Biomineralization is a complex process in which
inorganic ions are deposited on organic matter under the action of
proteins, hormones and enzymes (1,2).
It has a critical role in a variety of physiological and
pathological processes, such as bone and cardiovascular health, and
lithiasis. Bone requires mineralization to maintain its strength
and function. Insufficient mineralization and calcium (Ca) loss
from bone may cause osteochondrosis and osteoporosis; however,
excessive mineralization may lead to sclerosteosis (3). In addition, the excessive
deposition of inorganic ions in soft tissues, such as blood
vessels, joints and internal organs, is pathological, resulting in
gall-stones, kidney stones and vascular calcification (4). Certain studies have reported that
multivitamins, such as vitamin D (VD), vitamin C (VC) and vitamin K
(VK), have important roles in maintaining bone and blood vessel
health and preventing pathological calcium deposition or loss
(5).
VD affects mineralization mainly by influencing Ca
and phosphorus (P) metabolism and osteoclast activity (6). VC promotes bone mass via
anti-oxidation, promoting collagen formation and inhibiting
osteoclast activity (7). VK is a
pivotal factor in the biomineralization balance. VK maintains blood
vessel and bone health by promoting osteoblast differentiation,
anti-oxidation and inhibiting cell autophagy and ferroptosis
(8-10). In addition, the partial role of
VK in biomineralization is thought to be associated with
VK-dependent proteins (VKDPs), including osteocalcin, matrix
γ-carboxyglutamic acid (Gla) protein (MGP), Gla-rich protein (GRP)
and growth arrest specific 6 (Gas6) (11). The current review introduces the
role of VK and several VKDPs in physiological and pathological bone
mineralization.
VK, a fat-soluble vitamin, was discovered in 1929 by
the Danish biochemist Henrik Dam as being essential for blood
coagulation. VK is a general term for compounds with different
structural forms, including VK1, VK2,
VK3 and VK4 (Fig. 1A), which share the same menadione
structure (the 2-methyl-1,4-naphthoquinone core) (12). These VKs contain different
numbers of isoprenoid side chains at the 3-position of the
naphthoquinone ring (Fig. 1A),
which is the reason for their different solubilities.
VK1, consisting of a side chain containing three
isoprenes (liposoluble), is exclusively synthesized by algae and
green plants. The different VK2 (lip solubility) members
are called menaquinones (MKn) according to the amount of isoprene
in their side chain (MK-4 to MK-13) and are mainly found in fish,
chicken, Japanese natto and cheese. In addition, certain components
of the intestinal flora are able to produce MK-6, MK-8 and MK-11,
the form and concentration of which are influenced by the
composition of the intestinal microbiota. VK3, without a
side chain, is an artificially synthesized water-soluble vitamin.
VK4, a synthetic vitamin, is an oxidized form of
VK3 (12). To date,
several international institutions have published a recommended
daily intake (RDI) dose because of the important role of VK in
physiological functions; however, the recommended RDIs are not
consistent. The National Academy of Medicine recommends an adequate
intake of 120 μg per day for an adult male and 90 μg
per day for an adult female. The World Health Organization and the
Food and Agriculture Organization recommend a VK dose of 65
μg per day for an adult male and 55 μg per day for an
adult female (13,14).
The different types of VK vary in their absorption,
distribution and metabolism. As indicated in Fig. 1B, VK1 and
VK2 from food are absorbed in the small intestine by
forming a mixture containing bile salts, pancreatic lipolysis
products and other dietary lipids, and then spread throughout the
body via chylomicrons because of their fat-soluble nature. In
addition, the absorption of VK1 is related to three
protein transporters, Neimann-Pick C1 like 1, scavenger receptor
class B type I and CD36 (12,15). VK3 and VK4
are generally used for intramuscular injection and are absorbed in
the intestines, without bile water solubility. In blood
transportation, VK1 and VK2 are different.
VK1 is transported by triglyceride-rich lipoproteins,
while VK2 is mainly transported by low-density
lipoproteins. Previous research reported that VK1 is
highly enriched in the liver and VK2 is more widely
distributed in the body's extrahepatic tissues. However, recent
research has demonstrated that MK-4 is the major VK form in
mammalian tissues, regardless of the dietary input of
VK1 or VK2, which results from the action of
the enzyme UbiA prenyltransferase domain-containing protein 1,
which converts phylloquinone to MK-4. VK1 and
VK2 are catabolized in the liver and excreted through a
common degradation pathway. The two types of VK are reduced to
hydroquinone in the liver and excreted after combining with
glucuronic acid and sulfuric acid (Fig. 1C). In addition, a recent study
found that ATP-binding cassette protein G5 (ABCG5)/ABCG8, a
heterodimer exporting cholesterol, participates in the excretion of
VK from the intestines (16).
Although VK is well known as an anticoagulant, the
function of VK in other physiological processes is being
increasingly recognized. For instance, VK is able to regulate the
immune response, maintain intestinal health and inhibit cancer
growth (17,18). Clinically, VK has been used to
treat and prevent numerous diseases, such as osteoporosis,
atherosclerosis, intestinal diseases and cholestatic liver disease
(17,19,20). Some of the mechanisms by which VK
can treat and prevent these diseases are partly known. For
instance, MK-4, converted from VK, interacts with the cell surface
VK-binding nuclear receptor steroid and xenobiotic receptor to
improve bone quality. In addition, VK exerts anti-inflammatory
effects by inhibiting nuclear factor κB (NF-κB) signaling and
antioxidant effects by blocking the production of reactive oxygen
species (21). A recent study
reported that reduced forms of VK, including menadione and
chloroquinone, are potent anti-ferroptosis agents (22).
The most important role of VK is associated with the
VK cycle. In this cycle, VK is firstly reduced to hydroquinone VK
(KH2) under the action of VK reductase (VKR) or
VK-2,3-epoxidoreductase (VKOR). Subsequently, KH2, as a
coenzyme, assists carboxyglutamyl carboxylase (GGCX) to carboxylate
VKDPs [specific glutamyl (Glu) residues in VKDPs are converted to
Gla], while KH2 is oxidized to epoxide VK (KO). Finally,
KO is reduced to VK under the action of VKOR (23) (see Fig. 2). Currently, 17 members of the
Gla protein family of VKDPs have been identified. These include S
prothrombin, factor VII, factor IX, factor X, protein C, protein S
and protein Z, which are crucial in maintaining the delicate
balance of blood coagulation. In addition, there are MGP,
osteocalcin, Gas6, GRP, periostin and periostin-like factors, which
have significant roles in biomineralization. Furthermore, the
family consists of two amino acid-rich Gla proteins and two
transmembrane Gla proteins (24,25). When Glu residues are carboxylated
to Gla residues, these proteins gain a higher calcium-binding
ability, which is why VK has an important role in blood coagulation
and biomineralization. The American Health Association recommends
that VK is injected into newborns to reduce the risk of bleeding
because of low levels of VK in their bodies. Warfarin is used to
prevent and treat coagulation by decreasing the activity of VKOR.
However, the long-term use of warfarin is associated with
calcification of blood vessels, heart valves and osteoporosis
(26-28). Verma et al (29) found that VK antagonism impairs
the bone marrow microenvironment and bone density, and increases
osteoclast activity.
Biomineralization is a physical process that
includes the maintenance of bone and teeth. However, inappropriate
mineralization may cause several diseases, such as osteoporosis,
vascular calcification and renal calculi.
Bone is composed of organic matter (type I collagen,
proteins), inorganic matter and cells (osteoblasts, osteocytes and
osteoclasts). Phosphate-calcium (crystalline hydroxyapatite)
combines with type I collagen to enhance bone strength through
proteins (including VKDPs) and enzymes (30,31). Loss of Ca and the insufficient
ability of Ca to bind collagen may cause osteoporosis and fracture.
Excessive deposition of Ca/P may cause osteosclerosis, which
increases bone fragility. At present, it is inconclusive whether VK
can improve osteoporosis and reduce the fracture rate. A clinical
study showed that VK deficiency is associated with osteoporosis and
vascular calcification (25). A
meta-analysis by Ma et al (32) showed that VK increased the bone
mineral density (BMD) in the lumbar spine. In addition, VK can
reduce bone loss by increasing osteoprotegerin levels (33). However, a meta-analysis by Salma
et al (34) reported that
VK supplementation reduced the fracture rate but did not improve
the BMD in the femur and tibia. The notion that VK has a positive
effect on bone mineralization is dominant, which is recognized by
the European Food Safety Authority Association (35).
Vascular calcification, common in the elderly, and
in patients with diabetes and chronic kidney disease (CKD), is
caused by excessive Ca/P deposition in aortic elastin, which is the
main component of the intermediate elastic fibers (4). Vascular calcification is affected
by numerous factors. High expression of bone-related genes such as
RUNX family transcription factor 2 (RUNX2) and bone morphogenetic
protein-2 (BMP2) in vascular smooth muscle cells leads to the
phenotypic transformation to osteoblasts (36). After phenotypic transformation,
the high expression of alkaline phosphatase (ALP), osteopontin,
osteocalcin and MGP induced by RUNX2 result in extracellular
deposition of hydroxyapatite in a blood vessel, leading to
atherosclerosis. Endoplasmic reticulum stress, mitochondrial damage
and hyperphosphate blood damage vascular smooth muscle cells during
vascular calcification (37,38). In addition, the lack of
mineralization inhibitors (VKDPs) results in excessive deposition
of hydroxyapatite crystals on blood vessels, which is also a key
factor in vascular calcification (39). Treatment with VK antagonists for
anticoagulation carries the risk of concomitant vascular or
valvular calcification for patients (40,41). MK-7 supplementation may reduce
the progression of vascular calcification in patients with coronary
artery disease. VK may also reduce vascular calcification by
decreasing the production of inflammatory cytokines. VK inhibits
the production of tumor necrosis factor (TNF) in macrophages. TNF
can promote the osteogenic differentiation of vascular smooth
muscle cells under stimulation by high phosphate, oxidative stress
and high glucose (42).
Urolithiasis is a common disease including stones of
the kidney, bladder, ureter and urethra. It is affected by numerous
factors, including hypercalcemia, dietary habits, obesity, diabetes
and kidney disease. The most common types of stones are calcium
oxalate, calcium phosphate and uric acid. Certain studies have
demonstrated that the VK cycle and its dependent proteins are
related to the formation of various urinary stones (43,44).
In general, there is a close relationship between VK
and biomineralization. The effects of VKDPs on biomineralization
are mainly described in the following sections.
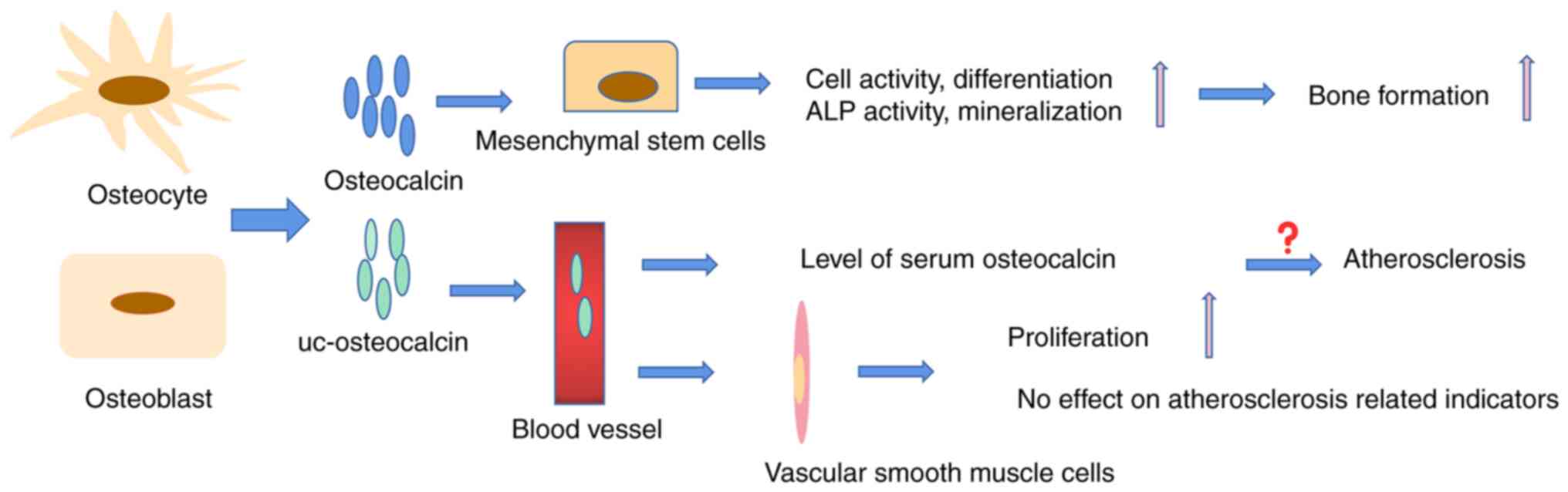
The levels of uc-osteocalcin in the blood are used
as a maker for the clinical detection of osteoporosis and VK
deficiency. However, clinical trials concerning the relationship
between osteocalcin and osteoporosis are not consistent. Most
clinical research reported that high serum osteocalcin
(uc-osteocalcin and osteocalcin) levels are closely associated with
a lower BMD and higher fracture risk (49,50). However, other studies determined
that the serum osteocalcin level was not associated with BMD
(32,51). A reasonable explanation is that
the reagents for detecting serum osteocalcin are not uniform, i.e.,
antibodies cannot distinguish uc-osteocalcin from osteocalcin.
However, the effect of osteocalcin on bone has also been
controversial in animal studies. It was reported that the bone
biophysical properties in osteocalcin-deficient mice were altered,
with fewer hydroxyapatite crystals, resulting in increased bone
fragility and fracture rates. A recent study reported increased
bone fragility and altered energy dissipation mechanisms in
osteocalcin-deficient mice and highlighted the role of glycation of
osteocalcin in bone (52). The
biological apatite (BAp) c-axis of hydroxyapatite parallel to the
collagen fiber is necessary for optimal bone strength. Microbeam
X-ray diffraction system analysis showed that osteocalcin-knockout
mice have a disrupted BAp c-axis orientation, which demonstrated
that osteocalcin is necessary for the alignment of the BAp c-axis
parallel to collagen fibers. The results of a nanoindentation test
showed that the Young's modulus of the femur in
osteocalcin-knockout mice was significantly lower than that of
wild-type mice (Fig. 3)
(53). However, the
osteocalcin-deficient mice were found have a higher bone mass and
BMD, which resulted in osteocalcin being identified as a negative
regulator of bone formation (54,55). Of note, in subsequent studies,
the same research group reported that knockdown of osteocalcin in
B6 mice resulted in a lower BMD and bone strength (Fig. 3) (56). This opposite effect was caused by
different genetic backgrounds. Although warfarin (a VK circulation
inhibitor) has a controversial role in BMD, certain studies suggest
that it has a negative effect on bone. Impaired osteoblast function
and osteoporosis have been described in mice receiving warfarin
(29). In addition, warfarin was
able to further reduce the bone calcium loss caused by 1,25(OH) D3,
which is thought to be related to osteocalcin. In an in
vitro study, osteocalcin promoted the proliferation, ALP
activity and mineral deposition of human osteoblast-like MG63 cells
(57). Similarly, Tsao et
al (58) also confirmed that
osteocalcin has an important role in osteogenic differentiation and
mineralization processes of mesenchymal stem cells. Overall,
osteocalcin has a positive effect on bone and osteoblasts, but the
opposite result may occur in different genetic backgrounds.
More than a decade ago, a strong association between
osteocalcin and atherosclerosis was reported (59). This finding was subsequently
confirmed in clinical investigations (60,61). For instance, in a five-year study
of 9,413 patients with type 2 diabetes, patients with lower serum
osteocalcin levels had a higher risk of all-cause and
cardiovascular mortality (62).
A study of 59 patients with atherosclerosis identified osteocalcin
in any form as a biomarker of vascular risk (63). Guo et al (64) demonstrated that higher serum
osteocalcin levels were associated with severe arteriosclerosis in
patients with kidney disease. In vitro, osteocalcin is also
used as an important indicator to detect the transformation of
vascular smooth muscle cells into osteoblasts, as it is highly
expressed in the early differentiation of osteoblasts (65). However, recently, certain
clinical investigations found that there was no association between
osteocalcin and atherosclerosis (Fig. 4) (66-68). Research by Millar et al
(66) demonstrated that
osteocalcin has no effect on the calcification of vascular smooth
muscle cells, suggesting that osteocalcin is not directly involved
in atherosclerosis or vascular disease. Certain researchers even
proposed that osteocalcin has a good protective effect on vascular
calcification in animal experiments (70,71). A long-term high-sugar and -fat
diet is a factor in the development of atherosclerosis. Huang et
al (70) found that
osteocalcin improved the protective effect against the induction of
arteriosclerosis in diabetic rat models by affecting glucose
levels, insulin sensitivity and lipid metabolites. Osteocalcin had
an endothelial-protective effect in mouse thoracic aortic
atherosclerosis caused by apolipoprotein E (ApoE) knockout
(71). Therefore, it may be
speculated that the high serum level of osteocalcin in patients
with atherosclerosis may be the body's attempt to produce more
osteocalcin to prevent the process of atherosclerosis.
MGP (a 14 kDa protein) is a calcification inhibitor
that was first isolated from bovine bone matrix in 1985 (72). The MGP gene consists of four
exons separated by three large intervening sequences; in addition,
the typical TATA and CAT boxes, and the binding sites of retinoic
acid and VD, were identified at regions of the MGP gene promoter
(73). It is expressed in
chondrocytes, fibroblasts and vascular smooth muscle cells. The
structural features of MGP are similar to those of osteocalcin,
both sharing a common historic ancestor. As for osteocalcin, MGP
contains a signaling peptide in primary structures; in addition, it
contains five Glu residues and three serine phosphorylation sites
at its N-terminus (72,74). MGP Gla residues provide binding
sites for Ca and hydroxyapatite. In addition, phosphorylation is
important for its function and influences its protein structure
(74). Therefore, there are four
forms of MGP in the body, including MGP, carboxylated but
underphosphorylated MGP, phosphorylated but undercarboxylated MGP,
and completely inactivated dephosphorylated and undercarboxylated
MGP (dpucMGP) (Fig. 5A).
MGP has an important role in the activity of bone
cells, bone calcification and osteoarthritis (OA). MGP gene
mutations and the long-term use of VK antagonists are associated
with an increased risk of OA and a decrease in BMD (75,76). Osteoporosis was found in MGP
knockout mice because of osteoblast dysfunction. Recently, Laurent
et al (77) demonstrated
that the femur size of MGP-deficient mice was smaller in the early
stages of life. The Wnt/β-Catenin signaling pathway is important in
osteoblast differentiation, promoting the expression of osteogenic
differentiation genes through RUNX2. Zhang et al (78) demonstrated that MGP promotes the
proliferation and mineralization of MG63 osteoblasts and improves
osteoporosis caused by ovariectomy through the Wnt by/β-Catenin
signaling pathway. The effect of MGP on bone cells is shown in
Fig. 5B. A high-fat diet of
pregnant mice resulted in a decrease in bone structure of their
offspring 6 weeks after birth, which is thought to be related to
the level of MGP gene expression (79). Further experiments in MGP
knockout mice confirmed the role of MGP in high-fat diet-induced
bone mass loss (80).
Conversely, another view is that MGP is a bone calcification
inhibitor. MGP inhibits bone matrix formation and hydroxyapatite
deposition (81). MGP inhibits
the formation and function of osteoclasts through nuclear factor of
activated T cells, cytoplasmic 1 and steroid receptor
coactivator/ras-related C3 botulinum toxin substrate 1 signaling
(82). Certain studies have
indicated that overexpression of MGP inhibits cartilage
mineralization and endochondral ossification. MGP-deficient mice
present with inappropriate calcification of cartilage, short
stature, osteopenia and fractures (83). In addition, the expression of MGP
in chondrocytes is regulated by extracellular inorganic phosphorus,
which may represent a feedback regulatory mechanism to inhibit
inappropriate calcification in cartilage (84). Of note, ectopic expression of MGP
in human osteosarcoma (OS) cells significantly increased cancer
cell metastasis to the lung in patients with OS, which may lead to
poor prognosis (88). In
general, MGP maintains bone health by affecting cartilage
mineralization, osteoblast function and osteoclast activity.
MGP was the first identified vascular calcification
inhibitor, which combines with Ca/P to prevent its deposition in
blood vessels. Several studies have indicated that the serum level
of MGP is associated with vascular diseases. A prospective study
reported that the levels of plasma dpucMGP were strongly associated
with vascular death, from an average of 15.5 years of follow-up
among 684 elderly patients aged 50-89 years (86). In an observational study of 7,066
adults, a significant association between high levels of ucMGP and
arterial stiffness was observed. In a subsequent experimental
study, it was reported that MGP heterozygous mice showed arterial
stiffness (87). In animal
studies, MGP-deficient mice showed severe vascular calcification
and premature bone mineralization, and died within two months
because of vascular rupture caused by arterial calcification
(83). In addition, rats treated
with warfarin for a long time developed extensive vascular
calcification, indicating that carboxylation of MGP has a key role
in inhibiting vascular calcification. However, a recent study
suggested something different. Parashar et al (88) constructed MGP mutant mice and
found that the serine residues at the MGP N-terminus and glutamate
at the C-terminus had a synergistic effect to inhibit vascular
tissue calcification; however, the serine residues had a more
critical role. In addition, a study used immunohistochemistry to
confirm that cMGP and pMGP have anti-calcification effects in veins
(89). Based on its high
anti-calcification ability, MGP has been considered an agent with
great potential in preventing calcic aortic valve disease (90).
The polymorphism of the MGP gene is related to
kidney stones in Chinese Han and Japanese populations (91,92). Li et al (43) confirmed that VK1
reduced the crystal deposition in HK2 cells by promoting MGP
expression. In rats with hyperoxaluria induced by 0.75% ethylene
glycol, crystals were only deposited in the damaged renal tubules
lacking MGP expression, indicating that MGP has a protective role
in maintaining cell survival and inhibiting crystal retention
(93). Goiko et al
(94) detected the effect of MGP
on hydroxyapatite formation and calcium oxalate crystallization
using dynamic light scattering and scanning electron microscopy.
The results showed that MGP inhibited the formation of calcium
oxalate monohydrate, whether the polypeptide was in the modified or
unmodified form after translation. In addition, high concentrations
of Ca significantly inhibited the expression of MGP and then
promoted the mineralization of NRK-52E cells. However, Castiglione
et al (95) found no
significant difference in serum dpucMGP levels among 498 cases of
calculous formers and 395 cases of non-calculous formers after the
evaluation of symptomatic patients with recurrent kidney stones
within 5 years, indicating that the increase in serum dpucMGP was
not related to recurrent renal stone events.
The role of GRP in bone is controversial in
different species. GRP gene deficiency experiments in zebrafish
showed that a lack of GRP and inhibition of GRP carboxylation
resulted in severe growth retardation and bone development
disorder, which indicated the important role of GRP in bone
development (103). However,
this was not observed in mice. GRP-deficient mice did not show any
obvious defects in bone and cartilage, indicating that GRP was not
necessary for mouse bone development (Fig. 6A). In cells, previous studies
reported that GRP is an inhibitor of osteogenic differentiation;
however, two studies by Lee et al (104,105) found the opposite result, as
they suggested that GRP is regulated by RUNX2 and OSX (osteogenic
differentiation proteins) and recombinant GRP promotes MC3T3 cell
osteogenic differentiation and mineralization, suggesting that it
is a candidate bone mineralization promoter (Fig. 6A). OA is a movement-limiting
joint disease and is characterized by loss of articular cartilage,
tissue inflammation, abnormal bone formation and extracellular
matrix mineralization. Studies have reported that the serum GRP
concentration is positively correlated with OA; however, it may be
a compensatory response mechanism. ucGRP/GRP reduced the mineral
deposition of chondrocytes and synovial cells derived from OA,
which may be associated with inhibition of aggregase activity and
the anti-inflammatory effect of GRP with or without γ-carboxylation
(106). Thus, GRP has been
proposed as a potential therapeutic candidate in OA (106).
Gas6, a secreted protein with a high degree of amino
acid conservation, was found in mouse fibroblasts (112), and was then successively
identified in myeloid progenitor cells, endothelial cells, vascular
smooth muscle cells and macrophages. The Gas6 gene, containing 15
exons, was isolated from the human chromosomal location 13q34
(113). Gas6 has high homology
(44%) with anticoagulant protein S, including an extensively
γ-carboxylated amino-terminal, four epidermal growth factor-like
motifs and a large carboxy-terminal region, known as the D domain;
however, it has no direct effect on blood coagulation (112). Gas6 has a Gla amino acid domain
at its N-terminus and is a member of the VKDPs. Although Gla
residues are important for Gas6 binding to its receptor, Gas6 does
not act as a promoter or inhibitor of calcification by combining
with Ca/P, as would have been expected. Gas6 is a ligand for
tyrosine kinase receptors, including tyrosine-protein kinase
receptor 3 (Tyro3), AXL receptor tyrosine kinase (Axl) and c-mer
proto-oncogene tyrosine kinase (Mer). Axl has the highest affinity
for Gas6 among the three receptors (112). Gas6 is mainly involved in the
pathogenesis of inflammation, atherosclerosis and cancer through
the Axl receptor. In addition, Gas6 is also considered a potential
target for the treatment of malignant tumors because of the role of
the Axl receptor in a variety of human malignant tumors (114).
Gas6 promotes the absorptive activity of osteoclasts
by promoting the autophosphorylation of the Tyro3 receptor on
osteoclasts (115). The GAS6
mRNA level in bone marrow increased after ovariectomy, suggesting
that it may be related to bone loss caused by estrogen deficiency
(115). However, this is
inconsistent with the treatment of osteoporosis in postmenopausal
women with VK. Gas6 inhibited the mRNA expression of collagen 2 and
aggrecan, which are the building blocks of cartilage. In addition,
Gas6 inhibits chondrocyte differentiation through the Erk1/2 and
serine/threonine kinase 39/JNK pathways (115). However, Hutchison et al
(116) reported the opposite
result: Collagen type II α1 chain (Col2a1) was upregulated after
long-term Gas6 treatment, whereas short-term Gas6 treatment
decreased the Col2a1 level.
Pericytes, a component of the microvasculature, have
a potential role in osteogenic differentiation (117), and are similar to bone marrow
stromal cells. An in vitro study found that inhibition of
the Gas6/Axl signaling pathway increased the rate of pericytes,
which could be alleviated using recombinant Gas6 (118). Inflammation, age,
hyperphosphatemia and CKD are the main factors of vascular
calcification. Gas6 has a role in the antagonism of adiponectin
toward TNF, thereby inducing vascular calcification (119). Postmenopausal women have a
higher prevalence of vascular calcification, indicating the
protective effect of estrogen. Nanao-Hamai et al (120) reported that estradiol inhibits
vascular smooth muscle cell calcification through estrogen receptor
α-mediated transactivation of Gas6. In older men, testosterone
levels decrease with age. There is an association between
testosterone levels and atherosclerosis (120). Testosterone delays the
progression of vascular smooth muscle cells through the Gas6/Axl
axis (122). The apoptosis of
aortic smooth muscle cells induced by high phosphorus is a major
inducer of vascular calcification. The level of Gas6 decreased in
apoptotic cells and restoring the Gas6/Axl signaling axis could
alleviate calcification through an anti-apoptotic effect, which is
the mechanism by which VK2 inhibits vascular smooth muscle cell
calcification (123). In
addition, studies have shown that several drugs inhibit vascular
smooth muscle cell calcification via this mechanism, such as
statins (124), and α-lipoic
acid and taurine (125). In
general, Gas6 can resist vascular mineralization complications
mainly by activating Axl downstream signals. However, the Ca
contents of wild-type and Gas6-/-mouse aortas were similar
(125). This indicated that
Gas6/Axl may have other, as-yet-unknown mechanisms in pathological
conditions, which require further investigation.
In general, VK and VKDPs have an important role in
biomineralization; however, their involvement and mechanisms are
complex and controversial. Clinical investigations of the effect of
VK on bone quality and cardiovascular health in different regions
reported inconsistent results, which may be related to different
dietary habits, geographical environments and genetic backgrounds
(20,34,126). In addition, different VKDPs
have opposite effects on the same tissue, which may also be one of
the reasons for the different results of clinical investigations of
VK on bone quality (VK deficiency or supplementation affects the
carboxylation of total VKDPs). Similarly, in animal studies on the
effects of VKDPs on bone quality, a VKDP may have different or even
opposite results for the same tissue under different genetic
backgrounds. Therefore, when studying the effect of VKDPs on bone
quality, it is necessary to select experimental animals with an
appropriate genetic background. In addition, warfarin exhibits wide
inter-individual differences in its pharmacodynamic effects,
resulting from polymorphisms in genes involved in the uptake of VK,
including ApoE, VKORC1 and GGCX (127,128). Without any doubt, these gene
polymorphisms also result in individual differences in the quality
of bone and blood vessels under circumstances such as VK deficiency
or warfarin treatment.
Not applicable.
MZ collected the data and wrote the manuscript. QZ
and PD selected and classified studies in the literature search. YZ
was involved in the study design. YZ and XC gave important
suggestions and revised the manuscript during the writing process.
All authors have read and approved the final manuscript. Data
authentication is not applicable.
Not applicable.
Not applicable.
The authors declare that they have no competing
interests.
Not applicable.
This work was supported by the Priority Academic Program
Development of Jiangsu Higher Education Institutions.
|
1
|
Guibert C and Landoulsi J: Enzymatic
approach in calcium phosphate biomineralization: A contribution to
reconcile the physicochemical with the physiological view. Int J
Mol Sci. 22:129572021.
|
|
2
|
Arnold A, Dennison E, Kovacs CS, Mannstadt
M, Rizzoli R, Brandi ML, Clarke B and Thakker RV: Hormonal
regulation of biomineralization. Nat Rev Endocrinol. 17:261–275.
2021.
|
|
3
|
Tang S, Dong Z, Ke X, Luo J and Li J:
Advances in biomineralization-inspired materials for hard tissue
repair. Int J Oral Sci. 13:422021.
|
|
4
|
Villa-Bellosta R: Vascular calcification:
Key roles of phosphate and pyrophosphate. Int J Mol Sci.
22:135362021.
|
|
5
|
Ziemińska M, Sieklucka B and Pawlak K:
Vitamin K and D supplementation and bone health in chronic kidney
disease-apart or together? Nutrients. 13:8092021.
|
|
6
|
Bouillon R, Marcocci C, Carmeliet G, Bikle
D, White JH, Dawson-Hughes B, Lips P, Munns CF, Lazaretti-Castro M,
Giustina A and Bilezikian J: Skeletal and extraskeletal actions of
vitamin D: Current evidence and outstanding questions. Endocr Rev.
40:1109–1151. 2019.
|
|
7
|
Brzezińska O, Łukasik Z, Makowska J and
Walczak K: Role of vitamin C in osteoporosis development and
treatment-a literature review. Nutrients. 12:23942020.
|
|
8
|
Jin C, Tan K, Yao Z, Lin BH, Zhang DP,
Chen WK, Mao SM, Zhang W, Chen L, Lin Z, et al: A novel
anti-osteoporosis mechanism of VK2: Interfering with ferroptosis
via AMPK/SIRT1 pathway in type 2 diabetic osteoporosis. J Agric
Food Chem. 71:2745–2761. 2023.
|
|
9
|
Wang H, Li L, Zhang N and Ma Y: Vitamin K2
improves osteogenic differentiation by inhibiting STAT1 via the
Bcl-6 and IL-6/JAK in C3H10 T1/2 clone 8 cells. Nutrients.
14:29342022.
|
|
10
|
Akbulut AC, Wasilewski GB, Rapp N, Forin
F, Singer H, Czogalla-Nitsche KJ and Schurgers LJ: Menaquinone-7
supplementation improves osteogenesis in pluripotent stem cell
derived mesenchymal stem cells. Front Cell Dev Biol.
8:6187602021.
|
|
11
|
Stock M and Schett G: Vitamin K-dependent
proteins in skeletal development and disease. Int J Mol Sci.
22:93282021.
|
|
12
|
Mladěnka P, Macáková K, Kujovská Krčmová
L, Javorská L, Mrštná K, Carazo A, Protti M, Remião F and Nováková
L; OEMONOM researchers and collaborators: Vitamin K-sources,
physiological role, kinetics, deficiency, detection, therapeutic
use, and toxicity. Nutr Rev. 80:677–698. 2022.
|
|
13
|
National Research Council: Dietary
reference intakes for vitamin A, vitamin K, arsenic, boron,
chromium, copper, iodine, iron, manganese, molybdenum, nickel,
silicon, vanadium, and zinc. National Academy Press; Washington,
DC, USA: pp. 162–196. 2000
|
|
14
|
World Health Organization and Food and
Agriculture Organization of the United Nations: Vitamin K. Vitamin
and mineral requirements in human nutrition. 2nd. World Health
Organization; Geneva, Switzerland: pp. 108–129. 2004
|
|
15
|
Takada T, Yamanashi Y, Konishi K, Yamamoto
T, Toyoda Y, Masuo Y, Yamamoto H and Suzuki H: NPC1L1 is a key
regulator of intestinal vitamin K absorption and a modulator of
warfarin therapy. Sci Transl Med. 7:275ra232015.
|
|
16
|
Matsuo M, Ogata Y, Yamanashi Y and Takada
T: ABCG5 and ABCG8 Are involved in vitamin K transport. Nutrients.
15:9982023.
|
|
17
|
Lai Y, Masatoshi H, Ma Y, Guo Y and Zhang
B: Role of vitamin K in intestinal health. Front Immunol.
12:7915652022.
|
|
18
|
Welsh J, Bak MJ and Narvaez CJ: New
insights into vitamin K biology with relevance to cancer. Trends
Mol Med. 28:864–881. 2022.
|
|
19
|
Sultana H, Komai M and Shirakawa H: The
role of vitamin K in cholestatic liver disease. Nutrients.
13:25152021.
|
|
20
|
Kaesler N, Schurgers LJ and Floege J:
Vitamin K and cardiovascular complications in chronic kidney
disease patients. Kidney Int. 100:1023–1036. 2021.
|
|
21
|
Regulska-Ilow B, Różańska D, Zatońska K
and Szuba A: Estimation of vitamin K content and its sources in the
diet of the polish participants of the PURE study. Nutrients.
14:19172022.
|
|
22
|
Mishima E, Ito J, Wu Z, Nakamura T, Wahida
A, Doll S, Tonnus W, Nepachalovich P, Eggenhofer E, Aldrovandi M,
et al: A non-canonical vitamin K cycle is a potent ferroptosis
suppressor. Nature. 608:778–783. 2022.
|
|
23
|
Shearer MJ and Okano T: Key pathways and
regulators of vitamin K function and intermediary metabolism. Annu
Rev Nutr. 38:127–151. 2018.
|
|
24
|
Dahms SO, Demir F, Huesgen PF, Thorn K and
Brandstetter H: Sirtilins-the new old members of the vitamin
K-dependent coagulation factor family. J Thromb Haemost.
17:470–481. 2019.
|
|
25
|
Fusaro M, Tripepi G, Plebani M, Politi C,
Aghi A, Taddei F, Schileo E, Zaninotto M, La Manna G, Cianciolo G,
et al: The vessels-bone axis: Iliac artery calcifications,
vertebral fractures and vitamin K from VIKI study. Nutrients.
13:35672021.
|
|
26
|
Nalevaiko JZ, Marques JVO, Oliveira MF,
Raetsch AWP, Marques GL, Petterle RR, Moreira CA and Borba VZC:
Bone density and quality in patients treated with direct-acting
oral anticoagulants versus warfarin. Bone. 150:1160002021.
|
|
27
|
Poterucha TJ and Goldhaber SZ: Warfarin
and vascular calcification. Am J Med. 129:635.e1–e4. 2016.
|
|
28
|
Tantisattamo E, Han KH and O'Neill WC:
Increased vascular calcification in patients receiving warfarin.
Arterioscler Thromb Vasc Biol. 35:237–242. 2015.
|
|
29
|
Verma D, Kumar R, Pereira RS, Karantanou
C, Zanetti C, Minciacchi VR, Fulzele K, Kunz K, Hoelper S,
Zia-Chahabi S, et al: Vitamin K antagonism impairs the bone marrow
microenvironment and hematopoiesis. Blood. 134:227–238. 2019.
|
|
30
|
Vimalraj S: Alkaline phosphatase:
Structure, expression and its function in bone mineralization.
Gene. 754:1448552020.
|
|
31
|
Murshed M: Mechanism of bone
mineralization. Cold Spring Harb Perspect Med. 8:a0312292018.
|
|
32
|
Ma ML, Ma ZJ, He YL, Sun H, Yang B, Ruan
BJ, Zhan WD, Li SX, Dong H and Wang YX: Efficacy of vitamin K2 in
the prevention and treatment of postmenopausal osteoporosis: A
systematic review and meta-analysis of randomized controlled
trials. Front Public Health. 10:9796492022.
|
|
33
|
Jadhav N, Ajgaonkar S, Saha P, Gurav P,
Pandey A, Basudkar V, Gada Y, Panda S, Jadhav S, Mehta D and Nair
S: Molecular pathways and roles for vitamin K2-7 as a
health-beneficial nutraceutical: Challenges and opportunities.
Front Pharmacol. 13:8969202022.
|
|
34
|
Salma, Ahmad SS, Karim S, Ibrahim IM,
Alkreathy HM, Alsieni M and Khan MA: Effect of vitamin K on bone
mineral density and fracture risk in adults: Systematic review and
meta-analysis. Biomedicines. 10:10482022.
|
|
35
|
Knapen MHJ, Drummen NE, Smit E, Vermeer C
and Theuwissen E: Three-year low-dose menaquinone-7 supplementation
helps decrease bone loss in healthy postmenopausal women.
Osteoporos Int. 24:2499–2507. 2013.
|
|
36
|
Li X, Yang HY and Giachelli CM: BMP-2
promotes phosphate uptake, phenotypic modulation, and calcification
of human vascular smooth muscle cells. Atherosclerosis.
199:271–277. 2008.
|
|
37
|
Ciccarelli G, Conte S, Cimmino G, Maiorano
P, Morrione A and Giordano A: Mitochondrial dysfunction: The hidden
player in the pathogenesis of atherosclerosis? Int J Mol Sci.
24:10862023.
|
|
38
|
Rao Z, Zheng Y, Xu L, Wang Z, Zhou Y, Chen
M, Dong N, Cai Z and Li F: Endoplasmic reticulum stress and
pathogenesis of vascular calcification. Front Cardiovasc Med.
9:9180562022.
|
|
39
|
Siltari A and Vapaatalo H: Vascular
calcification, vitamin K and warfarin therapy-possible or plausible
connection? Basic Clin Pharmacol Toxicol. 122:19–24. 2018.
|
|
40
|
Kosciuszek ND, Kalta D, Singh M and
Savinova OV: Vitamin K antagonists and cardiovascular
calcification: A systematic review and meta-analysis. Front
Cardiovasc Med. 9:9385672022.
|
|
41
|
Levy DS, Grewal R and Le TH: Vitamin K
deficiency: an emerging player in the pathogenesis of vascular
calcification and an iatrogenic consequence of therapies in
advanced renal disease. Am J Physiol Renal Physiol. 319:F618–F623.
2020.
|
|
42
|
Shioi A, Morioka T, Shoji T and Emoto M:
The inhibitory roles of vitamin K in progression of vascular
calcification. Nutrients. 12:5832020.
|
|
43
|
Li Y, Lu X, Yang B, Mao J, Jiang S, Yu D,
Pan J, Cai T, Yasui T and Gao B: Vitamin K1 inhibition of renal
crystal formation through matrix Gla protein in the kidney. Kidney
Blood Press Res. 44:1392–1403. 2019.
|
|
44
|
Hu B, Wang T, Liu Z, Guo X, Yang J, Liu J,
Wang S and Ye Z: Decreased expression of vitamin K epoxide
reductase complex subunit 1 in kidney of patients with calcium
oxalate urolithiasis. J Huazhong Univ Sci Technolog Med Sci.
31:807–814. 2011.
|
|
45
|
Hewett-Emmett D: Amino acid sequence
homology and the vitamin K-dependent proteins. Bibl Haematol.
44:94–104. 1977.
|
|
46
|
Barille S, Pellat-Deceunynck C, Bataille R
and Amiot M: Ectopic secretion of osteocalcin, the major
non-collagenous bone protein, by the myeloma cell line NCI-H929. J
Bone Miner Res. 11:466–471. 1996.
|
|
47
|
Cancela ML, Laizé V and Conceição N:
Matrix Gla protein and osteocalcin: From gene duplication to
neofunctionalization. Arch Biochem Biophys. 561:56–63. 2014.
|
|
48
|
Hauschka PV, Lian JB, Cole DE and Gundberg
CM: Osteocalcin and matrix Gla protein: Vitamin K-dependent
proteins in bone. Physiol Rev. 69:990–1047. 1989.
|
|
49
|
Xu Y, Shen L, Liu L, Zhang Z and Hu W:
Undercarboxylated osteocalcin and its associations with bone
mineral density, bone turnover markers, and prevalence of
osteopenia and osteoporosis in chinese population: A
cross-sectional study. Front Endocrinol (Lausanne).
13:8439122022.
|
|
50
|
Li R, Zhu X, Zhang M, Zong G and Zhang K:
Association of serum periostin level with classical bone turnover
markers and bone mineral density in Shanghai Chinese postmenopausal
women with osteoporosis. Int J Gen Med. 14:7639–7646. 2021.
|
|
51
|
Lateef M, Baig M and Azhar A: Estimation
of serum osteocalcin and telopeptide-C in postmenopausal
osteoporotic females. Osteoporos Int. 21:751–755. 2010.
|
|
52
|
Bailey S, Poundarik AA, Sroga GE and
Vashishth D: Structural role of osteocalcin and its modification in
bone fracture. Appl Phys Rev. 10:0114102023.
|
|
53
|
Kavukcuoglu NB, Patterson-Buckendahl P and
Mann AB: Effect of osteocalcin deficiency on the nanomechanics and
chemistry of mouse bones. J Mech Behav Biomed Mater. 2:348–354.
2009.
|
|
54
|
Ducy P, Desbois C, Boyce B, Pinero G,
Story B, Dunstan C, Smith E, Bonadio J, Goldstein S, Gundberg C, et
al: Increased bone formation in osteocalcin-deficient mice. Nature.
382:448–452. 1996.
|
|
55
|
Bucay N, Sarosi I, Dunstan CR, Morony S,
Tarpley J, Capparelli C, Scully S, Tan HL, Xu W, Lacey DL, et al:
Osteoprotegerin-deficient mice develop early onset osteoporosis and
arterial calcification. Genes Dev. 12:1260–1268. 1988.
|
|
56
|
Berezovska O, Yildirim G, Budell WC,
Yagerman S, Pidhaynyy B, Bastien C, van der Meulen MCH and Dowd TL:
Osteocalcin affects bone mineral and mechanical properties in
female mice. Bone. 128:1150312019.
|
|
57
|
Hosseini S, Naderi-Manesh H, Vali H,
Baghaban Eslaminejad M, Azam Sayahpour F, Sheibani S and Faghihi S:
Contribution of osteocalcin-mimetic peptide enhances osteogenic
activity and extracellular matrix mineralization of human
osteoblast-like cells. Colloids Surf B Biointerfaces. 173:662–671.
2019.
|
|
58
|
Tsao YT, Huang YJ, Wu HH, Liu YA, Liu YS
and Lee OK: Osteocalcin Mediates biomineralization during
osteogenic maturation in human mesenchymal stromal cells. Int J Mol
Sci. 18:1592017.
|
|
59
|
Gössl M, Mödder UI, Atkinson EJ, Lerman A
and Khosla S: Osteocalcin expression by circulating endothelial
progenitor cells in patients with coronary atherosclerosis. J Am
Coll Cardiol. 52:1314–1325. 2008.
|
|
60
|
Flammer AJ, Gössl M, Widmer RJ, Reriani M,
Lennon R, Loeffler D, Shonyo S, Simari RD, Lerman LO, Khosla S and
Lerman A: Osteocalcin positive CD133+/CD34-/KDR+ progenitor cells
as an independent marker for unstable atherosclerosis. Eur Heart J.
33:2963–2939. 2012.
|
|
61
|
Pal SN, Rush C, Parr A, Van Campenhout A
and Golledge J: Osteocalcin positive mononuclear cells are
associated with the severity of aortic calcification.
Atherosclerosis. 210:88–93. 2010.
|
|
62
|
Shen Y, Chen L, Zhou J, Wang C, Gao F, Zhu
W, Hu G, Ma X, Xia H and Bao Y: Low total osteocalcin levels are
associated with all-cause and cardiovascular mortality among
patients with type 2 diabetes: A real-world study. Cardiovasc
Diabetol. 21:982022.
|
|
63
|
Shahrour HE, Al Fahom S, Al-Massarani G,
AlSaadi AR and Magni P: Osteocalcin-expressing endothelial
progenitor cells and serum osteocalcin forms are independent
biomarkers of coronary atherosclerotic disease severit in male and
female patients. J Endocrinol Invest. 45:1173–1180. 2022.
|
|
64
|
Guo X, Li Y, Zhou Y, Zhang C, Liang S,
Zheng Y, Chen X and Cai G: Osteocalcin association with vascular
function in chronic kidney disease. J Clin Hypertens (Greenwich).
24:928–936. 2022.
|
|
65
|
Chai S, Chen Y, Xin S, Yuan N, Liu Y, Sun
J, Meng X and Qi Y: Positive association of leptin and artery
calcification of lower extremity in patients with type 2 diabetes
mellitus: A pilot study. Front Endocrinol (Lausanne).
12:5835752021.
|
|
66
|
Millar SA, John SG, McIntyre CW, Ralevic
V, Anderson SI and O'Sullivan SE: An investigation into the role of
osteocalcin in human arterial smooth muscle cell calcification.
Front Endocrinol (Lausanne). 11:3692020.
|
|
67
|
Keryakos HKH, Okaily NI, Boulis MAY and
Salama AMS: Osteocalcin and vascular calcification in hemodialysis
patients: An observational cohort study. Int Urol Nephrol.
53:1015–1023. 2021.
|
|
68
|
Hwang YC, Kang M, Cho IJ, Jeong IK, Ahn
KJ, Chung HY and Lee MK: Association between the circulating total
osteocalcin level and the development of cardiovascular disease in
middle-aged men: A mean 8.7-year longitudinal follow-up study. J
Atheroscler Thromb. 22:136–143. 2015.
|
|
69
|
Millar SA, Anderson SI and O'sullivan SE:
Human vascular cell responses to the circulating bone hormone
osteocalcin. J Cell Physiol. 234:21039–21048. 2019.
|
|
70
|
Huang L, Yang L, Luo L, Wu P and Yan S:
Osteocalcin improves metabolic profiles, body composition and
arterial stiffening in an induced diabetic rat model. Exp Clin
Endocrinol Diabetes. 125:234–240. 2017.
|
|
71
|
Dou J, Li H, Ma X, Zhang M, Fang Q, Nie M,
Bao Y and Jia W: Osteocalcin attenuates high fat diet-induced
impairment of endothelium-dependent relaxation through
Akt/eNOS-dependent pathway. Cardiovasc Diabetol. 13:742014.
|
|
72
|
Price PA and Williamson MK: Primary
structure of bovine matrix Gla protein, a new vitamin K-dependent
bone protein. J Biol Chem. 260:14971–14975. 1985.
|
|
73
|
Cancela L, Hsieh CL, Francke U and Price
PA: Molecular structure, chromosome assignment, and promoter
organization of the human matrix Gla protein gene. J Biol Chem.
265:15040–15048. 1990.
|
|
74
|
Price PA, Rice JS and Williamson MK:
Conserved phosphorylation of serines in the Ser-X-Glu/Ser(P)
sequences of the vitamin K-dependent matrix Gla protein from shark,
lamb, rat, cow, and human. Protein Sci. 3:822–830. 1994.
|
|
75
|
Boer CG, Szilagyi I, Nguyen NL, Neogi T,
Meulenbelt I, Ikram MA, Uitterlinden AG, Bierma-Zeinstra S,
Stricker BH and van Meurs JB: Vitamin K antagonist anticoagulant
usage is associated with increased incidence and progression of
osteoarthritis. Ann Rheum Dis. 80:598–604. 2021.
|
|
76
|
Houtman E, Coutinho de Almeida R,
Tuerlings M, Suchiman HED, Broekhuis D, Nelissen RGHH, Ramos YFM,
van Meurs JBJ and Meulenbelt I: Characterization of dynamic changes
in matrix Gla protein (MGP) gene expression as function of genetic
risk alleles, osteoarthritis relevant stimuli, and the vitamin K
inhibitor warfarin. Osteoarthritis Cartilage. 29:1193–1202.
2021.
|
|
77
|
Laurent C, Marano A, Baldit A, Ferrari M,
Perrin JC, Perroud O, Bianchi A and Kempf H: A preliminary study
exploring the mechanical properties of normal and Mgp-deficient
mouse femurs during early growth. Proc Inst Mech Eng H.
236:1106–1117. 2022.
|
|
78
|
Zhang J, Ma Z, Yan K, Wang Y, Yang Y and
Wu X: Matrix Gla protein promotes the bone formation by
up-regulating Wnt/β-catenin signaling pathway. Front Endocrinol
(Lausanne). 10:8912019.
|
|
79
|
Lanham SS, Cagampang FR and Oreffo ROC:
Maternal high-fat diet and offspring expression levels of vitamin
K-dependent proteins. Endocrinology. 155:4749–4761. 2014.
|
|
80
|
Lanham SA, Cagampang FR and Oreffo ROC:
The influence of a high fat diet on bone and soft tissue formation
in Matrix Gla Protein knockout mice. Sci Rep. 8:36352018.
|
|
81
|
Julien M, Khoshniat S, Lacreusette A,
Gatius M, Bozec A, Wagner EF, Wittrant Y, Masson M, Weiss P, Beck
L, et al: Phosphate-dependent regulation of MGP in osteoblasts:
Role of ERK1/2 and Fra-1. J Bone Miner Res. 24:1856–1868. 2009.
|
|
82
|
Zhang Y, Zhao L, Wang N, Li J, He F, Li X
and Wu S: Unexpected role of matrix Gla protein in osteoclasts:
Inhibiting osteoclast differentiation and bone resorption. Mol Cell
Biol. 39:e00012–19. 2019.
|
|
83
|
Luo G, Ducy P, McKee MD, Pinero GJ, Loyer
E, Behringer RR and Karsenty G: Spontaneous calcification of
arteries and cartilage in mice lacking matrix GLA protein. Nature.
386:78–81. 1997.
|
|
84
|
Julien M, Magne D, Masson M,
Rolli-Derkinderen M, Chassande O, Cario-Toumaniantz C, Cherel Y,
Weiss P and Guicheux J: Phosphate stimulates matrix Gla protein
expression in chondrocytes through the extracellular signal
regulated kinase signaling pathway. Endocrinology. 148:530–537.
2007.
|
|
85
|
Zandueta C, Ormazábal C, Perurena N,
Martínez-Canarias S, Zalacaín M, Julián MS, Grigoriadis AE,
Valencia K, Campos-Laborie FJ, Rivas Jde L, et al: Matrix-Gla
protein promotes osteosarcoma lung metastasis and associates with
poor prognosis. J Pathol. 239:438–449. 2016.
|
|
86
|
Willeit K, Santer P, Tschiderer L,
Pechlaner R, Vermeer C, Willeit J and Kiechl S: Association of
desphospho-uncarboxylated matrix gla protein with incident
cardiovascular disease and all-cause mortality: Results from the
prospective Bruneck study. Atherosclerosis. 353:20–27. 2022.
|
|
87
|
Malhotra R, Nicholson CJ, Wang D,
Bhambhani V, Paniagua S, Slocum C, Sigurslid HH, Lino Cardenas CL,
Li R, Boerboom SL, et al: Matrix Gla protein levels are associated
with arterial stiffness and incident heart failure with preserved
ejection fraction. Arterioscler Thromb Vasc Biol. 42:e61–e73.
2022.
|
|
88
|
Parashar A, Bak K and Murshed M:
Prevention of arterial elastocalcinosis: Differential roles of the
conserved glutamic acid and serine residues of matrix Gla protein.
Arterioscler Thromb Vasc Biol. 42:e155–e167. 2022.
|
|
89
|
Gheorghe SR, Vermeer C, Olteanu G, Silaghi
CN and Crăciun AM: The active isoforms of MGP are expressed in
healthy and varicose veins without calcification. J Clin Med.
10:58962021.
|
|
90
|
Chiyoya M, Seya K, Yu Z, Daitoku K,
Motomura S, Imaizumi T, Fukuda I and Furukawa KI: Matrix Gla
protein negatively regulates calcification of human aortic valve
interstitial cells isolated from calcified aortic valves. J
Pharmacol Sci. 136:257–265. 2018.
|
|
91
|
Lu X, Gao B, Liu Z, Tian X, Mao X,
Emmanuel N, Zhu Q and Xiao C: A polymorphism of matrix Gla protein
gene is associated with kidney stone in the Chinese Han population.
Gene. 511:127–130. 2012.
|
|
92
|
Gao B, Yasui T, Itoh Y, Tozawa K, Hayashi
Y and Kohri K: A polymorphism of matrix Gla protein gene is
associated with kidney stones. J Urol. 177:2361–2365. 2007.
|
|
93
|
Lu X, Gao B, Yasui T, Li Y, Liu T, Mao X,
Hirose M, Wu Y, Yu D, Zhu Q, et al: Matrix Gla protein is involved
in crystal formation in kidney of hyperoxaluric rats. Kidney Blood
Press Res. 37:15–23. 2013.
|
|
94
|
Goiko M, Dierolf J, Gleberzon JS, Liao Y,
Grohe B, Goldberg HA, de Bruyn JR and Hunter GK: Peptides of matrix
Gla protein inhibit nucleation and growth of hydroxyapatite and
calcium oxalate monohydrate crystals. PLoS One. 8:e803442013.
|
|
95
|
Castiglione V, Pottel H, Lieske JC, Lukas
P, Cavalier E, Delanaye P and Rule AD: Evaluation of inactive
matrix-Gla-Protein (MGP) as a biomarker for incident and recurrent
kidney stones. J Nephrol. 33:101–107. 2020.
|
|
96
|
Viegas CS, Simes DC, Laizé V, Williamson
MK, Price PA and Cancela ML: Gla-rich protein (GRP), a new vitamin
K-dependent protein identified from sturgeon cartilage and highly
conserved in vertebrates. J Biol Chem. 283:36655–3664. 2008.
|
|
97
|
Le Jeune M, Tomavo N, Tian TV, Flourens A,
Marchand N, Camuzeaux B, Mallein-Gerin F and Duterque-Coquillaud M:
Identification of four alternatively spliced transcripts of the
Ucma/GRP gene, encoding a new Gla-containing protein. Exp Cell Res.
316:203–215. 2010.
|
|
98
|
Tagariello A, Luther J, Streiter M,
Didt-Koziel L, Wuelling M, Surmann-Schmitt C, Stock M, Adam N,
Vortkamp A and Winterpacht A: Ucma-A novel secreted factor
represents a highly specific marker for distal chondrocytes. Matrix
Biol. 27:3–11. 2008.
|
|
99
|
Cancela ML, Conceição N and Laizé V:
Gla-rich protein, a new player in tissue calcification? Adv Nutr.
3:174–181. 2012.
|
|
100
|
Conceição N, Fazenda C and Cancela ML:
Comparative gene promoter analysis: An in silico strategy to
identify candidate regulatory factors for Gla rich protein. J Appl
Ichthyol. 28:372–376. 2012.
|
|
101
|
Viegas CS, Cavaco S, Neves PL, Ferreira A,
João A, Williamson MK, Price PA, Cancela ML and Simes DC: Gla-rich
protein is a novel vitamin K-dependent protein present in serum
that accumulates at sites of pathological calcifications. Am J
Pathol. 175:2288–2298. 2009.
|
|
102
|
Viegas CS, Herfs M, Rafael MS, Enriquez
JL, Teixeira A, Luís IM, van 't Hoofd CM, João A, Maria VL, Cavaco
S, et al: Gla-rich protein is a potential new vitamin K target in
cancer: Evidences for a direct GRP-mineral interaction. Biomed Res
Int. 2014:3402162014.
|
|
103
|
Neacsu CD, Grosch M, Tejada M, Winterpacht
A, Paulsson M, Wagener R and Tagariello A: Ucmaa (Grp-2) is
required for zebrafish skeletal development. Evidence for a
functional role of its glutamate γ-carboxylation. Matrix Biol.
30:369–378. 2011.
|
|
104
|
Lee YJ, Park SY, Lee SJ, Boo YC, Choi JY
and Kim JE: Ucma, a direct transcriptional target of Runx2 and
Osterix, promotes osteoblast differentiation and nodule formation.
Osteoarthritis Cartilage. 23:1421–1431. 2015.
|
|
105
|
Lee YJ, Ju HY, Park SY, Ihn HJ, Park EK
and Kim JE: Recombinant unique cartilage matrix-associated protein
potentiates osteogenic differentiation and mineralization of
MC3T3-E1 cells. Curr Mol Med. 22:747–754. 2022.
|
|
106
|
Cavaco S, Viegas CS, Rafael MS, Ramos A,
Magalhães J, Blanco FJ, Vermeer C and Simes DC: Gla-rich protein is
involved in the cross-talk between calcification and inflammation
in osteoarthritis. Cell Mol Life Sci. 73:1051–1065. 2016.
|
|
107
|
Bordoloi J, Dihingia A, Kalita J and Manna
P: Implication of a novel vitamin K dependent protein, GRP/Ucma in
the pathophysiological conditions associated with vascular and soft
tissue calcification, osteoarthritis, inflammation, and carcinoma.
Int J Biol Macromol. 113:309–316. 2018.
|
|
108
|
Viegas CSB, Rafael MS, Enriquez JL,
Teixeira A, Vitorino R, Luís IM, Costa RM, Santos S, Cavaco S,
Neves J, et al: Gla-rich protein acts as a calcification inhibitor
in the human cardiovascular system. Arterioscler Thromb Vasc Biol.
35:399–408. 2015.
|
|
109
|
Willems BA, Furmanik M, Caron MMJ, Chatrou
MLL, Kusters DHM, Welting TJM, Stock M, Rafael MS, Viegas CSB,
Simes DC, et al: Ucma/GRP inhibits phosphate-induced vascular
smooth muscle cell calcification via SMAD-dependent BMP signalling.
Sci Rep. 8:49612018.
|
|
110
|
Viegas CSB, Santos L, Macedo AL, Matos AA,
Silva AP, Neves PL, Staes A, Gevaert K, Morais R, Vermeer C, et al:
Chronic kidney disease circulating calciprotein particles and
extracellular vesicles promote vascular calcification: A role for
GRP (Gla-rich protein). Arterioscler Thromb Vasc Biol. 38:575–587.
2018.
|
|
111
|
Viegas CSB, Araújo N, Carreira J, Pontes
JF, Macedo AL, Vinhas M, Moreira AS, Faria TQ, Grenha A, de Matos
AA, et al: Nanoencapsulation of Gla-rich protein (GRP) as a novel
approach to target inflammation. Int J Mol Sci. 23:48132022.
|
|
112
|
Nagata K, Ohashi K, Nakano T, Arita H,
Zong C, Hanafusa H and Mizuno K: Identification of the product of
growth arrest-specific gene 6 as a common ligand for Axl, Sky, and
Mer receptor tyrosine kinases. J Biol Chem. 271:30022–30027.
1996.
|
|
113
|
Muñoz X, Sumoy L, Ramírez-Lorca R, Villar
J, de Frutos PG and Sala N: Human vitamin K-dependent GAS6: Gene
structure, allelic variation, and association with stroke. Hum
Mutat. 23:506–512. 2004.
|
|
114
|
Zhu C, Wei Y and Wei X: AXL receptor
tyrosine kinase as a promising anti-cancer approach: Functions,
molecular mechanisms and clinical applications. Mol Cancer.
18:1532019.
|
|
115
|
Nakamura YS, Hakeda Y, Takakura N, Kameda
T, Hamaguchi I, Miyamoto T, Kakudo S, Nakano T, Kumegawa M and Suda
T: Tyro 3 receptor tyrosine kinase and its ligand, Gas6, stimulate
the function of osteoclasts. Stem Cells. 16:229–238. 1998.
|
|
116
|
Hutchison MR, Bassett MH and White PC:
SCF, BDNF, and Gas6 are regulators of growth plate chondrocyte
proliferation and differentiation. Mol Endocrinol. 24:193–203.
2010.
|
|
117
|
Sweeney MD, Ayyadurai S and Zlokovic BV:
Pericytes of the neurovascular unit: Key functions and signaling
pathways. Nat Neurosci. 19:771–783. 2016.
|
|
118
|
Collett G, Wood A, Alexander MY, Varnum
BC, Boot-Handford RP, Ohanian V, Ohanian J, Fridell YW and Canfield
AE: Receptor tyrosine kinase Axl modulates the osteogenic
differentiation of pericytes. Circ Res. 92:1123–1129. 2003.
|
|
119
|
Son BK and Akishita M: Vascular
calcification and anti-aging. Clin Calcium. 18:912–917. 2008.In
Japanese.
|
|
120
|
Nanao-Hamai M, Son BK, Hashizume T, Ogawa
S and Akishita M: Protective effects of estrogen against vascular
calcification via estrogen receptor α-dependent growth
arrest-specific gene 6 transactivation. Biochem Biophys Res Commun.
480:429–435. 2016.
|
|
121
|
Srinath R, Gottesman RF, Hill Golden S,
Carson KA and Dobs A: Association between endogenous testosterone
and cerebrovascular disease in the ARIC study (atherosclerosis risk
in communities). Stroke. 47:2682–2688. 2016.
|
|
122
|
Son BK, Akishita M, Iijima K, Ogawa S,
Maemura K, Yu J, Takeyama K, Kato S, Eto M and Ouchi Y: Androgen
receptor-dependent transactivation of growth arrest-specific gene 6
mediates inhibitory effects of testosterone on vascular
calcification. J Biol Chem. 285:7537–7544. 2010.
|
|
123
|
Qiu C, Zheng H, Tao H, Yu W, Jiang X, Li
A, Jin H, Lv A and Li H: Vitamin K2 inhibits rat vascular smooth
muscle cell calcification by restoring the Gas6/Axl/Akt
anti-apoptotic pathway. Mol Cell Biochem. 433:149–159. 2017.
|
|
124
|
Kraler S, Blaser MC, Aikawa E, Camici GG
and Lüscher TF: Calcific aortic valve disease: From molecular and
cellular mechanisms to medical therapy. Eur Heart J. 43:683–697.
2022.
|
|
125
|
Kim H, Kim HJ, Lee K, Kim JM, Kim HS, Kim
JR, Ha CM, Choi YK, Lee SJ, Kim JY, et al: α-Lipoic acid attenuates
vascular calcification via reversal of mitochondrial function and
restoration of Gas6/Axl/Akt survival pathway. J Cell Mol Med.
16:273–286. 2012.
|
|
126
|
Hu L, Ji J, Li D, Meng J and Yu B: The
combined effect of vitamin K and calcium on bone mineral density in
humans: A meta-analysis of randomized controlled trials. J Orthop
Surg Res. 16:5922021.
|
|
127
|
Huang SW, Xiang DK, Wu HL, Chen BL, An BQ
and Li GF: Impact of five genetic polymorphisms on inter-individual
variation in warfarin maintenance dose. Zhonghua Yi Xue Yi Chuan
Xue Za Zhi. 28:661–665. 2011.In Chinese.
|
|
128
|
Yuan HY, Chen JJ, Lee MT, Wung JC, Chen
YF, Charng MJ, Lu MJ, Hung CR, Wei CY, Chen CH, et al: A novel
functional VKORC1 promoter polymorphism is associated with
inter-individual and inter-ethnic differences in warfarin
sensitivity. Hum Mol Genet. 14:1745–1751. 2005.
|