Introduction
Chronic kidney disease (CKD) is a major public
health problem worldwide (1). The
global prevalence of CKD is 9.1% (1), with rates up to 15% in the USA and
9.2% in China (2,3), causing a high economic burden. Renal
tubulointerstitial fibrosis (TIF) is considered a common stage in
the progression of CKD to end-stage renal disease; it is
accompanied by inflammatory cell infiltration and excessive
deposition of extracellular matrix (ECM) in the renal interstitium
(4). Moreover, excessive
activation of the renin-angiotensin-aldosterone system (RAAS)
results in glomerular sclerosis and TIF, serving a pivotal role in
CKD progression (5). Aldosterone
(ALD) can affect the renal system and contribute to tissue
inflammation, injury, glomerulosclerosis and interstitial fibrosis
(6). Our previous studies have
shown that increased ALD levels promote podocyte damage, glomerular
sclerosis, renal tubular epithelial cell damage, renal interstitial
inflammation and fibrosis (7-9).
At present, the treatments for TIF, such as renal replacement
therapy, dialysis and kidney transplantation, are limited.
MicroRNAs (miRNAs) are highly conserved, endogenous,
single-stranded non-coding RNA molecules with a length of ~22 nt,
which can regulate target genes expression by degrading mRNAs or
inhibiting post-transcriptional translation (10). Moreover, >30% of protein
expression in mammals is regulated by miRNAs, including that of
most kidney fibrosis-related proteins (11). A number of studies have
demonstrated that several miRNAs, including miR-21, miR-26, miR-29,
miR-141 and miR-200, are associated with renal fibrotic processes
(12,13). Our previous study has shown that
miR-26a exerts an antifibrotic effect in obstructive kidney disease
(14). Additionally, miR-26a
serves a considerable role in angiotensin-II-induced cardiac
fibrosis (15). Moreover, it has
been confirmed that miR-26a is downregulated in fibrotic diseases
of the lung, lens and liver (16-18). These data suggest that miR-26a
could act as an ideal anti-fibrosis candidate molecule. However,
the effect of miR-26a on ALD-induced TIF remains unclear. Moreover,
circulating miRNAs are extremely unstable and can be easily
degraded by ribonucleases in the plasma (19). Coupled with the complex structure
of the kidney, whose damage can lead to functional problems, and
the variety of kidney cells, whose morphology, gene expressions and
physiological functions varies, the lack of high-efficiency media
to introduce miRNAs into the kidney cells is a bottleneck that is
difficult to overcome in this field (20-22). Thus, it is important to identify
new, safe and efficient gene therapy vectors for TIF.
Exosomes are cup-shaped microvesicles with a
diameter of ~30-100 nm that are secreted by almost all cell types
(23). Exosomes are found in
blood, cerebrospinal fluid, urine and other body fluids; they
deliver proteins, miRNAs, long non-coding RNAs and mRNAs produced
by cells in different states throughout the body (24). In recent years, exosomes have
received extensive attention owing to their role in communication
between cells and tissues (25).
The various mechanisms involved in exosome biogenesis affect the
content of exosomes, including various receptors, proteins, genetic
materials (DNA, mRNAs and miRNA) and lipids to target cells
(23). Furthermore, exosomes
derived from human endothelial cells deliver miR-486 to the kidneys
to reduce ischemic kidney injury (26). Thus, an exosome
encapsulation-based miRNA delivery system may be an attractive
candidate for manipulating miR-26a for treating TIF in CKD
(14).
In the present study, we hypothesized that the
exogenous delivery of exosome encapsulated (Exo)-miR-26a to injured
kidneys could ameliorate ALD-induced TIF. Moreover, results from
the current study may provide a basis for not only clinical
research on the mechanism of TIF in CKD but also the use of
exosomes for the treatment of CKD.
Materials and methods
Generation and isolation of exosomes with
miR-26a
When 293 cells (cat. no. CL-0005; Procell Life
Science &Technology Co., Ltd.) grew to 60-70% confluence, they
were transfected with miR-26a or empty vector adenoviruses
(ADV1-U6-GFP-mmu-miR-26a and ADV1-NC, respectively; Shanghai
GenePharma Co., Ltd.). The 293 cells were cultured in serum-free
DMEM containing 0.1% DiD far-red plasma membrane fluorescent probe
(cat. no. C1039; Beyotime Institute of Biotechnology) at 37°C for
20 min. After washed by PBS, the 293 cells were cultured in DMEM
containing 10% FBS and miR-26a or empty vector adenoviruses (final
titer 1×106 PFU/ml), and the exosomes were allowed to be
secreted at 37°C for 48 h. The supernatant of the cell culture
medium was then collected and centrifuged at 2,000 × g at 4°C for
20 min. The supernatant was collected and centrifuged again at
13,500 × g at 4°C for 20 min. The supernatant was harvested and
centrifuged at 200,000 × g at 4°C for 2 h. Finally, the miR-26a
enriched exosomes and empty vector exosomes were resuspended in
PBS. The morphology and size of the exosomes were analyzed using
transmission electron microscopy (TEM). The identity of the
exosomes was confirmed by western blot analysis of exosomal marker
proteins CD63, CD81 and Alix, as described below. Mice were
injected with exosomes through the caudal vein, as described below;
at the end of the study, kidneys were excised, and fluorescence
images were captured using the IVIS Spectrum in vivo Image
System (PerkinElmer, Inc.).
TEM
The exosome pellets were re-suspended in 50
µl 2% paraformaldehyde (density 4.9×107
particles/ml), and 5 µl of the suspension was placed on a
sheet of parafilm. A carbon-coated copper grid was floated on the
drop for 20 min at room temperature. The grid was washed by PBS for
3 min twice and excess liquid was drained by touching the grid edge
against a piece of clean filter paper. The grid was placed in 1%
glutaraldehyde at room temperature for 5 min. The grid was stained
with uranyl acetate (UA) dye solution with pH 7.0 at room
temperature for 10 min, and then coated in a methyl cellulose-UA
suspension on ice for 10 min. The grid was removed and excess
liquid was drained off. The grid was allowed to dry at room
temperature for 5-10 min and then observed under the electron
microscopy at 80 kilo electron volts.
Animal model
Male C57BL/6J mice (n=20; age 6-8 weeks; weight,
20-22 g) were purchased from the Beijing Vital River Laboratory
Animal Technology Co., Ltd., housed with five mice per cage and
maintained at 19-21°C on a 12-h light/dark cycle with free access
to food and water. Following 1 week of adaptive feeding, an osmotic
mini-pump was implanted subcutaneously in the interscapular region
to administer 1X PBS (100 µl) or ALD (100 µl at 0.75
µg/h) after mice were anesthetized using intraperitoneal
(i.p.) injection with the 50 mg/kg 1% pentobarbital sodium.
Subsequently, the mice were randomly divided into four groups (n=5
mice/group): i) sham group; ii) ALD group; iii) ALD + Exo-negative
control (NC) group; and iv) ALD + Exo-miR-26a group. All mice were
given 1% NaCl drinking water throughout the experimental period.
The ALD + Exo-miR-26a group mice received 100 µg Exo-miR-26a
or Exo-NC injected into the tail vein every 7 days starting the day
the mini-pump was implanted. At the end of the 28-day infusion
period, all mice were anaesthetized using 50 mg/kg 1% pentobarbital
sodium and euthanized by i.p. injection of pentobarbital sodium
(100 mg/kg). Death was confirmed by the following criteria:
Respiratory arrest, cardiac arrest, dilation of the pupils and
disappearance of nerve reflex. The mice were placed in a metabolic
cage for 24 h before euthanasia to obtain urine samples. A total of
100 µl blood was collected from the orbital venous sinus of
anesthetized mice, prior to euthanasia, at the Animal Experiment
Center of Nanjing Medical University. Kidneys were harvested after
mice were sacrificed. The study protocols were reviewed and
approved by the Institutional Animal Care and Use Committee of
Nanjing Medical University (ref. no. IACUC-2107049).
Cell culture
Mouse tubular epithelial cells (mTECs; cat. no.
CRL-3361) were purchased from the American Type Culture Collection
and cultured in DMEM containing 10% FBS and 5 mM glucose at 37°C
and 5% CO2. To determine the effects of ALD on mTECs,
cells were divided into control and ALD groups; equal numbers
(0.3×106 cells) of growth-arrested mTECs (preincubated
in serum-free DMEM for 6 h) were incubated in medium (with 10% FBS)
containing either PBS (control) or ALD (1×10−6 M) at
37°C for 48 h when the density reached 60-70%. Cells were harvested
at the end of the incubation period for further experiments. 293
cells were cultured in DMEM containing 10% FBS and 25 mM glucose at
37°C and 5% CO2; 293 cells were used for exosome
production and luciferase binding experiments.
Reverse transcription-quantitative PCR
(RT-qPCR)
To measure miRNA or mRNA expression levels, total
RNA was extracted from the kidney tissue (10 mg) and mTECs
(1×106 cells) using TRIzol® reagent
(Invitrogen; Thermo Fisher Scientific, Inc.) For miR-26a, 1,000 ng
of total RNA enriched in small RNAs was reverse transcribed using
the All-in-One MicroRNA Assay kit (iGene Biotechnology Co., Inc.).
The mouse U6 gene was used as the loading control. For mRNA
expression, cDNA was synthesized using HIScript III RT SuperMix
(Vazyme Biotech Co., Ltd.); β-actin was used as an internal
reference. All primers were purchased from GeneRay Biotech Co.,
Ltd. Primer sequences are listed in Table I; a reverse primer was used for
miR-26a. miRNA and mRNA expression levels were calculated as the
difference between the threshold values of the control and
experimental genes using the 2−ΔΔcq method (27).
 | Table IPrimer sequences used for reverse
transcription-quantitative PCR. |
Table I
Primer sequences used for reverse
transcription-quantitative PCR.
| Gene | Primer sequence
(5′-3′) |
|---|
| β-actin | F:
CATCCGTAAAGACCTCTATGCCAAC |
| R:
ATGGAGCCACCGATCCACA |
| Collagen I | F:
GTCAGACCTGTGTGTTCCCTACTCA |
| R:
TCTCTCCAAACCAGACGTGCTTC |
| α-SMA | F:
CAGCAAACAGGAATACGACGAA |
| R:
AACCACGAGTAACAAATCAAAGC |
| LCN2 | F:
GCCCTGAGTGTCATGTGTCT |
| R:
GAACTGATCGCTCCGGAAGT |
| CTGF | F:
GGGCCTCTTCTGCGATTTC |
| R:
ATCCAGGCAAGTGCATTGGTA |
| miR-26a | F:
ACACTCCAGCTGGGTTCAAGTAATCCAGGA |
| R:
TGGTGTCGTGGAGTCG |
| U6 | F:
CTCGCTTCGGCAGCACATATACT |
| R:
ACGCTTCACGAATTTGCGTGTC |
Western blot
Western blot was performed as described previously
(14). RIPA lysis buffer (cat.
no. P0013B; Beyotime Institute of Biotechnology) and PMSF protease
inhibitor (cat. no. ST506; Beyotime Institute of Biotechnology)
were added to the kidney tissue (10 mg; crushed using a tissue
grinder) and mTECs (1×106 cells) at a ratio of 100:1.
The suspension was placed on ice, and tissues and cells were lysed
for 1 h. The suspension was centrifuged at 12,000 × g at 4°C for 30
min. The supernatant was collected, and the protein concentration
was then measured using enhanced BCA protein assay kit (cat. no.
P0010; Beyotime Institute of Biotechnology). Proteins (50
µg) were separated by 10% SDS-PAGE and transferred to PVDF
membranes (MilliporeSigma; cat. no. HATF09025). The PVDF membranes
were incubated with the primary antibodies at 4°C overnight; the
primary antibodies (all 1:1,000) used are as follows: Anti-α-smooth
muscle actin (α-SMA; cat. no. BF9212; Affinity Biosciences, Ltd.),
collagen I (cat. no. ab34710; Abcam), connective tissue growth
factor (CTGF; cat. no. ab6992; Abcam), lipocalin 2 (LCN2; cat. no.
DF6816; Affinity Biosciences, Ltd.), E-cadherin (cat. no. AF0131;
Affinity Biosciences, Ltd.), SMAD3 (cat. no. AF6362; Affinity
Biosciences, Ltd.), phosphorylated (p)-SMAD3 (cat. no. AF3362;
Affinity Biosciences, Ltd.), CD63 (cat. no. sc5275, Santa Cruz
Biotechnology, Inc.), CD81 (cat. no. P35762; Cell Signaling
Technology, Inc.), Alix (cat. no. sc53540; Santa Cruz
Biotechnology, Inc.), β-tubulin (cat. no. T0023; Affinity
Biosciences, Ltd.) and GAPDH (cat. no. T0004; Affinity Biosciences,
Ltd.). The PVDF membranes were incubated with the secondary
antibodies at room temperature for 1 h; the secondary antibodies
(all 1:2,000) used were: HRP-conjugated goat anti-mouse (cat. no.
L3032; Signalway Antibody LLC) and HRP-conjugated goat anti-rabbit
(cat. no. L3012; Signalway Antibody LLC). Protein bands were
visualized using an enhanced chemiluminescence reagent (Immobilon
Western HRP Substrate; cat. no. WBKLS0100; Merck KGaA) and a gel
imaging analysis system. ImageJ software 1.8.0 (National Institutes
of Health) was used to semi-quantify expression of proteins
normalized to GAPDH or β-tubulin.
Cell transfection
The miR-26a mimic, miR-26a inhibitor, CTGF small
interfering (si)RNA, CTGF overexpression (oe) plasmids
[pEX-3(pGCMV/MCS/Neo)], and their respective NCs were obtained from
Shanghai GenePharma Co., Ltd.; the sequences are listed in Table II. mTECs or 293 cells were
maintained in 6- or 12-well plates before transfection. When the
cell density reached 30-50%, they were transfected with
Lipofectamine® 2000 (cat. no. 11668019; Thermo Fisher
Scientific, Inc.) according to the manufacturer's instructions. For
the co-transfection of oe-CTGF plasmids and miR-26a mimic (or
si-CTGF and miR-26a inhibitor), plasmids (100 ng) and miR-26a mimic
(20 µM), si-CTGF (20 µM) or miR-26a inhibitor (20
µM) were used in serum-reduced medium (Opti-MEM). The cells
were incubated with the mimic or inhibitor (final concentration 50
nM)/Lipofectamine mixture at 37°C for 6 h, and then the Opti-MEM
medium was replaced with a complete medium. For single
transfections, oe-CTGF plasmids (100 ng), miR-26a mimic (20
µM), si-CTGF (20 µM) or miR-26a inhibitor (20
µM) was used in serum-reduced medium (Opti-MEM). The cells
were incubated with the mimic (or inhibitor or si-CTGF or
plasmids)/Lipofectamine mixture at 37°C for 6 h, and then the
Opti-MEM medium was replaced with a complete medium. After 24-48 h,
cells were collected for further analysis. The viability of cells
after treating with siRNA and inhibitors is presented in Fig. S1.
| Table IISequences of the RNA oligonucleotides
used for cell transfection. |
Table II
Sequences of the RNA oligonucleotides
used for cell transfection.
| Name | Sequence
(5′-3′) |
|---|
| miR-26a mimic | Sense:
UUCAAGUAAUCCAGGAUAGGCU |
| Antisense:
CCUAUCCUGGAUUACUUGAAUU |
| NC mimic | Sense:
UUCUCCGAACGUGUCACGUTT |
| Antisense:
ACGUGACACGUUCGGAGA ATT |
| miR-26a
inhibitor | Sense:
AGCCUAUCCUGGAUUACUUGAA |
| NC inhibitor | Sense:
CAGUACUUUUGUGUAGUACAA |
| CTGF siRNA | Sense:
CCCUGUACUACAGGAAGAUTT |
| Antisense:
AUCUUCCUGUAGUACAGGGTT |
Masson's trichrome staining
Fresh kidney tissue was fixed in 4% paraformaldehyde
at room temperature for 24 h, then soaked in 75% ethanol overnight.
The tissues were dehydrated in an ascending ethanol gradient. Then,
the tissues were cleared with xylene, and the transparent tissues
were immersed in paraffin; the embedded wax blocks were
subsequently sectioned to a thickness of 3 µm. After the
sections were straightened and flattened in a 40°C water bath, they
were collected by anti-slip slides and baked at 65°C overnight. The
sections were stained with hematoxylin at room temperature for 5-10
min and rinsed with water. The tissues were differentiated with an
ethanol hydrochloric acid differentiation solution and stained with
vincristine 3R at room temperature for 5-10 min. Next, the sections
were immersed in 1% phosphomolybdate solution for about 5 min at
room temperature until the collagen fibers or background were
colorless and the myelin sheath was red. Sections were then
counterstained with aniline blue for 5 min at room temperature,
soaked in 1% acetic acid at room temperature for 1 min, and then
dehydrated with 95% ethanol. The sections were dehydrated with
absolute ethanol, cleared with xylene for 10 min at room
temperature, and sealed with neutral resin. Finally, the sections
were observed under an inverted light microscope.
Immunofluorescence
Paraffin-embedded sections of the mouse kidney were
prepared as aforementioned. After baking at 65°C for 2 h, the
sections were dewaxed in xylene I (20 min) and xylene Ⅱ (20 min) at
room temperature, and then rehydrated through a descending series
of ethanol solutions (100-70%) at room temperature for 5 min
respectively. Antigen retrieval was followed by boiling in citrate
buffer and cooling to room temperature. After blocking with 5% BSA
(cat. no. 4240; Guangzhou Saiguo Biotech Co., Ltd.) for 1 h, the
primary antibody was added dropwise, and the sections were
incubated at 4°C overnight. Sections were then stained with
secondary antibodies for 1 h at room temperature, and the nuclei
were labeled with DAPI at room temperature for 10 min. The sections
in NC group were only incubated with secondary antibodies. The
primary antibodies (all 1:200 in 1% BSA) used were: Anti-E-cadherin
(cat. no. AF0131; Affinity Biosciences, Ltd.), anti-fibronectin
(cat. no. AF5335; Affinity Biosciences, Ltd.), anti-α-SMA (cat. no.
BF9212; Affinity Biosciences, Ltd.); and the secondary antibodies
(all 1:500, diluted with 1%BSA) used were: Alexa Fluor
647-conjugated goat anti-mouse IgG (cat. no. S0014; Affinity
Biosciences, Ltd.) and Alexa Fluor® 488-conjugated goat
anti-rabbit IgG (cat. no. ab150077; Abcam). After sealing the
tissues with coverslips and an antifade mounting medium (cat. no.
P0126; Beyotime Institute of Biotechnology), the sections were
observed under a fluorescence microscope.
Luciferase reporter assay
Previous studies have confirmed that CTGF is the
downstream target of miR-26a (14,15,28); thus, the targeting relationship
between them was verified in the present study. As CTGF was a
predicted target of miR-26a, luciferase reporter constructs in
which the luciferase coding sequence was fused to the 3′-UTRs of
CTGF (pLuc.miR-26a-CTGF-3′-UTR) were generated by Shanghai
GenePharma Co., Ltd. The wild-type (WT; 100 ng) or mutant (MUT; 100
ng) plasmids were co-transfected with NC mimic (20 µM) or
miR-26a mimic (20 µM) into 293 with Lipofectamine 2000
transfection reagent when the cells reached 30-50% density
(0.1×106 cells). After transfection at 37°C for 48 h,
the cells were harvested and lysed, and the luciferase activity was
measured using a dual luciferase reporter gene assay kit (Shanghai
GenePharma Co., Ltd.). Firefly luciferase activities were
normalized to Renilla luciferase activities.
Cell Counting Kit-8 (CCK-8)
Equal numbers of mTECs or 293 (5,000 cells/well)
were inoculated into a 96-well plate and incubated in DMEM
containing 10% FBS at 37°C and 5% CO2 for 24 h. siRNA
(10 µl) and miR-26a inhibitors (50 nM) were added to the
plate, and the cells were incubated at 37°C and 5% CO2
for 48 h. Subsequently, 10 µl of CCK-8 solution (cat. no.
K1018; APExBIO Technology LLC) was added to each well, and the
plate was incubated at 37°C and 5% CO2 for 1-4 h. The
absorbance of the plate at 450 nm was measured using a microplate
reader.
Serum creatinine (sCr) test
The collected blood was centrifuged at 956 × g at
4°C for 10 min. The supernatant was harvested for the sCr test
using a creatinine assay kit (sarcosine oxidase) (cat. no.
C011-2-1; Nanjing Jiancheng Bioengineering Institute). A total of 6
µl serum (test group), creatinine standard (standard group)
or ddH2O (blank group) were added to individual wells of
96-well plate. Enzyme solution (180 µl) was then added to
each well and the plate was then incubated at 37°C for 5 min. The
absorbance (A1) of the plate at 546 nm was then measured using a
microplate reader. The plate was incubated at 37°C for 5 min after
adding 60 µl of enzyme solution B to each well, and the
absorbance (A2) at 546 nm was measured again. The sCr content of
samples were calculated according to the calculation formula in the
protocol.
Statistical analysis
Data are presented as the mean ± standard deviation
of at least three independent experiments. Unpaired Student's
t-test was used to compare two groups; one-way ANOVA followed by
Tukey's post hoc test was used to compare the data from more than
two groups. Statistical analyses were assessed with the SPSS v25
statistical software package (IBM Corp). P<0.05 was considered
to indicate a statistically significant difference.
Results
miR-26a downregulation is associated with
TIF in ALD- induced mice
To explore whether ALD can cause renal fibrosis,
mice were randomly assigned to receive PBS or ALD treatment for 4
weeks. TIF was detected using Masson's trichrome staining, which
showed obvious fibrosis in the ALD group (Fig. 1A). sCr levels were also higher in
the ALD group compared with levels in the sham group (Table III). Compared with the sham
group, miR-26a expression levels were significantly reduced in the
kidney tissue of mice in the ALD group (Fig. 1B). The mRNA expression levels of
collagen I, α-SMA and the tubular injury marker LCN2 were higher in
the kidney tissues of mice in the ALD group compared with levels in
the sham group (Fig. 1C). Western
blotting results revealed that ALD treatment increased the protein
expression levels of collagen I, α-SMA, CTGF and LCN2 (Fig. 1D). Immunofluorescence results
showed that the expression of α-SMA and fibronectin were notably
increased in the kidneys of mice in the ALD group (Fig. 1E). Conversely, E-cadherin
expression was lower in ALD-induced mice compared with the sham
group (Fig. 1D and E). The
immunofluorescence NC experiments shown in Fig. S2A demonstrate no notable differ-
ence between NC in Fig. S2A and
the sham group in Fig. 1E. These
results suggested that ALD-induced TIF may be related to reduced
miR-26a expression levels.
 | Figure 1miR-26a expression is downregulated
in the kidneys of ALD-induced mice. (A) Representative images of
Masson's trichrome stained kidney tissues of mice in the sham and
ALD groups. Scale bar, 50 µm. (B) RT-qPCR analysis of
miR-26a expression levels in the kidney tissues of mice in the sham
and ALD groups; U6 was used for normalization. (C) RT-qPCR analysis
of collagen I, α-SMA and LCN2 mRNA expression levels in the kidney
tissues of mice from the sham and ALD groups; β-actin was used for
normalization. (D) Representative western blotting images and
semi-quantitative analysis of E-cadherin, collagen I, α-SMA, CTGF
and LCN2 protein expression levels in the kidney tissue of mice in
the sham and ALD groups. (E) Immunohistochemical analysis of
E-cadherin (green), α-SMA (red) and fibronectin (green) in the
kidney tissue of mice in the sham and ALD groups; DAPI (blue) was
used to stain the nuclei. Scale bar, 50 µm. Data are
presented as the mean ± SD; n=5 mice/group; *P<0.05
vs. sham. α-SMA, α-smooth muscle actin2; CTGF, connective tissue
growth factor; LCN2, lipocalin; RT-qPCR, reverse
transcription-quantitative PCR. |
| Table IIIsCr levels in ALD-induced model
mice. |
Table III
sCr levels in ALD-induced model
mice.
| Group | sCr,
µmol/l |
|---|
| Sham | 17.09±6.95 |
| ALD | 58.49±11.81a |
| ALD + Exo-NC | 57.06±12.75 |
| ALD +
Exo-miR-26a | 30.24±11.19b |
Exo-miR-26a alleviates TIF in mice
To characterize the exosomes derived from 293 cells,
their morphology and size were examined by TEM, which showed a
lipid bilayer structure with a diameter of about 100 nm (Fig. 2A). The identity of the exosomes
was confirmed by western blot analysis, which showed that the
protein expression levels of the exosomal marker proteins CD63,
CD81 and Alix in the 293-Exo group was increased compared with that
in the 293-cell group, confirming that exosomes were harvested from
the supernatant (Fig. 2B).
Moreover, RT-qPCR analysis revealed that compared with that in the
exosomes extracted from 293 cells transfected with the empty
vector, the expression level of miR-26a in the exosomes of the
Exo-miR-26a group was increased (Fig.
2C).
 | Figure 2Exo-miR-26a alleviates ALD-induced
renal fibrosis in vivo. (A) Transmission electron micrograph
of exosomes extracted by ultra-high-speed differential
centrifugation. Scale bar, 200 nm. (B) Representative western
blotting images and semi-quantitative analysis of Alix, CD63 and
CD81 protein levels in the exosomes. The protein of 293-cell group
is the protein extracted from the adherent cells in the lower
layer, the protein of 293-Exo group is the protein extracted from
the exosomes isolated from the supernatant. (C) RT-qPCR analysis of
miR-26a expression in the exosomes. Data are presented as the mean
± SD; *P<0.05 vs. Exo-NC. (D) DiD fluorescence
imaging of exosomes in excised kidneys. The sham mice (left) were
not injected and the ALD mice (right) were injected with exosomes
through the caudal vein. (E) RT-qPCR analysis of miR-26a in the
kidney of mice in the sham, ALD, ALD + Exo-NC and ALD + Exo-miR-26a
groups; U6 was used for normalization. (F) Representative images of
Masson's trichrome staining in the kidney tissues of mice in the
different groups. Scale bar, 50 µm. (G) RT-qPCR analysis of
collagen I, α-SMA and LCN2 mRNA expression levels in the kidney
tissues of mice in the different groups; β-actin was used for
normalization. (H) Representative western blotting images and
semi-quantitative analysis of E-cadherin, collagen I, α-SMA, CTGF
and LCN2 protein expression levels in the kidney tissues of mice
from each group. Data are presented as the mean ± SD; n=5
mice/group; *P<0.05 vs. sham; #P<0.05
vs. ALD + Exo-NC. (I) Immunofluorescence analysis of E-cadherin
(green), α-SMA (red) and fibronectin (green) in the kidney tissue
of mice in the sham, ALD, ALD + Exo-NC and ALD + Exo-miR-26a
groups; DAPI (blue) was used to stain the nuclei. Scale bar, 50
µm. α-SMA, α-smooth muscle actin; ALD, aldosterone; CTGF,
connective tissue growth factor; Exo, exosome encapsulated; LCN2,
lipocalin 2; miR, microRNA; NC, negative control; RT-qPCR, reverse
transcription-quantitative PCR. |
ALD-treated mice were injected with Exo-miR-26a or
Exo-NC through the caudal vein. DiD fluorescence imaging results
showed that the concentration of exosomes in the excised kidneys of
mice injected with Exo-miR-26a was higher compared with that in the
sham group, confirming that exosomes can deliver miR-26a into the
kidneys (Fig. 2D). The results of
RT-qPCR analysis showed that miR-26a expression levels in the
kidneys of mice in the ALD + Exo-miR-26a group was significantly
higher compared with that in the ALD and ALD + Exo-NC groups,
whereas there was no significant difference between the ALD and ALD
+ Exo-NC groups (Fig. 2E).
Masson's trichrome staining was used to determine
the degree of TIF. The results of the pathological examination of
the mouse kidney sections indicated that collagen deposition in the
renal interstitium of mice in the ALD + Exo-miR-26a group was
reduced compared with that in the ALD group, whereas that in the
ALD + Exo-NC group showed no difference with the ALD-only group
(Fig. 2F). Compared with the ALD
group, the sCr levels of the mice in the ALD + Exo-miR-26a group
were significantly decreased, whereas those in the ALD + Exo-NC
group was similar to that of the ALD group (Table III).
Moreover, the RT-qPCR results showed that the mRNA
expression levels of collagen I, α-SMA and LCN2 in the kidney
tissue of mice in the ALD + Exo-miR-26a group was decreased
compared with that in the ALD and ALD + Exo-NC groups (Fig. 2G); no significant difference was
detected between the ALD and ALD + Exo-NC groups (Fig. 2G). Western blot analysis revealed
that compared with the ALD and ALD + Exo-NC groups, the ALD +
Exo-miR-26a group exhibited significantly lower expression levels
of collagen I, α-SMA, CTGF and LCN2 in the kidneys (Fig. 2H). Immunofluorescence results
showed that α-SMA and fibronectin expression levels were notably
reduced in the ALD + Exo-miR-26a group compared with those in the
ALD group (Fig. 2I), whereas
E-cadherin expression showed the opposite trend (Fig. 2H and I). The immunofluorescence
NCs are shown in Fig. S2B; there
was no notable difference between NC in Fig. S2B and sham in Fig. 2I. These data suggested that
miR-26a overexpression may reduce TIF in mice.
miR-26a overexpression alleviates
ALD-induced epithelial-mesenchymal transition (EMT) and ECM
deposition in mTECs
To explore whether ALD induces EMT and ECM
deposition in mTECs, the cells were divided into two groups treated
with either PBS or ALD for 48 h. The miR-26a expression level was
determined by RT-qPCR, which revealed a decrease in the ALD group
compared with the control (Fig.
3A). Moreover, the mRNA expression levels of collagen I, α-SMA
and LCN2 in the control group were significantly lower compared
with those in the ALD group (Fig.
3B). The results of western blot analysis revealed that
collagen I, α-SMA, CTGF and LCN2 protein expression levels in the
ALD group was upregulated compared with that in the control group,
whereas the E-cadherin expression exhibited the opposite trend
(Fig. 3C). Immunofluorescence
results revealed that the α-SMA expression level in the ALD group
was higher compared with that in the control group (Fig. 3G). These results suggested that
ALD may induce EMT and ECM deposition in mTECs in vitro.
 | Figure 3miR-26a overexpression alleviates
ALD-induced EMT and ECM deposition in mTECs. RT-qPCR analysis of
(A) miR-26a expression levels and (B) collagen I, α-SMA and LCN2
mRNA expression levels in mTECs treated with buffer (control) or
ALD (1×10−6 M) for 48 h. Results were normalized to
β-actin. (C) Representative western blotting images and
semi-quantitative analysis of E-cadherin, collagen I, α-SMA, CTGF
and LCN2 protein expression levels in control- and ALD-treated
mTECs. RT-qPCR analysis of (D) miR-26a expression and (E) collagen
I, α-SMA and LCN2 mRNA expression levels in mTECs treated with
buffer (control), ALD, ALD + Exo-NC and ALD + Exo-miR-26a. Results
were normalized to β-actin. (F) Representative western blotting
images and semi-quantitative analysis of E-cadherin, collagen I,
α-SMA, CTGF and LCN2 protein expression levels in the variously
treated mTECs. (G) Immunofluorescence analysis of α-SMA (red) in
mTECs treated with buffer (control), ALD, ALD + Exo-NC or
ALD+Exo-miR-26a; DAPI (blue) was used to stain the nuclei. Scale
bar, 50 µm. Data are presented as the mean ± SD;
*P<0.05 vs. Control; #P<0.05 vs. ALD +
Exo-NC. A-SMA, α-smooth muscle actin; ALD, aldosterone; CTGF,
connective tissue growth factor; ECM, extracellular matrix; EMT,
epithelial-mesenchymal transition; Exo, exosome encapsulated; LCN2,
lipocalin 2; miR, microRNA; mTEC, mouse tubular epithelial cells;
NC, negative control; RT-qPCR, reverse transcription-quantitative
PCR. |
The role of miR-26a in EMT and ECM deposition was
further investigated in mTECs treated with Exo-miR-26a. RT-qPCR
results showed that the miR-26a expression levels were
significantly increased in the ALD + Exo-miR-26a group compared
with that in the ALD + Exo-NC group (Fig. 3D). Furthermore, the results from
RT-qPCR and western blot analysis confirmed that compared with the
ALD + Exo-NC group, Exo-miR-26a co-treatment inhibited the mRNA and
protein expression levels, respectively, of collagen I, α-SMA, CTGF
and LCN2 (Fig. 3E and F).
However, E-cadherin protein expression level was higher in the ALD
+ Exo-miR-26a group compared with that in the ALD + Exo-NC group
(Fig. 3F). The results of
immunofluorescence showed that Exo-miR-26a co-treatment notably
decreased the expression of α-SMA in comparison with that in the
ALD + Exo-NC group (Fig. 3G). The
immunofluorescence NC shown in Fig.
S2C indicates no difference compared with control group in
Fig. 3G. Taken together, miR-26a
overexpression reduced ALD-induced EMT and ECM deposition in
mTECs.
miR-26a directly targets CTGF and
negatively regulates its expression
Previous studies have confirmed that CTGF is the
downstream target of miR-26a (14,15,28); the target site between miR-26a and
3′UTR of CTGF is shown in Fig.
4A, and it was verified using the luciferase reporter assay in
293 cells. Luciferase activity was decreased in response to
co-transfection of miR-26a mimic and WT-CTGF-3′UTR, whereas no
changes were observed in the cells treated with miR-26a mimic and
MUT-CTGF-3′UTR (Fig. 4B). To
further verify that miR-26a inhibits CTGF, miR-26a mimic or miR-26a
inhibitor was co-transfected into 293 cells with oe-CTGF or CTGF
siRNA, respectively, and the expression levels of miR-26a and CTGF
were determined using RT-qPCR and western blot analysis. The
results revealed a significant increase in miR-26a expression in
oe-NC + miR-26a mimic group compared with oe-NC + NC mimic group,
and in oe-CTGF + miR-26a mimic group compared with oe-CTGF + NC
mimic group, whereas the miR-26a expression was decreased in
oe-CTGF + NC mimic group compared with oe-NC + NC mimic group
(Fig. 4C). RT-qPCR and western
blotting analyses showed that the CTGF mRNA and protein expression
levels, respectively, were decreased in the group transfected with
oe-NC + miR-26a mimic compared with the group transfected with
oe-NC + NC mimic, and in the group transfected with oe-CTGF +
miR-26a mimic compared with the group transfected with oe-CTGF + NC
mimic, whereas it was increased in the group transfected with
oe-CTGF + NC mimic compared with the group transfected with oe-NC +
NC mimic (Fig. 4D and E).
Moreover, miR-26a expression was reduced in si-NC + miR-26a
inhibitor group compared with si-NC + NC inhibitor group, and in
si-CTGF + miR-26a inhibitor group compared with si-CTGF + NC
inhibitor group, whereas it showed the opposite trend in si-CTGF +
NC inhibitor group compared with si-NC + NC inhibitor group
(Fig. 4F). CTGF mRNA and protein
expression levels were upregulated in the group transfected with
si-NC + miR-26a inhibitor compared with the group transfected with
si-NC + NC inhibitor, and in the group transfected with si-CTGF +
miR-26a inhibitor compared with the group transfected with si-CTGF
+ NC inhibitor, whereas it showed the opposite trend in the group
transfected with si-CTGF + NC inhibitor compared with the group
transfected with si-NC + NC inhibitor (Fig. 4G and H). These results further
confirmed that miR-26a may attenuate TIF by inhibiting CTGF
expression in mTECs.
 | Figure 4miR-26a directly targets CTGF and
negatively regulates its expression. (A) miR-26a target site in the
3′UTR of CTGF. (B) Relative luciferase activity in 293 cells
co-transfected with plasmids containing CTGF-WT or -Mut 3′-UTR and
miR-26a mimic or NC mimic for 48 h. *P<0.05 vs. NC
mimic. RT-qPCR analysis of (C) miR-26a expression and (D) CTGF mRNA
expression levels in 293 cells co-transfected with oe-CTGF or oe-NC
and miR-26a mimic or NC mimic. Results were normalized to U6 or
β-actin, respectively. (E) Representative western blotting images
and semi-quantitative analysis of CTGF protein expression levels in
293 cells co-transfected with oe-CTGF or oe-NC and miR-26a mimic or
NC mimic. RT-qPCR analysis of (F) miR-26a expression and (G) CTGF
mRNA expression levels in 293 cells co-transfected with si-CTGF or
si-NC and miR-26a inhibitor or NC inhibitor. Results were
normalized to U6 or β-actin, respectively. (H) Representative
western blotting images and semi-quantitative analysis of CTGF
protein levels in 293 cells co-transfected with si-CTGF or si-NC
and miR-26a inhibitor or NC inhibitor. Data are presented as the
mean ± SD; *P<0.05, **P<0.01,
***P<0.001; #P<0.05,
##P<0.01. CTGF, connective tissue growth factor; miR,
microRNA; Mut, mutant; NC, negative control; oe, overexpression;
RT-qPCR, reverse transcription-quantitative PCR; si, small
interfering RNA; UTR, untranslated region; WT, wild-type. |
CTGF knockdown alleviates ALD-induced EMT
and ECM deposition in mTECs
To further elucidate the role of CTGF in EMT and ECM
deposition, ALD-treated mTECs were transfected with si-CTGF. After
transfection with si-CTGF, miR-26a expression increased (Fig. 5A). In addition, the results of
RT-qPCR and western blot analysis revealed that the mRNA and
protein expression levels, respectively, of collagen I, α-SMA and
LCN2 were reduced in the ALD + si-CTGF group compared with those in
the ALD group (Fig. 5B and C);
the protein expression of CTGF showed the same trend (Fig. 5C), whereas the E-cadherin protein
expression level was the opposite (Fig. 5C). Immunofluorescence results
showed that CTGF knockdown resulted in decreased α-SMA expression
levels in ALD-treated mTECs (Fig.
5D); the immunofluorescence NC shown in Fig. S2D exhibits no notable difference
compared with control group in Fig.
5D. Collectively, these results indicated that CTGF knockdown
alleviates ALD-induced EMT and ECM deposition in mTECs.
 | Figure 5CTGF knockdown alleviates ALD-induced
EMT and ECM deposition in mTECs. Cells were transfected with si-NC
or si-CTGF for 6 h and then treated with ALD (1×10−6 M)
for 48 h. RT-qPCR analysis of (A) miR-26a expression and (B)
collagen I, α-SMA and LCN2 mRNA expression levels in mTECs treated
with buffer (control), ALD, ALD + si-NC or ALD + si-CTGF. Results
were normalized to U6 or β-actin, respectively. (C) Representative
western blotting images and semi-quantitative analysis of
E-cadherin, collagen I, α-SMA, CTGF and LCN2 protein levels in the
variously treated mTECs. (D) Immunofluorescence analysis of α-SMA
(red) in the treated mTECs; DAPI (blue) was used to stain the
nucleus. Scale bar, 50 µm. Data are presented as the mean ±
SD; *P<0.05 vs. control, #P<0.05 vs.
ALD + si-NC. α-SMA, α-smooth muscle actin; ALD, aldosterone; CTGF,
connective tissue growth factor; ECM, extracellular matrix; EMT,
epithelial-mesenchymal transition; LCN2, lipocalin 2; mTEC, mouse
tubular epithelial cells; NC, negative control; RT-qPCR, reverse
transcription-quantitative PCR; si, small interfering RNA. |
miR-26a/CTGF axis inhibits SMAD3
activation
SMAD proteins have been reported to be associated
with TIF, where SMAD3 plays a pathogenic role and SMAD2 and SMAD7
are protective (29). We
hypothesized that miR-26a/CTGF might attenuate ALD-induced EMT and
ECM deposition in mTECs by inhibiting SMAD3 activation. To test
this, SMAD3 and p-SMAD3 protein expression levels were examined in
the kidneys of ALD-treated mice. Western blot analysis revealed
that p-SMAD3 expression in the ALD + Exo-NC group was higher
compared with that in the ALD + Exo-miR-26a group, whereas the
expression level of SMAD3 did not differ (Fig. 6A). In vitro, p-SMAD3
expression was increased in cells transfected with NC mimic and
oe-CTGF plasmids compared with cells transfected with NC mimic and
oe-NC plasmids, whereas it was decreased in cells transfected with
oe-NC + miR-26a mimic compared with cells transfected with oe-NC +
NC mimic and in cells transfected with oe-CTGF + miR-26a mimic
compared with cells transfected with oe-CTGF + NC mimic (Fig. 6B); there was no difference in
SMAD3 expression between the groups. These data suggested that
miR-26a may inhibit and CTGF may increase SMAD3 activation.
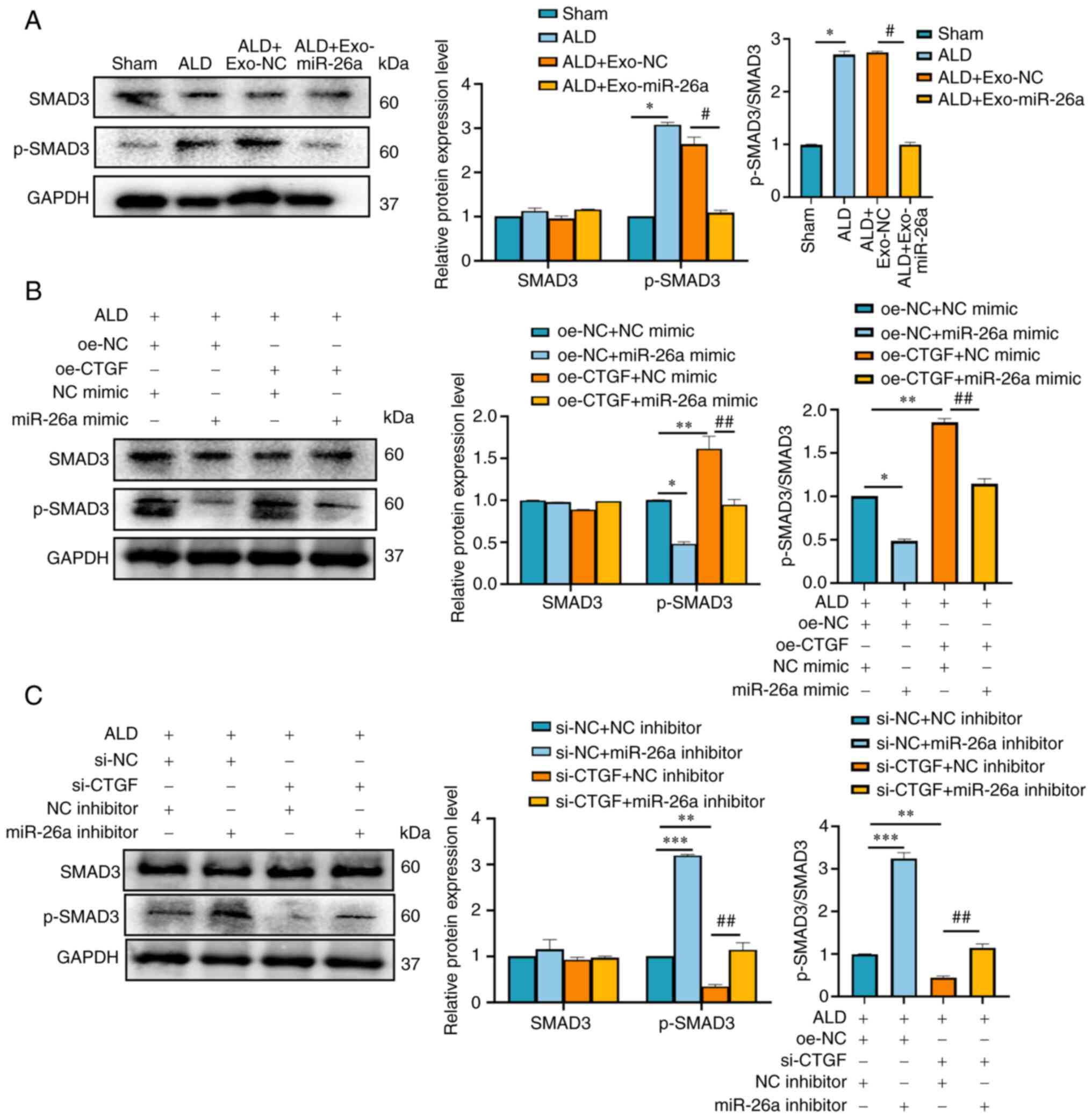 | Figure 6miR-26a/CTGF inhibits SMAD3
activation. (A) Western blot analysis of SMAD3 and p-SMAD3 protein
expression levels in the kidneys of mice in the sham, ALD, ALD +
Exo-NC and ALD + Exo-miR-26a groups. (B) Western blot analysis of
SMAD3 and p-SMAD3 protein expression levels in mTECs co-transfected
with oe-CTGF or oe-NC and miR-26a mimic or NC mimic for 6 h, and
then treated with ALD (1×10−6 M) for 48 h. (C) Western
blot analysis of SMAD3 and p-SMAD3 protein levels in mTECs
co-transfected with si-CTGF or si-NC and miR-26a inhibitor or NC
inhibitor for 6 h, and then treated with ALD (1×10−6 M)
for 48 h. Data are presented as mean ± SD; Data are presented as
the mean ± SD; *P<0.05, **P<0.01,
***P<0.001; #P<0.05,
##P<0.01. ALD, aldosterone; CTGF, connective tissue
growth factor; Exo, exosome encapsulated; miR, microRNA; mTEC,
mouse tubular epithelial cells; NC, negative control;
p-phosphorylated. |
To further explore the underlying mechanism, miR-26a
inhibitor and CTGF siRNA were transfected into ALD-treated mTECs.
Western blot analysis demonstrated that the expression of p-SMAD3
was reduced in cells transfected with si-CTGF + NC inhibitor
compared with cells transfected with si-NC + NC inhibitor, whereas
the expression was elevated in cells transfected with si-NC +
miR-26a inhibitor compared with NC inhibitor and si-NC, and in
cells transfected with si-CTGF + miR-26a inhibitor compared with
cells transfected with si-CTGF + NC inhibitor (Fig. 6C); no obvious changes were
observed in SMAD3 expression between the groups.
In summary, miR-26a may inhibit p-SMAD3 expression
by binding to CTGF to rescue ALD-mediated TIF in vivo, as
well as EMT and ECM deposition in vitro.
Discussion
TIF is a serious consequence of CKD, leading to
disease progression to end-stage renal disease (30). In the present study, it was
demonstrated that injection of miR-26a-rich exosomes through the
tail vein in mice effectively alleviated ALD-induced TIF in
vivo, and miR-26a mimic transfection rescued EMT and ECM
deposition in mTECs in vitro by inhibiting the activation of
the CTGF/SMAD signaling pathway. Results from this study may
provide a potential avenue for the treatment of TIF in CKD using
Exo-miR-26a.
ALD, a member of the RAAS, has been demonstrated to
act on the heart, blood vessels and kidneys, and is reported to
damage the cardiovascular and renal systems by promoting tissue
inflammation, injury, glomerulosclerosis and interstitial fibrosis
(31,32). Our previous studies have also
confirmed that hyperaldosteronism promotes podocyte damage,
glomerular sclerosis, renal tubular epithelial cell damage, renal
interstitial inflammation and fibrosis (7-9).
The present study demonstrated that ALD promotes the upregulation
of α-SMA, collagen I and LCN2 and the downregulation of E-cadherin
in vivo and in vitro. The increased expression of
collagen I suggested the deposition of ECM in the
tubulointerstitium. The increase in α-SMA, a hallmark of mature
myofibroblasts (33), and LCN2, a
crucial injury response factor (34), and the decrease in E-cadherin, a
marker protein of tubular epithelium, indicated that EMT occurred
in the kidney and mTECs.
Exosomes are an important and universal form of
intercellular communication, and can mediate communication between
organs (35). In addition,
exosomes can be used as therapeutic carriers and serve as
biomarkers to diagnose several diseases in a non-invasive manner
(36). Our previous study
demonstrated that Exo-miR-26a can not only prevent muscle atrophy
and limit TIF in a mouse model of unilateral ureteral obstruction
(UUO), but also attenuate skeletal muscle atrophy and ameliorate
uremic cardiomyopathy in a 5/6 nephrectomy mouse model (14,37). The present study verified that
exosomes can transport miR-26a into the kidney and rescue
ALD-induced TIF using DiD fluorescence imaging experiments,
indicating that exosomes may be an effective tool for delivering
miRNA into the kidney.
Increasing evidence suggests that miR-26a is
associated with the fibrosis process in various organs. For
example, miR-26a was shown to inhibit EMT of lens epithelial cells
and lens fibrosis in an injury-induced anterior subcapsular
cataract (ASC) model and the mouse lens anterior capsular injury
model in vivo and in a TGFβ2-induced ASC model in
vitro (17). Another previous
study reported that miR-26a overexpression can suppress
cardiomyocyte apoptosis caused by ST-elevation myocardial
infarction and oxygen-glucose deprivation, and it can ameliorate
cardiac dysfunction and fibrosis in cardiac remodeling (38). Furthermore, miR-26a can alleviate
fibrosis in hypertensive myocardial fibrosis (39), diabetic cardiomyopathy (40), and idiopathic pulmonary fibrosis
(16). In addition, our previous
studies have demonstrated that miR-26a can limit muscle atrophy and
alleviate cardiomyopathy and TIF (14,37). The present study also revealed
that miR-26a expression was reduced in the kidney of the
ALD-treated mice and mTECs. By contrast, miR-26a overexpression
rescued the expression levels of α-SMA, collagen I, CTGF and
E-cadherin in ALD-induced mice and mTECs. These results suggested
that increased miR-26a levels may serve an important role in
alleviating TIF.
In addition, the present study verified that miR-26a
targets CTGF to reduce its expression level. CTGF is mainly
expressed and secreted by proximal tubular epithelial cells and
renal interstitial fibroblasts in the kidney (41), and it is a pro-fibrotic factor
involved in many chronic diseases. CTGF is involved in the fibrosis
of various organ systems, including the liver, lungs, skin and
kidneys (42,43). Increasing evidence shows that
increased expression of CTGF in CKD kidney tissue is closely
related to TIF (14,37,44). Our previous studies confirmed that
CTGF was upregulated in the mouse model of UUO and 5/6 nephrectomy
(14,37). The current study also revealed
that CTGF was elevated in the ALD-treated mice. Moreover, in an
experiment to verify the binding relationship between miR-26a and
CTGF, the results showed that miR-26a expression is associated with
CTGF expression, and vice versa. Given the above functions of CTGF,
miR-26a may be an ideal factor to alleviate fibrosis, owing to its
ability to inhibit the expression of CTGF.
SMAD protein family members are involved in organ
fibrosis, and previous studies showed that deletion of SMAD3
inhibits renal fibrosis induced by almost all etiologies (45,46), and deletion of SMAD2 significantly
enhances renal fibrosis (29),
which indicates SMAD3 as a pathogenic protein. The present study
revealed that p-SMAD3 expression in the kidneys of ALD-treated mice
was increased compared with that in sham-treated mice, and the
expression levels in the ALD + Exo-miR-26a mice were decreased
compared with the ALD + Exo-NC mice. In vitro, miR-26a mimic
and si-CTGF transfection alleviated EMT and ECM deposition in mTECs
compared with miR-26a inhibitor and CTGF overexpression plasmids.
These findings indicated that miR-26a restrained the activation of
SMAD3 to rescue fibrosis.
In the present study, however, the expression status
of miR-26a was not detected in tissues in TIF patients owing to the
lack of tissue samples, which is an area of future research. Since
injection through the tail vein is not targeted delivery, further
experiment should be conducted to determine how to make exosomes
able to target and transport miR-26a into the kidneys to achieve a
better treatment plan. Owing to the different role of SMAD2 and
SMAD3 in the regulation of pro-fibrotic TGF-β1 responses in human
proximal-tubule epithelial cells, investigations into the role of
SMAD2 and the regulatory relationship between SMAD2 and SMAD3 may
increase our understanding of TGF-β1 in follow-up experiments.
In conclusion, the present study results
demonstrated that the downregulation of miR-26a serves a
significant role in the occurrence and development of TIF, and
exosomes containing abundant miR-26a effectively alleviated TIF.
These findings may provide us with a theoretical basis for the
treatment of TIF.
Supplementary Data
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
AZ designed the study. HZ and JJ performed the
experiments and wrote the manuscript. TZ and EW contributed to the
data analysis. HZ and JJ confirm the authenticity of all the raw
data. All authors have read and approved the final manuscript.
Ethics approval and consent to
participate
The experimental procedures in the present study
were approved by Nanjing Medical University (Nanjing, China; ref.
no. IACUC-2107049).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
Not applicable.
Funding
This work was supported by The National Natural Science
Foundation of China (grant no. 81970664), The Natural Science
Foundation of Jiangsu Province (grant no. BK2021022945), and the
789 Outstanding Talent Program of SAHNMU (grants nos.
789ZYRC202080119 and 789ZYRC202090251).
Abbreviations:
|
α-SMA
|
α-smooth muscle actin
|
|
ALD
|
aldosterone
|
|
CKD
|
chronic kidney disease
|
|
CTGF
|
connective tissue growth factor
|
|
ECM
|
extracellular matrix
|
|
EMT
|
epithelial-mesenchymal transition
|
|
Exo
|
exosome encapsulated
|
|
LCN2
|
lipocalin 2
|
|
mTEC
|
mouse tubular epithelial cells
|
|
NC
|
negative control
|
|
oe-
|
overexpression
|
|
TIF
|
tubulointerstitial fibrosis
|
References
|
1
|
GBD Chronic Kidney Disease Collaboration:
Global, regional, and national burden of chronic kidney disease,
1990-2017: A systematic analysis for the global burden of disease
study 2017. Lancet. 395:709–733. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kramer H: Diet and chronic kidney disease.
Adv Nutr. 10(Suppl 4): S367–S379. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Liyanage T, Toyama T, Hockham C, Ninomiya
T, Perkovic V, Woodward M, Fukagawa M, Matsushita K,
Praditpornsilpa K, Hooi LS, et al: Prevalence of chronic kidney
disease in Asia: A systematic review and analysis. BMJ Glob Health.
7:e0075252022. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Qi R and Yang C: Renal tubular epithelial
cells: The neglected mediator of tubulointerstitial fibrosis after
injury. Cell Death Dis. 9:11262018. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Lerman LO: Imaging: BOLD
assessment-effects of RAAS inhibition in CKD. Nat Rev Nephrol.
10:247–248. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Blasi ER, Rocha R, Rudolph AE, Blomme EAG,
Polly ML and McMahon EG: Aldosterone/salt induces renal
inflammation and fibrosis in hypertensive rats. Kidney Int.
63:1791–1800. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Yuan Y, Zhang A, Qi J, Wang H, Liu X, Zhao
M, Duan S, Huang Z, Zhang C, Wu L, et al: p53/Drp1-dependent
mitochondrial fission mediates aldosterone-induced podocyte injury
and mitochondrial dysfunction. Am J Physiol Renal Physiol.
314:F798–F808. 2018. View Article : Google Scholar
|
|
8
|
Shi H, Zhang A, He Y, Yang M and Gan W:
Effects of p53 on aldosterone-induced mesangial cell apoptosis in
vivo and in vitro. Mol Med Rep. 13:5102–5108. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Qu G, Shi H, Wang B, Li S, Zhang A and Gan
W: Alterations in the long noncoding RNA transcriptome in mesangial
cells treated with aldosterone in vitro. Mol Med Rep. 16:6004–6012.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Bartel DP: Metazoan MicroRNAs. Cell.
173:20–51. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Chandrasekaran K, Karolina DS, Sepramaniam
S, Armugam A, Wintour EM, Bertram JF and Jeyaseelan K: Role of
microRNAs in kidney homeostasis and disease. Kidney Int.
81:617–627. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zheng Z, Guan M, Jia Y, Wang D, Pang R, Lv
F, Xiao Z, Wang L, Zhang H and Xue Y: The coordinated roles of
miR-26a and miR-30c in regulating TGFbeta1-induced
epithelial-to-mesenchymal transition in diabetic nephropathy. Sci
Rep. 6:374922016. View Article : Google Scholar
|
|
13
|
Wang H, Wang B, Zhang A, Hassounah F, Seow
Y, Wood M, Ma F, Klein JD, Price SR and Wang XH: Exosome-mediated
miR-29 transfer reduces muscle atrophy and kidney fibrosis in mice.
Mol Ther. 27:571–583. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zhang A, Wang H, Wang B, Yuan Y, Klein JD
and Wang XH: Exogenous miR-26a suppresses muscle wasting and renal
fibrosis in obstructive kidney disease. FASEB J. 33:13590–13601.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wei C, Kim IK, Kumar S, Jayasinghe S, Hong
N, Castoldi G, Catalucci D, Jones WK and Gupta S: NF-kappaB
mediated miR-26a regulation in cardiac fibrosis. J Cell Physiol.
228:1433–1442. 2013. View Article : Google Scholar
|
|
16
|
Kadota T, Fujita Y, Araya J, Watanabe N,
Fujimoto S, Kawamoto H, Minagawa S, Hara H, Ohtsuka T, Yamamoto Y,
et al: Human bronchial epithelial cell-derived extracellular
vesicle therapy for pulmonary fibrosis via inhibition of
TGF-beta-WNT crosstalk. J Extracell Vesicles. 10:e121242021.
View Article : Google Scholar
|
|
17
|
Chen X, Xiao W, Chen W, Liu X, Wu M, Bo Q,
Luo Y, Ye S, Cao Y and Liu Y: MicroRNA-26a and -26b inhibit lens
fibrosis and cataract by negatively regulating Jagged-1/Notch
signaling pathway. Cell Death Differ. 24:1431–1442. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Jiang S, Jiang W, Xu Y, Wang X, Mu Y and
Liu P: Serum miR-21 and miR-26a levels negatively correlate with
severity of cirrhosis in patients with chronic hepatitis B.
Microrna. 8:86–92. 2019. View Article : Google Scholar
|
|
19
|
Smyth T, Kullberg M, Malik N, Smith-Jones
P, Graner MW and Anchordoquy TJ: Biodistribution and delivery
efficiency of unmodified tumor-derived exosomes. J Control Release.
199:145–155. 2015. View Article : Google Scholar :
|
|
20
|
Mahtal N, Lenoir O, Tinel C, Anglicheau D
and Tharaux PL: MicroRNAs in kidney injury and disease. Nat Rev
Nephrol. 18:643–662. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Srivastava SP, Koya D and Kanasaki K:
MicroRNAs in kidney fibrosis and diabetic nephropathy: Roles on EMT
and EndMT. Biomed Res Int. 2013:1254692013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Trionfini P, Benigni A and Remuzzi G:
MicroRNAs in kidney physiology and disease. Nat Rev Nephrol.
11:23–33. 2015. View Article : Google Scholar
|
|
23
|
Colombo M, Raposo G and Thery C:
Biogenesis, secretion, and intercellular interactions of exosomes
and other extracellular vesicles. Annu Rev Cell Dev Biol.
30:255–289. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Skog J, Würdinger T, van Rijn S, Meijer
DH, Gainche L, Sena-Esteves M, Curry WT Jr, Carter BS, Krichevsky
AM and Breakefield XO: Glioblastoma microvesicles transport RNA and
proteins that promote tumour growth and provide diagnostic
biomarkers. Nat Cell Biol. 10:1470–1476. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Jalabert A, Vial G, Guay C, Wiklander OPB,
Nordin JZ, Aswad H, Forterre A, Meugnier E, Pesenti S, Regazzi R,
et al: Exosome-like vesicles released from lipid-induced
insulin-resistant muscles modulate gene expression and
proliferation of beta recipient cells in mice. Diabetologia.
59:1049–1058. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Vinas JL, Burger D, Zimpelmann J, Haneef
R, Knoll W, Campbell P, Gutsol A, Carter A, Allan DS and Burns KD:
Transfer of microRNA-486-5p from human endothelial colony forming
cell-derived exosomes reduces ischemic kidney injury. Kidney Int.
90:1238–1250. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
28
|
Liang H, Xu C, Pan Z, Zhang Y, Xu Z, Chen
Y, Li T, Li X, Liu Y, Huangfu L, et al: The antifibrotic effects
and mechanisms of microRNA-26a action in idiopathic pulmonary
fibrosis. Mol Ther. 22:1122–1133. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Tang PM, Zhang YY, Mak TSK, Tang PCT,
Huang XR and Lan HY: Transforming growth factor-beta signalling in
renal fibrosis: From smads to non-coding RNAs. J Physiol.
596:3493–3503. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Li ZL and Liu BC: Hypoxia and renal
tubulointerstitial fibrosis. Adv Exp Med Biol. 1165:467–485. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Epstein M: Aldosterone blockade: An
emerging strategy for abrogating progressive renal disease. Am J
Med. 119:912–919. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Hostetter TH and Ibrahim HN: Aldosterone
in chronic kidney and cardiac disease. J Am Soc Nephrol.
14:2395–2401. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Shinde AV, Humeres C and Frangogiannis NG:
The role of alpha-smooth muscle actin in fibroblast-mediated matrix
contraction and remodeling. Biochim Biophys Acta Mol Basis Dis.
1863:298–309. 2017. View Article : Google Scholar
|
|
34
|
Rudman-Melnick V, Adam M, Potter A,
Chokshi SM, Ma Q, Drake KA, Schuh MP, Kofron JM, Devarajan P and
Potter SS: Single-cell profiling of AKI in a murine model reveals
novel transcriptional signatures, profibrotic phenotype, and
epithelial-to-stromal crosstalk. J Am Soc Nephrol. 31:2793–2814.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Shen AR, Zhong X, Tang TT, Wang C, Jing J,
Liu BC and Lv LL: Integrin, exosome and kidney disease. Front
Physiol. 11:6278002020. View Article : Google Scholar
|
|
36
|
Kalluri R and LeBleu VS: The biology,
function, and biomedical applications of exosomes. Science.
367:eaau69772020. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Wang B, Zhang A, Wang H, Klein JD, Tan L,
Wang ZM, Du J, Naqvi N, Liu BC and Wang XH: miR-26a limits muscle
wasting and cardiac fibrosis through exosome-mediated microrna
transfer in chronic kidney disease. Theranostics. 9:1864–1877.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Chiang MH, Liang CJ, Lin LC, Yang YF,
Huang CC, Chen YH, Kao HL, Chen YC, Ke SR and Lee CW: miR-26a
attenuates cardiac apoptosis and fibrosis by targeting
ataxia-telangiectasia mutated in myocardial infarction. J Cell
Physiol. 235:6085–6102. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Zhang W, Wang Q, Feng Y, Chen X, Yang L,
Xu M, Wang X, Li W, Niu X and Gao D: MicroRNA-26a protects the
heart against hypertension-induced myocardial fibrosis. J Am Heart
Assoc. 9:e0179702020. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Zhu C, Zhang H, Wei D and Sun Z: Silencing
lncRNA GAS5 alleviates apoptosis and fibrosis in diabetic
cardiomyopathy by targeting miR-26a/b-5p. Acta Diabetol.
58:1491–1501. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Ito Y, Aten J, Bende RJ, Oemar BS,
Rabelink TJ, Weening JJ and Goldschmeding R: Expression of
connective tissue growth factor in human renal fibrosis. Kidney
Int. 53:853–861. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Perbal B: CCN proteins: Multifunctional
signalling regulators. Lancet. 363:62–64. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Wang S, Denichilo M, Brubaker C and
Hirschberg R: Connective tissue growth factor in tubulointerstitial
injury of diabetic nephropathy. Kidney Int. 60:96–105. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Okada H, Kikuta T, Kobayashi T, Inoue T,
Kanno Y, Takigawa M, Sugaya T, Kopp JB and Suzuki H: Connective
tissue growth factor expressed in tubular epithelium plays a
pivotal role in renal fibrogenesis. J Am Soc Nephrol. 16:133–143.
2005. View Article : Google Scholar
|
|
45
|
Lan HY: Transforming growth
factor-beta/Smad signalling in diabetic nephropathy. Clin Exp
Pharmacol Physiol. 39:731–738. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Sato M, Muragaki Y, Saika S, Roberts AB
and Ooshima A: Targeted disruption of TGF-beta1/Smad3 signaling
protects against renal tubulointerstitial fibrosis induced by
unilateral ureteral obstruction. J Clin Invest. 112:1486–1494.
2003. View Article : Google Scholar : PubMed/NCBI
|














