu M, Lin QW, Cao AL, Yu X, Dong
JH, Wang JP, Zhang JH, Wang M, Guo HP, et al: Th17 cells contribute
to viral replication in coxsackievirus B3-induced acute viral
myocarditis. J Immunol. 185:4004–4010. 2010. View Article : Google Scholar
|
|
42
|
Kurdi M, Zgheib C and Booz GW: Recent
developments on the crosstalk Between STAT3 and inflammation in
heart function and disease. Front Immunol. 9:30292018. View Article : Google Scholar
|
|
43
|
Turkson J and Jove R: STAT proteins: Novel
molecular targets for cancer drug discovery. Oncogene.
19:6613–6626. 2000. View Article : Google Scholar
|
|
44
|
Yang Y, Wang Y, Che X, Hou K, Wu J, Zheng
C, Cheng Y, Liu Y, Hu X and Zhang J: Integrin α5 promotes migration
and invasion through the FAK/STAT3/AKT signaling pathway in
icotinib-resistant non-small cell lung cancer cells. Oncol Lett.
22:5562021. View Article : Google Scholar
|
|
45
|
Hu X, Jiao F, Zhang L and Jiang Y:
Dihydrotanshinone Inhibits hepatocellular carcinoma by suppressing
the JAK2/STAT3 pathway. Front Pharmacol. 12:6549862021. View Article : Google Scholar
|
|
46
|
Liu X, Zhou F, Wang W, Chen G, Zhang Q, Lv
R, Zhao Z, Li X, Yu Q, Meves JM, et al: IL-9-triggered lncRNA
Gm13568 regulates Notch1 in astrocytes through interaction with
CBP/P300: Contribute to the pathogenesis of experimental autoimmune
encephalomyelitis. J Neuroinflammation. 18:1082021. View Article : Google Scholar
|
|
47
|
Camporeale A and Poli V: IL-6, IL-17 and
STAT3: A holy trinity in auto-immunity? Front Biosci (Landmark Ed).
17:2306–2326. 2012. View
Article : Google Scholar
|
|
48
|
Wu J, Niu P, Zhao Y, Cheng Y, Chen W, Lin
L, Lu J, Cheng X and Xu Z: Impact of miR-223-3p and miR-2909 on
inflammatory factors IL-6, IL-1ß, and TNF-α, and the
TLR4/TLR2/NF-κB/STAT3 signaling pathway induced by
lipopolysaccharide in human adipose stem cells. PLoS One.
14:e02120632019. View Article : Google Scholar
|
|
49
|
Bonetto A, Aydogdu T, Jin X, Zhang Z, Zhan
R, Puzis L, Koniaris LG and Zimmers TA: JAK/STAT3 pathway
inhibition blocks skeletal muscle wasting downstream of IL-6 and in
experimental cancer cachexia. Am J Physiol Endocrinol Metab.
303:E410–E421. 2012. View Article : Google Scholar
|
|
50
|
Damasceno LE, Prado DS, Veras FP, Fonseca
MM, Toller-Kawahisa JE, Rosa MH, Públio GA, Martins TV, Ramalho FS,
Waisman A, et al: PKM2 promotes Th17 cell differentiation and
autoimmune inflammation by fine-tuning STAT3 activation. J Exp Med.
217:e201906132020. View Article : Google Scholar
|
|
51
|
Shui X, Chen S, Lin J, Kong J, Zhou C and
Wu J: Knockdown of lncRNA NEAT1 inhibits Th17/CD4+ T cell
differentiation through reducing the STAT3 protein level. J Cell
Physiol. 234:22477–22484. 2019. View Article : Google Scholar
|
|
52
|
Jiang L, Wei XF, Yi DH, Xu P, Liu H, Chang
Q, Yang SM, Li ZF, Gao HB and Hao GJ: Synergistic effects of cyclic
strain and Th1-like cytokines on tenascin-C production by rheumatic
aortic valve interstitial cells. Clin Exp Immunol. 155:216–223.
2009. View Article : Google Scholar
|
|
53
|
Li M, Yi XIN, Ma L and Zhou Y: Hepatocyte
growth factor and basic fibroblast growth factor regulate atrial
fibrosis in patients with atrial fibrillation and rheumatic heart
disease via the mitogen-activated protein kinase signaling pathway.
Exp Ther Med. 6:1121–1126. 2013. View Article : Google Scholar
|
|
54
|
Zhang P, Wang W, Wang X, Wang X, Song Y,
Zhang J and Zhao H: Focal adhesion kinase mediates atrial fibrosis
via the AKT/S6K signaling pathway in chronic atrial fibrillation
patients with rheumatic mitral valve disease. Int J Cardiol.
168:3200–3207. 2013. View Article : Google Scholar
|
|
55
|
Zhang L, Zhang N, Tang X, Liu F, Luo S and
Xiao H: Increased α-actinin-2 expression in the atrial myocardium
of patients with atrial fibrillation related to rheumatic heart
disease. Cardiology. 135:151–159. 2016. View Article : Google Scholar
|
|
56
|
Guo F, Yi X, Li M, Fu J and Li S: Snail1
is positively correlated with atrial fibrosis in patients with
atrial fibrillation and rheumatic heart disease. Exp Ther Med.
14:4231–4237. 2017.
|
|
57
|
Wu Y, Xu M, Bao H and Zhang JH:
Sitagliptin inhibits EndMT in vitro and improves cardiac function
of diabetic rats through the SDF-1α/PKA pathway. Eur Rev Med
Pharmacol Sci. 23:841–848. 2019.
|
|
58
|
Remenyi B, ElGuindy A, Smith SC Jr, Yacoub
M and Holmes DR Jr: Valvular aspects of rheumatic heart disease.
Lancet. 387:1335–1346. 2016. View Article : Google Scholar
|