Introduction
Cell death is an essential physiological process
that is key in tissue homeostasis, aging, development and various
pathological states (1).
Ferroptosis is a newly identified type of cell death that is
characterized by the iron-dependent accumulation of lipid peroxides
to lethal levels, which is genetically, biochemically and
morphologically distinct from autophagy, necroptosis and apoptosis
(2). Ferroptosis can be
prevented or reversed by iron chelation (3) and has been previously shown to be
involved in the pathological progression of carcinogenesis
(4), renal ischemia/reperfusion
injury (5) and oxidative
stress-induced cell death (6).
Herceptin (trastuzumab) is a chemotherapeutic agent
that targets HER2 and is commonly utilized for the treatment of
patients with HER2+ breast cancer (7). However, the clinical use of
Herceptin is limited due to its reported cardiotoxic effects,
including congestive heart failure and irreversible degenerative
cardiomyopathy (8-10). Death of terminally differentiated
cardiomyocytes is one of the major pathogenic causes of heart
injury (11). Therefore,
preventing cardiomyocyte cell death is proposed to be an effective
cardioprotective approach (11).
Iron homeostasis is sustained by several mechanisms,
including hepcidin and iron regulatory proteins (IRP), including
IRP1 and IRP2, at systemic and cellular levels (12). Disruption of iron homeostasis
leads to excessive intracellular iron accumulation, thereby causing
the oxidation and modification of lipids, proteins and DNA through
the production of free radicals and oxidative stress (13). Aberrant dysregulation of iron
metabolism is associated with in a number diseases, including
neurodegenerative diseases (Alzheimer's disease, Parkinson's
disease and Huntington's disease) (14,15), cancer (lung cancer, colon cancer
and breast cancer) (16,17) and cardiovascular diseases
(atherosclerosis and myocardial ischemia-reperfusion injury)
(18,19). Dysregulation of iron homeostasis
results in excessive iron deposition in several organs, leading to
progressive tissue damage (20).
Iron overload serves a pathogenic role in cardiomyopathy by
generating ROS via the Fenton reaction and exerts harmful
influences (18,21). In 2012, iron-dependent cell death
(ferroptosis) was discovered for the first time by Dixon et
al (2). In animal models of
cardiomyopathy and patients with heart failure, iron overload has
been revealed to mediate the pathogenesis of cardiotoxicity under
numerous cardiopathic conditions, where doxorubicin toxicity
typically occurs by the preferential accumulation of iron
specifically in the mitochondria independent of the
topoisomerase-2β (a well-known target of doxorubicin) pathway
(22,23). However, little is known regarding
the underlying mechanism of iron accumulation or its toxicity. The
present study therefore aimed to investigate the effects of
targeting ferroptosis on Herceptin-induced heart failure in an
in vitro model.
Materials and methods
Cell culture and in vitro treatment
H9c2 rat cardiomyocytes were purchased from The Cell
Bank of Type Culture Collection of The Chinese Academy of Sciences.
Cells were incubated in DMEM (Gibco; Thermo Fisher Scientific,
Inc.) containing 10% heat-inactivated FBS (Gibco; Thermo Fisher
Scientific, Inc.) and 1% penicillin-streptomycin (Gibco; Thermo
Fisher Scientific, Inc.) at 37°C in a humidified atmosphere
containing 5% CO2.
Herceptin [0, 0.2, 0.5, 1 and 10 µM;
Genentech Inc.; Roche Pharma (Schweiz) Ltd.] was administrated for
12, 24 or 48 h at 37°C (24).
The ferroptosis inhibitor, ferrostatin-1 (Fer-1; Sigma-Aldrich;
Merck KGaA), a lipid reactive oxygen species (ROS) scavenger, was
initially dissolved in DMSO and diluted to a final concentration of
10 µM (25). Deferoxamine
(DFO; cat. no. HY-B0988; MedChem Express), which is an iron
chelator, was diluted to a final concentration of 100 µM
(26). H9c2 rat cardiomyocytes
were randomly divided into the following four groups: i) Control;
ii) Herceptin; iii) Herceptin + Fer-1; and iv) Herceptin + DFO.
Fer-1 or DFO was co-administrated with Herceptin. Samples were
collected at the indicated time points after Herceptin
treatment.
Cell viability
Cell viability was assessed using MTT assay.
Briefly, H9c2 cells (3×104 cells/well) were seeded into
96-well plates and incubated at 37°C for 24 h. Following the
aforementioned treatments, H9c2 cells were added with a MTT
solution (5 mg/ml) and incubated for 4 h at 37°C. Next, DMSO was
added into each well to lyse the cells and dissolve the formazan
crystals after the removal of supernatant. The absorbance at 570 nm
was recorded in each well with a Flexstation® 3
microplate reader (Molecular Devices, LLC). The optical density
value was reported as the ratio to control group (set as 100%).
Measurement of intracellular and
mitochondrial ROS
The intracellular and mitochondrial ROS levels were
quantified by measuring the fluorescence of DCFH-DA-A (cat. no.
S0033; Beyotime Institute of Biotechnology) and MitoSOX™ Red
(Mitochondrial Superoxide Indicator; cat. no. M36008; Thermo Fisher
Scientific, Inc.), respectively. After the aforementioned
treatments, H9c2 cells were collected and washed three times with
1X PBS, followed by incubation with 5 µM DCFH-DA-A or
MitoSOX™ Red for 45 min at 37°C in the dark. H9c2 cells were then
washed three times with 1X PBS and detached with trypsin/EDTA. The
relative level of cellular fluorescence was quantified by flow
cytometry (BD FACSCanto™; BD Biosciences). Data were analyzed using
FlowJo version ×0.7 (FlowJo LLC).
Measurement of glutathione (GSH)/oxidized
glutathione (GSSG) ratio
H9c2 cells (5×104 cells/well) were seeded
into 96-well white-walled multiwall luminometer plates. After
treatment with Herceptin (0, 0.2, 0.5, 1 and 10 µM) for 12,
24 and 48 h at 37°C, the GSH/GSSG ratio was detected using a
GSH/GSSG Ratio Detection Assay kit (cat. no. ab138881; Abcam),
following the manufacturer's protocols.
Western blot analysis
Total protein was extracted from H9c2 cells via
homogenization in RIPA buffer (cat. no. P0013B; Beyotime Institute
of Biotechnology) containing protease inhibitors. Protein
concentration was evaluated using a Pierce BCA protein assay kit
(Thermo Fisher Scientific, Inc.). In total, 20 µg protein
was separated by 10-12% SDS-PAGE and subsequently transferred onto
nitrocellulose membranes. The membranes were blocked with 5% BSA
(Thermo Fisher Scientific, Inc.) in TBS containing 0.2% Tween-20
for 1 h, followed by an overnight incubation at 4°C with primary
antibodies against glutathione peroxidase (GPX)4 (cat. no. DF6701;
dilution 1:1,000; Affinity Biosciences), recombinant solute carrier
family 7 member 11 (SLC7A11; cat. no. A13685; dilution 1:1,000;
ABclonal Biotech Co., Ltd.), acyl-CoA synthetase long chain family
member 4 (ACSL4; cat. no. DF12141; dilution 1:1,000; Affinity
Biosciences), voltage-dependent anion-selective channel (VDAC2/3;
cat. nos. 11663-1-AP and 14451-1-AP; dilution 1:1,000; Proteintech
Group, Inc.), cytochrome c (Cyto c; cat. no.
10993-1-AP; dilution 1:1,000; Proteintech Group, Inc.),
mitochondrial fission 1 protein (fis1; cat. no. 10956-1-AP;
dilution 1:1,000; Proteintech Group, Inc.), mitofusin (Mfn1/2; cat.
no. 13798-4-AP; dilution 1:1,000; Proteintech Group, Inc.),
dynamin-related protein (Drp1; cat. no. ab184247; dilution 1:1,000;
Abcam), mitochondrial optic atrophy (OPA1-1/2; cat. no. 27733-1-AP;
dilution 1:2,000; Proteintech Group, Inc.), mitochondrial ferritin
(Mtf; cat. no. LS0-C293861; dilution 1:500; Lifespan BioSciences,
Inc.) and GAPDH (cat. no. AB-P-R001; dilution 1:1,000; Hangzhou
Xianzhi Biological Technology Co., Ltd.). The membranes were then
incubated for 1 h at room temperature with an HRP-conjugated
secondary antibody (cat. no. BA1054; dilution 1:5,000; Boster
Biological Technology). Finally, the bands were detected using the
Pierce ECL system (Thermo Fisher Scientific, Inc.) and band
intensity quantified using the ImageJ software (version 1.8.0;
National Institutes of Health).
Measurement of intracellular labile iron
pool (LIP)
H9c2 cells (1×104 cells/well) were seeded
into a 96-well plate and grown overnight. The H9c2 cells were then
harvested, washed and resuspended in a buffer containing 140 mM
NaCl, 5 mM KCl, 1 mM MgCl2, 5.6 mM glucose, 1.5 mM
CaCl2 and 20 mM HEPES (pH 7.4), followed by the addition
of 0.25 mM Calcein-AM (cat. no. C2012; Beyotime Institute of
Biotechnology). This reaction mixture was incubated for 30 min at
37°C and washed three times with the buffer aforementioned prior to
the resuspension of H9c2 cells. The fluorescence intensity of
Calcein-AM was quantified using a microplate reader (Synergy H4;
488 nm excitation and 525 nm emission; BioTek China). After the
baseline became stable, salicylaldehyde isonicotinoyl hydrazone
(SIH, 100 µM) which was synthetized by Schiff base
condensation between 2-hydroxybenzaldehyde and isonicotinic acid
hydrazide (Shanghai Nafu Biotechnology Co., Ltd.; https://nayuansu.company.lookchem.cn/)
was added. The reaction mixture was subsequently incubated for 30
min at 37°C before the fluorescence intensity was detected.
Calcein-AM itself has no fluorescence. After entering the H9c2
cells, it is hydrolyzed by endogenous esterase in the H9c2 cells to
produce Calcein, a polar molecule with strong negative charge that
cannot penetrate the cell membrane, which is retained in the cell
and emit strong green fluorescence. The increase in fluorescence
intensity indicates the level of Calcein-bound iron. The results
were presented as fluorescence intensity/mg protein.
Measurement of mitochondrial iron
Mitochondria were isolated from H9c2 cells
(1×106 cells/dish) using a Mitochondria Isolation kit
for Cultured Cells (Thermo Fisher Scientific, Inc.) according to
the manufacturer's protocol. The levels of iron in the mitochondria
was then determined using an Iron Colorimetric assay kit (cat. no.
BIV-K390-100; BioVision, Inc.) according to the manufacturer's
protocol. The absorbance was measured at 593 nm using a TECAN
infinite M200 microplate reader (Tecan Group, Ltd.).
5,5′,6,6′-Tetrachloro-1,1′,3,3′-tetraethylbenzimidazolocarbo-
cyanine iodide (JC-1) staining
Following JC-1 staining (cat. no. C2006; Beyotime
Institute of Biotechnology) at 37°C for 10 min, H9c2 cells
(5×104/well) were washed with the JC-1 staining buffer
three times and detected via flow cytometry (BD FACSCanto™; BD
Biosciences). Data were analyzed using FlowJo version ×0.7 (FlowJo
LLC). In the healthy mitochondria, JC-1 aggregated to form a
polymer in the mitochondrial matrix, which emits intense red
fluorescence (Excitation, 585 nm; Emission, 590 nm). In the
unhealthy mitochondria, JC-1 monomers presented in the cytoplasm
due to the decline/loss of mitochondrial membrane potential, which
generate green fluorescence (Excitation, 514 nm; Emission, 529 nm).
Therefore, changes in ratio of H1-UL (red)/H1-UR (green) reflected
the change in mitochondrial membrane potential.
ATP content
H9c2 cells (1×104 cells/well) were lysed
by ATP lysis buffer on ice, exposed to the ATP substrate solution
and cellular ATP content was determined by bioluminescent assay kit
(cat. no. ab113849; Abcam).
Statistical analysis
Data were repeated ≥ three times independently,
analyzed using GraphPad Prism software (version 6.01, GraphPad
Software, Inc.) and are presented as the mean ± SD. Groups were
compared using one-way ANOVA followed by Tukey's post hoc test.
P<0.05 was considered to indicate a statistically significant
difference.
Results
Herceptin induces cell injury and
oxidative stress in H9c2 cells
Herceptin (0, 0.2, 0.5, 1 and 10 µM) was
found to reduce H9c2 cell viability in a dose-dependent manner
(Fig. 1A). In addition, 10
µM Herceptin reduced H9c2 cell viability in a time-dependent
manner from 12 to 48 h (Fig.
1B). Herceptin (0, 0.2, 0.5, 1 and 10 µM) also
dose-dependently increased intracellular (Fig. 1C) and mitochondrial (Fig. 1E) ROS levels in H9c2 cells.
Furthermore, Herceptin (10 µM) increased intracellular
(Fig. 1D) and mitochondrial
(Fig. 1F) ROS levels in H9c2
cells from 12 to 48 h, peaking at 24 h.
Herceptin induces ferroptosis in H9c2
cells
Herceptin (0, 0.2, 0.5, 1 and 10 µM)
dose-dependently reduced the protein expression of GPX4 in H9c2
cells (Fig. 2A), whilst at 10
µM Herceptin also inhibited GPX4 protein expression in H9c2
cells in a time-dependent manner from 12 to 48 h (Fig. 2B). Herceptin (0, 0.2, 0.5, 1 and
10 µM) decreased GSH content (Fig. 2C) and increased the GSSG content
(Fig. 2D), thereby decreasing
the GSH/GSSG ratio in H9c2 cells in a dose-dependent manner
(Fig. 2E). Furthermore,
Herceptin at 10 µM decreased the GSH content (Fig. 2F) whilst increasing that of GSSG
(Fig. 2G), which resulted in the
reduction in the GSH/GSSG ratio in H9c2 cells in a time-dependent
manner from 12 to 48 h (Fig.
2H).
Fer-1 protects H9c2 cells against
Herceptin-induced cell injury and ferroptosis
Fer-1 treatment was found to reverse the
Herceptin-induced reduction in cell viability (Fig. 3A), restored GPX4 and SLC7A11
expression that was previously suppressed by Herceptin whilst
reversing the Herceptin-induced increase in ACSL14 expression
(Fig. 3B). Fer-1 was also
revealed to partially restore GSH production that was previously
suppressed by Herceptin without affecting the GSSG content, which
in turn lead to the reversal of the Herceptin-induced reduction in
GSH/GSSG ratio in H9c2 cells (Fig.
3C-E). Additionally, Fer-1 reversed the Herceptin-induced
increase in intracellular (Fig.
3F) and mitochondrial (Fig.
3G) iron levels in H9c2 cells. However, compared with those
mediated by DFO, the effects of Fer-1 were less potent.
 | Figure 3Fer-1 protects H9c2 cells against
Herceptin-induced cell injury and ferroptosis. Fer-1 and DFO
reversed the (A) Herceptin-induced reduction in cell viability, (B)
Herceptin-induced decrease in GPX4 and SLC7A11 protein expression
and Herceptin-induced increase in ACSL4 protein expression. Fer-1
and DFO reversed the Herceptin-induced (C) reduction in GSH
content. (D) Fer-1 and DFO did not affect GSSG content. (E) Fer-1
and DFO reversed the Herceptin-induced reduction in the ratio of
GSH/GSSG in H9c2 cells. Fer-1 and DFO reversed the
Herceptin-induced increase in (F) intracellular and (G)
mitochondrial iron levels in H9c2 cells. However, compared with
DFO, the effects of Fer-1 were less potent. **P<0.01
and ***P<0.001 vs. NC. #P<0.05 and
##P<0.01 vs. Herceptin (10 µM). Fer-1, ferrostatin-1;
GPX4, glutathione peroxidase 4; SLC7A11, recombinant solute carrier
family 7 member 11; ACSL4, acyl-CoA synthetase long chain family
member 4; GSH, reduced glutathione; GSSG, oxidized glutathione;
DFO, deferoxamine; OD, optical density. |
Fer-1 protects H9c2 cells from
Herceptin-induced mitochondrial dysfunction
Fer-1 was demonstrated to reverse the
Herceptin-induced reduction in ATP production (Fig. 4A). In addition, Fer-1 could
negate the Herceptin-mediated increases in mitochondrial ROS
production (Fig. 4B) whilst
reversing the Herceptin-induced decrease in mitochondrial membrane
potential (Fig. 4C). In terms of
protein expression, Herceptin increased Cyto c, VDAC2/3,
fis1, Drp1 and Mtf protein expression but decreased OPA1-1/2 and
Mfn1/2 expression (Fig. 4D), all
of which were reversed by Fer-1 co-treatment. However, compared
with DFO, the effects mediated by Fer-1 were not as potent.
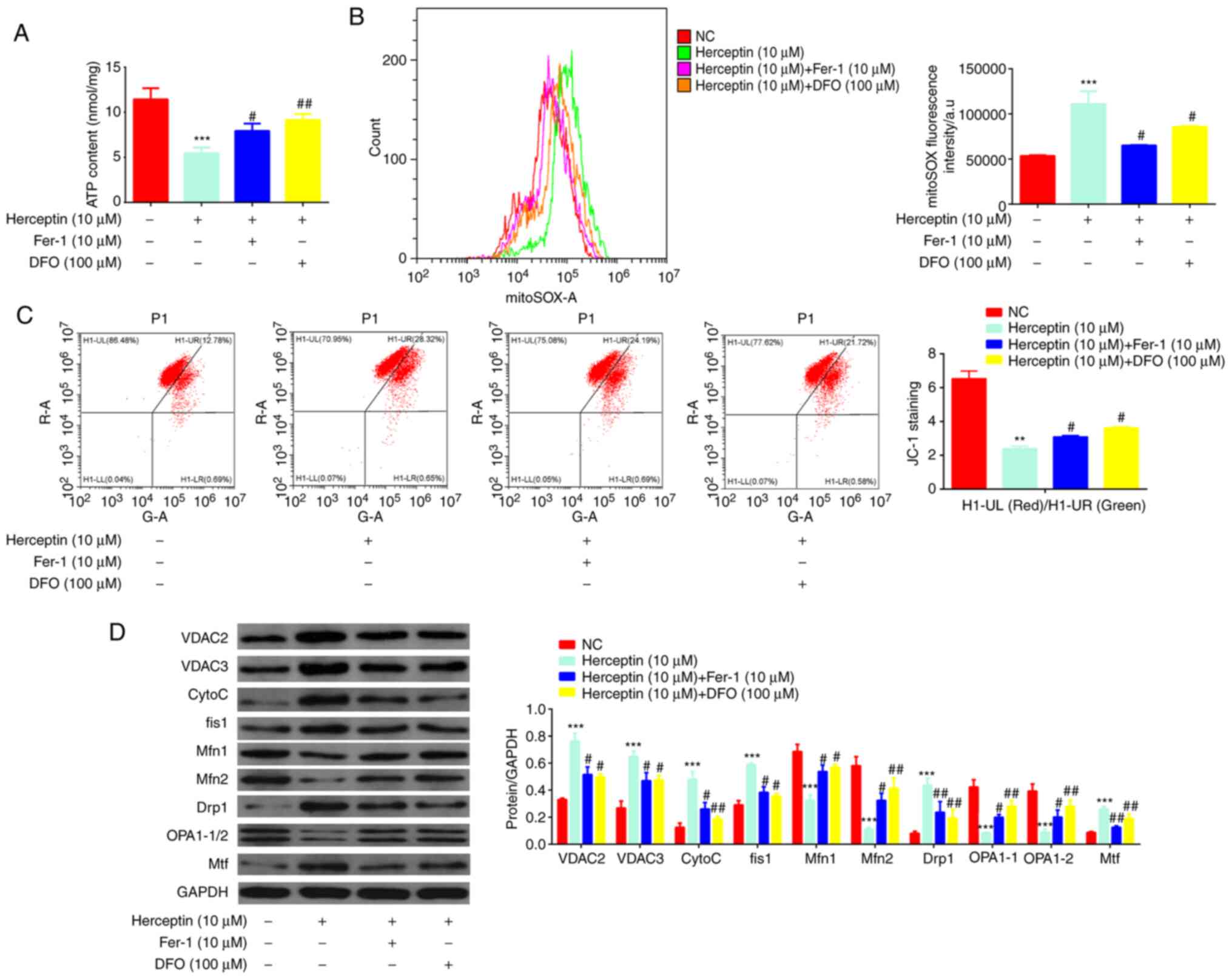 | Figure 4Fer-1 protects H9c2 cells from
Herceptin-induced mitochondrial dysfunction. (A) Fer-1 and DFO
reversed the Herceptin-induced reduction in ATP content. (B) Fer-1
and DFO reversed the Herceptin-induced increase in ROS levels. (C)
Fer-1 and DFO reversed the Herceptin-induced decrease in
mitochondrial membrane potential (UL, Red; UR, Green). (D) Fer-1
and DFO reversed the Herceptin-induced increase of Cyto c,
VDAC2/3, fis1, Drp1 and Mtf expression, whilst also reversing the
Herceptin-induced decrease in OPA1-1/2 and Mfn1/2 expression.
However, compared with those by DFO, the effects of Fer-1 were less
potent. **P<0.01 and ***P<0.001 vs. NC.
#P<0.05 and ##P<0.01 vs. Herceptin (10
µM). Fer-1, ferrostatin-1; ROS, reactive oxygen species;
DFO, deferoxamine; Cyto c, cytochrome C; VDAC2/3,
voltage-dependent anion-selective channels 2/3; fis1, mitochondrial
fission 1 protein; Drp1, dynamin-related protein 1; OPA1-1/2,
mitochondrial optic atrophy 1; Mfn1/2, Mitofusin 1/2; Mtf,
mitochondrial ferritin. |
Discussion
The present study discovered that Herceptin induced
injury, oxidative stress, mitochondrial dysfunction and ferroptosis
in H9c2 cells, which could be attenuated by Fer-1. Oxidative stress
has been frequently reported to mediate cellular and molecular
damage resulting from excessive ROS production caused by the
inhibition of antioxidant enzyme systems and/or insufficient
antioxidants production (27).
This can cause numerous diseases, including cardiovascular diseases
(27). The overproduction of ROS
leads to the dysregulation of cell cycle progression, breaking of
DNA strands and/or cell death (28). In the present study, it was
demonstrated that Herceptin reduced cell viability and increased
both intracellular and mitochondrial ROS generation in H9c2 cells.
This suggest that Herceptin induced cell injury and oxidative
stress in H9c2 cells, which is consistent with a previous report
that also showed the involvement of oxidative stress in
Herceptin-mediated cardiotoxicity (29).
Ferroptosis is characterized by the iron-dependent
accumulation of lipid peroxides to lethal levels (2). Excessive ROS accumulation can
induce damage in numerous cell types, which is associated with
decreased GSH content (30). GSH
is a water-soluble tripeptide that consists of the amino acid triad
of glutamine, cysteine and glycine, which exists in either the
oxidized GSSG state or the reduced GSH state inside the cell
(31). Therefore, it is crucial
to sustain optimal GSH/GSSG ratios in the cells for survival
(31). Mechanistically, GSH is a
key antioxidant enzyme that participates in the clearance of ROS
(32) and detoxification of
various electrophilic compounds and peroxides by glutathione
S-transferases and GPX through catalysis (31). In the present study, Herceptin
was observed to decrease GPX4 expression and the GSH/GSSG ratio in
H9c2 cells, suggesting that Herceptin induced ferroptosis in H9c2
cells, consistent with a previously published report that also
demonstrated that ferroptosis may mediate Herceptin-induced
cardiotoxicity (33).
Altered iron homeostasis results in excessive iron
deposition in the heart, leading to progressive tissue damage
(20). Excessive iron burden
promotes oxidative stress, which serves an important role in the
pathogenesis of iron overload-mediated heart disease (34), iron overload cardiomyopathy
(18,35), myocardial injury and heart
failure (36). However, to the
best of our knowledge, the association between Herceptin-induced
changes in iron homeostasis and ferroptosis in H9c2 cells has yet
to be elucidated. GPX4 and SLC7A11 acts as ferroptosis inhibitors
(37), GPX4 uses reduced
glutathione to convert lipid hydroperoxides to lipid alcohols,
thereby mitigating lipid peroxidation and inhibiting ferroptosis
(38). The majority of cells
obtain cysteine biologically through the import of extracellular
cystine transporter SLC7A11 (39). ACSL4 is a lipid metabolism enzyme
required for ferroptosis and can elevate lipid peroxidation and
ferroptosis (40). In detail,
inactivation of GPX4 or SLC7A11 through genetic or pharmacological
means has been previously found to induce ferroptosis (2). In addition, ferroptosis can be
attenuated by the inactivation of ACSL4 (41). Therefore these aforementioned
indicators of ferroptosis were detected in the present study. It
was demonstrated that Fer-1 treatment reversed the
Herceptin-induced reduction in H9c2 cell viability, reversed
Herceptin-mediated decrease in GPX4 and SLC7A11 protein expression
in addition to reversing the Herceptin-induced increase in ACSL4
protein expression. Furthermore, Fer-1 reversed the inhibitory
effects of Herceptin on the GSH/GSSG ratio in H9c2 cells, as well
as reversing the increase in intracellular and mitochondrial iron
levels in H9c2 cells induced by Herceptin. These findings suggest
that targeting ferroptosis can protect H9c2 cells from
Herceptin-induced cardiomyocyte injury and ferroptosis.
Oxidative stress can also result in mitochondrial
dysfunction (42). Since
reductions in the mitochondrial membrane potential is associated
with heart failure (43), the
present study also assessed the effects of Fer-1 on
Herceptin-induced mitochondrial dysfunction. Under physiological
conditions, adult cardiac tissues primarily rely on fatty acid
oxidation for the production of ATP, with ~70% ATP being generated
by fatty acid metabolism (44).
However, during heart failure, the overall production of ATP is
decreased (45). The present
study found that Fer-1 treatment restored mitochondrial membrane
potential and ATP production, both of which were previously
inhibited by Herceptin. In addition, Fer1-1 reversed the
Herceptin-induced increases in ROS production. Subsequently, the
expression levels of the associated proteins were also detected.
Drp1 (fission gene, which increases mitochondrial division), Fis1
(fission gene, which increases mitochondrial division), Mfn1/2
(fusion gene, which increases mitochondrial fusion and
mitochondrial connectivity) and Opa1-1/2 (fusion gene, which
increases mitochondrial fusion and mitochondrial connectivity) can
regulate, maintain and remodel mammalian mitochondria dynamics and
distribution (46). In addition,
mitochondrial release of cytochrome c is a hallmark of cell
death (47). ADP/ATP exchange is
realized by VDAC2/3 on the outer mitochondrial membrane (48). Iron loss triggers mitophagy by
induction of Mtf (49). It was
observed that Fer-1 reversed the Herceptin-induced reduction in
OPA1-1/2 and Mfn1/2 protein expression and Herceptin-induced
increase in Cyto c, VDAC2/3, fis1, Drp1 and Mtf protein
expression, suggesting that targeting ferroptosis protected H9c2
cells from Herceptin-induced mitochondrial dysfunction. However,
compared with DFO, the effects of Fer-1 were not as potent
effective.
Taken together, targeting ferroptosis can protect
the cardiomyocytes from Herceptin-induced toxicity in the present
H9c2 in vitro model. A previous study has shown that haploid
embryonic stem cells have been used for toxicology drug screening
(50), which may be useful for
finding additional ferroptosis inhibitors to protect myocardial
cells against chemotherapeutic drugs for cancer. The present study
proposed that iron-dependent ferroptosis is one of the pathological
processes underlying the development of Herceptin-induced
cardiomyopathy. Mechanistically, Herceptin contributed to the
release of free iron, which accumulated in the mitochondria.
Furthermore, targeting ferroptosis also protected H9c2 cells from
Herceptin-induced injury. Collectively, these findings provided
novel insights into the pathogenic mechanisms underlying iron
overload-induced cardiomyopathy and offer therapeutic targets for
the development of novel strategies.
However, several limitations remain in the present
study. There is a lack of in vivo evidence, rendering animal
experiments a necessity for future study to verify the present
findings, using techniques such as immunohistochemistry and
echocardiography. Additionally, only ferroptosis was focused upon
in the present study, such that other types of cell death,
including apoptosis or autophagy, were not detected. Simultaneous
observations of all four types of cell death should be detected in
future investigations.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
LS and QZ conceived the study, established the
initial design of the study and confirmed the authenticity of all
the raw data. LS, HW, SY, LZ and JJ performed the experiments and
analyzed the data. QZ prepared the manuscript. All authors read and
approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
Not applicable.
Funding
The present study was funded by the Natural Science Foundation
of Shaanxi Province (grant no. 2019JM-523) and the Fundamental
Research Funds for the Central Universities (grant no.
xzy012021059).
References
|
1
|
Fuchs Y and Steller H: Programmed cell
death in animal development and disease. Cell. 147:742–758. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Dixon SJ, Lemberg KM, Lamprecht MR, Skouta
R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS,
et al: Ferroptosis: An iron-dependent form of nonapoptotic cell
death. Cell. 149:1060–1072. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Angeli JPF, Shah R, Pratt DA and Conrad M:
Ferroptosis inhibition: Mechanisms and opportunities. Trends
Pharmacol Sci. 38:489–498. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Fang X, Wang H, Han D, Xie E, Yang X, Wei
J, Gu S, Gao F, Zhu N, Yin X, et al: Ferroptosis as a target for
protection against cardiomyopathy. Proc Natl Acad Sci USA.
116:2672–2680. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Linkermann A, Skouta R, Himmerkus N, Mulay
SR, Dewitz C, De Zen F, Prokai A, Zuchtriegel G, Krombach F, Welz
PS, et al: Synchronized renal tubular cell death involves
ferroptosis. Proc Natl Acad Sci USA. 111:16836–16841. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Yang WS and Stockwell BR: Ferroptosis:
Death by lipid peroxidation. Trends Cell Biol. 26:165–176. 2016.
View Article : Google Scholar :
|
|
7
|
Loibl S and Gianni L: HER2-positive breast
cancer. Lancet. 389:2415–2429. 2017. View Article : Google Scholar
|
|
8
|
Slamon D, Eiermann W, Robert N, Pienkowski
T, Martin M, Press M, Mackey J, Glaspy J, Chan A, Pawlicki M, et
al: Adjuvant trastuzumab in HER2-positive breast cancer. N Engl J
Med. 365:1273–1283. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Jerusalem G, Lancellotti P and Kim SB:
HER2+ breast cancer treatment and cardiotoxicity:
Monitoring and management. Breast Cancer Res Treat. 177:237–250.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Barish R, Gates E and Barac A:
Trastuzumab-induced cardiomyopathy. Cardiol Clin. 37:407–418. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Whelan RS, Kaplinskiy V and Kitsis RN:
Cell death in the pathogenesis of heart disease: Mechanisms and
significance. Annu Rev Physiol. 72:19–44. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Pantopoulos K, Porwal SK, Tartakoff A and
Devireddy L: Mechanisms of mammalian iron homeostasis.
Biochemistry. 51:5705–5724. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ward RJ, Zucca FA, Duyn JH, Crichton RR
and Zecca L: The role of iron in brain ageing and neurodegenerative
disorders. Lancet Neurol. 13:1045–1060. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Rouault TA: Iron metabolism in the CNS:
Implications for neurodegenerative diseases. Nat Rev Neurosci.
14:551–564. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
15
|
Reichert CO, de Freitas FA, Sampaio-Silva
J, Rokita-Rosa L, Barros PL, Levy D and Bydlowski SP: Ferroptosis
mechanisms involved in neurodegenerative diseases. Int J Mol Sci.
21:87652020. View Article : Google Scholar :
|
|
16
|
Torti SV and Torti FM: Iron and cancer:
More ore to be mined. Nat Rev Cancer. 13:342–355. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Chen Y, Fan Z, Yang Y and Gu C: Iron
metabolism and its contribution to cancer (Review). Int J Oncol.
54:1143–1154. 2019.PubMed/NCBI
|
|
18
|
Lapice E, Masulli M and Vaccaro O: Iron
deficiency and cardiovascular disease: An updated review of the
evidence. Curr Atheroscler Rep. 15:3582013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kobayashi M, Suhara T, Baba Y, Kawasaki
NK, Higa JK and Matsui T: Pathological roles of iron in
cardiovascular disease. Curr Drug Targets. 19:1068–1076. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wood JC: History and current impact of
cardiac magnetic resonance imaging on the management of iron
overload. Circulation. 120:1937–1939. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Valko M, Jomova K, Rhodes CJ, Kuča K and
Musílek K: Redox- and non-redox-metal-induced formation of free
radicals and their role in human disease. Arch Toxicol. 90:1–37.
2016. View Article : Google Scholar
|
|
22
|
Ichikawa Y, Ghanefar M, Bayeva M, Wu R,
Khechaduri A, Naga Prasad SV, Mutharasan RK, Naik TJ and Ardehali
H: Cardiotoxicity of doxorubicin is mediated through mitochondrial
iron accumulation. J Clin Invest. 124:617–630. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Gujja P, Rosing DR, Tripodi DJ and
Shizukuda Y: Iron overload cardiomyopathy: Better understanding of
an increasing disorder. J Am Coll Cardiol. 56:1001–1012. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kurokawa YK, Shang MR, Yin RT and George
SC: Modeling trastuzumab-related cardiotoxicity in vitro using
human stem cell-derived cardiomyocytes. Toxicol Lett. 285:74–80.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Baba Y, Higa JK, Shimada BK, Horiuchi KM,
Suhara T, Kobayashi M, Woo JD, Aoyagi H, Marh KS, Kitaoka H and
Matsui T: Protective effects of the mechanistic target of rapamycin
against excess iron and ferroptosis in cardiomyocytes. Am J Physiol
Heart Circ Physiol. 314:H659–H668. 2018. View Article : Google Scholar :
|
|
26
|
Wu C, Zhao W, Yu J, Li S, Lin L and Chen
X: Induction of ferroptosis and mitochondrial dysfunction by
oxidative stress in PC12 cells. Sci Rep. 8:5742018. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Xu T, Ding W, Ji X, Ao X, Liu Y, Yu W and
Wang J: Oxidative stress in cell death and cardiovascular diseases.
Oxid Med Cell Longev. 2019:90305632019. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Chen S, Zhang Z, Qing T, Ren Z, Yu D,
Couch L, Ning B, Mei N, Shi L, Tolleson WH and Guo L: Activation of
the Nrf2 signaling pathway in usnic acid-induced toxicity in HepG2
cells. Arch Toxicol. 91:1293–1307. 2017. View Article : Google Scholar :
|
|
29
|
Mohan N, Jiang J and Wu WJ: Implication of
autophagy and oxidative stress in trastuzumab-mediated cardiac
toxicities. Austin Pharmacol Pharm. 2:10052017.
|
|
30
|
Lee H, Ki J, Lee SY, Park JH and Hwang GS:
Processed panax ginseng, sun ginseng, decreases oxidative damage
induced by tert-butyl hydroperoxide via regulation of antioxidant
enzyme and anti-apoptotic molecules in HepG2 cells. J Ginseng Res.
3:248–255. 2012. View Article : Google Scholar
|
|
31
|
Anderson ME: Glutathione: An overview of
biosynthesis and modulation. Chem Biol Interact. 111-112:1–14.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Townsend DM, Tew KD and Tapiero H: The
importance of glutathione in human disease. Biomed Pharmacother.
57:145–155. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Ma W, Wei S, Zhang B and Li W: Molecular
mechanisms of cardiomyocyte death in drug-induced cardiotoxicity.
Front Cell Dev Biol. 8:4342020. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Oudit GY, Sun H, Trivieri MG, Koch SE,
Dawood F, Ackerley C, Yazdanpanah M, Wilson GJ, Schwartz A, Liu PP
and Backx PH: L-type Ca2+ channels provide a major
pathway for iron entry into cardiomyocytes in iron-overload
cardiomyopathy. Nat Med. 9:1187–1194. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Pennell DJ, Udelson JE, Arai AE, Bozkurt
B, Cohen AR, Galanello R, Hoffman TM, Kiernan MS, Lerakis S, Piga
A, et al: Cardiovascular function and treatment in beta-thalassemia
major: A consensus statement from the American heart association.
Circulation. 128:281–308. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Munzel T, Gori T, Keaney JF Jr, Maack C
and Daiber A: Pathophysiological role of oxidative stress in
systolic and diastolic heart failure and its therapeutic
implications. Eur Heart J. 36:2555–2564. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Lei G, Zhang Y, Koppula P, Liu X, Zhang J,
Lin SH, Ajani JA, Xiao Q, Liao Z, Wang H and Gan B: The role of
ferroptosis in ionizing radiation-induced cell death and tumor
suppression. Cell Res. 30:146–162. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Seibt TM, Proneth B and Conrad M: Role of
GPX4 in ferroptosis and its pharmacological implication. Free Radic
Biol Med. 133:144–152. 2019. View Article : Google Scholar
|
|
39
|
Koppula P, Zhang Y, Zhuang L and Gan B:
Amino acid transporter SLC7A11/xCT at the crossroads of regulating
redox homeostasis and nutrient dependency of cancer. Cancer Commun
(Lond). 38:122018. View Article : Google Scholar
|
|
40
|
Cheng J, Fan YQ, Liu BH, Zhou H, Wang JM
and Chen QX: ACSL4 suppresses glioma cells proliferation via
activating ferroptosis. Oncol Rep. 43:147–158. 2020.
|
|
41
|
Yuan H, Li X, Zhang X, Kang R and Tang D:
Identification of ACSL4 as a biomarker and contributor of
ferroptosis. Biochem Biophys Res Commun. 478:1338–1343. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Aon MA, Bhatt N and Cortassa SC:
Mitochondrial and cellular mechanisms for managing lipid excess.
Front Physiol. 5:2822014. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Kadenbach B, Ramzan R, Moosdorf R and Vogt
S: The role of mitochondrial membrane potential in ischemic heart
failure. Mitochondrion. 11:700–706. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Opie LH: Metabolism of the heart in health
and disease. I Am Heart J. 76:685–698. 1968. View Article : Google Scholar
|
|
45
|
Ventura-Clapier R, Garnier A and Veksler
V: Energy metabolism in heart failure. J Physiol. 555:1–13. 2004.
View Article : Google Scholar
|
|
46
|
Chen H and Chan DC: Mitochondrial
dynamics-fusion, fission, movement, and mitophagy-in
neurodegenerative diseases. Hum Mol Genet. 18:R169–R176. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Nguyen D, Alavi MV, Kim KY, Kang T, Scott
RT, Noh YH, Lindsey JD, Wissinger B, Ellisman MH, Weinreb RN, et
al: A new vicious cycle involving glutamate excitotoxicity,
oxidative stress and mitochondrial dynamics. Cell Death Dis.
2:e2402011. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Varikmaa M, Bagur R, Kaambre T, Grichine
A, Timohhina N, Tepp K, Shevchuk I, Chekulayev V, Metsis M, Boucher
F, et al: Role of mitochondria-cytoskeleton interactions in
respiration regulation and mitochondrial organization in striated
muscles. Biochim Biophys Acta. 1837:232–245. 2014. View Article : Google Scholar
|
|
49
|
Hara Y, Yanatori I, Tanaka A, Kishi F,
Lemasters JJ, Nishina S, Sasaki K and Hino K: Iron loss triggers
mitophagy through induction of mitochondrial ferritin. EMBO Rep.
21:e502022020. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Wang H, Zhang W, Yu J, Wu C, Gao Q, Li X,
Li Y, Zhang J, Tian Y, Tan T, et al: Genetic screening and
multipotency in rhesus monkey haploid neural progenitor cells.
Development. 145:dev1605312018. View Article : Google Scholar : PubMed/NCBI
|
















