Osteoporosis is as a skeletal disorder characterized
by reduced bone mineralization and strength, leading to an
increased risk of fractures (1).
The overall prevalence of osteoporosis worldwide has been estimated
at 18.3%, with an almost 2-fold higher prevalence in females
(23.1%) than males (11.7%) (2).
Osteoporosis is also characterized by high geographic differences,
with the highest prevalence in Africa (26.9%) (3). Yet, even in developed countries,
the economic burden of osteoporosis-related fractures is
significant, with annual costs of 17.9 billion USD and 4 billion
GBP in the USA and UK, respectively (4). The geographic heterogeneity of
osteoporosis is mediated by the distinct prevalence of risk
factors, including genetic patterns, environmental factors,
sedentary lifestyle, smoking, alcohol use, medications
(glucocorticoids), morbidities (hyperparathyroidism, rheumatoid
arthritis, diabetes mellitus, cancer), as well as nutritional
deficiencies (5).
The objective of the present review was to highlight
the molecular mechanisms of the effects of vitamin groups A, C, E,
K and B on bone and their potential role in the development of
osteoporosis. To the best of our knowledge, this is the first
comprehensive review focusing on the association between the intake
of vitamins A, C, E and K, and group B vitamins and osteoporosis
since the article by Ahmadieh and Arabi (16) published over than a decade ago
and focusing mainly on epidemiological data. Since the publication
of the aforementioned study (16) significant progress has been made
in understanding the molecular mechanisms of vitamin functions in
bone has been achieved, while epidemiological studies provided
additional evidence on the association between vitamin status and
osteoporosis. Therefore, in the present review, the role of vitamin
forms and doses and their biological effects on bone tissue are
discussed in detail, with particular focus on the most recent
findings. Given the high prevalence of osteoporosis and vitamin
deficiency worldwide, the further understanding of the role of
vitamins as osteoprotective agents may markedly improve the
prevention of and treatment strategies for osteoporosis, as well as
prevent adverse effects of excessive supplementation.
Vitamin E (VE) is a fat-soluble vitamin with
antioxidant activity that is present in the form of tocopherols
(α-, β-, γ- and δ-) and tocotrienols (α-, β-, γ- and δ-) (17). VE is considered as
bone-protecting due to its complex effects on bone physiology that
are not limited to its antioxidant activity (18). A Mendelian randomization study
demonstrated a significant positive association between circulating
α-tocopherol levels and bone mineral density (BMD) (19). A low serum VE level has been
found to be associated with a reduced BMD, and has therefore been
considered a risk factor for osteoporosis in post-menopausal women
(20).
Correspondingly, low serum α-tocopherol
concentrations have been found to be associated with a 51 and 58%
increase in the hazard ratio of hip fractures in older Norwegians
(21) and Swedes (22). In turn, supplementation with
tocotrienol, a form of VE, for 12 weeks was shown to decrease
oxidative stress and bone resorption in post-menopausal women with
osteopenia (23,24).
Despite a positive association between serum
α-tocopherol and femoral neck BMD observed in the Aberdeen
Prospective Osteoporosis Screening Study, the authors considered
this association to lack biological significance (25). However, the analysis of NHANES
2005-2006 data demonstrated an inverse association between the
serum α-tocopherol levels and femoral neck BMD following adjustment
for confounders (26).
Notably, serum α-tocopherol, but not γ-tocopherol,
has been found to be inversely associated with bone formation
marker, procollagen type 1 amino-terminal propeptide, in
post-menopausal women (27).
These findings generally corroborate the earlier observed inverse
relationship between α-tocopherol intake and γ-tocopherol levels
(28).
In addition, tocotrienol supplementation has been
shown to improve bone calcination in testosterone
deficiency-associated osteoporosis (31). In ovariectomy-induced
osteoporotic fractures, α-tocopherol supplementation has been found
to significantly improve fracture healing, although it does not
increase callous bone volume in rats (32), nor does it improve bone strength
(33). It has been shown that
both an intraperitoneal (34)
and intramuscular (35)
injection with α-tocopherol significantly increases BMD and
osteogenesis, as well as osteoblast activity in a rabbit model of
distraction osteogenesis.
Correspondingly, VE deficiency has been shown to
alter exercise-induced plasma membrane disruptions, membrane repair
and the survival of osteocytes (36). The co-administration of Se and
vitamin C (VC) with VE significantly increases its efficiency in
the improvement of bone structure (37). In turn, excessive VE intake has
failed to induce bone loss in an animal model of
ovariectomy-induced osteoporosis (38), as well as in normal female rats
(39).
The association between VE intake and bone health
established in the aforementioned epidemiological studies is
mediated by the influence of tocopherols and tocotrienols on bone
physiology.
In agreement with the role of VE as an antioxidant,
tocopherol has been shown to promote the osteogenic differentiation
and oxidative stress resistance of rat bone marrow-derived
mesenchymal stem cells by inhibiting
H2O2-induced ferroptosis by increasing the
phosphorylation of PI3K, Akt and mammalian target of rapamycin
(mTOR) (40).
α-tocopherol-stimulated osteoblastogenesis has been shown to be
associated with the upregulation of alkaline phosphatase (ALP)2,
TGF1β, fibroblast growth factor receptor 1, MMP-2, muscle segment
homeobox 2, bone morphogenetic protein (BMP)-1, VEGF-B, Runx2,
Smad2 and other genes, whereas the expression of
osteopetrosis-associated transmembrane protein 1,
microphthalmia-associated transcription factor (MITF) and EGFR
genes is downregulated (41). VE
has been shown to reduce osteocyte apoptosis in a model of
steroid-induced osteonecrosis through inhibition of caspase-3
expression and upregulation of Bcl-2 (42). At the same time, α-tocopherol and
δ-tocopherol may also inhibit osteoblast differentiation from the
early stages of osteogenesis to the osteoid-producing stage
(43). At the same time, both
α-tocopherol (100 and 200 μM) and δ-tocopherol (2 and 20
μM) significantly reduces osteoblast differentiation
(43).
In addition to the promotion of osteoblast
differentiation, tocopherol has been shown to inhibit IL-1-induced
osteoclastogenesis through the downregulation of receptor activator
of nuclear factor kappa-B ligand (RANKL) mRNA expression (44). The VE-induced inhibition of
osteoclastogenesis may also be associated with reduced monocyte and
lymphocyte production (45). In
addition, treatment with 10-20 μM α-tocopherol has been
shown to result in reduced bone mass by upregulating osteoclast
fusion via p38 MAPK and MITF activation (46).
It has also been demonstrated that another form of
VE, tocotrienol, may also significantly modulate bone formation and
resorption (47) in a distinct
manner of that observed for tocopherols (48). γ-tocotrienol significantly
promotes Runx2-dependent osteoblastogenesis with the upregulation
of ALP, osteocalcin (OCN) and type I collagen (49). Annatto-derived tocotrienol has
been found to significantly increase osteoblast differentiation, as
evidenced by increased osterix (OSX), COL1α1, ALP and OCN gene
expression, and enhanced mineralization (50).
Tocotrienol also significantly increases
mineralization in osteoblasts by increasing BMP-2 protein
expression in association with the downregulation of RhoA
activation and HMG-CoA reductase gene expression (51). The tocotrienol-induced
upregulation of BMP-2 and BMP-4 gene expression has also been shown
to be associated with the stimulation of Wnt/β-catenin signaling
(52). d-δ-tocotrienol (0-25
μmol/l) has been shown to induce MC3T3-E1 preosteoblast
differentiation through the upregulation of BMP-2 and the
inhibition of HMG-CoA reductase expression, resulting in
mineralized nodule formation (53).
δ-tocotrienol also promotes osteoblast migration
through an increase in Akt phosphorylation and Wnt/β-catenin
signaling activation (54).
Notably, at low doses, γ-tocotrienol has been shown to exert
protective effects on osteoblasts against
H2O2-induced oxidative stress and apoptosis,
whereas high doses are cytotoxic and induce apoptotic cell death
(55). It has also been
demonstrated that δ-tocotrienol protects osteoblastic MC3T3-E1 and
MLO-Y4 cells from oxidative stress and subsequent apoptosis through
the upregulation of glutathione production and the upregulation of
the PI3K/Akt and nuclear factor-erythroid factor 2-related factor 2
(Nrf2) signaling pathways (56).
The osteogenic effects of γ-tocotrienol on human bone
marrow-derived mesenchymal stem cells have been shown to be
mediated by the promotion of p-AMPK and p-Smad1 phosphorylation
(57).
α-Tocotrienol, but not α-tocopherol, has been shown
to reduce osteoclastogenesis (58) through the inhibition of RANKL
expression along with the downregulation of c-Fos expression
(59). Specifically,
γ-tocotrienol has been shown to inhibit RANKL mRNA expression,
while increasing osteoprotegerin (OPG) mRNA expression in human
bone-derived cells, whereas α-tocopherol is capable of only
upregulating OPG expression (60). Tocotrienol has also been shown to
inhibit IL-17-induced osteoclastogenesis in rheumatoid arthritis
fibroblast-like synoviocytes through the downregulation of mTOR,
ERK and IκB phosphorylation, and the inhibition of RANKL mRNA
expression, while increasing AMPK phosphorylation (61). In a model of metabolic
syndrome-associated osteoporosis supplementation with tocotrienol,
there was a significant reduction in RANKL and FGF-23 expression,
as well as a reduction in Dickkopf-related protein (DKK)-1 levels,
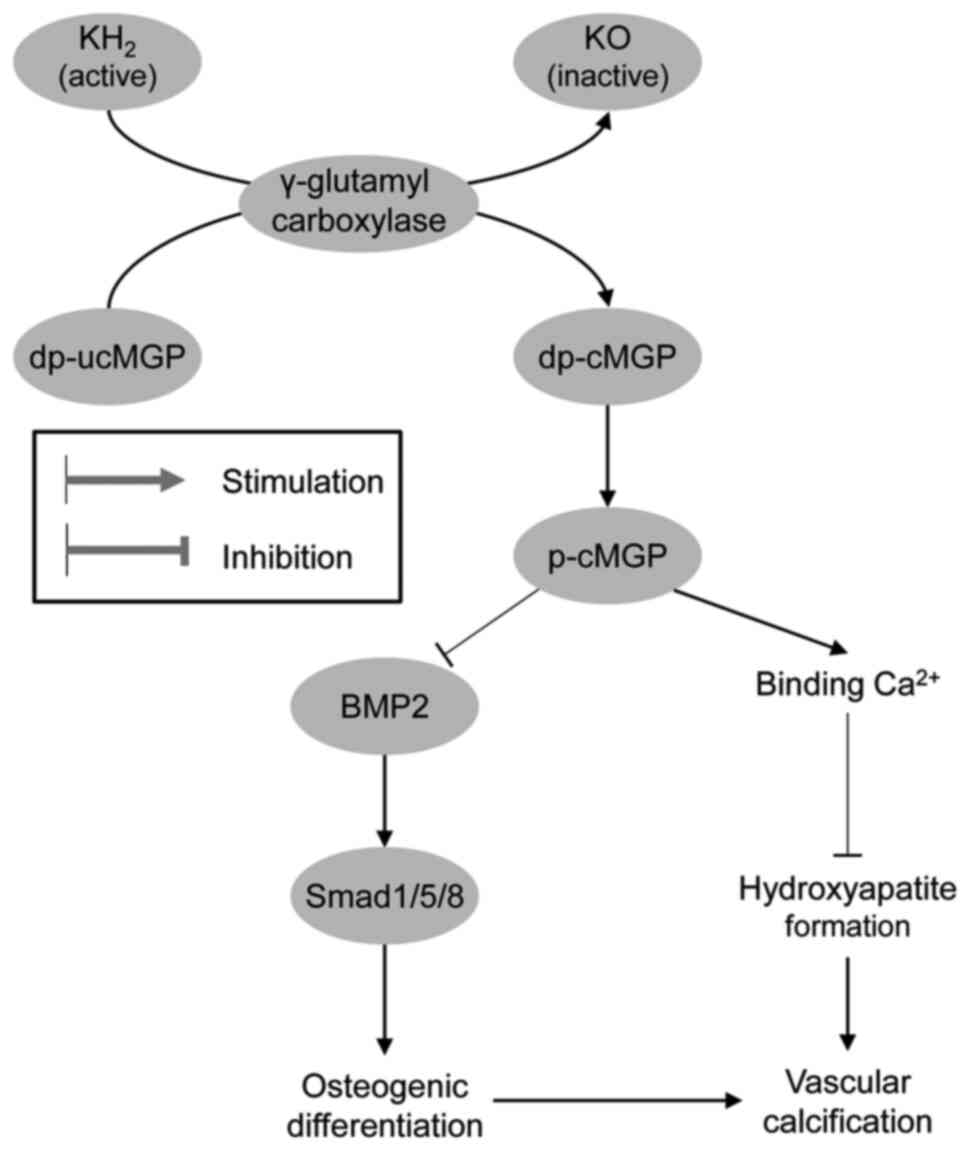
being indicative of Wnt pathway activation (62) (Fig. 1).
Annatto bean-derived tocotrienol has also been shown
to prevent bone resorption in testosterone-deficiency-associated
osteoporosis in rats (63).
γ-tocotrienol also reduces ovariectomy-induced bone loss in mice
through HMG-CoA reductase inhibition (64). Moreover, palm oil-derived
tocotrienols have been shown to prevent bone loss in ovariectomized
rats more effectively than Ca2+ (65). The inhibition of skeletal
sclerostin expression may be also responsible for the
anti-osteoporotic effects of annatto tocotrienol in ovariectomized
rats in parallel with the reduction of the RANKL/OPG ratio
(66). According to the positive
role of tocotrienols in the prevention of bone resorption, these
were considered as the potential treatment strategy for
menopause-associated osteoporosis (67).
In general, VE may be considered as an
osteoprotective agent, although the biological effects are strongly
dependent on the specific forms. Epidemiological studies have
demonstrated that the serum α-tocopherol level is significantly
associated with BMD, whereas its deficiency is related to an
increased risk of fractures, although certain inconsistencies
exist. Both tocopherol and tocotrienol isomers significantly
increase bone quality and promote regeneration in animal models of
osteoporosis. α-tocopherol has been shown to exert osteogenic
effects due to its antioxidant effects, the inhibition of
osteoblast ferroptosis and apoptosis, as well as the activation of
the TGF1β/Smad and PI3K/Akt pathways. Even more potent osteogenic
effects have been demonstrated for tocotrienol that promote BMP-2
and Wnt/β-catenin signaling, also activating Akt and protecting the
cells from oxidative stress and apoptosis. The inhibitory effects
of both tocopherol and tocotrienol on osteoclast formation have
been shown to be mediated by the inhibition of
inflammation-associated RANKL-induced osteoclastogenesis.
Therefore, dietary VE as tocotrienol, has been shown to exert
osteoprotective effects in laboratory studies, although
epidemiological data are available only for tocopherol.
VK has been shown to be a cost-effective strategy
for preventing fractures in older women (70). A recent meta-analysis of 16
randomized controlled trials with 6,425 subjects involved
demonstrated that VK2 supplementation significantly
improved BMD and reduced the risk of fractures (71), as well as undercarboxylated OCN
levels (72) in post-menopausal
women. Similarly, other meta-analyses have demonstrated positive
impact of vitamin K on BMD and fracture risk (73). Correspondingly, in 10-year
follow-up studies, a higher dietary intake of VK was shown to be
associated with a 24% decrease in the relative fracture risk
(74). Each 1 μg/l
increase in serum VK1 (phylloquinone) levels was associated with a
45% reduction in fracture risk in post-menopausal osteoporosis due
to an increase in hip strength (75). However, no significant effects of
phylloquinone intake on bone turnover or bone mass were observed in
adult patients with Crohn's disease (76). In turn, low plasma phylloquinone
levels were associated with a higher incidence of vertebral
fractures, although no significant difference in BMD in subjects
with low and high plasma K1 levels was observed (77). VK intake was also shown to be
inversely associated with undercarboxylated OCN that was negatively
associated with lumbar BMD and was directly interrelated with
urinary type-I collagen cross-linked-N-telopeptide levels, a marker
for bone resorption (78).
A previous meta-analysis demonstrated that the
combination of vitamin D with VK significantly increased total BMD
with the more profound effect observed in VK2 users
(79). The co-supplementation of
phylloquinone with vitamin D3 and calcium has been shown to
increase BMD and bone mineral content (BMC) at the ultradistal
radius (80). The combined
administration of VK and Ca2+ also possessed positive
effect on BMD, as evidenced by a recent meta-analysis (81). Correspondingly, a low dietary
Ca2+ and VK intake was considered a risk factor for
osteoporotic fractures in women (82). In a previous study, a 3-year
low-dose MK-7 supplementation in healthy post-menopausal women
significantly reduced the aging-associated decrease in lumbar spine
and femoral neck BMD and BMC, vertebral height and bone strength
(83). The administration of 375
μg MK-7 for 12 months prevented an increase in trabecular
spacing and the reduction of trabecular number in post-menopausal
women with osteopenia (84). The
results of a 24-month trial demonstrated a significant reduction in
the incidence of fractures in patients with osteoporosis
supplemented with MK-4 when compared to the control groups
(85). Consistently, the results
from a meta-analysis demonstrated that MK-4 intake significantly
improved BMD and decreased the risk for vertebral fractures as
compared to treatment with the placebo (86).
In animal models of osteoporosis, VK has also been
shown to exert osteoprotective effects. Specifically, VK
supplementation was even shown to be more effective in the
improvement of bone characteristics in a model of immobilization
osteoporosis as compared to combined Ca2+ and vitamin D
administration (91). A similar
protective effect of VK2 (menatetrenone) was observed in
a model of glucocorticoid- (92,93) and hyperglycemia-induced (94) bone loss. MK-7 has been shown to
promote diaphyseal and metaphyseal Ca2+ deposition due
to increased osteoblastic proliferation and differentiation
(95). Moreover, MK-7, but not
MK-4 intake, has also bees shown to improve bone microstructure
characterized by higher trabecular number, improved trabecular
architecture and greater bone volume in ovariectomized rats
(96).
The results obtained from laboratory studies are
generally consistent with those from the epidemiological studies,
also demonstrating the osteogenic effects of VK, although the
specific effects and underlying mechanisms have been shown to be
greatly dependent on the forms and homologues of VK.
MK-7 has been shown to promote MC3T3E1 cell
differentiation characterized by an increased OCN, OPG and RANKL
mRNA expression (97).
Menaquinone-7 treatment also increases osteoblast migration and
activity along with the downregulation of Runx2 expression,
indicative of promotion of cell maturation (98). MK-7-induced osteogenesis has also
been found to be associated with a significant increase in BMP-2
mRNA expression, tenascin C gene expression and increased p-Smad1
levels in MC3T3E1 cells (99).
MK-7 promotes vitamin D3-induced osteogenesis that may be at least
partially mediated by the enhanced expression of genes, including
growth differentiation factor-10 (GDF10), IGF1, VEGFA and
fms-related tyrosine kinase 1 (FLT1) (100). Concomitantly,
hydrophobins-modified menaquinone-7 has been shown to be more
effective in increasing osteoblast differentiation, while reducing
osteoclastogenesis in MC3T3-E1 cells, as compared to native MK-7
(101). It has also been
demonstrated that MK-7 inhibits basal and cytokine-induced NF-κB
signaling through an increase in IκB mRNA expression, and
ameliorates TNFα-induced inhibition of SMAD signaling (102). These findings generally
resemble the earlier observed amelioration of inhibitory effect of
inflammation on osteogenesis through down-regulation of
IL-6-induced JAK/STAT signaling upon VK2 treatment
(103).
MK-4 has been shown to be the most potent promotor
of bone formation compared to estrogen, icariin, lactoferrin and
lithium chloride (104). It has
been sown that menaquinone 4 inhibits ovariectomy-induced bone loss
by increasing osteoblast activity with the stimulation of BMP-2 and
Runx2 signaling, and the downregulation of osteoclast
differentiation (105).
Correspondingly, the osteogenic effect of MK-4 has been shown to be
mediated by the activation of the Wnt/β-catenin signaling pathway
(106). In addition to
increased osteoblast proliferation, the osteogenic effect of MK-4
may be associated with the inhibition of Fas-induced osteoblast
apoptosis (107).
Correspondingly, MK-4 also prevents osteoblast apoptosis through
the upregulation of FoxO signaling and the reduction of reactive
oxygen species (ROS) production (108), in agreement with the observed
upregulation of SIRT1 signaling and the inhibition of mitochondrial
dysfunction and endoplasmic reticulum stress (ERS) (109). At the same time, MK-4 reduces
excessive bone mineralization induced by Mg deficiency (110). It is also notable that in
vascular smooth muscle cells, MK-4 reduces
β-glycerophosphate-induced calcification by downregulating BMP-2
and Smad1 expression (111).
Several studies have demonstrated that VK is capable
of inducing osteoblast formation, while inhibiting osteoclast
differentiation and bone-resorbing activity. Specifically, in a
culture of bone marrow cells, MK-4 was found to significantly
inhibit adipogenic and osteoclastogenic differentiation, while
promoting osteoblast differentiation (118). It has been demonstrated that,
in comparison to VK1 and VK3, MK7 and particularly MK4, are more
effective in the promotion of osteoblast activity and the
inhibition of osteoclastic bone resorption (119), although another study
demonstrated a higher anti-osteoclast activity for MK7 (120). Both phylloquinone (VK1) and
menaquinone-4 have been shown to promote osteogenesis, as evidenced
by increased OCN and OPG levels in parallel with decreased
circulating RANKL levels in a model of high-fat-induced obesity
(121). Both MK-4 and VK1
significantly reduce dihydroxyvitamin D3-induced osteoclastogenesis
mainly by reducing RANKL expression (observed at 1.0 μM),
whereas the upregulation of OPG expression has been observed at
higher exposure levels (10 μM) (122). MK-4 also reduced
1,25(OH)2D3-induced formation of multinucleated osteoclasts
(123). It has been also
demonstrated that MK-7 ameliorated parathyroid hormone (PTH) and
prostaglandin E2 (PGE2)-induced bone resorption by osteoclasts
(124,125).
The inhibition of RANKL-induced osteoclastogenesis
by menaquinone 4 and 7 has been found to be dose-dependent
(126). The MK-4-induced
inhibition of RANKL signaling has been shown to result in the
subsequent reduction of nuclear factor of activated T-cells 1
(NFATc1), osteoclast-associated receptor and cathepsin K mRNA
expression (127). In addition
to the downregulation of RANKL signaling, menaquinone 4 or VK1 have
been shown to inhibit macrophage colony stimulating factor
(M-CSF)-induced osteoclast differentiation in a dose-dependent
manner (128).
Although OCN is an abundant protein of bone
extracellular matrix, its functioning has been shown to not be
responsible for the regulation of bone development; rather, it
plays a crucial role in the improvement of bone strength by
adjustment of biological apatite parallel to collagen fibrils
(131), as well as carbohydrate
metabolism regulation in its uncarboxylated form (132). At the same time, the VK-induced
decrease in the level of undercarboxylated OCN did not induce
insulin resistance, and the change in percentage of
undercarboxylated OCN is directly associated with the improvement
of glucose sensitivity (133).
Moreover, VK treatment has been shown to increase OCN gene
expression, resulting in an improvement of β-cell proliferation and
adiponectin production, thus exerting a hypoglycemic effect
(134). In agreement with this,
insulin signaling in osteoblasts has been shown to result in
reduced OCN γ-carboxylation, thus increasing its hypoglycemic
effect (135).
Analogous to OCN, matrix Gla protein has been shown
to be activated by VK-dependent carboxylation and phosphorylation,
exerting a significant inhibitory effect on vascular calcification
(136), and the level of
non-phosphorylated uncarboxylated matrix Gla protein (MGP) may be
considered as a biomarker of VK status (137). Therefore, VK deficiency is
associated with a reduced Ca2+ deposition in bones and
an increase in vascular calcification (138). Menaquinone-4 insufficiency is
also considered as a predictor of aortic calcification (139). Moreover, the administration of
VK antagonists has been shown to significantly increase
dephosphorylated and uncarboxylated matrix Gla protein levels,
which directly correlated with vascular calcification (140). Correspondingly, MK-4 has been
shown to inhibit the osteogenic transdifferentiation of vascular
smooth muscle cells and preserve a contractile phenotype in
spontaneously hypertensive rats (141). Active MGP has been shown to
inhibit osteogenic stimuli by binding to BMP-2 and reducing
mineralization, whereas inactive MGP is unable to inhibit the
osteogenic differentiation of vascular smooth muscle cells
(142). The protective effects
of VK on vascular calcification may also be mediated by its
influence on Gla-rich protein and growth arrest-specific gene 6
protein expression (143)
(Fig. 2).
Taken together, the existing clinical and laboratory
data demonstrate that VK supplementation effectively improves BMD
and reduces the risk of fractures in post-menopausal women. In
addition it enhances the anti-osteoporotic effects of vitamin D and
Ca2+ supplementation. The osteogenic effect of
VK2 as MK4 and MK7 has been shown to be attributed to
the activation of BMP-2 and Wnt/β-catenin signaling, the promotion
of autophagy and the amelioration of the inhibitory effects of
pro-inflammatory cytokines on SMAD signaling. VK2 also
exerts protective effects in osteoblast culture by preventing
apoptosis and ferroptosis. In addition to the osteogenic effect, it
also inhibits bone resorption by inhibiting osteoclastogenesis and
activation by downregulating RANKL signaling with a shift to OPG
activation. VK has also been shown to prevent vascular
calcification by activating MGP, therefore directing
Ca2+ from the vascular wall to its deposition in bones.
Therefore, VK may be considered protective, not only against
osteoporosis, but also vascular calcification and associated
cardiovascular disease.
Vitamin A (VA) has been shown to be involved in the
regulation of bone physiology through retinoic acid receptor (RAR)
signaling (144), although the
existing data on the association between VA intake or accumulation
and BMD remain controversial due to the distinct effects of
different doses (145).
In osteoporotic untreated post-menopausal women,
serum retinol has been shown to be directly associated with the
risk of low bone mass at the lumbar spine and femoral neck
(146). The association between
high retinol levels and osteoporosis is aggravated in subjects with
vitamin D deficiency (147).
Concomitantly, a U-shaped association between the plasma retinol
concentration and BMD has been observed, with both deficiency and
excess being associated with a lower BMD in children (148). These findings generally
corroborate an observation of improved bone formation following the
reduction of VA in children with high vitamin stores (149). The results of a meta-analysis
demonstrated that the intake of VA and retinol, but not β-carotene,
was associated with the risk of hip fractures, although serum
retinol levels were characterized by a U-shaped association with
the risk of hip fractures (14).
It is noteworthy that, in non-supplemented subjects
with a low dietary VA intake, plasma levels of retinol and
carotenoids were inversely associated with osteoporosis (150,151). Moreover, the maternal plasma
retinol level is directly associated with adult offspring spine BMD
and trabecular bone score following adjustment for multiple
covariates, including vitamin D levels (152).
Recent findings have demonstrated that the
association between the VA status and the risk of fractures as the
outcome of osteoporosis is not significant. Specifically, no
association between high serum retinol levels and an increased risk
of fractures was observed in the elderly involved in Norwegian
Epidemiologic Osteoporosis Studies (153). The results of a meta-analysis
demonstrated that an increased VA intake was not associated with a
risk of fractures (154). No
association between VA intake with BMD or the risk of fractures was
observed in pre-menopausal women with a lower baseline VA intake
level (155). It is proposed
that the association between a high VA intake and an increased risk
of fractures may be mediated by an increased body mass index
(156).
The VA status is also tightly associated with the
dietary intake of provitamin A carotenoids that may also have a
significant effect on bone health (157). A high dietary total carotenoid
intake (Q1 vs. Q4) has been shown to be associated with a 39% lower
risk of hip fractures in males, whereas no association was observed
in females (158). Another
study also demonstrated reduced odds of hip fractures with a high
dietary intake of both total carotenoid and individual β-carotene,
β-cryptoxanthin, and lutein/zeaxanthin intake, while the intake of
α-carotene and lycopene was not associated with the risk of hip
fractures (159).
Correspondingly, a meta-analysis of epidemiological studies
involving 140,265 subjects demonstrated that a high total
carotenoid, as well as β-carotene intake was associated with a 28%
lower risk of hip fractures, while no association between
circulating carotenoid and fracture risk was shown (160). Correspondingly, the
meta-analysis by Gao and Zhao (161) demonstrated a significant
association between the dietary β-carotene intake and a reduction
in the risk of developing osteoporosis.
Serum β-cryptoxanthin, lycopene and α-carotene
levels have been found to be associated with a
concentration-dependent increase in BMD in Chinese adults, with a
more pronounced association in females (162). Correspondingly, in the European
Prospective Investigation into Cancer and Nutrition (EPIC)-Norfolk
cohort, plasma α and β-carotene levels were inversely associated
with a risk of hip fractures in males (163).
These findings demonstrate that VA, as well as
provitamin A carotenoid intake is associated with bone health,
although this association appears to be non-linear. Despite being
rather contradictory, the existing epidemiological data demonstrate
that both the deficiency and excess of VA may promote the risk of
bone loss. The laboratory findings also demonstrate that VA
metabolites may possess distinct effect on mechanisms associated
with bone formation.
A number of studies have demonstrated that the VA
metabolite, ATRA, significantly increases osteoblastogenesis and
osteogenesis. Specifically, in rat bone marrow-derived mesenchymal
stem cells, exposure to 10 μM ATRA was shown to promote
osteogenic differentiation through the pregulation of osteogenic
(ALP, BMP-2, OSX, Runx2, OPN and OCN) and angiogenic [VEGF,
hypoxia-inducible factor-1, Fms related receptor tyrosine kinase 3,
angiotensin (ANG)-2 and ANG-4] gene mRNA expression, while in an
in vivo model, ATRA injection (10 μM, 100 μl)
into the distraction gap significantly improved bone consolidation
and its properties (168). The
administration of 10 μm ATRA has been shown to promote the
Wnt3a-induced osteogenic differentiation of mesenchymal stem cells
through the activation of PI3K/AKT/GSK3β pathway (169). Both ATRA and 9-cis retinoic
acid at the concentrations of 5-20 μM have been shown to
promote the in vitro osteogenic differentiation induced by
BMP-9 in mesenchymal progenitor cells (170). The osteogenic differentiation
of retinoic acid-treated murine induced pluripotent stem cells has
also shown to be at least partially mediated by Notch signaling
(171).
It has also been demonstrated that ATRA promotes a
shift from adipogenic to osteogenic differentiation. Specifically,
1 μM retinoic acid-induced osteoblastogenesis and the
inhibition of adipogenesis in mesenchymal stem cells have been
shown to be dependent on Smad3 upregulation with the subsequent
replacement of C/EBPβ from the Runx2 promoter (172), in agreement with earlier
observation of the C/EBPβ-induced inhibition of the 1 μM
ATRA-induced osteoblastogenesis in C3H10T1/2 cells (173). It has been also demonstrated
that 1 μM RA promotes BMP-2-induced osteogenesis, while
inhibiting BMP-2-induced adipogenesis with the suppression of
adipogenic transcription factors, PPARγ and C/EBPs, thus being a
key factor regulating the commitment of mesenchymal stem cells into
osteoblasts and adipocytes (174). Moreover, 2.5 μM retinoic
acid has been shown to enhance the osteogenic effect of BMP-2 in
human adipose-derived stem cells (175). ATRA (1 μM) has been
shown to promote the BMP-9-induced osteogenic transdifferentiation
of 3T3-L1 preadipocytes through the activation of BMP/Smad and
Wnt/β-catenin signaling (176).
It has been shown that 1 μM retinoic acid promotes the
BMP-2-induced osteoblastic differentiation of preadipocytes through
BMP-RIA and BMP-RIB signaling (177). Correspondingly, retinoic acid
has been shown to induce the osteogenic differentiation of stromal
cells from both visceral and subcutaneous adipose tissue depots
(178). Moreover, as previously
demonstrated, in mouse embryonic fibroblasts, 0.4 μM ATRA
promotes a shift to osteogenesis from rosiglitazone-induced
adipogenic differentiation through the upregulation of Smad1/5/8
phosphorylation and Smad6 expression, resulting in the activation
of BMP/Smad pathway (179). At
the same time, it has also been observed that pharmacological
concentrations of 1-10 μM ATRA inhibit osteoblast
proliferation, while increasing its differentiation (180). However, it is notable that
premature osteoblast-to-preosteocyte transitioning induced by 1
μM ATRA may result in altered bone formation (181).
The osteogenic effects of retinoic acid have also
been shown to be associated with RAR activation. Specifically,
retinoic acid (1 μM) has been shown to promote the
osteogenesis of human induced pluripotent stem cells, a process
dependent on RARa and RARb, but not on RARy signaling (182). Correspondingly, it has been
demonstrated that treatment with 20 μM ATRA increases the
spreading of pre-osteoblasts on bio-inert glass surfaces and its
osteogenic activity through RARα and RARβ signaling (183). At the same time, Karakida et
al (184) demonstrated that
ATRA promoted the osteogenic transdifferentiation of myoblastic
C2C12 cells by BMP-2 in a concentration-dependent manner at a range
of 8-2,000 nM, while this effect was ameliorated by RARγ, but not
RARα or RARβ inhibition.
In contrast to previously discussed observations,
several laboratory studies have demonstrated the inhibitory effects
of ATRA on osteogenesis. In particular, 1 μM ATRA has been
shown to inhibit the osteoblastogenesis of the MC3T3-E1
pre-osteoblast cell line (185). Furthermore, 0.5 μM
retinoic acid has been found to significantly inhibit MC-3T3 cell
mineralization through the increased expression of the Wnt
inhibitors, DKK-1 and DKK-2 (186), resulting in the downregulation
of Wnt signaling (187). It has
also been shown that 1 μM ATRA inhibits osteoblastogenesis
induced by BMP-2, BMP-7 or heterodimer BMP-2/7, with the latter
being a more potent activator as compared to homodimers (188). In addition, the inhibition of
the osteogenic differentiation of mouse embryonic palate
mesenchymal cells by 1 μM ATRA has been shown to be
associated with the inhibition of BMPR-IB and Smad5 mRNA expression
(189,190).
The upregulation of IL-1β expression through NF-κB
activation and inflammasome formation may also contribute to the
anti-osteogenic effects of 1-10 μM ATRA (194). These findings correspond to the
observation that IL-6 overproduction by human osteoblasts occurs
even upon exposure to physiological (10 nM) and higher (up to 10
μM) ATRA concentrations (195).
Taken together, the existing studies demonstrate
that ATRA at various concentrations can both promote and inhibit
osteogenesis, with ATRA at nanomolar concentrations inhibiting, and
at micromolar concentrations activating osteoblasts (15). However, it has been suggested
that the inhibitory effects on osteogenesis occur at higher
exposure levels (196).
Therefore, further studies are required to clarify the
mode-of-action of ATRA in osteogenesis and to provide a solid
rationale for adequate VA intake in vivo.
In addition to its impact on osteoblast physiology,
VA is also involved in the regulation of bone resorption through
the modulation of osteoclast activity. Specifically, retinoic acid
has been shown to increase the proliferation of osteoclast
progenitors, while inhibiting osteoclast differentiation by
suppressing RANK/RANKL signaling (197) with the downregulation of NFATc1
(198), NFAT2, c-Fos and MafB
(199). These effects were
shown to de dependent on RAR activation, with RARα signaling being
the most effective (198). In
another study, 1 μM ATRA significantly inhibited
BMP2/7-induced osteoclastogenesis through the downregulation of
RANK and Nfatc1 expression (200). It is also notable that not only
ATRA, but also 9-cis retinoic acid at a concentration of 1 nM,
significantly inhibited calcitriol-induced bone resorption
(201).
In another study, retinoic acid was shown to
increase periosteal bone resorption by increasing osteoclast
differentiation through the RARα-dependent increase in the
RANKL/OPG ratio (202). The
stimulation of osteoclast activity by retinoic acid was associated
with an increased expression of cathepsin K (203). In addition to osteoclast
activation, retinoic acid-induced bone damage has been shown to be
associated with osteocytic osteolysis, as evidenced by a reduction
in mature osteoblast/osteocyte-specific genes (Bglap2 and Ibsp),
without any significant alteration of Runx2 mRNA expression
(204). Therefore, these
findings demonstrate that analogous to osteoblasts, the effects of
ATRA on osteoclast proliferation, differentiation and functioning
are likely bimodal.
In addition to VA and its metabolites, carotenoids
have also been shown to promote osteoblast proliferation and
differentiation (157).
β-cryptoxanthin has been shown to exert osteoprotective effects by
promoting osteoblastogenesis and inhibiting osteoclastic bone
resorption (205). It has been
shown that β-cryptoxanthin significantly increases the osteoblastic
differentiation of MC3T3-E1 cells with a significant increase in
Runx2 mRNA expression (206).
β-cryptoxanthin-induced osteoblast differentiation has been shown
to be mediated by the activation of TGF-β1-induced Smad activation,
being independent of BMP2-Smad signaling (207). Both β-cryptoxanthin and
p-hydroxycinnamic acid have been shown to inhibit basal NF-κB
activity in MC3T3 pre-osteoblasts, whereas only p-hydroxycinnamic
acid significantly suppresses TNF-induced NF-κB activity (208). p-Hydroxycinnamic acid
ameliorates inhibitory effects of TNF-α-induced NF-κB signaling on
Smad-mediated TGF-β and BMP-2 signaling (209).
Osteoclastogenesis is also considered as the target
for carotenoid effects on bone health. Specifically, it has been
shown that 0.1-1 μM β-cryptoxanthin significantly inhibits
PTH, PGE2-, 1,25-dihydroxyvitamin D3-, lipopolysaccharide-, or
TNFα-induced osteoclastogenesis through the downregulation of RANKL
and M-CSF signaling (217). The
downregulation of RANKL-mediated osteoclastogenesis by 5 μM
β-cryptoxanthin has been shown to be dependent on the suppression
of the inhibitor of NF-κB kinase β (IKK β) activity, suppressing
NF-κB activation (218). The
anti-osteoclastogenic effect of β-cryptoxanthin has also been shown
to b associated with the promotion of caspase-3-mediated apoptosis
(219). Correspondingly, in
another study, dietary β-cryptoxanthin intake prevented
osteoclastic bone resorption in ovariectomized mice through
interference with the RANKL pathway (220). A similar protective effect was
observed against inflammatory bone resorption in a mouse model of
periodontitis (221).
In addition to β-cryptoxanthin, other carotenoids
have also been shown to modulate osteoclast functioning. It has
been shown that β-carotene (0.2 μM) significantly
ameliorates RANKL-induced NFATc1, c-Fos and CTSK expression, as
well as osteoclastic bone resorption through the inhibition of IκB
phosphorylation, whereas ERK, JNK and p38 expression remain
unaltered (222). Similarly, 50
μg/ml astaxanthin has been shown to inhibit
Nε-carboxymethyllysine-induced osteoclastogenesis
through the inhibition of NF-κB activation and subsequent
downregulation of NFATc1 expression (223). In bone marrow cells, treatment
with 30 μM lutein was shown to inhibit IL-1-induced
RANKL-mediated osteoclastogenesis, while promoting osteogenesis in
an osteoblast culture by increasing BMP-2 and decreasing sclerostin
mRNA expression (224).
The role of VA in osteoporosis thus appears
unclear. While multiple studies have demonstrated that the
excessive dietary intake of VA and its accumulation in the organism
is associated with a reduced BMD and osteoporosis, observations in
children and VA-depleted subjects have demonstrated that its
deficiency may also exert adverse effects on bone physiology. In
vivo studies have demonstrated adverse effects of the excessive
VA intake on bone health, while in vitro studies have been
inconsistent with the epidemiological findings, indicating positive
effects of VA at micromolar doses on osteogenesis, whereas lower
nanomolar doses exert inhibitory effects. Specifically, it has been
demonstrated that the effects of VA on bone formation are mediated
by the modulation of BMP-2 and Wnt/β-catenin-mediated osteogenesis.
Other targets for the effects of VA in bones include the GH/IGF-1
axis, RAR and Notch signaling, as well as the modulation of
NF-κB-mediated inflammation. Similarly, the effects of VA on
osteoclast formation and activity vary significantly from
inhibition to stimulation, due to the differential modulation of
RANKL signaling. These findings demonstrate that VA intake needs to
be carefully monitored in subjects who are at risk in order to
avoid the hazardous effects of both hypo- and hypervitaminosis on
bone health.
VC plays a crucial role in bone physiology,
exerting beneficial effects on trabecular bone formation, thereby
being considered as a potential treatment modality for osteoporosis
(226).
VC supplementation in post-menopausal women has
been shown to be associated with an almost 3% increase in BMD in
multiple sites, while the highest BMD was observed in women using
VC, estrogen and Ca2+ (227). A higher VC intake has also been
shown to be associated with a 33% lower risk of developing
osteoporosis (228).
These findings corroborate the results of a more
recent meta-analysis, demonstrating that a higher frequency of
dietary VC intake was associated with a 34% lower prevalence of hip
fractures (229). It has been
shown that a 50 mg/day increase in VC intake is associated with a
5% decrease in the risk of hip fractures (230). The results of a 17-year
follow-up demonstrated that VC supplementation resulted in lower
rates of hip fractures (231).
The results from the KNHANES IV (2009) study demonstrated a
significant association between dietary VC intake and BMD only in
vitamin D-deficient elderly individuals (232).
Epidemiological findings have also demonstrated a
positive association between VC intake, circulating ascorbate
levels and BMD (233). In turn,
a suboptimal plasma VC level is considered as a significant
predictor of a low BMD in males (234). Despite the lack of significant
effects of dietary VC intake, a normal plasma VC concentration has
been shown to be associated with a higher BMD in post-menopausal
Puerto Rican women without estrogen therapy (235).
The promotion of bone formation by VC appears to be
mediated by the modulatory effects of VC on osteoblast
differentiation and activity. Specifically, VC significantly
increases osteoblast differentiation in a suspension of mononuclear
cells (242), in association
with increased type I collagen production and extracellular matrix
mineralization (243). VC
promotes both the proliferation and osteoblastic differentiation of
MC3T3-E1 type pre-osteoblast cells (244). VC-induced osteogenic
differentiation has been shown to affect the expression of
>15,000 genes that are related to cell growth, morphogenesis,
metabolism, cell communication and cell death in addition to
osteoblast-specific genes (245). It is also notable that VC
increases the phosphate-induced osteoblastic transformation of
vascular smooth muscle cells by promoting intracellular
Ca2+ deposition (246), thus increasing the risk of
vascular calcification.
It has been demonstrated that low doses of VC
significantly promote osteoblast differentiation through the
upregulation of RUNX2 and SPP1 gene expression in MG-63 cells,
whereas high doses of VC induce apoptotic cell death (247). The osteogenic effects of VC
have been shown to involve the activation of BMP-2 and
Wnt/β-Catenin/ATF4 signaling (248). VC also reduces the number of
senescent cells by increasing the proportion of cells with
proliferative capacity (249).
The activation of casein kinase 2 involved in the regulation of
bone formation may also be involved in the osteogenic effects of
ascorbate osteoblast-like (MG63) cells (250). Osteoblastogenesis has been
shown to be mediated by VC-induced OSX expression through the
activation of PHD and subsequent proteasomal degradation of OSX
gene transcriptional repressors (251). The activation of osteogenesis
by VC has been shown to involve its direct interaction with PHD2
(252).
The osteogenic effects of VC are also dependent on
microtubule plus-end-binding protein 1 expression with the
subsequent activation of β-catenin expression (253). Of note, VC has been found to
exert osteogenic effects at the beginning of bone formation,
although at later periods (9 days) it may exert adverse effects
(254). It has also been
demonstrated that VC induces a shift to osteogenesis and myogenesis
from adipogenesis in mesoderm-derived stem cells, at least
partially through the p38MAPK/CREB pathway (255). A similar effect mediated by the
depletion of the cAMP pool was observed in the OP9 mesenchymal cell
line (256).
Ascorbic acid 2-phosphate, a long-acting VC
derivative, has been shown to promote osteoblast differentiation,
in contrast to the inhibitory effects of VC in a culture of MG-63
cells (257). Correspondingly,
ascorbate-2-phosphate has been shown to increase the expression of
MMP-2 and MMP-13, whereas the ascorbic acid-induced expression of
membrane type1-MMP has been observed only at the early stages of
differentiation (258).
Epigenetic mechanisms may also underlie the
modulatory effects of VC on osteogenesis. Specifically, VC-induced
osteogenic differentiation is tightly associated with H3K9me3 and
H3K27me3 demethylation and 5-hydroxy-methyl-cytosine levels
(259).
VC also significantly modulates bone resorption
through the regulation of osteoclastogenesis and osteoclast
activity. Specifically, VC has been shown to reduce RANKL-induced
osteoclastogenesis in vitro (260) through the redox-dependent
inhibition of NF-κB signaling (261). Correspondingly, it has been
shown that VC significantly inhibits the RANKL and NF-κB
expression-associated increase in osteoclast differentiation in
rats fed a high-cholesterol diet (262). In turn, VC deficiency has been
shown to increase bone resorption and osteoclastogenesis via the
ERK-dependent upregulation of RANK, c-jun and c-fos expression
(263).
VC has also been shown to be essential for
appropriate osteoclastogenesis by increasing RANKL mRNA expres-
sion (264,265). VC has been shown to be
essential for osteoclast differentiation by increasing
preosteoclast maturation and improvement in cell viability
(266). In addition, VC
promotes glycerophosphate-induced osteoclast differentiation by
increasing RANKL-induced NFATc1, c-fos and COX-1 expression
(267). It is notable that VC
promotes osteoclast formation only at earlier stage of
osteoclastogenesis, whereas at the late stage, it increases
osteoclast death (268).
The existing epidemiological studies demonstrate
that a higher VC intake is associated with a lower risk of
osteoporosis and fractures, that may be mediated by the osteogenic
effects of VC via the activation of BMP-2 and Wnt/β-catenin
signaling. Epigenetic effects may also underlie the positive
effects of VC on osteoblast differentiation. Despite the
observation of inhibitory effects of VC on RANKL and
NF-κB-associated osteoclastogenesis, VC has been shown to be
essential for appropriate osteoclast formation.
Group B vitamins represent a group of structurally
heterogeneous water-soluble molecules performing cofactor roles for
a plethora of enzymes involved in human energy metabolism (269), including bone physiology and
protection against osteoporosis (270). However, certain contradictions
regarding the protective effects of group B vitamins exist
(271).
An analysis of the Framingham Offspring
Osteoporosis Study (1996-2001) data demonstrated that males and
females with plasma vitamin B12 levels <148 pM are
characterized by decreased hip and spine BMD, respectively
(271). Correspondingly, an
insufficient B12 intake has been considered as a risk
factor for osteoporosis in vegans (272). A meta-analysis study by Zhang
et al (273)
demonstrated that both homocysteine (Hcy) and B12 levels
were found to be elevated in post-menopausal osteoporotic women. In
addition, in Moroccan women, plasma B12 levels, as well
as the circulating Hcy concentration, were inversely associated
with hip BMD (274).
Folic acid levels have been found to be
significantly associated with BMD following adjustment for Hcy
concentrations and other confounders (278). It is considered that
supplementation with folic acid at a dose of 0.5-5 mg may be useful
for the improvement of BMD in patients with low folic acid levels
or hyperhomocysteinemia (279).
Several studies have investigated combined group B
vitamin supplementation. It was previously demonstrated shown that
the 2-year group B vitamin (folic acid, B6,
B12, B2) supplementation in subjects with a
low B12 status prevented a significant reduction in BMD
at the femoral neck and hip (280). In turn, circulating plasma
folic acid and B12 levels have been shown to be directly associated
with BMD and bone strength in post-menopausal Chinese-Singaporean
women, respectively (281). Low
serum folic acid and B6, but not B12 levels,
have been shown to be associated with lower bone trabecular number
and thickness in subjects who underwent hip arthroplasty (282).
The results of a recent meta-analysis demonstrated
that severe folic acid, but not B6 or B12 deficiency, was
associated with an increased risk of fractures in the elderly
(283). Other studies have
failed to reveal an association between serum B12 or
folic acid levels with BMD (284,285) or the vertebral fracture rate
(286), although a reduction in
the Hcy concentration has been observed (287).
Hcy affects the efficacy of group B vitamin
supplementation. Specifically, although long-term vitamin
B12 and folic acid supplementation do not reduce the
risk of osteoporotic fractures (288) or improve BMD (289), in the general cohort of the
B-PROOF trial, vitamin supplementation reduced the number of
fractures in subjects with hyperhomocysteinemia (288). Nonetheless, no effect of folic
acid, vitamin B6 and B12 supplementation on
fracture risk or bone turnover biomarkers in hyperhomocysteinemic
subjects has been observed (290).
Genetic factors also significantly modulate the
association between the group B vitamin status and bone health. Ahn
et al (291)
demonstrated that 3'-UTR polymorphisms of vitamin B-related genes,
transcobalamin II, reduced folate carrier protein 1 and thiamine
carrier 1, and particularly CD320 (transcobalamin II receptor),
were associated with osteoporosis and osteoporotic spinal fractures
in post-menopausal women. The association between vitamin B levels
and BMD was also shown to be modified by genetic variants in the
1-carbon methylation pathway (292).
Laboratory studies have also demonstrated that
group B vitamins have a significant impact on bone physiology and
osteoporosis. Specifically, folic acid has been shown to
significantly improve bone architecture and prevent bone loss
through the reduction of osteoclast number via AMPK activation and
the upregulation of Nrf2 signaling in high-fat diet-induced
osteoporosis (293). It has
been shown that folic acid supplementation significantly reduces
the inhibitory effects of dexamethasone on vertebral osteogenesis
through the upregulation of the TGF-β signaling pathway, with a
subsequent increase in p-Smad2/3, Runx2 and Osterix expression in
chick embryos (294). Similar
beneficial effect of FA supplementation on bone density was
observed in a model of cyclosporine-induced bone loss (295). Folic acid also ameliorated the
adverse effect of homocysteine on osteoblast proliferation,
differentiation and mineralization through inhibition of
PERK-activated ERS (296).
Folic acid potentiated osteoblastogenic effect of hydroxyapatite
nanoparticles, as evidenced by a more profound RUNX2 expression in
human mesenchymal stem cells (297). At the same time, high maternal
folic acid intake was shown to reduce BMD in the offspring
(298).
Taken together, although B group vitamins have been
shown to play a crucial role in bone physiology, as demonstrated in
deficiency models, epidemiological data on the efficiency of
vitamin supplementation are inconclusive. However, the beneficial
effects of folic acid and B12 supplementation on bone
quality have been reported to be critical in subjects with
insufficient vitamin intake.
Existing data demonstrate that an adequate vitamin
intake is essential for bone health, while vitamin deficiency is
associated with an increased risk of developing osteoporosis.
Specifically, the intake of vitamins E, K2 and C has
been shown to be associated with increased BMD and a reduced risk
of fractures. In turn, the excessive intake of vitamins can also
have adverse effect on bone health and osteoporosis, as clearly
demonstrated for VA. The observed effects of vitamins on the risk
of osteoporosis have been shown to be mediated via mechanisms that
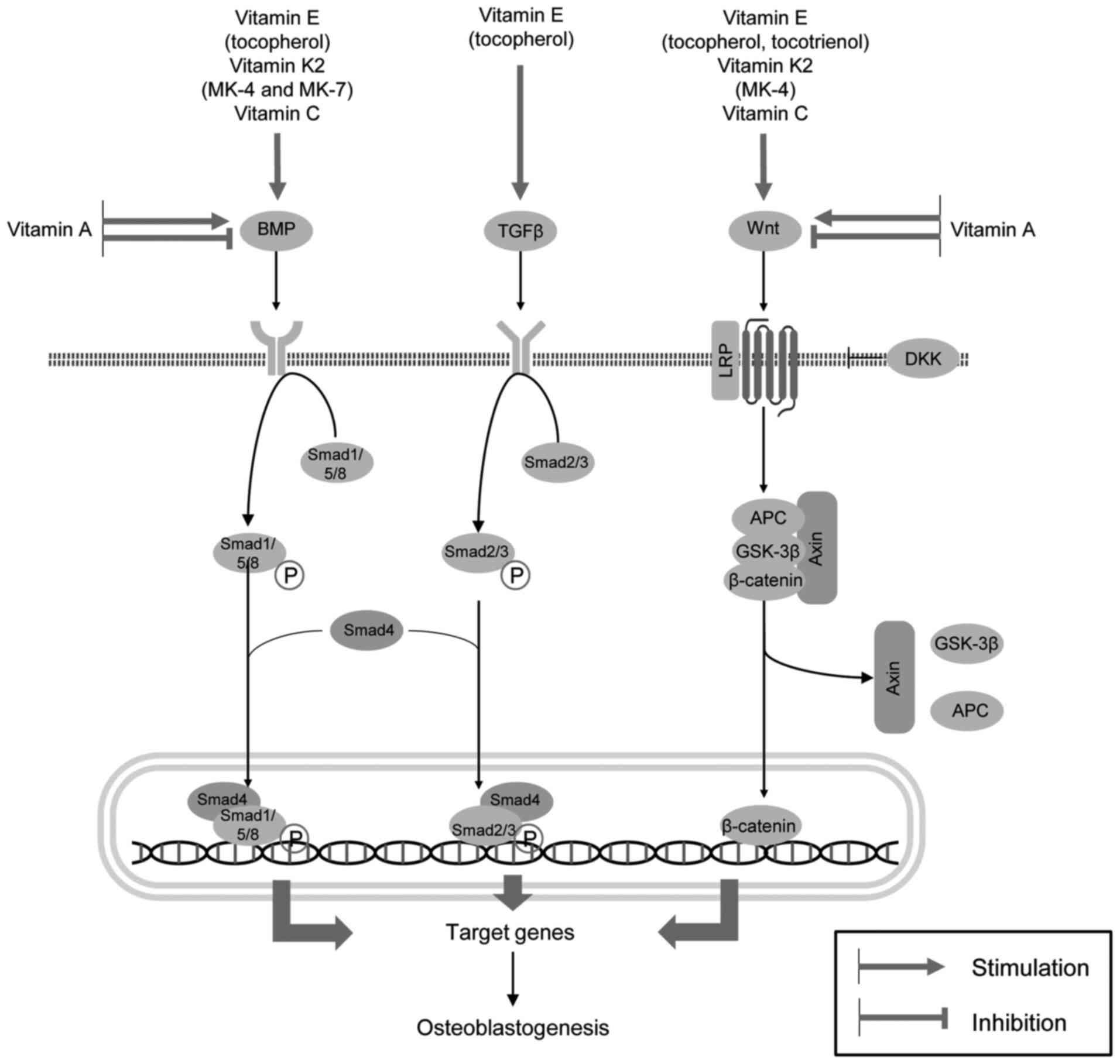
regulate bone formation and resorption. VE (tocopherols and
tocotrienols), VK2 (menaquinones 4 and 7) and VC have
been shown to promote osteoblast development via the upregulation
of BMP/Smad and Wnt/β-catenin signaling. Tocopherol also
contributes to osteoblastogenesis through the stimulation of the
TGFβ/Smad pathway. The VA metabolite (ATRA) appears to exert both
inhibitory and stimulatory effects on BMP- and
Wnt/β-catenin-mediated osteogenesis at nanomolar and micromolar
concentrations, respectively (Fig.
3). However, these observations are contradictory to those of
epidemiological studies demonstrating adverse effects of the
excessive intake of VA on bone health. In addition to these
mechanisms, the upregulation of PI3K/Akt/mTOR signaling, the
inhibition of osteoblast apoptosis and ferroptosis, the improvement
of redox homeostasis through SIRT1/Nrf2 and other pathways, as well
as the inhibition of NF-κB signaling, may contribute to higher
osteoblast viability and osteogenesis. In addition, the osteogenic
effects of certain vitamins have been shown to be mediated by the
modulation of the effects of hormones, including insulin, GH and
PTH on bone physiology.
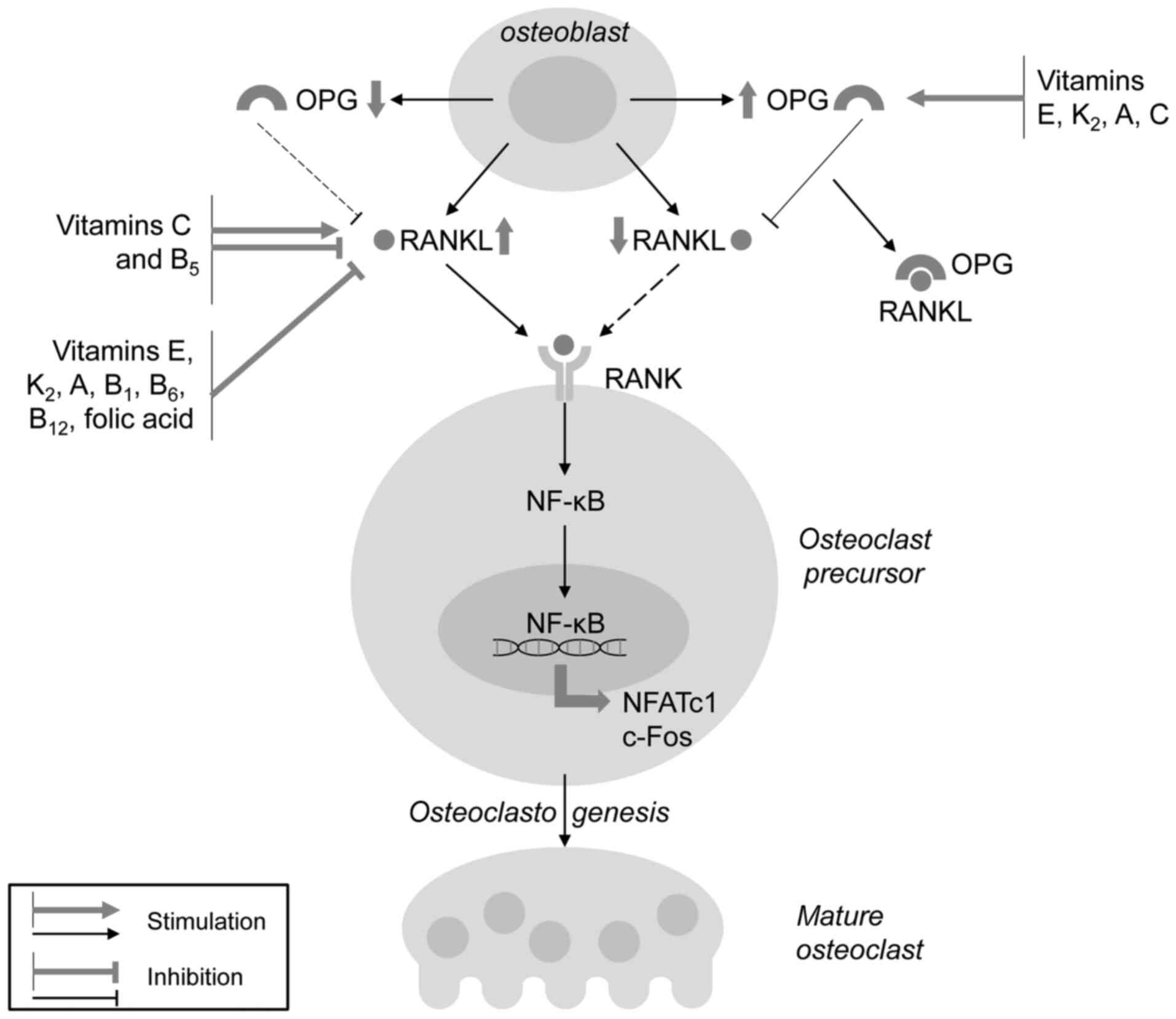
In addition to increased osteoblast proliferation
and differentiation, vitamins are involved in the regulation of
bone resorption through the modulation of osteoclast development
and activity (Fig. 4), thus
increasing the ratio between osteoblast and osteoclast activity.
Both lipid-soluble vitamins E, K2, A, and water-soluble
vitamins B1, B6, B12, C and folic
acid significantly reduce RANKL production, thus reducing the
RANKL/OPG ratio and RANKL/RANK signaling with a subsequent
anti-osteoclastogenic effect. Notably, VC has been shown to be
essential for osteoclast development, and its effect on
osteoclastogenesis has been shown to be dependent on the dose and
the stage of cell development, as also observed for vitamin
B5. In addition, VK2 has been shown to
prevent vascular calcification by activating MGP through its
carboxylation, thereby directing Ca from the vascular wall to its
deposition in bones.
In view of the epidemiological and laboratory
findings, it appears that antioxidant group E vitamins,
particularly in the form of α-tocopherol and VC should be
considered as effective micronutrients for the reduction of
osteoporosis and to lower the risk of adverse effects. Although
VK2 exerts a positive effect on bone formation through
the modulation of both osteoblast and osteoclast activity, as well
as a reduction in vascular calcification and the promotion of
calcium deposition in bones, its intake should be closely monitored
in subjects at a higher risk of hypercoagulation due to its role in
blood clotting. It appears that the therapeutic window of VA for
improved bone health and quality is rather narrow, and both
insufficient and excessive VA intake reduces bone quality; thus, it
should be supplemented only in subjects with VA deficiency. The
beneficial effects of folic acid and B12 supplementation
on bone health are also likely to be inherent to subjects with
insufficient vitamin intake, thus maintaining optimal B group
vitamin dietary intake is also essential for prevention of
osteoporosis. In view of the existing data, further studies are
required to unravel the effects and mechanisms underlying the
impact of various forms and doses of vitamins on bone physiology,
as well as dependence of these effects on baseline vitamin
status.
Not applicable.
AVS, MA and AAT were involved in the
conceptualization of the study. MA, AT, JBTR, AS, DAS, ACM, RL,
TVK, WC, JSC, JCJC and CL were involved the investigation/search of
the literature for the purposes of the review. MA, AT, JBTR, AS,
DAS, ACM, RL, TVK, WC, JSC, JCJC, CL and AAT were involved in the
data curation. AAT was involved in figure preparation. ACM, RL,
TVK, WC, JSC, JCJC, CL and AAT were involved in the writing and
preparation of the original draft. AVS, MA, AT, JBTR, AS and DAS
were involved in the writing, reviewing and editing of the
manuscript. AVS and MA supervised the study. All authors have read
and agreed to the published version of the manuscript.
Not applicable.
Not applicable.
DAS is the Editor-in-Chief for the journal, but had
no personal involvement in the reviewing process, or any influence
in terms of adjudicating on the final decision, for this article.
The other authors declare that they have no competing
interests.
Not applicable.
The present study was supported by the RUDN University Strategic
Academic Leadership Program (award no. 202713-0-000 'Development of
a scientifically based methodology for the ecological adaptation of
foreign students to the new environmental conditions').
|
1
|
Lorentzon M and Cummings SR: Osteoporosis:
The evolution of a diagnosis. J Intern Med. 277:650–661. 2015.
|
|
2
|
Salari N, Ghasemi H, Mohammadi L, Behzadi
MH, Rabieenia E, Shohaimi S and Mohammadi M: The global prevalence
of osteoporosis in the world: A comprehensive systematic review and
meta-analysis. J Orthop Surg Res. 16:6092021.
|
|
3
|
Xiao PL, Cui AY, Hsu CJ, Peng R, Jiang N,
Xu XH, Ma YG, Liu D and Lu HD: Global, regional prevalence, and
risk factors of osteoporosis according to the World Health
Organization diagnostic criteria: A systematic review and
meta-analysis. Osteoporos Int. 33:2137–2153. 2022.
|
|
4
|
Clynes MA, Harvey NC, Curtis EM, Fuggle
NR, Dennison EM and Cooper C: The epidemiology of osteoporosis. Br
Med Bull. 133:105–117. 2020.
|
|
5
|
Pouresmaeili F, Kamalidehghan B, Kamarehei
M and Goh YM: A comprehensive overview on osteoporosis and its risk
factors. Ther Clin Risk Manag. 14:2029–2049. 2018.
|
|
6
|
Levis S and Lagari VS: The role of diet in
osteoporosis prevention and management. Curr Osteoporos Rep.
10:296–302. 2012.
|
|
7
|
Muñoz-Garach A, García-Fontana B and
Muñoz-Torres M: Nutrients and dietary patterns related to
osteoporosis. Nutrients. 12:19862020.
|
|
8
|
Brincat M, Gambin J, Brincat M and
Calleja-Agius J: The role of vitamin D in osteoporosis. Maturitas.
80:329–332. 2015.
|
|
9
|
Goltzman D: Functions of vitamin D in
bone. Histochem Cell Biol. 149:305–312. 2018.
|
|
10
|
Ratajczak AE, Rychter AM, Zawada A,
Dobrowolska A and Krela-Kaźmierczak I: Do only calcium and vitamin
D matter? Micronutrients in the diet of inflammatory bowel diseases
patients and the risk of osteoporosis. Nutrients. 13:5252021.
|
|
11
|
Martiniakova M, Babikova M, Mondockova V,
Blahova J, Kovacova V and Omelka R: The role of macronutrients,
micronutrients and flavonoid polyphenols in the prevention and
treatment of osteoporosis. Nutrients. 14:5232022.
|
|
12
|
Heaney RP: Nutrition and risk for
osteoporosis. Osteoporosis. Acadmic Press; pp. 669–700. 2001
|
|
13
|
Nazrun AS, Norazlina M, Norliza M and
Nirwana SI: Comparison of the effects of tocopherol and tocotrienol
on osteoporosis in animal models. Int J Pharmacol. 6:561–568.
2010.
|
|
14
|
Wu AM, Huang CQ, Lin ZK, Tian NF, Ni WF,
Wang XY, Xu HZ and Chi YL: The relationship between vitamin A and
risk of fracture: Meta-analysis of prospective studies. J Bone
Miner Res. 29:2032–2039. 2014.
|
|
15
|
Henning P, Conaway HH and Lerner UH:
Retinoid receptors in bone and their role in bone remodeling. Front
Endocrinol (Lausanne). 6:312015.
|
|
16
|
Ahmadieh H and Arabi A: Vitamins and bone
health: Beyond calcium and vitamin D. Nutr Rev. 69:584–598.
2011.
|
|
17
|
Szewczyk K, Chojnacka A and Górnicka M:
Tocopherols and tocotrienols-bioactive dietary compounds; What is
certain, what is doubt? Int J Mol Sci. 22:62222021.
|
|
18
|
Wong SK, Mohamad NV, Ibrahim N', Chin KY,
Shuid AN and Ima-Nirwana S: The molecular mechanism of vitamin E as
a bone-protecting agent: A review on current evidence. Int J Mol
Sci. 20:14532019.
|
|
19
|
Michaëlsson K and Larsson SC: Circulating
alpha-tocopherol levels, bone mineral density, and fracture:
Mendelian randomization study. Nutrients. 13:19402021.
|
|
20
|
Mata-Granados JM, Cuenca-Acebedo R, Luque
de Castro MD and Quesada Gómez JM: Lower vitamin E serum levels are
associated with osteoporosis in early postmenopausal women: A
cross-sectional study. J Bone Miner Metab. 31:455–460. 2013.
|
|
21
|
Holvik K, Gjesdal CG, Tell GS, Grimnes G,
Schei B, Apalset EM, Samuelsen SO, Blomhoff R, Michaëlsson K and
Meyer HE: Low serum concentrations of alpha-tocopherol are
associated with increased risk of hip fracture. A NOREPOS study.
Osteoporos Int. 25:2545–2554. 2014.
|
|
22
|
Michaëlsson K, Wolk A, Byberg L, Ärnlöv J
and Melhus H: Intake and serum concentrations of α-tocopherol in
relation to fractures in elderly women and men: 2 Cohort studies.
Am J Clin Nutr. 99:107–114. 2014.
|
|
23
|
Shen CL, Yang S, Tomison MD, Romero AW,
Felton CK and Mo H: Tocotrienol supplementation suppressed bone
resorption and oxidative stress in postmenopausal osteopenic women:
A 12-week randomized double-blinded placebo-controlled trial.
Osteoporos Int. 29:881–891. 2018.
|
|
24
|
Vallibhakara SAO, Nakpalat K,
Sophonsritsuk A, Tantitham C and Vallibhakara O: Effect of vitamin
E supplement on bone turnover markers in postmenopausal osteopenic
women: A double-blind, randomized, placebo-controlled trial.
Nutrients. 13:42262021.
|
|
25
|
Yang TC, Duthie GG, Aucott LS and
Macdonald HM: Vitamin E homologues α- and γ-tocopherol are not
associated with bone turnover markers or bone mineral density in
peri-menopausal and post-menopausal women. Osteoporos Int.
27:2281–2290. 2016.
|
|
26
|
Zhang J, Hu X and Zhang J: Associations
between serum vitamin E concentration and bone mineral density in
the US elderly population. Osteoporos Int. 28:1245–1253. 2017.
|
|
27
|
Hampson G, Edwards S, Sankaralingam A,
Harrington DJ, Voong K, Fogelman I and Frost ML: Circulating
concentrations of vitamin E isomers: Association with bone turnover
and arterial stiffness in post-menopausal women. Bone. 81:407–412.
2015.
|
|
28
|
Hamidi MS, Corey PN and Cheung AM: Effects
of vitamin E on bone turnover markers among US postmenopausal
women. J Bone Miner Res. 27:1368–1380. 2012.
|
|
29
|
Mehat MZ, Shuid AN, Mohamed N, Muhammad N
and Soelaiman IN: Beneficial effects of vitamin E isomer
supplementation on static and dynamic bone histomorphometry
parameters in normal male rats. J Bone Miner Metab. 28:503–509.
2010.
|
|
30
|
Muhammad N, Luke DA, Shuid AN, Mohamed N
and Soelaiman IN: Two different isomers of vitamin E prevent bone
loss in postmenopausal osteoporosis rat model. Evid Based
Complement Alternat Med. 2012:1615272012.
|
|
31
|
Chin KY, Gengatharan D, Mohd Nasru FS,
Khairussam RA, Ern SL, Aminuddin SA and Ima-Nirwana S: The effects
of annatto tocotrienol on bone biomechanical strength and bone
calcium content in an animal model of osteoporosis due to
testosterone deficiency. Nutrients. 8:8082016.
|
|
32
|
Shuid AN, Mohamad S, Muhammad N, Fadzilah
FM, Mokhtar SA, Mohamed N and Soelaiman IN: Effects of α-tocopherol
on the early phase of osteoporotic fracture healing. J Orthop Res.
29:1732–1738. 2011.
|
|
33
|
Mohamad S, Shuid AN, Mohamed N, Fadzilah
FM, Mokhtar SA, Abdullah S, Othman F, Suhaimi F, Muhammad N and
Soelaiman IN: The effects of alpha-tocopherol supplementation on
fracture healing in a postmenopausal osteoporotic rat model.
Clinics (São Paulo). 67:1077–1085. 2012.
|
|
34
|
Akçay H, Kuru K, Tatar B and Şimşek F:
Vitamin E promotes bone formation in a distraction osteogenesis
model. J Craniofac Surg. 30:2315–2318. 2019.
|
|
35
|
Kurklu M, Yildiz C, Kose O, Yurttas Y,
Karacalioglu O, Serdar M and Deveci S: Effect of alpha-tocopherol
on bone formation during distraction osteogenesis: A rabbit model.
J Orthop Traumatol. 12:153–158. 2011.
|
|
36
|
Hagan ML, Bahraini A, Pierce JL, Bass SM,
Yu K, Elsayed R, Elsalanty M, Johnson MH, McNeil A, McNeil PL and
McGee-Lawrence ME: Inhibition of osteocyte membrane repair activity
via dietary vitamin E deprivation impairs osteocyte survival.
Calcif Tissue Int. 104:224–234. 2019.
|
|
37
|
Turan B, Can B and Delilbasi E: Selenium
combined with vitamin E and vitamin C restores structural
alterations of bones in heparin-induced osteoporosis. Clin
Rheumatol. 22:432–436. 2003.
|
|
38
|
Ikegami H, Kawawa R, Ichi I, Ishikawa T,
Koike T, Aoki Y and Fujiwara Y: Excessive vitamin E intake does not
cause bone loss in male or ovariectomized female mice fed normal or
high-fat diets. J Nutr. 147:1932–1937. 2017.
|
|
39
|
Kasai S, Ito A, Shindo K, Toyoshi T and
Bando M: High-dose α-tocopherol supplementation does not induce
bone loss in normal rats. PLoS One. 10:e01320592015.
|
|
40
|
Lan D, Yao C, Li X, Liu H, Wang D, Wang Y
and Qi S: Tocopherol attenuates the oxidative stress of BMSCs by
inhibiting ferroptosis through the PI3k/AKT/mTOR pathway. Front
Bioeng Biotechnol. 10:9385202022.
|
|
41
|
Ahn KH, Jung HK, Jung SE, Yi KW, Park HT,
Shin JH, Kim YT, Hur JY, Kim SH and Kim T: Microarray analysis of
gene expression during differentiation of human mesenchymal stem
cells treated with vitamin E in vitro into osteoblasts. Korean J
Bone Metab. 18:23–32. 2011.
|
|
42
|
Jia YB, Jiang DM, Ren YZ, Liang ZH, Zhao
ZQ and Wang YX: Inhibitory effects of vitamin E on osteocyte
apoptosis and DNA oxidative damage in bone marrow hemopoietic cells
at early stage of steroid-induced femoral head necrosis. Mol Med
Rep. 15:1585–1592. 2017.
|
|
43
|
Soeta S, Higuchi M, Yoshimura I, Itoh R,
Kimura N and Aamsaki H: Effects of vitamin E on the osteoblast
differentiation. J Vet Med Sci. 72:951–957. 2010.
|
|
44
|
Kim HN, Lee JH, Jin WJ and Lee ZH:
α-Tocopheryl succinate inhibits osteoclast formation by suppressing
receptor activator of nuclear factor-kappaB ligand (RANKL)
expression and bone resorption. J Bone Metab. 19:111–120. 2012.
|
|
45
|
Johnson SA, Feresin RG, Soungdo Y, Elam ML
and Arjmandi BH: Vitamin E suppresses ex vivo osteoclastogenesis in
ovariectomized rats. Food Funct. 7:1628–1633. 2016.
|
|
46
|
Fujita K, Iwasaki M, Ochi H, Fukuda T, Ma
C, Miyamoto T, Takitani K, Negishi-Koga T, Sunamura S, Kodama T, et
al: Vitamin E decreases bone mass by stimulating osteoclast fusion.
Nat Med. 18:589–594. 2012.
|
|
47
|
Chin KY and Ima-Nirwana S: The biological
effects of tocotrienol on bone: A review on evidence from rodent
models. Drug Des Devel Ther. 9:2049–2061. 2015.
|
|
48
|
Shen CL, Klein A, Chin KY, Mo H, Tsai P,
Yang RS, Chyu MC and Ima-Nirwana S: Tocotrienols for bone health: A
translational approach. Ann N Y Acad Sci. 1401:150–165. 2017.
|
|
49
|
Xu W, He P, He S, Cui P, Mi Y, Yang Y, Li
Y and Zhou S: Gamma-tocotrienol stimulates the proliferation,
differentiation, and mineralization in osteoblastic MC3T3-E1 cells.
J Chem. 2018:38059322018.
|
|
50
|
Wan Hasan WN, Abd Ghafar N, Chin KY and
Ima-Nirwana S: Annatto-derived tocotrienol stimulates osteogenic
activity in preosteoblastic MC3T3-E1 cells: A temporal sequential
study. Drug Des Devel Ther. 12:1715–1726. 2018.
|
|
51
|
Wan Hasan WN, Chin KY, Abd Ghafar N and
Soelaiman IN: Annatto-derived tocotrienol promotes mineralization
of MC3T3-E1 cells by enhancing BMP-2 protein expression via
inhibiting RhoA activation and HMG-CoA reductase gene expression.
Drug Des Devel Ther. 14:969–976. 2020.
|
|
52
|
Xu W, Li Y, Feng R, He P and Zhang Y:
γ-Tocotrienol induced the proliferation and differentiation of
MC3T3-E1 cells through the stimulation of the Wnt/β-catenin
signaling pathway. Food Funct. 13:398–410. 2022.
|
|
53
|
Shah AK and Yeganehjoo H: The stimulatory
impact of d-δ-Tocotrienol on the differentiation of murine MC3T3-E1
preosteoblasts. Mol Cell Biochem. 462:173–183. 2019.
|
|
54
|
Casati L, Pagani F, Maggi R, Ferrucci F
and Sibilia V: Food for bone: Evidence for a role for
delta-tocotrienol in the physiological control of osteoblast
migration. Int J Mol Sci. 21:46612020.
|
|
55
|
Abd Manan N, Mohamed N and Shuid AN:
Effects of low-dose versus high-dose γ-tocotrienol on the bone
cells exposed to the hydrogen peroxide-induced oxidative stress and
apoptosis. Evid Based Complement Alternat Med. 2012:6808342012.
|
|
56
|
Casati L, Pagani F, Limonta P, Vanetti C,
Stancari G and Sibilia V: Beneficial effects of δ-tocotrienol
against oxidative stress in osteoblastic cells: Studies on the
mechanisms of action. Eur J Nutr. 59:1975–1987. 2020.
|
|
57
|
Cai J, Tian X, Ren J, Lu S and Guo J:
Synergistic effect of sesamin and γ-Tocotrienol on promoting
osteoblast differentiation via AMPK signaling. Nat Prod Commun.
17:1–8. 2022.
|
|
58
|
Radzi NFM, Ismail NAS and Alias E:
Tocotrienols regulate bone loss through suppression on osteoclast
differentiation and activity: A systematic review. Curr Drug
Targets. 19:1095–1107. 2018.
|
|
59
|
Ha H, Lee JH, Kim HN and Lee ZH:
α-Tocotrienol inhibits osteoclastic bone resorption by suppressing
RANKL expression and signaling and bone resorbing activity. Biochem
Biophys Res Commun. 406:546–551. 2011.
|
|
60
|
Ormsby RT, Hosaka K, Evdokiou A, Odysseos
A, Findlay DM, Solomon LB and Atkins GJ: The effects of vitamin E
analogues α-Tocopherol and γ-Tocotrienol on the human osteocyte
response to ultra-high molecular weight polyethylene wear
particles. Prosthesis. 4:480–489. 2022.
|
|
61
|
Kim KW, Kim BM, Won JY, Min HK, Lee SJ,
Lee SH and Kim HR: Tocotrienol regulates osteoclastogenesis in
rheumatoid arthritis. Korean J Intern Med. 36(Suppl 1): S273–S282.
2021.
|
|
62
|
Wong SK, Chin KY and Ima-Nirwana S: The
effects of tocotrienol on bone peptides in a rat model of
osteoporosis induced by metabolic syndrome: The possible
communication between bone cells. Int J Environ Res Public Health.
16:33132019.
|
|
63
|
Chin KY, Abdul-Majeed S, Fozi NF and
Ima-Nirwana S: Annatto tocotrienol improves indices of bone static
histomorphometry in osteoporosis due to testosterone deficiency in
rats. Nutrients. 6:4974–4983. 2014.
|
|
64
|
Deng L, Ding Y, Peng Y, Wu Y, Fan J, Li W,
Yang R, Yang M and Fu Q: γ-Tocotrienol protects against
ovariectomy-induced bone loss via mevalonate pathway as HMG-CoA
reductase inhibitor. Bone. 67:200–207. 2014.
|
|
65
|
Soelaiman IN, Ming W, Abu Bakar R, Hashnan
NA, Mohd Ali H, Mohamed N, Muhammad N and Shuid AN: Palm
tocotrienol supplementation enhanced bone formation in
oestrogen-deficient rats. Int J Endocrinol. 2012:5328622012.
|
|
66
|
Mohamad NV, Ima-Nirwana S and Chin KY:
Self-emulsified annatto tocotrienol improves bone histomorphometric
parameters in a rat model of oestrogen deficiency through
suppression of skeletal sclerostin level and RANKL/OPG ratio. Int J
Med Sci. 18:3665–3673. 2021.
|
|
67
|
Liang G, Kow ASF, Tham CL, Ho YC and Lee
MT: Ameliorative effect of tocotrienols on
perimenopausal-associated osteoporosis-a review. Antioxidants
(Basel). 11:21792022.
|
|
68
|
Bus K and Szterk A: Relationship between
structure and biological activity of various vitamin K forms.
Foods. 10:31362021.
|
|
69
|
Myneni VD and Mezey E: Regulation of bone
remodeling by vitamin K2. Oral Dis. 23:1021–1028. 2017.
|
|
70
|
Stevenson M, Lloyd-Jones M and Papaioannou
D: Vitamin K to prevent fractures in older women: Systematic review
and economic evaluation. Health Technol Assess. 13:iii–xi. 1–134.
2009.
|
|
71
|
Ma ML, Ma ZJ, He YL, Sun H, Yang B, Ruan
BJ, Zhan WD, Li SX, Dong H and Wang YX: Efficacy of vitamin K2 in
the prevention and treatment of postmenopausal osteoporosis: A
systematic review and meta-analysis of randomized controlled
trials. Front Public Health. 10:9796492022.
|
|
72
|
Zhou M, Han S, Zhang W and Wu D: Efficacy
and safety of vitamin K2 for postmenopausal women with osteoporosis
at a long-term follow-up: Meta-analysis and systematic review. J
Bone Miner Metab. 40:763–772. 2022.
|
|
73
|
Salma, Ahmad SS, Karim S, Ibrahim IM,
Alkreathy HM, Alsieni M and Khan MA: Effect of vitamin K on bone
mineral density and fracture risk in adults: Systematic review and
meta-analysis. Biomedicines. 10:10482022.
|
|
74
|
Hao G, Zhang B, Gu M, Chen C, Zhang Q,
Zhang G and Cao X: Vitamin K intake and the risk of fractures: A
meta-analysis. Medicine (Baltimore). 96:e67252017.
|
|
75
|
Moore AE, Kim E, Dulnoan D, Dolan AL,
Voong K, Ahmad I, Gorska R, Harrington DJ and Hampson G: Serum
vitamin K1 (phylloquinone) is associated with fracture
risk and hip strength in post-menopausal osteoporosis: A
cross-sectional study. Bone. 141:1156302020.
|
|
76
|
O'Connor EM, Grealy G, McCarthy J, Desmond
A, Craig O, Shanahan F and Cashman KD: Effect of phylloquinone
(vitamin K1) supplementation for 12 months on the indices of
vitamin K status and bone health in adult patients with Crohn's
disease. Br J Nutr. 112:1163–1174. 2014.
|
|
77
|
Tsugawa N, Shiraki M, Suhara Y, Kamao M,
Ozaki R, Tanaka K and Okano T: Low plasma phylloquinone
concentration is associated with high incidence of vertebral
fracture in Japanese women. J Bone Miner Metab. 26:79–85. 2008.
|
|
78
|
Yamauchi M, Yamaguchi T, Nawata K, Takaoka
S and Sugimoto T: Relationships between undercarboxylated
osteocalcin and vitamin K intakes, bone turnover, and bone mineral
density in healthy women. Clin Nutr. 29:761–765. 2010.
|
|
79
|
Kuang X, Liu C, Guo X, Li K, Deng Q and Li
D: The combination effect of vitamin K and vitamin D on human bone
quality: A meta-analysis of randomized controlled trials. Food
Funct. 11:3280–3297. 2020.
|
|
80
|
Bolton-Smith C, McMurdo ME, Paterson CR,
Mole PA, Harvey JM, Fenton ST, Prynne CJ, Mishra GD and Shearer MJ:
Two-year randomized controlled trial of vitamin K1 (phylloquinone)
and vitamin D3 plus calcium on the bone health of older women. J
Bone Miner Res. 22:509–519. 2007.
|
|
81
|
Hu L, Ji J, Li D, Meng J and Yu B: The
combined effect of vitamin K and calcium on bone mineral density in
humans: A meta-analysis of randomized controlled trials. J Orthop
Surg Res. 16:5922021.
|
|
82
|
Platonova K, Kitamura K, Watanabe Y,
Takachi R, Saito T, Kabasawa K, Takahashi A, Kobayashi R, Oshiki R,
Solovev A, et al: Dietary calcium and vitamin K are associated with
osteoporotic fracture risk in middle-aged and elderly Japanese
women, but not men: The Murakami cohort study. Br J Nutr.
125:319–328. 2021.
|
|
83
|
Knapen MHJ, Drummen NE, Smit E, Vermeer C
and Theuwissen E: Three-year low-dose menaquinone-7 supplementation
helps decrease bone loss in healthy postmenopausal women.
Osteoporos Int. 24:2499–2507. 2013.
|
|
84
|
Rønn SH, Harsløf T, Pedersen SB and
Langdahl BL: Vitamin K2 (menaquinone-7) prevents age-related
deterioration of trabecular bone microarchitecture at the tibia in
postmenopausal women. Eur J Endocrinol. 175:541–549. 2016.
|
|
85
|
Shiraki M, Shiraki Y, Aoki C and Miura M:
Vitamin K2 (menatetrenone) effectively prevents fractures and
sustains lumbar bone mineral density in osteoporosis. J Bone Miner
Res. 15:515–521. 2000.
|
|
86
|
Su S, He N, Men P, Song C and Zhai S: The
efficacy and safety of menatetrenone in the management of
osteoporosis: A systematic review and meta-analysis of randomized
controlled trials. Osteoporos Int. 30:1175–1186. 2019.
|
|
87
|
Abdel Aziz DM, Saleh HA, Taha NM and
Elbadawy MA: Relation between circulating vitamin K2 level and
osteoporosis in post-menopausal women. QJM: Int J Med. 114(Suppl
1): hcab116–002. 2021.
|
|
88
|
Heiss C, Hoesel LM, Wehr U, Keller T,
Horas U, Meyer C, Rambeck W and Schnettler R: Vitamin K in
combination with other biochemical markers to diagnose
osteoporosis. Biomarkers. 9:479–488. 2004.
|
|
89
|
Li C, Liang C, Kong Z, Su Y, Ren W, Dong
H, Wu Y, Yang N, Liu R, Wu J and Zheng Y: Determination of vitamin
K1, MK-4, MK-7, and D levels in human serum of postmenopausal
osteoporosis women based on high stability LC-MS/MS: MK-7 may be a
new marker of bone metabolism. Ann Nutr Metab. 79:334–342.
2023.
|
|
90
|
Kawana K, Takahashi M, Hoshino H and
Kushida K: Circulating levels of vitamin K1, menaquinone-4, and
menaquinone-7 in healthy elderly Japanese women and patients with
vertebral fractures and patients with hip fractures. Endocr Res.
27:337–343. 2001.
|
|
91
|
El-Morsy AS, Beshir SR, Farrag KAER,
Mohamed MS and Hamam GG: Comparative study on the effect of vitamin
K versus combined Ca and vitamin D administration on the prevention
of experimentally-induced osteoporosis in adult male albino rats.
Egypt J Histol. 34:5–14. 2011.
|
|
92
|
Hara K, Kobayashi M and Akiyama Y: Vitamin
K2 (menatetrenone) inhibits bone loss induced by prednisolone
partly through enhancement of bone formation in rats. Bone.
31:575–581. 2002.
|
|
93
|
Sasaki N, Kusano E, Takahashi H, Ando Y,
Yano K, Tsuda E and Asano Y: Vitamin K2 inhibits
glucocorticoid-induced bone loss partly by preventing the reduction
of osteoprotegerin (OPG). J Bone Miner Metab. 23:41–47. 2005.
|
|
94
|
Jin C, Tan K, Yao Z, Lin BH, Zhang DP,
Chen WK, Mao SM, Zhang W, Chen L, Lin Z, et al: A novel
anti-osteoporosis mechanism of VK2: Interfering with ferroptosis
via AMPK/SIRT1 pathway in Type 2 diabetic osteoporosis. J Agric
Food Chem. 71:2745–2761. 2023.
|
|
95
|
Yamaguchi M, Sugimoto E and Hachiya S:
Stimulatory effect of menaquinone-7 (vitamin K2) on osteoblastic
bone formation in vitro. Mol Cell Biochem. 223:131–137. 2001.
|
|
96
|
Iwamoto D, Masaki C, Shibata Y, Watanabe
C, Nodai T, Munemasa T, Mukaibo T, Kondo Y and Hosokawa R:
Microstructural and mechanical recovery of bone in ovariectomized
rats: The effects of menaquinone-7. J Mech Behav Biomed Mater.
120:1045712021.
|
|
97
|
Katsuyama H, Otsuki T, Tomita M, Fukunaga
M, Fukunaga T, Suzuki N, Saijoh K, Fushimi S and Sunami S:
Menaquinone-7 regulates the expressions of osteocalcin, OPG, RANKL
and RANK in osteoblastic MC3T3E1 cells. Int J Mol Med. 15:231–236.
2005.
|
|
98
|
Akbulut AC, Wasilewski GB, Rapp N, Forin
F, Singer H, Czogalla-Nitsche KJ and Schurgers LJ: Menaquinone-7
supplementation improves osteogenesis in pluripotent stem cell
derived mesenchymal stem cells. Front Cell Dev Biol.
8:6187602021.
|
|
99
|
Katsuyama H, Saijoh K, Otsuki T, Tomita M,
Fukunaga M and Sunami S: Menaquinone-7 regulates gene expression in
osteoblastic MC3T3E1 cells. Int J Mol Med. 19:279–284. 2007.
|
|
100
|
Gigante A, Brugè F, Cecconi S, Manzotti S,
Littarru GP and Tiano L: Vitamin MK-7 enhances vitamin D3-induced
osteogenesis in hMSCs: Modulation of key effectors in
mineralization and vascularization. J Tissue Eng Regen Med.
9:691–701. 2015.
|
|
101
|
Tang H, Zhu Z, Zheng Z, Wang H, Li C, Wang
L, Zhao G and Wang P: A study of hydrophobins-modified
menaquinone-7 on osteoblastic cells differentiation. Mol Cell
Biochem. 476:1939–1948. 2021.
|
|
102
|
Yamaguchi M and Weitzmann MN: Vitamin K2
stimulates osteoblastogenesis and suppresses osteoclastogenesis by
suppressing NF-κB activation. Int J Mol Med. 27:3–14. 2011.
|
|
103
|
Wang H, Li L, Zhang N and Ma Y: Vitamin K2
improves osteogenic differentiation by inhibiting STAT1 via the
Bcl-6 and IL-6/JAK in C3H10 T1/2 clone 8 cells. Nutrients.
14:29342022.
|
|
104
|
Owen R, Bahmaee H, Claeyssens F and Reilly
GC: Comparison of the anabolic effects of reported osteogenic
compounds on human mesenchymal progenitor-derived osteoblasts.
Bioengineering (Basel). 7:122020.
|
|
105
|
Wang H, Zhang N, Li L, Yang P and Ma Y:
Menaquinone 4 reduces bone loss in ovariectomized mice through dual
regulation of bone remodeling. Nutrients. 13:25702021.
|
|
106
|
Cui Q, Li N, Nie F, Yang F, Li H and Zhang
J: Vitamin K2 promotes the osteogenic differentiation of
periodontal ligament stem cells via the Wnt/β-catenin signaling
pathway. Arch Oral Biol. 124:1050572021.
|
|
107
|
Urayama S, Kawakami A, Nakashima T, Tsuboi
M, Yamasaki S, Hida A, Ichinose Y, Nakamura H, Ejima E, Aoyagi T,
et al: Effect of vitamin K2 on osteoblast apoptosis: Vitamin K2
inhibits apoptotic cell death of human osteoblasts induced by Fas,
proteasome inhibitor, etoposide, and staurosporine. J Lab Clin Med.
136:181–193. 2000.
|
|
108
|
Jiang Y, Lin L, Xin H, Jin Y, Jiang Y and
Xue L: Study on the protective effect of menatetrenone against the
oxidative stress of osteoblasts. J Pharm Pract Serv. 38:523–527.
2020.
|
|
109
|
Cui Y, Zhang W, Yang P, Zhu S, Luo S and
Li M: Menaquinone-4 prevents medication-related osteonecrosis of
the jaw through the SIRT1 signaling-mediated inhibition of cellular
metabolic stresses-induced osteoblast apoptosis. Free Radic Biol
Med. 206:33–49. 2023.
|
|
110
|
Amizuka N, Li M and Maeda T: The interplay
of magnesium and vitamin K2 on bone mineralization. Clin Calcium.
15:57–61. 2005.In Japanese.
|
|
111
|
Cui L, Xu J, Zhang J, Zhang M, Zhang S and
Bai Y: Menaquinone-4 modulates the expression levels of
calcification-associated factors to inhibit calcification of rat
aortic vascular smooth muscle cells in a dose-dependent manner. Exp
Ther Med. 16:3172–3178. 2018.
|
|
112
|
Li W, Zhang S, Liu J, Liu Y and Liang Q:
Vitamin K2 stimulates MC3T3-E1 osteoblast differentiation and
mineralization through autophagy induction. Mol Med Rep.
19:3676–3684. 2019.
|
|
113
|
Chen L, Shi X, Weng SJ, Xie J, Tang JH,
Yan DY, Wang BZ, Xie ZJ, Wu ZY and Yang L: Vitamin K2 can rescue
the dexamethasone-induced downregulation of osteoblast autophagy
and mitophagy thereby restoring osteoblast function in vitro and in
vivo. Front Pharmacol. 11:12092020.
|
|
114
|
Fusaro M, Cianciolo G, Brandi ML, Ferrari
S, Nickolas TL, Tripepi G, Plebani M, Zaninotto M, Iervasi G, La
Manna G, et al: Vitamin K and osteoporosis. Nutrients.
12:36252020.
|
|
115
|
Tabb MM, Sun A, Zhou C, Grün F, Errandi J,
Romero K, Pham H, Inoue S, Mallick S, Lin M, et al: Vitamin K2
regulation of bone homeostasis is mediated by the steroid and
xenobiotic receptor SXR. J Biol Chem. 278:43919–43927. 2003.
|
|
116
|
Ichikawa T, Horie-Inoue K, Ikeda K,
Blumberg B and Inoue S: Steroid and xenobiotic receptor SXR
mediates vitamin K2-activated transcription of extracellular
matrix-related genes and collagen accumulation in osteoblastic
cells. J Biol Chem. 281:16927–16934. 2006.
|
|
117
|
Zhang Y, Weng S, Yin J, Ding H, Zhang C
and Gao Y: Vitamin K2 promotes mesenchymal stem cell
differentiation by inhibiting miR-133a expression. Mol Med Rep.
15:2473–2480. 2017.
|
|
118
|
Takeuchi Y, Suzawa M, Fukumoto S and
Fujita T: Vitamin K(2) inhibits adipogenesis, osteoclastogenesis,
and ODF/RANK ligand expression in murine bone marrow cell cultures.
Bone. 27:769–776. 2000.
|
|
119
|
Jiang Y, Xia T, Xin H, Jin Y, Jiang Y and
Xue L: Effects of vitamin K on osteoblastic bone formation and
osteoclastic bone absorption. J Pharm Pract. 340–345. 2020.
|
|
120
|
Wu WJ, Gao H, Jin JS and Ahn BY: A
comparatively study of menaquinone-7 isolated from Cheonggukjang
with vitamin K1 and menaquinone-4 on osteoblastic cells
differentiation and mineralization. Food Chem Toxicol.
131:1105402019.
|
|
121
|
Kim M, Na W and Sohn C: Vitamin K1
(phylloquinone) and K2 (menaquinone-4) supplementation improves
bone formation in a high-fat diet-induced obese mice. J Clin
Biochem Nutr. 53:108–113. 2013.
|
|
122
|
Koshihara Y, Hoshi K, Okawara R, Ishibashi
H and Yamamoto S: Vitamin K stimulates osteoblastogenesis and
inhibits osteoclastogenesis in human bone marrow cell culture. J
Endocrinol. 176:339–348. 2003.
|
|
123
|
Akiyama Y, Hara K, Tajima T, Murota S and
Morita I: Effect of vitamin K2 (menatetrenone) on osteoclast-like
cell formation in mouse bone marrow cultures. Eur J Pharmacol.
263:181–185. 1994.
|
|
124
|
Yamaguchi M and Ma ZJ: Inhibitory effect
of menaquinone-7 (vitamin K2) on osteoclast-like cell formation and
osteoclastic bone resorption in rat bone tissues in vitro. Mol Cell
Biochem. 228:39–47. 2001.
|
|
125
|
Tsukamoto Y: Studies on action of
menaquinone-7 in regulation of bone metabolism and its preventive
role of osteoporosis. Biofactors. 22:5–19. 2004.
|
|
126
|
Wu WJ, Kim MS and Ahn BY: The inhibitory
effect of vitamin K on RANKL-induced osteoclast differentiation and
bone resorption. Food Funct. 6:3351–3358. 2015.
|
|
127
|
Lee AS, Sung MJ, Son SJ, Han AR, Hong SM
and Lee SH: Effect of menaquinone-4 on receptor activator of
nuclear factor κB ligand-induced osteoclast differentiation and
ovariectomy-induced bone loss. J Med Food. 26:128–134. 2023.
|
|
128
|
Taira H, Fujikawa Y, Kudo O, Itonaga I and
Torisu T: Menatetrenone (vitamin K2) acts directly on circulating
human osteoclast precursors. Calcif Tissue Int. 73:78–85. 2003.
|
|
129
|
Stock M and Schett G: Vitamin K-dependent
proteins in skeletal development and disease. Int J Mol Sci.
22:93282021.
|
|
130
|
Alonso N, Meinitzer A, Fritz-Petrin E,
Enko D and Herrmann M: Role of Vitamin K in bone and muscle
metabolism. Calcif Tissue Int. 112:178–196. 2023.
|
|
131
|
Komori T: Functions of osteocalcin in
bone, pancreas, testis, and muscle. Int J Mol Sci. 21:75132020.
|
|
132
|
Lacombe J and Ferron M:
Gamma-carboxylation regulates osteocalcin function. Oncotarget.
6:19924–19925. 2015.
|
|
133
|
Rasekhi H, Karandish M, Jalali MT,
Mohammad-Shahi M, Zarei M, Saki A and Shahbazian H: The effect of
vitamin K1 supplementation on sensitivity and insulin resistance
via osteocalcin in prediabetic women: A double-blind randomized
controlled clinical trial. Eur J Clin Nutr. 69:891–895. 2015.
|
|
134
|
Hussein AG, Mohamed RH, Shalaby SM and Abd
El Motteleb DM: Vitamin K2 alleviates type 2 diabetes in
rats by induction of osteocalcin gene expression. Nutrition.
47:33–38. 2018.
|
|
135
|
Clemens TL and Karsenty G: The osteoblast:
An insulin target cell controlling glucose homeostasis. J Bone
Miner Res. 26:677–680. 2011.
|
|
136
|
Roumeliotis S, Dounousi E, Eleftheriadis T
and Liakopoulos V: Association of the inactive circulating matrix
Gla protein with vitamin K Intake, calcification, mortality, and
cardiovascular disease: A review. Int J Mol Sci. 20:6282019.
|
|
137
|
Dalmeijer GW, van der Schouw YT, Vermeer
C, Magdeleyns EJ, Schurgers LJ and Beulens JW: Circulating matrix
Gla protein is associated with coronary artery calcification and
vitamin K status in healthy women. J Nutr Biochem. 24:624–628.
2013.
|
|
138
|
Mandatori D, Pelusi L, Schiavone V, Pipino
C, Di Pietro N and Pandolfi A: The dual role of vitamin K2 in
'bone-vascular crosstalk': Opposite effects on bone loss and
vascular calcification. Nutrients. 13:12222021.
|
|
139
|
Fusaro M, Noale M, Viola V, Galli F,
Tripepi G, Vajente N, Plebani M, Zaninotto M, Guglielmi G, Miotto
D, et al: Vitamin K, vertebral fractures, vascular calcifications,
and mortality: VItamin K Italian (VIKI) dialysis study. J Bone
Miner Res. 27:2271–2278. 2012.
|
|
140
|
Delanaye P, Krzesinski JM, Warling X,
Moonen M, Smelten N, Médart L, Pottel H and Cavalier E:
Dephosphorylated-uncarboxylated Matrix Gla protein concentration is
predictive of vitamin K status and is correlated with vascular
calcification in a cohort of hemodialysis patients. BMC Nephrol.
15:1452014.
|
|
141
|
Mandatori D, Pipino C, Di Tomo P,
Schiavone V, Ranieri A, Pantalone S, Di Silvestre S, Di
Pietrantonio N, Ucci M, Palmerini C, et al: Osteogenic
transdifferentiation of vascular smooth muscle cells isolated from
spontaneously hypertensive rats and potential menaquinone-4
inhibiting effect. J Cell Physiol. 234:19761–19773. 2019.
|
|
142
|
Schurgers LJ, Uitto J and Reutelingsperger
CP: Vitamin K-dependent carboxylation of matrix Gla-protein: A
crucial switch to control ectopic mineralization. Trends Mol Med.
19:217–226. 2013.
|
|
143
|
Tesfamariam B: Involvement of vitamin
K-dependent proteins in vascular calcification. J Cardiovasc
Pharmacol Ther. 24:323–333. 2019.
|
|
144
|
Yee MMF, Chin KY, Ima-Nirwana S and Wong
SK: Vitamin A and bone health: A review on current evidence.
Molecules. 26:17572021.
|
|
145
|
Burckhardt P: Vitamin A and bone health.
Nutrition and bone health. Humana Press; New York, NY: pp. 409–421.
2015
|
|
146
|
Navarro-Valverde C, Caballero-Villarraso
J, Mata-Granados JM, Casado-Díaz A, Sosa-Henríquez M, Malouf-Sierra
J, Nogués-Solán X, Rodríguez-Mañas L, Cortés-Gil X,
Delgadillo-Duarte J and Quesada-Gómez JM: High serum retinol as a
relevant contributor to low bone mineral density in postmenopausal
osteoporotic women. Calcif Tissue Int. 102:651–656. 2018.
|
|
147
|
Mata-Granados JM, Cuenca-Acevedo JR, Luque
de Castro MD, Holick MF and Quesada-Gómez JM: Vitamin D
insufficiency together with high serum levels of vitamin A
increases the risk for osteoporosis in postmenopausal women. Arch
Osteoporos. 8:1242013.
|
|
148
|
Zhang X, Huang J, Zhou Y, Hong Z, Lin X,
Chen S, Ye Y and Zhang Z: Vitamin A nutritional status is a key
determinant of bone mass in children. Nutrients. 14:46942022.
|
|
149
|
Tanumihardjo SA, Gannon BM, Kaliwile C,
Chileshe J and Binkley NC: Restricting vitamin A intake increases
bone formation in Zambian children with high liver stores of
vitamin. Arch Osteoporos. 14:722019.
|
|
150
|
Maggio D, Polidori MC, Barabani M, Tufi A,
Ruggiero C, Cecchetti R, Aisa MC, Stahl W and Cherubini A: Low
levels of carotenoids and retinol in involutional osteoporosis.
Bone. 38:244–248. 2006.
|
|
151
|
Yang Z, Zhang Z, Penniston KL, Binkley N
and Tanumihardjo SA: Serum carotenoid concentrations in
postmenopausal women from the United States with and without
osteoporosis. Int J Vitam Nutr Res. 78:105–111. 2008.
|
|
152
|
Balasuriya CND, Larose TL, Mosti MP,
Evensen KAI, Jacobsen GW, Thorsby PM, Stunes AK and Syversen U:
Maternal serum retinol, 25(OH)D and 1,25(OH)2D concentrations
during pregnancy and peak bone mass and trabecular bone score in
adult offspring at 26-year follow-up. PLoS One.
14:e02227122019.
|
|
153
|
Holvik K, Ahmed LA, Forsmo S, Gjesdal CG,
Grimnes G, Samuelsen SO, Schei B, Blomhoff R, Tell GS and Meyer HE:
No increase in risk of hip fracture at high serum retinol
concentrations in community-dwelling older Norwegians: The
Norwegian epidemiologic osteoporosis studies. Am J Clin Nutr.
102:1289–1296. 2015.
|
|
154
|
Zhou P, Shao R, Wang H, Miao J and Wang X:
Dietary vitamin A, C, and E intake and subsequent fracture risk at
various sites: A meta-analysis of prospective cohort studies.
Medicine (Baltimore). 99:e208412020.
|
|
155
|
Rejnmark L, Vestergaard P, Charles P,
Hermann AP, Brot C, Eiken P and Mosekilde L: No effect of vitamin A
intake on bone mineral density and fracture risk in perimenopausal
women. Osteoporos Int. 15:872–880. 2004.
|
|
156
|
de Jonge EA, Kiefte-de Jong JC,
Campos-Obando N, Booij L, Franco OH, Hofman A, Uitterlinden AG,
Rivadeneira F and Zillikens MC: Dietary vitamin A intake and bone
health in the elderly: The Rotterdam study. Eur J Clin Nutr.
69:1360–1368. 2015.
|
|
157
|
Zia-Ul-Haq M, Riaz M and Modhi AO:
Carotenoids and bone health. In: Carotenoids: Structure and
Function in the Human Body. Springer Cham. 697–713. 2021.
|
|
158
|
Dai Z, Wang R, Ang LW, Low YL, Yuan JM and
Koh WP: Protective effects of dietary carotenoids on risk of hip
fracture in men: The Singapore Chinese health study. J Bone Miner
Res. 29:408–417. 2014.
|
|
159
|
Cao WT, Zeng FF, Li BL, Lin JS, Liang YY
and Chen YM: Higher dietary carotenoid intake associated with lower
risk of hip fracture in middle-aged and elderly Chinese: A matched
case-control study. Bone. 111:116–122. 2018.
|
|
160
|
Xu J, Song C, Song X, Zhang X and Li X:
Carotenoids and risk of fracture: A meta-analysis of observational
studies. Oncotarget. 8:2391–2399. 2017.
|
|
161
|
Gao SS and Zhao Y: The effects of
β-carotene on osteoporosis: A systematic review and meta-analysis
of observational studies. Osteoporos Int. 34:627–639. 2023.
|
|
162
|
Zhang ZQ, Cao WT, Liu J, Cao Y, Su YX and
Chen YM: Greater serum carotenoid concentration associated with
higher bone mineral density in Chinese adults. Osteoporos Int.
27:1593–1601. 2016.
|
|
163
|
Hayhoe RPG, Lentjes MAH, Mulligan AA,
Luben RN, Khaw KT and Welch AA: Carotenoid dietary intakes and
plasma concentrations are associated with heel bone ultrasound
attenuation and osteoporotic fracture risk in the European
prospective investigation into cancer and nutrition (EPIC)-Norfolk
cohort. Br J Nutr. 117:1439–1453. 2017.
|
|
164
|
Tanaka K, Tanaka S, Sakai A, Ninomiya T,
Arai Y and Nakamura T: Deficiency of vitamin A delays bone healing
process in association with reduced BMP2 expression after
drill-hole injury in mice. Bone. 47:1006–1012. 2010.
|
|
165
|
Shen Q, Wang X, Bai H, Tan X and Liu X:
Effects of high-dose all-trans retinoic acid on longitudinal bone
growth of young rats. Growth Horm IGF Res. 62:1014462022.
|
|
166
|
Broulík PD, Raška I and Brouliková K:
Prolonged overdose of all-trans retinoic acid enhances bone
sensitivity in castrated mice. Nutrition. 29:1166–1169. 2013.
|
|
167
|
Lionikaite V, Henning P, Drevinge C, Shah
FA, Palmquist A, Wikström P, Windahl SH and Lerner UH: Vitamin A
decreases the anabolic bone response to mechanical loading by
suppressing bone formation. FASEB J. 33:5237–5247. 2019.
|
|
168
|
Weng Z, Wang C, Zhang C, Xu J, Chai Y, Jia
Y, Han P and Wen G: All-trans retinoic acid promotes osteogenic
differentiation and bone consolidation in a rat distraction
osteogenesis model. Calcif Tissue Int. 104:320–330. 2019.
|
|
169
|
Zhang S, Chen X, Hu Y, Wu J, Cao Q, Chen S
and Gao Y: All-trans retinoic acid modulates Wnt3A-induced
osteogenic differentiation of mesenchymal stem cells via activating
the PI3K/AKT/GSK3β signalling pathway. Mol Cell Endocrinol.
422:243–253. 2016.
|
|
170
|
Zhang W, Deng ZL, Chen L, Zuo GW, Luo Q,
Shi Q, Zhang BQ, Wagner ER, Rastegar F, Kim SH, et al: Retinoic
acids potentiate BMP9-induced osteogenic differentiation of
mesenchymal progenitor cells. PLoS One. 5:e119172010.
|
|
171
|
Osathanon T, Manokawinchoke J, Egusa H and
Pavasant P: Notch signaling partly regulates the osteogenic
differentiation of retinoic acid-treated murine induced pluripotent
stem cells. J Oral Sci. 59:405–413. 2017.
|
|
172
|
Dingwall M, Marchildon F, Gunanayagam A,
Louis CS and Wiper-Bergeron N: Retinoic acid-induced Smad3
expression is required for the induction of osteoblastogenesis of
mesenchymal stem cells. Differentiation. 82:57–65. 2011.
|
|
173
|
Wiper-Bergeron N, St-Louis C and Lee JM:
CCAAT/Enhancer binding protein beta abrogates retinoic acid-induced
osteoblast differentiation via repression of Runx2 transcription.
Mol Endocrinol. 21:2124–2135. 2007.
|
|
174
|
Hisada K, Hata K, Ichida F, Matsubara T,
Orimo H, Nakano T, Yatani H, Nishimura R and Yoneda T: Retinoic
acid regulates commitment of undifferentiated mesenchymal stem
cells into osteoblasts and adipocytes. J Bone Miner Metab.
31:53–63. 2013.
|
|
175
|
Cruz ACC, Cardozo FTGS, Magini RS and
Simões CMO: Retinoic acid increases the effect of bone
morphogenetic protein type 2 on osteogenic differentiation of human
adipose-derived stem cells. J Appl Oral Sci. 27:e201803172019.
|
|
176
|
Liu Y, Liu Y, Zhang R, Wang X, Huang F,
Yan Z, Nie M, Huang J, Wang Y, Wang Y, et al: All-trans retinoic
acid modulates bone morphogenic protein 9-induced osteogenesis and
adipogenesis of preadipocytes through BMP/Smad and Wnt/β-catenin
signaling pathways. Int J Biochem Cell Biol. 47:47–56. 2014.
|
|
177
|
Skillington J, Choy L and Derynck R: Bone
morphogenetic protein and retinoic acid signaling cooperate to
induce osteoblast differentiation of preadipocytes. J Cell Biol.
159:135–146. 2002.
|
|
178
|
Ferreira-Baptista C, Queirós A, Ferreira
R, Fernandes MH, Gomes PS and Colaço B: Retinoic acid induces the
osteogenic differentiation of cat adipose tissue-derived stromal
cells from distinct anatomical sites. J Anat. 242:277–288.
2023.
|
|
179
|
Shao Y, Chen QZ, Zeng YH, Li Y, Ren WY,
Zhou LY, Liu RX, Wu K, Yang JQ, Deng ZL, et al: All-trans retinoic
acid shifts rosiglitazone-induced adipogenic differentiation to
osteogenic differentiation in mouse embryonic fibroblasts. Int J
Mol Med. 38:1693–1702. 2016.
|
|
180
|
Song HM, Nacamuli RP, Xia W, Bari AS, Shi
YY, Fang TD and Longaker MT: High-dose retinoic acid modulates rat
calvarial osteoblast biology. J Cell Physiol. 202:255–262.
2005.
|
|
181
|
Jeradi S and Hammerschmidt M: Retinoic
acid-induced premature osteoblast-to-preosteocyte transitioning has
multiple effects on calvarial development. Development.
143:1205–1216. 2016.
|
|
182
|
Jacobsen C and Craft AM:
Retinoic-acid-induced osteogenesis of hiPSCs. Nat Biomed Eng.
3:504–506. 2019.
|
|
183
|
Sun W, Shi A, Ma D, Bolscher JGM, Nazmi K,
Veerman ECI, Bikker FJ, Lin H and Wu G: All-trans retinoic acid and
human salivary histatin-1 promote the spreading and osteogenic
activities of pre-osteoblasts in vitro. FEBS Open Bio. 10:396–406.
2020.
|
|
184
|
Karakida T, Yui R, Suzuki T, Fukae M and
Oida S: Retinoic acid receptor γ-dependent signaling cooperates
with BMP2 to induce osteoblastic differentiation of C2C12 cells.
Connect Tissue Res. 52:365–372. 2011.
|
|
185
|
Bi W, Gu Z, Zheng Y, Zhang X, Guo J and Wu
G: Heterodimeric BMP-2/7 antagonizes the inhibition of all-trans
retinoic acid and promotes the osteoblastogenesis. PLoS One.
8:e781982013.
|
|
186
|
Roa LA, Bloemen M, Carels CEL, Wagener
FADTG and Von den Hoff JW: Retinoic acid disrupts osteogenesis in
pre-osteoblasts by down-regulating WNT signaling. Int J Biochem
Cell Biol. 116:1055972019.
|
|
187
|
Krutzen CLJM, Roa LA, Bloemen M and Von
den Hoff JW: Excess vitamin a might contribute to submucous
clefting by inhibiting WNT-mediated bone formation. Orthod
Craniofac Res. 26:132–139. 2023.
|
|
188
|
Liu Y, Ma X, Guo J, Lin Z, Zhou M, Bi W,
Liu J, Wang J, Lu H and Wu G: All-trans retinoic acid can
antagonize osteoblastogenesis induced by different BMPs
irrespective of their dimerization types and dose-efficiencies.
Drug Des Devel Ther. 12:3419–3430. 2018.
|
|
189
|
Chen M, Huang HZ, Wang M and Wang AX:
Retinoic acid inhibits osteogenic differentiation of mouse
embryonic palate mesenchymal cells. Birth Defects Res A Clin Mol
Teratol. 88:965–970. 2010.
|
|
190
|
Chen M, Yang X, LI ZM, Liu X, Wang WC and
Huang HZ: Inhibitory effect of all-trans retinoic acid on
osteogenic differentiation of mouse embryonic palate mesenchymal
cells and its possible mechanism. Chin J Pharmacol Toxicol.
29:836–841. 2015.
|
|
191
|
Wang S, Bi W, Liu Y, Cheng J, Sun W, Wu G
and Xu X: The antagonist of retinoic acid receptor α, ER-50891
antagonizes the inhibitive effect of all-trans retinoic acid and
rescues bone morphogenetic protein 2-induced osteoblastogenic
differentiation. Drug Des Devel Ther. 14:297–308. 2020.
|
|
192
|
Nuka S, Sawada N, Iba K, Chiba H, Ishii S
and Mori M: All-trans retinoic acid inhibits dexamethasone-induced
ALP activity and mineralization in human osteoblastic cell line SV
HFO. Cell Struct Funct. 22:27–32. 1997.
|
|
193
|
Ewendt F, Lehmann A, Wodak MF and Stangl
GI: All-trans retinoic acid and beta-carotene increase sclerostin
production in C2C12 myotubes. Biomedicines. 11:14322023.
|
|
194
|
Guo L, Zhang Y, Liu H, Cheng Q, Yang S and
Yang D: All-trans retinoic acid inhibits the osteogenesis of
periodontal ligament stem cells by promoting IL-1β production via
NF-κB signaling. Int Immunopharmacol. 108:1087572022.
|
|
195
|
Ahmed N, Sammons J, Khokher MA and Hassan
HT: Retinoic acid suppresses interleukin 6 production in normal
human osteoblasts. Cytokine. 12:289–293. 2000.
|
|
196
|
Shen CX and Bi WJ: Role of all-trans
retinoic acid in osteogenic differentiation. J Oral Sci Res.
34:1038–1041. 2018.
|
|
197
|
Hu L, Lind T, Sundqvist A, Jacobson A and
Melhus H: Retinoic acid increases proliferation of human osteoclast
progenitors and inhibits RANKL-stimulated osteoclast
differentiation by suppressing RANK. PLoS One. 5:e133052010.
|
|
198
|
Balkan W, Rodríguez-Gonzalez M, Pang M,
Fernandez I and Troen BR: Retinoic acid inhibits NFATc1 expression
and osteoclast differentiation. J Bone Miner Metab. 29:652–661.
2011.
|
|
199
|
Conaway HH, Persson E, Halén M, Granholm
S, Svensson O, Pettersson U, Lie A and Lerner UH: Retinoids inhibit
differentiation of hematopoietic osteoclast progenitors. FASEB J.
23:3526–3538. 2009.
|
|
200
|
Bi W, Liu Y, Guo J, Lin Z, Liu J, Zhou M,
Wismeijer D, Pathak JL and Wu G: All-trans retinoic-acid inhibits
heterodimeric bone morphogenetic protein 2/7-stimulated
osteoclastogenesis, and resorption activity. Cell Biosci.
8:482018.
|
|
201
|
Kindmark A, Melhus H, Ljunghall S and
Ljunggren O: Inhibitory effects of 9-cis and all-trans retinoic
acid on 1,25(OH)2 vitamin D3-induced bone resorption. Calcif Tissue
Int. 57:242–244. 1995.
|
|
202
|
Conaway HH, Pirhayati A, Persson E,
Pettersson U, Svensson O, Lindholm C, Henning P, Tuckermann J and
Lerner UH: Retinoids stimulate periosteal bone resorption by
enhancing the protein RANKL, a response inhibited by monomeric
glucocorticoid receptor. J Biol Chem. 286:31425–31436. 2011.
|
|
203
|
Saneshige S, Mano H, Tezuka K, Kakudo S,
Mori Y, Honda Y, Itabashi A, Yamada T, Miyata K, Hakeda Y, et al:
Retinoic acid directly stimulates osteoclastic bone resorption and
gene expression of cathepsin K/OC-2. Biochem J. 309:721–724.
1995.
|
|
204
|
Lind T, Öhman C, Calounova G, Rasmusson A,
Andersson G, Pejler G and Melhus H: Excessive dietary intake of
vitamin A reduces skull bone thickness in mice. PLoS One.
12:e01762172017.
|
|
205
|
Yamaguchi M: Role of carotenoid
β-cryptoxanthin in bone homeostasis. J Biomed Sci. 19:362012.
|
|
206
|
Uchiyama S and Yamaguchi M:
Beta-cryptoxanthin stimulates cell differentiation and
mineralization in osteoblastic MC3T3-E1 cells. J Cell Biochem.
95:1224–1234. 2005.
|
|
207
|
Yamaguchi M and Weitzmann MN: The bone
anabolic carotenoid beta-cryptoxanthin enhances transforming growth
factor-beta1-induced SMAD activation in MC3T3 preosteoblasts. Int J
Mol Med. 24:671–675. 2009.
|
|
208
|
Yamaguchi M and Weitzmann MN: The bone
anabolic carotenoids p-hydroxycinnamic acid and β-cryptoxanthin
antagonize NF-κB activation in MC3T3 preosteoblasts. Mol Med Rep.
2:641–644. 2009.
|
|
209
|
Yamaguchi M and Weitzmann MN: The bone
anabolic carotenoid p-hydroxycinnamic acid promotes osteoblast
mineralization and suppresses osteoclast differentiation by
antagonizing NF-κB activation. Int J Mol Med. 30:708–712. 2012.
|
|
210
|
Zhu K, Yang C, Dai H, Li J, Liu W, Luo Y,
Zhang X and Wang Q: Crocin inhibits titanium particle-induced
inflammation and promotes osteogenesis by regulating macrophage
polarization. Int Immunopharmacol. 76:1058652019.
|
|
211
|
Kalalinia F, Ghasim H, Amel Farzad S,
Pishavar E, Ramezani M and Hashemi M: Comparison of the effect of
crocin and crocetin, two major compounds extracted from saffron, on
osteogenic differentiation of mesenchymal stem cells. Life Sci.
208:262–267. 2018.
|
|
212
|
Russo C, Ferro Y, Maurotti S, Salvati MA,
Mazza E, Pujia R, Terracciano R, Maggisano G, Mare R, Giannini S,
et al: Lycopene and bone: An in vitro investigation and a pilot
prospective clinical study. J Transl Med. 18:432020.
|
|
213
|
Oliveira GR, Vargas-Sanchez PK, Fernandes
RR, Ricoldi MST, Semeghini MS, Pitol DL, de Sousa LG, Siessere S
and Bombonato-Prado KF: Lycopene influences osteoblast functional
activity and prevents femur bone loss in female rats submitted to
an experimental model of osteoporosis. J Bone Miner Metab.
37:658–667. 2019.
|
|
214
|
Semeghini MS, Scalize PH, Coelho MC,
Fernandes RR, Pitol DL, Tavares MS, de Sousa LG, Coppi AA, Siessere
S and Bombonato-Prado KF: Lycopene prevents bone loss in
ovariectomized rats and increases the number of osteocytes and
osteoblasts. J Anat. 241:729–740. 2022.
|
|
215
|
Odes-Barth S, Khanin M, Linnewiel-Hermoni
K, Miller Y, Abramov K, Levy J and Sharoni Y: Inhibition of
osteoclast differentiation by carotenoid derivatives through
inhibition of the NF-κB pathway. Antioxidants (Basel).
9:11672020.
|
|
216
|
Linnewiel-Hermoni K, Motro Y, Miller Y,
Levy J and Sharoni Y: Carotenoid derivatives inhibit nuclear factor
kappa B activity in bone and cancer cells by targeting key thiol
groups. Free Radic Biol Med. 75:105–120. 2014.
|
|
217
|
Uchiyama S and Yamaguchi M: Inhibitory
effect of beta-cryptoxanthin on osteoclast-like cell formation in
mouse marrow cultures. Biochem Pharmacol. 67:1297–1305. 2004.
|
|
218
|
Hirata N, Ichimaru R, Tominari T,
Matsumoto C, Watanabe K, Taniguchi K, Hirata M, Ma S, Suzuki K,
Grundler FMW, et al: Beta-cryptoxanthin inhibits
lipopolysaccharide-induced osteoclast differentiation and bone
resorption via the suppression of inhibitor of NF-κB kinase
activity. Nutrients. 11:3682019.
|
|
219
|
Uchiyama S and Yamaguchi M:
Beta-cryptoxanthin stimulates apoptotic cell death and suppresses
cell function in osteoclastic cells: Change in their related gene
expression. J Cell Biochem. 98:1185–1195. 2006.
|
|
220
|
Ozaki K, Okamoto M, Fukasawa K, Iezaki T,
Onishi Y, Yoneda Y, Sugiura M and Hinoi E: Daily intake of
β-cryptoxanthin prevents bone loss by preferential disturbance of
osteoclastic activation in ovariectomized mice. J Pharmacol Sci.
129:72–77. 2015.
|
|
221
|
Matsumoto C, Ashida N, Yokoyama S,
Tominari T, Hirata M, Ogawa K, Sugiura M, Yano M, Inada M and
Miyaura C: The protective effects of β-cryptoxanthin on
inflammatory bone resorption in a mouse experimental model of
periodontitis. Biosci Biotechnol Biochem. 77:860–862. 2013.
|
|
222
|
Wang F, Wang N, Gao Y, Zhou Z, Liu W, Pan
C, Yin P, Yu X and Tang M: β-Carotene suppresses osteoclastogenesis
and bone resorption by suppressing NF-κB signaling pathway. Life
Sci. 174:15–20. 2017.
|
|
223
|
Mamun-Or-Rashid ANM, Lucy TT, Yagi M and
Yonei Y: Inhibitory effects of astaxanthin on CML-HSA-induced
inflammatory and RANKL-induced osteoclastogenic gene expression in
RAW 264.7 Cells. Biomedicines. 10:542021.
|
|
224
|
Tominari T, Matsumoto C, Watanabe K,
Hirata M, Grundler FM, Inada M and Miyaura C: Lutein, a carotenoid,
suppresses osteoclastic bone resorption and stimulates bone
formation in cultures. Biosci Biotechnol Biochem. 81:302–306.
2017.
|
|
225
|
Das SK, Ren R, Hashimoto T and Kanazawa K:
Fucoxanthin induces apoptosis in osteoclast-like cells
differentiated from RAW264.7 cells. J Agric Food Chem.
58:6090–6095. 2010.
|
|
226
|
Aghajanian P, Hall S, Wongworawat MD and
Mohan S: The roles and mechanisms of actions of vitamin C in bone:
New developments. J Bone Miner Res. 30:1945–1955. 2015.
|
|
227
|
Morton DJ, Barrett-Connor EL and Schneider
DL: Vitamin C supplement use and bone mineral density in
postmenopausal women. J Bone Miner Res. 16:135–140. 2001.
|
|
228
|
Malmir H, Shab-Bidar S and Djafarian K:
Vitamin C intake in relation to bone mineral density and risk of
hip fracture and osteoporosis: A systematic review and
meta-analysis of observational studies. Br J Nutr. 119:847–858.
2018.
|
|
229
|
Zeng LF, Luo MH, Liang GH, Yang WY, Xiao
X, Wei X, Yu J, Guo D, Chen HY, Pan JK, et al: Can dietary intake
of vitamin C-oriented foods reduce the risk of osteoporosis,
fracture, and BMD loss? Systematic review with meta-analyses of
recent studies. Front Endocrinol (Lausanne). 10:8442020.
|
|
230
|
Sun Y, Liu C, Bo Y, You J, Zhu Y, Duan D,
Cui H and Lu Q: Dietary vitamin C intake and the risk of hip
fracture: A dose-response meta-analysis. Osteoporos Int. 29:79–87.
2018.
|
|
231
|
Sahni S, Hannan MT, Gagnon D, Blumberg J,
Cupples LA, Kiel DP and Tucker KL: Protective effect of total and
supplemental vitamin C intake on the risk of hip fracture-a 17-year
follow-up from the Framingham osteoporosis study. Osteoporos Int.
20:1853–1861. 2009.
|
|
232
|
Kim YA, Kim KM, Lim S, Choi SH, Moon JH,
Kim JH, Kim SW, Jang HC and Shin CS: Favorable effect of dietary
vitamin C on bone mineral density in postmenopausal women (KNHANES
IV, 2009): Discrepancies regarding skeletal sites, age, and vitamin
D status. Osteoporos Int. 26:2329–2337. 2015.
|
|
233
|
Rondanelli M, Peroni G, Fossari F, Vecchio
V, Faliva MA, Naso M, Perna S, D Paolo E, Riva A, Petrangolini G,
et al: Evidence of a positive link between consumption and
supplementation of ascorbic acid and bone mineral density.
Nutrients. 13:10122021.
|
|
234
|
Lan KM, Wang LK, Lin YT, Hung KC, Wu LC,
Ho CH, Chang CY and Chen JY: Suboptimal plasma vitamin C is
associated with lower bone mineral density in young and early
middle-aged men: A retrospective cross-sectional study. Nutrients.
14:35562022.
|
|
235
|
Mangano KM, Noel SE, Dawson-Hughes B and
Tucker KL: Sufficient plasma vitamin C is related to greater bone
mineral density among postmenopausal women from the Boston Puerto
Rican Health Study. J Nutr. 151:3764–3772. 2021.
|
|
236
|
Sakamoto Y and Takano Y: Morphological
influence of ascorbic acid deficiency on endochondral ossification
in osteogenic disorder Shionogi rat. Anat Rec. 268:93–104.
2002.
|
|
237
|
Hasegawa T, Li M, Hara K, Sasaki M, Tabata
C, de Freitas PH, Hongo H, Suzuki R, Kobayashi M, Inoue K, et al:
Morphological assessment of bone mineralization in tibial
metaphyses of ascorbic acid-deficient ODS rats. Biomed Res.
32:259–269. 2011.
|
|
238
|
Segawa T, Miyakoshi N, Kasukawa Y, Aonuma
H, Tsuchie H and Shimada Y: Combined treatment with minodronate and
vitamin C increases bone mineral density and strength in vitamin
C-deficient rats. Osteoporos Sarcopenia. 2:30–37. 2016.
|
|
239
|
Zhu LL, Cao J, Sun M, Yuen T, Zhou R, Li
J, Peng Y, Moonga SS, Guo L, Mechanick JI, et al: Vitamin C
prevents hypogonadal bone loss. PLoS One. 7:e470582012.
|
|
240
|
Deyhim F, Strong K, Deyhim N, Vandyousefi
S, Stamatikos A and Faraji B: Vitamin C reverses bone loss in an
osteopenic rat model of osteoporosis. Int J Vitam Nutr Res.
88:58–64. 2018.
|
|
241
|
Park JK, Lee EM, Kim AY, Lee EJ, Min CW,
Kang KK, Lee MM and Jeong KS: Vitamin C deficiency accelerates bone
loss inducing an increase in PPAR-γ expression in SMP30 knockout
mice. Int J Exp Pathol. 93:332–340. 2012.
|
|
242
|
Hadzir SN, Ibrahim SN, Abdul Wahab RM,
Zainol Abidin IZ, Senafi S, Ariffin ZZ, Abdul Razak M and Zainal
Ariffin SH: Ascorbic acid induces osteoblast differentiation of
human suspension mononuclear cells. Cytotherapy. 16:674–682.
2014.
|
|
243
|
Okajima LS, Martinez EF, Pinheiro IF,
Fonseca Silva AS and Demasi APD: Effect of sodium ascorbyl
phosphate on osteoblast viability and differentiation. J
Periodontal Res. 55:660–666. 2020.
|
|
244
|
Yang HM and Seo HS: Effects of ascorbic
acid on osteoblast differentiation in MC3T3-E1 cells. Soonchunhyang
Med Sci. 19:93–98. 2013.
|
|
245
|
Carinci F, Pezzetti F, Spina AM, Palmieri
A, Laino G, De Rosa A, Farina E, Illiano F, Stabellini G, Perrotti
V and Piattelli A: Effect of vitamin C on pre-osteoblast gene
expression. Arch Oral Biol. 50:481–496. 2005.
|
|
246
|
Ciceri P, Volpi E, Brenna I, Arnaboldi L,
Neri L, Brancaccio D and Cozzolino M: Combined effects of ascorbic
acid and phosphate on rat VSMC osteoblastic differentiation.
Nephrol Dial Transplant. 27:122–127. 2012.
|
|
247
|
Valenti MT, Zanatta M, Donatelli L,
Viviano G, Cavallini C, Scupoli MT and Dalle Carbonare L: Ascorbic
acid induces either differentiation or apoptosis in MG-63
osteosarcoma lineage. Anticancer Res. 34:1617–1627. 2014.
|
|
248
|
Choi HK, Kim GJ, Yoo HS, Song DH, Chung
KH, Lee KJ, Koo YT and An JH: Vitamin C activates
osteoblastogenesis and inhibits osteoclastogenesis via
Wnt/β-catenin/ATF4 signaling pathways. Nutrients. 11:5062019.
|
|
249
|
Burger MG, Steinitz A, Geurts J, Pippenger
BE, Schaefer DJ, Martin I, Barbero A and Pelttari K: Ascorbic acid
attenuates senescence of human osteoarthritic osteoblasts. Int J
Mol Sci. 18:25172017.
|
|
250
|
Son E, Do H, Joo HM and Pyo S: Induction
of alkaline phosphatase activity by L-ascorbic acid in human
osteoblastic cells: A potential role for CK2 and Ikaros. Nutrition.
23:745–753. 2007.
|
|
251
|
Xing W, Pourteymoor S and Mohan S:
Ascorbic acid regulates osterix expression in osteoblasts by
activation of prolyl hydroxylase and ubiquitination-mediated
proteosomal degradation pathway. Physiol Genomics. 43:749–757.
2011.
|
|
252
|
Rosadi I, Indrady FT, Karina K and Hariani
N: Evaluation effects of ascorbic acid leads to activate and induce
osteogenic protein marker expression: In silico and in-vitro study.
Biomed Res Ther. 9:4832–4841. 2022.
|
|
253
|
Pustylnik S, Fiorino C, Nabavi N,
Zappitelli T, da Silva R, Aubin JE and Harrison RE: EB1 levels are
elevated in ascorbic Acid (AA)-stimulated osteoblasts and mediate
cell-cell adhesion-induced osteoblast differentiation. J Biol Chem.
288:22096–22110. 2013.
|
|
254
|
Farhadian N, Miresmaeili A, Azar R,
Zargaran M, Moghimbeigi A and Soheilifar S: Effect of dietary
ascorbic acid on osteogenesis of expanding midpalatal suture in
rats. J Dent (Tehran). 12:39–48. 2015.
|
|
255
|
Rahman F, Bordignon B, Culerrier R,
Peiretti F, Spicuglia S, Djabali M, Landrier JF and Fontes M:
Ascorbic acid drives the differentiation of mesoderm-derived
embryonic stem cells. Involvement of p38 MAPK/CREB and SVCT2
transporter. Mol Nutr Food Res. 61:2017.
|
|
256
|
Rahman F, Al Frouh F, Bordignon B,
Fraterno M, Landrier JF, Peiretti F and Fontes M: Ascorbic acid is
a dose-dependent inhibitor of adipocyte differentiation, probably
by reducing cAMP pool. Front Cell Dev Biol. 2:292014.
|
|
257
|
Takamizawa S, Maehata Y, Imai K, Senoo H,
Sato S and Hata R: Effects of ascorbic acid and ascorbic acid
2-phosphate, a long-acting vitamin C derivative, on the
proliferation and differentiation of human osteoblast-like cells.
Cell Biol Int. 28:255–265. 2004.
|
|
258
|
Mizutani A, Sugiyama I, Kuno E, Matsunaga
S and Tsukagoshi N: Expression of matrix metalloproteinases during
ascorbate-induced differentiation of osteoblastic MC3T3-E1 cells. J
Bone Miner Res. 16:2043–2049. 2001.
|
|
259
|
Thaler R, Khani F, Sturmlechner I,
Dehghani SS, Denbeigh JM, Zhou X, Pichurin O, Dudakovic A, Jerez
SS, Zhong J, et al: Vitamin C epigenetically controls osteogenesis
and bone mineralization. Nat Commun. 13:58832022.
|
|
260
|
Xiao XH, Liao EY, Zhou HD, Dai RC, Yuan LQ
and Wu XP: Ascorbic acid inhibits osteoclastogenesis of RAW264.7
cells induced by receptor activated nuclear factor kappaB ligand
(RANKL) in vitro. J Endocrinol Invest. 28:253–260. 2005.
|
|
261
|
Takarada T, Hinoi E, Kambe Y, Sahara K,
Kurokawa S, Takahata Y and Yoneda Y: Osteoblast protects osteoclast
devoid of sodium-dependent vitamin C transporters from oxidative
cytotoxicity of ascorbic acid. Eur J Pharmacol. 575:1–11. 2007.
|
|
262
|
Sanbe T, Tomofuji T, Ekuni D, Azuma T,
Irie K, Tamaki N, Yamamoto T and Morita M: Vitamin C intake
inhibits serum lipid peroxidation and osteoclast differentiation on
alveolar bone in rats fed on a high-cholesterol diet. Arch Oral
Biol. 54:235–240. 2009.
|
|
263
|
Hie M and Tsukamoto I: Vitamin
C-deficiency stimulates osteoclastogenesis with an increase in RANK
expression. J Nutr Biochem. 22:164–171. 2011.
|
|
264
|
Otsuka E, Kato Y, Hirose S and Hagiwara H:
Role of ascorbic acid in the osteoclast formation: Induction of
osteoclast differentiation factor with formation of the
extracellular collagen matrix. Endocrinology. 141:3006–3011.
2000.
|
|
265
|
Tsuneto M, Yamazaki H, Yoshino M, Yamada T
and Hayashi S: Ascorbic acid promotes osteoclastogenesis from
embryonic stem cells. Biochem Biophys Res Commun. 335:1239–1246.
2005.
|
|
266
|
Ragab AA, Lavish SA, Banks MA, Goldberg VM
and Greenfield EM: Osteoclast differentiation requires ascorbic
acid. J Bone Miner Res. 13:970–977. 1998.
|
|
267
|
Noh AL and Yim M: Beta-glycerophosphate
accelerates RANKL-induced osteoclast formation in the presence of
ascorbic acid. Pharmazie. 66:195–200. 2011.
|
|
268
|
Le Nihouannen D, Barralet JE, Fong JE and
Komarova SV: Ascorbic acid accelerates osteoclast formation and
death. Bone. 46:1336–1343. 2010.
|
|
269
|
Rahman S and Baumgartner M: B vitamins:
Small molecules, big effects. J Inherit Metab Dis. 42:579–580.
2019.
|
|
270
|
Dai Z and Koh WP: B-vitamins and bone
health-a review of the current evidence. Nutrients. 7:3322–3346.
2015.
|
|
271
|
Tucker KL, Hannan MT, Qiao N, Jacques PF,
Selhub J, Cupples LA and Kiel DP: Low plasma vitamin B12 is
associated with lower BMD: The Framingham osteoporosis study. J
Bone Miner Res. 20:152–158. 2005.
|
|
272
|
Pawlak R: Vitamin B12 status is a risk
factor for bone fractures among vegans. Med Hypotheses.
153:1106252021.
|
|
273
|
Zhang H, Tao X and Wu J: Association of
homocysteine, vitamin B12, and folate with bone mineral density in
postmenopausal women: A meta-analysis. Arch Gynecol Obstet.
289:1003–1009. 2014.
|
|
274
|
Ouzzif Z, Oumghar K, Sbai K, Mounach A,
Derouiche M and El Maghraoui A: Relation of plasma total
homocysteine, folate and vitamin B12 levels to bone mineral density
in Moroccan healthy postmenopausal women. Rheumatol Int.
32:123–128. 2012.
|
|
275
|
Wang J, Chen L, Zhang Y, Li CG, Zhang H,
Wang Q, Qi X, Qiao L, Da WW, Cui XJ, et al: Association between
serum vitamin B6 concentration and risk of osteoporosis in the
middle-aged and older people in China: A cross-sectional study. BMJ
Open. 9:e0281292019.
|
|
276
|
Dai Z, Wang R, Ang LW, Yuan JM and Koh WP:
Dietary B vitamin intake and risk of hip fracture: The Singapore
Chinese health study. Osteoporos Int. 24:2049–2059. 2013.
|
|
277
|
Li Z, Zhang S, Wan L, Song X, Yuan D,
Zhang S, Wu D and Jiang J: Vitamin B6 as a novel risk biomarker of
fractured ankles. Medicine (Baltimore). 100:e274422021.
|
|
278
|
Baines M, Kredan MB, Usher J, Davison A,
Higgins G, Taylor W, West C, Fraser WD and Ranganath LR: The
association of homocysteine and its determinants MTHFR genotype,
folate, vitamin B12 and vitamin B6 with bone mineral density in
postmenopausal British women. Bone. 40:730–736. 2007.
|
|
279
|
Rondanelli M, Tartara A, Fossari F,
Vecchio V, Faliva MA, Naso M, Perna S, Nichetti M and Peroni G:
Adequate intake and supplementation of B vitamins, in particular
folic acid, can play a protective role in bone health. Curr Aging
Sci. 15:110–120. 2022.
|
|
280
|
Clements M, Heffernan M, Ward M, Hoey L,
Doherty LC, Hack Mendes R, Clarke MM, Hughes CF, Love I, Murphy S,
et al: A 2-year randomized controlled trial with low-dose B-vitamin
supplementation shows benefits on bone mineral density in adults
with lower B12 status. J Bone Miner Res. 37:2443–2455. 2022.
|
|
281
|
Kalimeri M, Leek F, Wang NX, Koh HR, Roy
NC, Cameron-Smith D, Kruger MC, Henry CJ and Totman JJ: Folate and
vitamin B-12 status is associated with bone mineral density and hip
strength of postmenopausal Chinese-Singaporean women. JBMR Plus.
4:e103992020.
|
|
282
|
Holstein JH, Herrmann M, Splett C,
Herrmann W, Garcia P, Histing T, Graeber S, Ong MF, Kurz K, Siebel
T, et al: Low serum folate and vitamin B6 are associated with an
altered cancellous bone structure in humans. Am J Clin Nutr.
90:1440–1445. 2009.
|
|
283
|
He T, Jin X, Koh YS, Zhang Q, Zhang C and
Liu F: The association of homocysteine, folate, vitamin B12, and
vitamin B6 with fracture incidence in older adults: A systematic
review and meta-analysis. Ann Transl Med. 9:11432021.
|
|
284
|
Haliloglu B, Aksungar FB, Ilter E, Peker
H, Akin FT, Mutlu N and Ozekici U: Relationship between bone
mineral density, bone turnover markers and homocysteine, folate and
vitamin B12 levels in postmenopausal women. Arch Gynecol Obstet.
281:663–668. 2010.
|
|
285
|
Haroon NN, Marwaha RK, Godbole MM and
Gupta SK: Role of B12 and homocysteine status in
determining BMD and bone turnover in young Indians. J Clin
Densitom. 15:366–373. 2012.
|
|
286
|
El Maghraoui A, Ghozlani I, Mounach A,
Rezqi A, Oumghar K, Achemlal L, Bezza A and Ouzzif Z: Homocysteine,
folate, and vitamin B12 levels and vertebral fracture risk in
postmenopausal women. J Clin Densitom. 15:328–333. 2012.
|
|
287
|
Keser I, Ilich JZ, Vrkić N, Giljević Z and
Colić Barić I: Folic acid and vitamin B(12) supplementation lowers
plasma homocysteine but has no effect on serum bone turnover
markers in elderly women: A randomized, double-blind,
placebo-controlled trial. Nutr Res. 33:211–219. 2013.
|
|
288
|
Oliai Araghi S, Kiefte-de Jong JC, van
Dijk SC, Swart KMA, Ploegmakers KJ, Zillikens MC, van Schoor NM, de
Groot LCPGM, Lips P, Stricker BH, et al: Long-term effects of folic
acid and vitamin-B12 supplementation on fracture risk and
cardiovascular disease: Extended follow-up of the B-PROOF trial.
Clin Nutr. 40:1199–1206. 2021.
|
|
289
|
Enneman AW, Swart KM, van Wijngaarden JP,
van Dijk SC, Ham AC, Brouwer-Brolsma EM, van der Zwaluw NL,
Dhonukshe-Rutten RA, van der Cammen TJ, de Groot LC, et al: Effect
of vitamin B12 and folic acid supplementation on bone mineral
density and quantitative ultrasound parameters in older people with
an elevated plasma homocysteine level: B-PROOF, a randomized
controlled trial. Calcif Tissue Int. 96:401–409. 2015.
|
|
290
|
Stone KL, Lui LY, Christen WG, Troen AM,
Bauer DC, Kado D, Schambach C, Cummings SR and Manson JE: Effect of
combination folic acid, vitamin B6, and vitamin
B12 supplementation on fracture risk in women: A
randomized, controlled trial. J Bone Miner Res. 32:2331–2338.
2017.
|
|
291
|
Ahn TK, Kim JO, An HJ, Park HS, Choi UY,
Sohn S, Kim KT, Kim NK and Han IB: 3'-UTR polymorphisms of vitamin
B-related genes are associated with osteoporosis and osteoporotic
vertebral compression fractures (OVCFs) in postmenopausal women.
Genes (Basel). 11:6122020.
|
|
292
|
Liu CT, Karasik D, Xu H, Zhou Y, Broe K,
Cupples LA, Cpgm de Groot L, Ham A, Hannan MT, Hsu YH, et al:
Genetic variants modify the associations of concentrations of
methylmalonic acid, vitamin B-12, vitamin B-6, and folate with bone
mineral density. Am J Clin Nutr. 114:578–587. 2021.
|
|
293
|
He H, Zhang Y, Sun Y, Zhang Y, Xu J, Yang
Y and Chen J: Folic acid attenuates high-fat diet-induced
osteoporosis through the AMPK signaling pathway. Front Cell Dev
Biol. 9:7918802022.
|
|
294
|
Cai H, Lin L, Wang G, Berman Z, Yang X and
Cheng X: Folic acid rescues corticosteroid-induced vertebral
malformations in chick embryos through targeting TGF-β signaling. J
Cell Physiol. 235:8626–8639. 2020.
|
|
295
|
Mohammadi A, Omrani L, Omrani LR, Kiani F,
Eshraghian A, Azizi Z and Omrani GR: Protective effect of folic
acid on cyclosporine-induced bone loss in rats. Transpl Int.
25:127–133. 2012.
|
|
296
|
Su S, Zhang D, Liu J, Zhao H, Tang X, Che
H, Wang Q, Ren W and Zhen D: Folate ameliorates
homocysteine-induced osteoblast dysfunction by reducing endoplasmic
reticulum stress-activated PERK/ATF-4/CHOP pathway in MC3T3-E1
cells. J Bone Miner Metab. 40:422–433. 2022.
|
|
297
|
Santos C, Gomes P, Duarte JA, Almeida MM,
Costa MEV and Fernandes MH: Development of hydroxyapatite
nanoparticles loaded with folic acid to induce osteoblastic
differentiation. Int J Pharm. 516:185–195. 2017.
|
|
298
|
Huot PS, Dodington DW, Mollard RC,
Reza-López SA, Sánchez-Hernández D, Cho CE, Kuk J, Ward WE and
Anderson GH: High folic acid intake during pregnancy lowers body
weight and reduces femoral area and strength in female rat
offspring. J Osteoporos. 2013:1541092013.
|
|
299
|
Singh P, Telnova S, Zhou B, Mohamed AD,
Mello V, Wackerhage H, Guo XE, Panda AK and Yadav VK: Maternal
vitamin B12 in mice positively regulates bone, but not
muscle mass and strength in post-weaning and mature offspring. Am J
Physiol Regul Integr Comp Physiol. 320:R984–R993. 2021.
|
|
300
|
Roman-Garcia P, Quiros-Gonzalez I, Mottram
L, Lieben L, Sharan K, Wangwiwatsin A, Tubio J, Lewis K, Wilkinson
D, Santhanam B, et al: Vitamin B12-dependent taurine
synthesis regulates growth and bone mass. J Clin Invest.
124:2988–3002. 2014.
|
|
301
|
Vaes BLT, Lute C, Blom HJ, Bravenboer N,
de Vries TJ, Everts V, Dhonukshe-Rutten RA, Müller M, de Groot
LCPGM and Steegenga WT: Vitamin B(12) deficiency stimulates
osteoclastogenesis via increased homocysteine and methylmalonic
acid. Calcif Tissue Int. 84:413–422. 2009.
|
|
302
|
Herrmann M, Widmann T, Colaianni G,
Colucci S, Zallone A and Herrmann W: Increased osteoclast activity
in the presence of increased homocysteine concentrations. Clin
Chem. 51:2348–2353. 2005.
|
|
303
|
Shiga T, Kimira Y, Mano H, Kawata T,
Tadokoro T, Suzuki T and Yamamoto Y: Vitamin B12
deficiency-induced increase of osteoclastic bone resorption caused
by abnormal renal resorption of inorganic phosphorus via Napi2a.
Biosci Biotechnol Biochem. 80:510–513. 2016.
|
|
304
|
Massé PG, Delvin EE, Hauschka PV, Donovan
SM, Grynpas MD, Mahuren JD, Watkins BA and Howell DS: Perturbations
in factors that modulate osteoblast functions in vitamin B6
deficiency. Can J Physiol Pharmacol. 78:904–911. 2000.
|
|
305
|
Narisawa S, Wennberg C and Millán JL:
Abnormal vitamin B6 metabolism in alkaline phosphatase knock-out
mice causes multiple abnormalities, but not the impaired bone
mineralization. J Pathol. 193:125–133. 2001.
|
|
306
|
Ma Q, Liang M, Tang X, Luo F and Dou C:
Vitamin B5 inhibit RANKL induced osteoclastogenesis and ovariectomy
induced osteoporosis by scavenging ROS generation. Am J Transl Res.
11:5008–5018. 2019.
|
|
307
|
Cicek B, Hacimuftuoglu A, Yeni Y, Danisman
B, Ozkaraca M, Mokhtare B, Kantarci M, Spanakis M, Nikitovic D,
Lazopoulos G, et al: Chlorogenic acid attenuates
doxorubicin-induced oxidative stress and marks of apoptosis in
cardiomyocytes via Nrf2/HO-1 and dityrosine signaling. J Pers Med.
13:6492023.
|
|
308
|
Ma Q, Liang M, Wang Y, Ding N, Wu Y, Duan
L, Yu T, Lu Y, Xu J, Kang F and Dou C: Non-coenzyme role of vitamin
B1 in RANKL-induced osteoclastogenesis and ovariectomy induced
osteoporosis. J Cell Biochem. 121:3526–3536. 2020.
|
|
309
|
Herrmann M, Schmidt J, Umanskaya N,
Colaianni G, Al Marrawi F, Widmann T, Zallone A, Wildemann B and
Herrmann W: Stimulation of osteoclast activity by low B-vitamin
concentrations. Bone. 41:584–591. 2007.
|