1. Introduction
As a common disease of urinary system, urolithiasis
mainly manifests by the formation of stones in the renal pelvis and
calyces. The incidence of urolithiasis is ~15%, and its occurrence
may be associated with dietary habits, inflammation,
oxidation/antioxidant imbalances, angiogenesis, lifestyle factors
(e.g., reduced physical activity), purine metabolism and urea cycle
disorders (1). Symptoms of
kidney stones include lower back pain, hematuria, difficulty
urinating and, in severe cases, the stones may lead to kidney
failure. By stone composition, urolithiasis can be divided into
calcium oxalate (CaOx; main), uric acid, calcium phosphate,
struvite, apatite, cystine and other types (2). The key processes of kidney stone
formation are supersaturation, nucleation, crystal growth and
adhesion retention in cells, as well as renal tubular cell damage
caused by high concentrations of oxalate and other factors, which
facilitate crystal adhesion and growth (3).
Based on functional differences, cell death can
generally be classified as accidental, triggered by unexpected
injurious stimuli, or regulatory, characterized by structured
signaling cascades involving effector molecules (4,5).
Common forms of cell death include apoptosis, lysosomal cell death,
necroptosis, pyroptosis, NETosis, immunogenic cell death, entosis,
ferroptosis, autosis, oxeiptosis, cuproptosis and disulfidptosis
(4,6,7).
Cell death plays key roles in the pathogenesis and treatment of a
variety of uropathies, including cancer, urinary tract infections
and urolithiasis (8). Apoptosis,
necroptosis, pyroptosis and ferroptosis induce acute kidney injury
(AKI) or chronic kidney injury, which is associated with
nephrolithiasis (9,10).
Cell death is a complex pathological process that
can be affected by crystal shape and structure, as well as other
physical and chemical properties; however, studies on the roles of
crystal characteristics in the mode of cell death are limited
(11). For example, calcium
oxalate monohydrate (COM) nanoparticles adhere more readily to
injured Vero cells than do CaOx dehydrate nanoparticles, although
both particle types aggravate Vero cell injury (12). In addition, the same stimulus can
induce different forms of regulatory cell death, depending on its
intensity or the presence of co-stimulatory factors; these forms
are characterized by multiple layers of interconnection, including
common triggers, molecular components and protective mechanisms
(13). The present review
summarizes the major advances made in the understanding of
regulated cell death and the progression of kidney stones.
2. Key factors for renal stone
formation
Oxalate is a non-essential metabolic end product
that can cause hyperoxaluria, and renal cells exposed to oxalate
stress produce reactive oxygen species (ROS), ultimately promoting
the formation of CaOx stones (14). In a previous study, the
examination of a spatially anchored transcriptome map of the human
renal papillae revealed the upregulation of a variety of cell
damage pathways in patients with stone-related disease,
characterized by immune activation, the oxidative stress response
and extracellular matrix tissue remodeling; matrix
metalloproteinase 7 and matrix metalloproteinase 9 were found to be
associated with active stones and mineralization (15). Thus, oxidative stress and
inflammatory responses are key factors in CaOx crystal-induced
kidney injury (16). The
underlying causes of kidney stone formation are multifactorial,
including environmental, dietary, hormonal and genetic factors,
leading to an imbalance in crystallization inhibitors and promoters
(17). There are inhibitors
(e.g., citrate, magnesium and pyrophosphate) and promoters
(Randall's plaques, cell injury, bacterial products and slow
urinary flow) of nephrolithiasis (18). In addition to their ability to
inhibit crystallization, citrate and magnesium can also prevent the
crystallization of CaOx by reducing its supersaturation (19). Among various theories about the
urolithiasis formation mechanism, the papillary calcification of
Randall's plaque is considered to be the origin of CaOx stone
formation (20). It has recently
been reported that the deficiency of pyrophosphate, a calcification
inhibitor, predisposes to cardiovascular and renal papillary
calcification, which can lead to the development of kidney stones
(21). In fact, the oral
administration of pyrophosphate inhibits connective tissue
calcification (22). In addition
to pyrophosphate, the dissolution effect of hexametaphosphate on
CaOx stones is 12-fold greater than that of citrate (23). Predisposing factors of
urolithiasis include a low urine volume, hypocitraturia,
hypercalciuria, hyperoxaluria, a low urinary pH (uric acid or
cystine stones), medullary sponge kidney, polycystic kidney disease
and hyperuricosuria (18,24).
The renal tubules are the main kidney components
injured by hypoxia, proteinuria, toxins, metabolic disorders,
aging, stone obstruction, silica, cholesterol, and calcium oxalate
(25,26). AKI is an epidemic syndrome
characterized by a rapid decline in kidney function (27). Recovery from crystalline or
obstruction-induced chronic kidney disease, on the other hand, is
characterized by remaining tissue damage, fibrosis and nephron
loss, rather than being reflected by standard measures of kidney
function (28). In addition, the
pathogenesis of crystalline nephropathy involves various forms of
cell death in the processes of tubule crystal deposition, tubule
obstruction and urinary tract infection.
Urinary tract obstruction caused by calculi is
another common symptom that induces mechanical stretching,
oxidative stress and inflammation that may lead to tubular cell
death and kidney injury via the key processes of apoptosis,
necroptosis and autophagy (8,29,30). Following ureteral obstruction,
renal tubule pyroptosis mediated by tumor necrosis factor
(TNF)-α/caspase 3/gasdermin (GSDM)E signaling triggers the release
of high mobility group box 1 and the activation of inflammasomes,
ultimately leading to tubule injury and promoting the development
of hydronephrosis, inflammatory responses and renal fibrosis
(31). The ferroptosis inhibitor
liproxstatin-1 reduces lipid peroxidation and inhibits the
downregulation of glutathione peroxidase 4 (GPX4) expression, the
ferroptosis of renal tubular epithelial cells induced by ureteral
obstruction, and the paracrine activity of profibrotic factors in
human proximal renal tubular cells, thereby alleviating renal
fibrosis in a mouse model (32).
During a urinary tract infection, invading bacteria
can promote or prevent host cell death by interfering with cell
death pathways (8).
Microorganisms, such as bacteria may be involved in kidney stone
formation through hyperoxaluria and CaOx supersaturation, biofilm
formation and crystal binding to promote aggregation, urothelial
injury and inflammation (33).
Uropathogenic Escherichia coli (UPEC), for example, can
promote the formation of CaOx stones by enhancing oxidative damage
and inflammation regulated by polyphosphate kinase 1/flagellin and
activating the nuclear factor κB (NF-κB)/p38 pathway (34). Within the urinary microbiome, the
role of urease-producing bacteria (i.e., Proteus mirabilis)
in stone formation is well-established (33).
Hyperuricemia or uric acid crystals can induce
pyroptosis by activating the NLR family pyrin domain containing 3
(NLRP3) inflammasome and promoting the release of a number of
pro-inflammatory molecules within the cells, thereby playing a
critical role in kidney disease (35). Cystine crystals can also activate
the NLRP3 inflammasome through the inducion of ROS production,
increase the expression of CD44 and osteopontin in HK-2 cells, and
promote cell apoptosis and crystal adhesion (36). The antioxidant, L-ergothioneine,
prevents cystine stones in solute carrier family 7
(Slc7)a9-/- mouse models by increasing urinary cystine
solubility and restoring renal glutathione (GSH) metabolism and
mitochondrial function (37). As
a common component of most CaOx stones and the core of Randall's
plaques, hydroxyapatite crystals cause oxidative stress, decrease
cell viability and mitochondrial membrane potential, and lead to
cell swelling and necrosis (38). In addition to being caused by
programmed pathway activation (i.e., apoptosis, necroptosis and
pyroptosis), cell death can also be caused by imbalances resulting
from the loss of cytoplasmic or cell membrane integrity, the
accumulation of misfolded proteins, excitatory toxicity, oxidative
stress and lipid peroxidation (39). Thus, the pathogenesis of
crystalline nephropathy involves various forms of cell death in the
processes of tubule crystal deposition, tubule obstruction and
urinary tract infection.
3. The various forms of cell death and
urolithiasis
Apoptosis and urolithiasis
Apoptosis can be triggered by the intrinsic
mitochondrial (BCL-2) pathway, which is regulated by pro-apoptotic
and anti-apoptotic members of the BCL-2 protein family, or by the
extrinsic death receptor pathway, which is activated by ligands of
members of the TNF receptor (TNFR) superfamily with intracellular
death domains (4). Overall,
apoptosis is performed by caspase-3 and caspase-7, which are
activated by upstream extrinsic apoptosis-related caspase-8 and
intrinsic stress-related caspase-9 molecules, respectively
(40). Endoplasmic reticulum
(ER) stress, oxidative stress, growth factor withdrawal and
microtubular alteration are intrinsic lethal stimuli for the
apoptotic pathway, and FASL/FAS, TNF/TNFR1 and TRAIL/TRAIL
receptors are the main extrinsic apoptotic drivers and activators
of inflammation (5). The level
of ER stress (ERS) is closely associated with the degree of HK-2
cell injury and apoptosis induced by CaOx crystals, and the latter
can be reduced significantly by inhibiting the former (41).
Ions, amino acids and their transporters and
channels, calcium-sensitive receptor signaling pathways, and the
metabolic pathways of vitamin D, oxalic acid, cysteine, purine and
uric acid are considered to play key roles in the etiology of
kidney stones (42). CaOx stones
formed due to hyperoxaluria account for approximately two-thirds of
all kidney stones (43). In HK-2
cells, oxalate activates ERS/ROS via NF-κB-dependent pathways,
causing autophagy, apoptosis and mitochondrial damage (44). The ER affects protein and lipid
synthesis, Ca2+ homeostasis regulation and subcellular
organelle crosstalk, and disruptions in homeostasis can cause toxic
protein and lipid accumulation and Ca2+ homeostasis
disorders, leading to cell damage and death and thereby promoting
the development of kidney disease (45). Hyperoxaluria causes ERS, which
leads to an unfolded protein response in rat kidney tissue and to
altered sigma-1 receptor protein expression in
mitochondria-associated ER membranes, resulting in mitochondrial
dysfunction, cell apoptosis, kidney injury and CaOx crystal
deposition (46). Oxalate
poisoning can directly induce the expression of the ERS markers,
glucose-regulated protein 78 and CHOP, upregulate transforming
growth factor β-1, activate ERS-mediated apoptosis, and induce
renal fibrosis (14).
Idiopathic hypercalciuria is another key risk factor
for the formation of calcium-containing kidney stones (47). By activating
Ca2+-sensing receptors, melamine can increase
intracellular Ca2+ concentrations, promote ROS
production, activate the apoptosis and necroptosis of renal
epithelial cells, lead to renal tubular cell injury, inflammation
and fibrosis, and promote the formation of kidney stones (48). Under co-exposure to melamine and
oxalate, the antioxidant capacity of nuclear factor erythroid
2-related factor (Nrf2) decrease and the levels of DNA oxidative
damage in HK-2 cells and kidney tissues, renal tubule cell
apoptosis, tubule atrophy and interstitial fibrosis increase
(49). In a previous study, more
calcium deposits were detected in the medullae of male mice than in
those of female and castrated male mice, and testosterone was found
to induce renal tubular epithelial cell apoptosis and necrosis via
the hypoxia-inducible factor 1α/BCL-2 interacting protein 3 pathway
(50). Crystal internalization
causes the transformation from receptor-operated Ca2+
entry to store-operated Ca2+ entry (SOCE), and the
prevention of SOCE can antagonize crystal-induced ERS and proximal
tubular cell death, thereby reversing pathological outcomes,
including cardiovascular calcification, in crystal-induced
environments (51).
Macrophages can clear CaOx crystals to a certain
extent; however, exosomes derived from macrophages via CaOx crystal
pretreatment can accelerate HK-2 cell apoptosis by increasing
autophagy, suggesting that they have an important function in
CaOx-induced injury to human proximal tubule cells (52). Idiopathic CaOx stones frequently
feature Randall's plaques on renal papillae surfaces; these plaques
are composed of calcium phosphate crystals mixed with a protein-and
lipid-rich organic matrix, associated with the presence of
classically activated pro-inflammatory M1 macrophages and the
downregulation of anti-inflammatory M2 macrophages in the
surrounding renal tissue (53).
Medulla macrophages in renal medullary tubules were recently found
to spontaneously form protrusions, penetrate epithelial cells to
'sample' urine contents depending on integrin β1, and even migrate
to the lumen and carry particles out with urine, suggesting that
urine flushing is not the only mechanism of urinary tract particle
removal (54). By inhibiting the
activation of NADPH oxidase, the production of ROS, and the
phosphorylation of p38 MAPK, M2 macrophages reduce oxidative stress
damage and apoptosis in HK-2 cells, thereby reducing the formation
of kidney stones (55).
Rosiglitazone, a macrophage polarization (Mp) regulator, can
significantly inhibit oxidative stress and inflammation through the
Nrf2/heme oxygenase-1 (HO-1) pathway and promote M2Mp, thereby
reducing renal tubule injury, apoptosis, and crystal adhesion
(56).
Long non-coding RNAs (lncRNAs) play crucial roles in
the regulation of CaOx crystal-induced kidney stone formation and
deposition. The expression of LINC01197 and sirtuin (SIRT)3 is
downregulated in patients with kidney stones, and LINC01197
knockdown promotes CaOx-induced cell adhesion and apoptosis via the
miR-516b-5p/SIRT3/forkhead box (FOX)O1 signaling pathway (57). The overexpression of SIRT3 may
lead to the activation of the NRF2/HO-1 signaling pathway in HK-2
cells, reduce oxidative stress and apoptosis induced by CaOx
crystals in renal tubular epithelial cells, and reduce crystal
adhesion on cell surfaces, thereby inhibiting the formation of
kidney stones (58). miR-200a
mimics can reduce COM-induced cell injury, apoptosis, inhibit
proliferation and changes in epithelial-mesenchymal transition, and
lncRNA-ATB can participate in the regulation of CaOx
crystal-induced kidney injury and apoptosis through sponge
adsorption by the miR-200 family (59). miR-21 can facilitate CaOx-induced
renal tubular cell damage by targeting PPAR-α, and its inhibition
can enhance the proliferation of HK-2 cells and reduce apoptosis
and lipid accumulation following COM exposure and in vivo,
suggesting that it is a therapeutic target for kidney stones
(60).
Esophageal cancer-related gene 4, a tumor suppressor
gene originally described in the esophagus, has recently been
proven to be associated with apoptosis, cell senescence, cell
migration and inflammation, and its loss may ameliorate
CaOx-induced nephropathy (61).
Enhancer of zeste homolog 2 (EZH2) inhibition can restore cell
viability, inhibit lactate dehydrogenase release and intracellular
ROS production via the regulation of the JNK/FOXO3a pathway, and
significantly reduce renal CaOx crystal deposition and oxidative
and inflammatory damage induced by hyperoxaluria in vivo
(62). Sodium butyrate may
partially reverse the oxidative stress, inflammation and apoptosis
induced by CaOx crystallization or nephrolithiasis by inhibiting
CYP2C9 (63). FKBP prolyl
isomerase 5, regarded as a predictor of kidney damage, promotes
cell-crystal adhesion, apoptosis, stone aggregation and kidney
injury in cells and mice (64).
Nox4-derived ROS induced by high calcium-protein kinase C levels
cause oxidative stress injury and the apoptosis of renal tubular
epithelial cells, as well as abnormal activation of bone
morphogenetic protein 2 through the MAPK signaling pathway, thereby
promoting calcium salt deposition and kidney stone formation
(65).
Glutamine can induce the transcriptional and
proteomic reprogramming of mouse renal tubular epithelial cells,
thereby reducing neutrophil recruitment, improving mitochondrial
function and oxidative phosphorylation, and reducing endogenous
apoptosis of mitochondria to alleviate kidney injury (66). The adhesion of crystals to cells
is a key initial step in kidney stone formation; ATPase
Na+/K+ transporting subunit alpha 1 (ATP1A1)
is involved in renal crystal formation via the Src/ROS/p38
signaling pathway, and the specific suppression of the ATP1A1/Src
complex with pNaKtide mitigates crystal-cell adhesion, apoptosis,
inflammation and oxidative stress (67). Resveratrol, a well-known
antioxidant, inhibits crystal deposition and kidney cell injury by
increasing the expression of SIRT1 (68). Thus, ROS-associated ERS,
inflammatory macrophage phenotype switching, and various other
factors induced by high oxalate, calcium and/or crystal levels are
involved in apoptosis.
Pyroptosis and urolithiasis
In contrast to ferroptosis and apoptosis, pyroptosis
is a pro-inflammatory form of cell death with unique morphological
and mechanistic characteristics; it involves the release of the
inflammatory cytokines IL-1β and IL-18 by gasdermin family members
(69). Whereas caspase-3/7 are
involved in apoptosis, inflammatory caspase-1/4/5/11 mediate
pyroptosis by cracking GSDMD (40). Various pathogenic microorganisms
and endogenous harmful substances can activate the NLRP3
inflammasome, which can assemble the intracytoplasmic innate immune
complex, activate the cysteine protease caspase-1, and then lyse
GSDMD, eventually leading to pyroptosis (70,71). As a sensor required for
inflammasome formation, NLRP3 plays a key role in
oxalate-associated renal failure (72). The NLRP3-GSDMD pathway is
involved in oxalate-induced pyroptosis in HK-2 cells, and the
inhibition of ROS production or silencing of NLRP3 can prevent
NLRP3 inflammasome formation, thereby reducing oxalate-induced
damage to membrane integrity and ultrastructural changes (73).
In cell and animal models, IL-22 has been shown to
reduce the sodium oxalate-induced NLRP3 inflammasome and mature
IL-1β expression in kidney tissue and ROS accumulation,
mitochondrial damage, and renal tubular epithelial cell death by
decreasing the serum levels of IL-1β, IL-18, TNF-α and other
cytokines (74). Hyperuricemia
induces a pro-inflammatory microenvironment with increased serum
levels of IL-1β, IL-6 and TNF-α, elevates renal expression of NLRP3
and cleaved caspase-1, and leads to microstructural kidney
disorders in mice; Simiao San alleviates hyperuricemia and renal
inflammation by inhibiting the NLRP3 inflammasome and the
JAK2/STAT3 signaling pathway (75).
Cytoplasmic and mitochondrial ROS production induced
by CaOx crystals can promote the initiation and activation of the
NLRP3 inflammasome, thereby stimulating the maturation and
activation of IL-18/1β; polydatin can reduce the resulting
inflammatory kidney damage and renal epithelial cell injury by
decreasing ROS production (16).
CaOx crystals can induce the expression of caspase-1, GSDMD-N,
IL-1β and IL-18 in renal tubular cells, thereby promoting
pyroptosis, and miR-141-3p can inhibit NLRP3-mediated pyroptosis by
inhibiting the expression of NLRP3, thereby protecting against
renal tubular cell injury (76).
Vitexin can also inhibit GSDMD-related pyroptosis, which is
involved in nephrolithiasis (77). The expression of the lncRNA
LINC00339 has been found to be elevated in HK-2 cells treated with
COM, and LINC00339 has been found to regulate the expression of
NLRP3 by sponging miR-22-3p, which contributes to pyroptosis
(78).
Human recombinant relaxin 3 can act on the
transmembrane receptor, relaxin family peptide receptor 1, to
produce cAMP, and then inhibit the NLRP3 inflammasome activated by
CaOx crystals through the consumption of ATP, thereby reducing
CaOx-induced inflammatory pyroptosis in the kidneys (79). In addition to CaOx lithiasis, the
NLRP3-mediated inflammasome and oxidative stress damage play
important roles in ceftriaxone calcium crystal-induced urinary
lithiasis, thereby promoting acute kidney injury (80). Similar to CaOx and ceftriaxone
calcium, cystine crystals are endogenous inflammasome activators;
thus, sodium urate, calcium phosphate, and other crystals can be
inferred to cause kidney damage through NLRP3-mediated inflammation
(72). NLRP12 is another key
cytosolic sensor in the activation of the inflammasome, the
PANoptosome, and cell death driven by heme plus PAMPs or TNF, whose
deletion protects mice from AKI and death (81).
Necroptosis and urolithiasis
Unlike apoptosis, necroptosis is morphologically
involved in cell swelling, membrane rupture, and the release of
cytoplasmic contents. It is a regulated inflammatory cell death
mechanism mediated by the cascade phosphorylation-induced
activation of receptor-interacting serine/threonine-protein kinase
(RIPK)1, RIPK3 and mixed lineage kinase domain like pseudokinase
(MLKL) (14). Extrinsic
apoptosis-inducing molecules, such as FASL/FAS can activate
caspase-3/7 by promoting the activity of caspase-8 to trigger
apoptosis; when caspase-8 is inhibited, the same cell
death-inducing factors trigger its oligomerization and membrane
destruction via RIPK/MLKL, leading to necroptosis (40). Intratubular crystal deposition
may result in tubular cell injury, obstruction, interstitial
inflammation, and crystal-induced renal colic, which are driven in
part by the NLRP3 inflammasome and necroptosis (82). Both necroptosis and pyroptosis
can cause kidney damage directly or through the recruitment of
immune cells and stimulation of an inflammatory response (13).
As a novel RIPK3 inhibitor, compound 42 alleviates
CaOx crystal-induced renal tubular epithelial cell damage by
inhibiting necroptosis and inflammation, while improving impaired
renal function and reducing intrarenal crystal deposition in mice
with renal calcification; thus, it achieves a better inhibition of
necroptosis than does the classical RIPK3 inhibitor dabrafenib
(83). Tubastatin A, an HDAC6
inhibitor, can inhibit acute oxalate nephropathy by modulating
kidney tubule IL-1β secretion and RIP kinase-mediated necroptosis
(84). 6,7-Dihydroxycoumarin was
shown to inhibit the phosphorylation of MLKL, protecting cells from
CaOx crystal–induced necroptosis, both in vitro and in
vivo (85). TNFR signaling
is essential for intrarenal crystallization-induced inflammation
and kidney cell necroptosis, and may influence CaOx crystal
adhesion to the renal tubule lumen by modulating the expression of
the crystal adhesion molecules CD44 and annexin II (86). Thus, various inhibitors of
RIPK/MLKL signaling have urolithiasis-inhibiting effects.
Ferroptosis and urolithiasis
Ferroptosis is a non-apoptotic form of cell death
characterized by abnormal iron homeostasis, lipid metabolism and
redox system regulation that plays crucial roles in organ injury
and degenerative diseases (87).
It has been observed in various forms of AKI, such as sepsis,
ischemia, and folate cisplatin, and oxalate nephropathies; it not
only is a mechanism of renal injury, but also alters the course of
AKI and inhibits recovery (10).
Ferroptosis, a novel iron- and ROS-dependent form of programmed
cell death, can be induced by various drugs (e.g., erastin,
cisplatin, sorafenib, artemisinin, and statins) through various
mechanisms (88).
Under ferroptosis regulation, renal tubular
epithelial cell damage has been found to significantly increase
with the ferroptosis level and vice versa, suggesting that
ferroptosis is essential for the injury caused by CaOx crystals
(89). The significant
activation of ferroptosis has been observed in patients with kidney
stones and in hyperoxaluric mice, and this activation can be
inhibited by p53 deacetylation, thereby mitigating CaOx
crystal-induced renal fibrosis (90). Oxalate has been found to induce
ferroptosis in HK-2 cells by activating NCOA4-mediated autophagy
(91). Ferrostatin-1, a
ferroptosis inhibitor, can mitigate oxalate-induced kidney tubular
epithelial cell damage, fibrosis, and CaOx lithogenesis by
suppressing ferroptosis (92).
It can also regulate abnormal kidney lipid metabolism enzymes in
AKI (93).
GPX4 is an antioxidant enzyme that uses GSH as a
cosubstrate to reduce lipid hydroperoxides, and GSH may replace
cysteine or homocysteine as a GPX4 cofactor in the evolution of
aerobic metabolism (94). AKI
repair ability differs significantly between males and females, as
GPX4 knockout leads to increased renal tubular epithelial cell
injury and ferroptosis in male, but not female, mice (95). OTU deubiquitinase 5 (OTUD5), a
protein that interacts with GPX4, can promote ferroptosis
resistance during ischemia/reperfusion injury by stabilizing GPX4
expression; in turn, hypoxia/ischemia-induced OTUD5 autophagy can
destabilize GPX4, leading to ferroptosis-dependent kidney injury
(96). Vitexin has been found to
increase GPX4 expression by activating the NRF2/HO-1 pathway,
inhibit the ferroptosis of renal tubule epithelial cells, and
significantly reduce renal tubule injury, interstitial fibrosis,
and renal inflammation in mice with unilateral ureteral obstruction
(97).
SLC7A11, a cellular transmembrane protein that makes
up the light chain of system Xc-, is a key pathway for REDOX
homeostasis, transporting extracellular cysteine into cells for
cysteine production and GSH biosynthesis and thereby maintaining
cellular GSH levels to antagonize cellular oxidative stress and
inhibit ferroptosis (98). SOX4
promotes ferroptosis in CaOx crystal-induced kidney injury by
modulating EZH2/H3K27Me3-mediated SLC7A11 inhibition. Ankyrin
repeat domain 1 is involved in CaOx kidney stone formation via the
activation of p53/SLC7A11-mediated ferroptosis (99). Von Hippel-Lindau, a critical
renal tumor suppressor gene, interacts with BICD2 and weakens
system Xc--mediated ferroptosis processes, which can be
disrupted by a BRAF inhibitor during severe ferroptosis and
nephrotoxicity (100).
Polyunsaturated fatty acids (PUFAs) are the main
targets of lipid peroxidation, and their incorporation into
phospholipids, a key event in lipid hydroperoxide-induced
ferroptosis, is dependent on acyl coenzyme A synthase long-chain
family member 4 (ACSL4) (101).
Instead of causing ferroptosis, PUFAs have been shown to reduce the
COM-induced apoptosis of HK-2 cells and diminish kidney tubular
damage in a renal-stone mouse model via the miR-93-5p/Pknox1 axis
(102). Yes-associated protein
can enhance ACSL4 expression, thereby inducing ferroptosis and
increasing CaOx crystal-induced renal fibrosis (103). FSP1-dependent non-canonical
vitamin K, a naphthoquinone group that includes
methylnaphthoquinone and phylloquinone, has powerful
anti-ferroptosis properties and can protect cells from detrimental
lipid peroxidation (104).
NRF2 regulates several genes that are critical; for
ferroptosis, including GPX4 (105). In erastinor oxalate-induced
HK-2 cells, schizandrin B modulates the expression of
ferroptosis-related proteins and reduces ferroptosis-related
cellular Fe2+ accumulation and lipid peroxidation by
facilitating Nrf2 nuclear translocation (106). Drugs and substances, such as
gallic acid (107), curcumin
(108) and dimethyl fumarate
(109), significantly
ameliorate CaOx crystal-induced renal injury via Nrf2 pathway
regulation, which involves antioxidant and antiapoptotic effects
and the inhibition of autophagy and inflammation to prevent
nephrolithiasis. Oxidative stress may promote and increase renal
crystal formation through the Keap1-Nrf2 pathway (110). Thus, Nrf2 is associated with
apoptosis, autophagy and ferroptosis through its numerous effects
(i.e., antioxidant effects).
Melatonin can increase the total antioxidant
capacity of HK-2 cells and decrease the ERS and apoptosis induced
by oxalate in a dose-dependent manner, depending partly on 5'
adenosine monophosphate-activated protein kinase (AMPK) activation
(111). It may also alleviate
oxalate-induced renal injury via the PTEN induced kinase 1/AMPK
pathway to restore mitophagy and subsequently inhibit ferroptosis
(112). Exosome Ambra1 can be
secreted by HK-2 cells damaged by supersaturated oxalate, inducing
mitochondrial and ferroptosis-related autophagy in normal HK-2
cells, which contributes to the occurrence of kidney stones
(113). In the context of
nephropathy induced by adenine and 8-dihydroxyadenine (DHA)
crystals, ferroptosis is the main mode of proximal tubular
epithelial cell necrosis, and baicalein is a potential therapeutic
tool for ferroptosis-related crystalline (e.g., DHA or oxalate)
nephropathy (114). Thus, a
number of ferroptosis-related molecules (e.g., GPX4 and NRF2) can
affect certain steps of urolithiasis and may participate in
apoptosis, autophagy, and other forms of cell death. A simplified
summary of apoptosis-, necroptosis-, pyroptosis-, ferroptosis- and
autophagy-related signaling pathways and their crosstalk in kidney
cells is shown in Fig. 1.
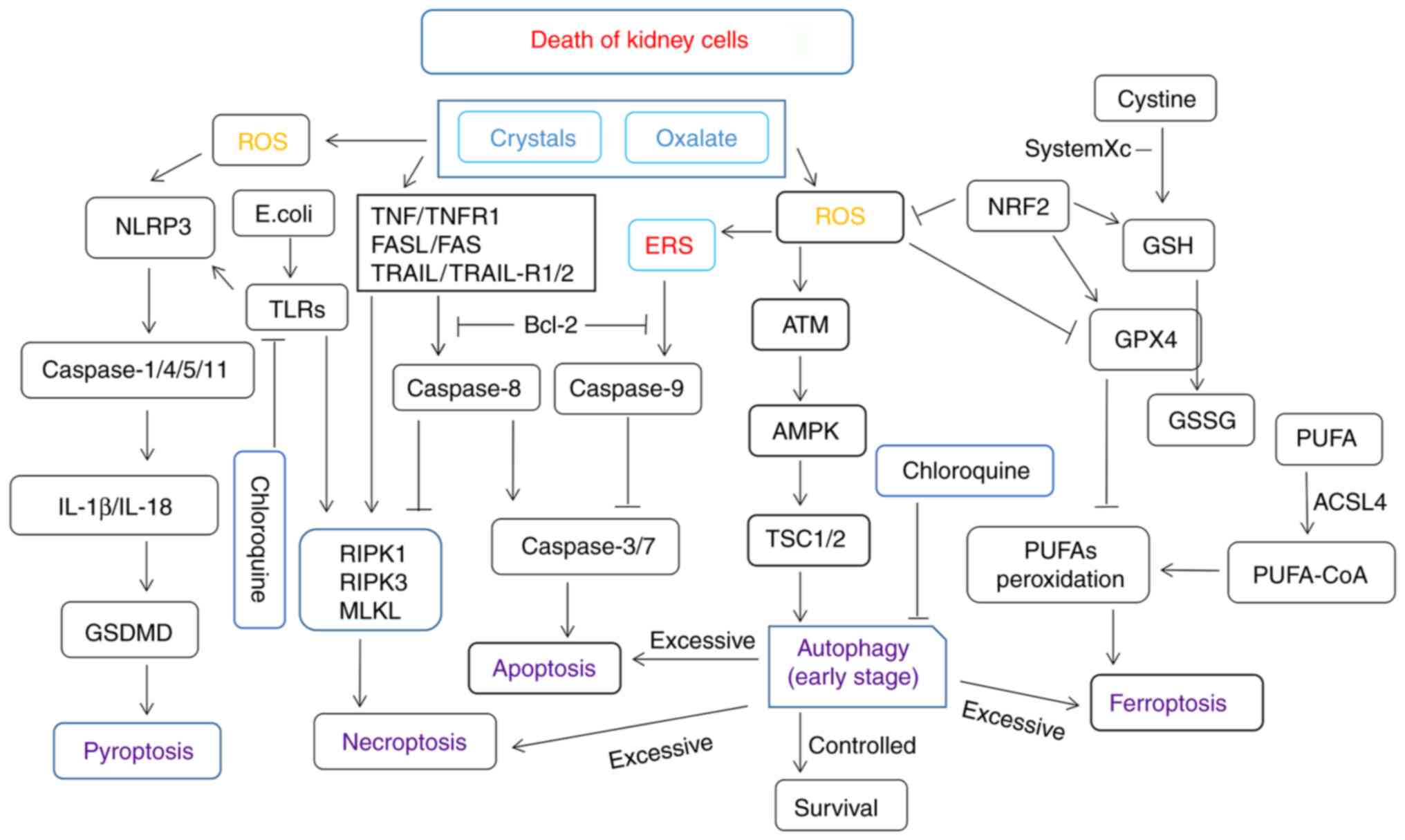 | Figure 1A simplified summary of apoptosis-,
necroptosis-, pyroptosis-, ferroptosis-, and autophagy-related
signaling pathways and crosstalk among them. Kidney cell injury
induced by high oxalate, crystal, and Escherichia coli
concentrations or other bacteria may promote the production of ROS
and various forms of cell death. Nrf2, nuclear factor erythroid
2-related factor; GPx, glutathione peroxidase; GSH, glutathione;
PUFAs, polyunsaturated fatty acids; ROS, reactive oxygen species;
NLRP3, NLR family pyrin domain containing 3; TNF, tumor necrosis
factor; TNFR, tumor necrosis factor receptor; ERS, endoplasmic
reticulum stress; GSDMD, gasdermin D; ACSL4, acyl coenzyme A
synthase long-chain family member 4; TLRs, Toll-like receptors;
ERS, endoplasmic reticulum stress; RIPK, receptor-interacting
serine/threonine-protein kinase; MLKL, mixed lineage kinase domain
like pseudokinase; TSC1/2, tuberous sclerosis proteins 1 and 2;
AMPK, 5' adenosine monophosphate-activated protein kinase; GSSG,
glutathione disulfide; CoA, coenzyme A. |
Autophagy and lysozyme cell death in
urolithiasis
Autophagy is a process through which cells maintain
homeostasis by degrading and recycling organelles and their
proteins. Autophagy often has a protective effect; however, the
disruption of its mechanisms or excessive autophagic flux induces
cell death; indeed, autophagy is involved in the regulation of
almost all cell death in various diseases (115). Such death can be
autophagy-dependent (or autophagic, i.e., ER-phagy, mitophagy and
autosis) or mediated (i.e., apoptosis, necroptosis and ferroptosis)
(115). Autophagy is considered
to be an enhancer that promotes ROS-dependent ferroptosis by
regulating iron-dependent lipid peroxidation, and excessive
autophagy and lysosomal activity can also promote ferroptosis
through iron accumulation (116). Thus, there are many different
types of molecular signaling crosstalk between autophagy and
various forms of cell death.
Autophagy and mitophagy play critical roles in cell
survival by preventing nutrient deprivation and regulating
oxidative stress; however, they can be dysregulated, leading to
cell death, by oxidative stress caused by sustained kidney injury
(117). The autophagy
antagonist, chloroquine, can significantly reduce oxalate-induced
autophagy activation and oxidative and mitochondrial damage in
renal tubule cells in vitro and in vivo, and inhibit
hyperoxaluria-induced renal CaOx crystal deposition, reflecting the
roles of autophagy in the regulation of oxalate-induced renal
oxidative damage and CaOx crystal deposition (118). ERS can mediate excessive
autophagy and regulate cell damage and apoptosis through the
PERK-eIF2α pathway, and the inhibition of ERS-mediated autophagy
can effectively protect renal function and prevent renal cell
apoptosis and kidney stone formation (119). The enhancement of superoxide
dismutase activity by atorvastatin can reduce the autophagy-ERS
response and CaOx kidney stone formation, and thus may be an option
for the prevention and treatment of nephrolithiasis (120).
Mitochondrial ROS and IL-10 are involved in
oxalate-induced metabolism and immune response impairment via the
disruption of monocyte and macrophage function (121). CaOx crystals significantly
induce lysosomal injury and subsequent transcription factor EB
(TFEB) activation in proximal renal tubule epithelial cells,
whereas tubule injury, renal function impairment, the expression of
the lysosome damage marker Gal-3, and cell apoptosis were
significantly increased in TFEB-deficient mice treated with
oxalates for 48 h, suggesting that TFEB activation can alleviate
subsequent tissue damage by promoting lysosomal damage repair
(122). Resveratrol reduces
oxalate-induced renal inflammation, oxidative damage and CaOx
crystal deposition by activating TFEB-induced autophagy (123). Trimethylamine N-oxide, a
metabolite derived from gut microbes, not only plays a key role in
the pathogenesis of atherosclerosis, diabetes and chronic kidney
disease, but also exacerbates kidney damage and renal tubule cell
autophagy, apoptosis and inflammation, and promotes
hyperoxaluria-induced CaOx crystal deposition by triggering the
PERK/ROS pathway (124).
Other forms of cell death and
urolithiasis: NETosis, cuproptosis and disulfidptosis
Recent research has demonstrated that NETosis, a
unique neutrophil death mechanism that can cause various
inflammatory and autoimmune conditions, is also involved in the
development and progression of kidney disease (125). Increased neutrophil
infiltration and NETosis have been found to differentiate patients
with brushite and CaOx stones, potentially explaining the apparent
increases in scarring and inflammation in the papillae in the
former, although the role of neutrophil activation in the increased
incidence of brushite stone formation needs to be investigated
further (126). Recent studies
have also shown that ferroptosis in renal parenchymal cells
initiates neutrophil migration to injury sites through C-X-C
chemokine receptor type 4, a surface receptor that binds to the
metformin-iron-NGAL complex, causing NETosis to aggravate AKI, and
that a reduction in iron concentration may protect against kidney
damage (127).
Malireddi et al (128) described PANoptosis as an
inflammatory programmed cell death type with key features of
pyroptosis, apoptosis and/or necroptosis that cannot be
characterized as any one of these death modes alone. As previously
demonstrated, NLRP12 drives the activation of the inflammasome and
PANoptosome, and its loss in a hemolysis model protected mice from
AKI and death (81). It has been
mentioned previously that urolithiasis-related processes can cause
pyroptosis, apoptosis, and necroptosis; whether they are associated
directly with PANoptosis requires further investigation. Other
regulatory pathways of non-apoptotic cell death (i.e., parthanatos,
lysozincrosis and disulfidptosis) have also been found to exist
(129), although their roles in
kidney stone progression remain to be determined.
4. Oxalate metabolism and the tricarboxylic
acid cycle in cell death
Oxalic acid is a toxic end-metabolite with no known
physiological function in humans (130). The metabolic precursors of
oxalic acid in vivo include glyoxylate, aromatic amino acids
glyoxal and vitamin C (131).
Oxalic acid in human blood comes from diet, ascorbic acid
degradation and liver synthesis (132). Therefore, the causes of
hyperoxaluria include the excessive intake of oxalic acid in the
daily diet, intestinal diseases leading to the excessive absorption
of oxalic acid in the intestine and the intestinal excretion of
oxalic acid, and metabolic disorders of the liver, leading to
abnormal increases in endogenous oxalates, such as primary
hyperoxaluria (133). A low
urine volume and dehydration are common risk factors for all types
of stones, while hyperoxaluria, high-calcium urea and low-citrate
urea are the main risk factors for calcium-containing stones
(130). The increased excretion
of urine citrate, a molecule involved in the tricarboxylic acid
(TCA) cycle, reduces the risk of stone development by inhibiting
calcium crystallization and complexing (134).
Metabolites in the TCA cycle include malate,
fumarate, pyruvate, alpha-ketoglutarate, succinate and
citrate/isocitrate, while low urinary TCA circulating organic
anions, particularly low methylmalonic acid, ethyl malonic acid and
citrate/isocitrate, are potential biomarkers of renal damage in
early diabetic nephropathy (135). By reducing Cu2+ to
Cu+, ferredoxin-1 promotes the abnormal oligomerization
of copper-dependent fatty acylated proteins in the TCA cycle and
reduces Fe-S cluster protein levels, leading to protein
aggregation, toxic stress, and ultimately, to cuproptosis (136). Fumarate, an intermediate in the
TCA cycle, succinacylates GSDMD and GSDME at specific cysteine
sites, inhibiting the formation of oligomers and thereby preventing
pyroptosis (137). Succinate
also reduces renal calcium deposition and damage in an ethylene
glycol-induced rat model via anti-inflammatory effects and the
inhibition of cell adhesion and osteogenic differentiation
(138). The TCA cycle also
affects ferroptosis by participating in the production of
O2·and NAD(P)H (139). However, the direct role of
cuproptosis in kidney stone development, particularly via the TCA
cycle, remains to be determined.
In a previous study, the preliminary analyses of
metabolic profiles in urine from 110 patients with kidney stones
and 106 healthy controls revealed that glycine, serine and
threonine metabolism, the TCA cycle, glyoxylate and dicarboxylate
metabolism, and phenylalanine metabolism were four metabolic
pathways closely related to the presence of kidney stones (140). Citric acid and malate, two TCA
cycle molecules, can also alkalize urine, particularly for uric
acid stone formation (141,142). Vinegar can affect urinary
citrate and calcium excretion through epigenetic regulation,
thereby preventing the formation of calcium crystals in the kidney
(143). The metabolite
α-ketoglutaric acid (α-KG) can promote GSDMC-dependent pyroptosis
via the death receptor 6-activated caspase-8, and its pyroptosis
inducing efficiency depends on the acidity of the environment
(144). In addition to
affecting REDOX homeostasis and cytokine signaling, oxalate can
also impair macrophage metabolism, leading to a reduced
antimicrobial response and increased infection (145). UPEC can use a novel
UPEC-associated two-component signaling system to facilitate the
utilization of the metabolite α-KG to adapt to life in the urinary
tract (146). However, in
cardiomyocytes α-KG inhibits ferroptosis and alleviates myocardial
cell injury by upregulating NAD+ levels and activating the SIRT1
signaling pathway (147). The
roles of small organic acids, TCA cycle-related molecules (i.e.,
citric acid, malate, fumarate and succinate), and cell death in
urolithiasis are illustrated in Fig.
2. In total, the main targets or molecules that have signaling
crosstalk with cell death and urolithiasis are mainly oxidative
stress (e.g., NRF2), inflammatory (e.g., NLRP3), adhesive (e.g.,
succinate), aggregation (e.g., citrate) or metabolic genes
(α-ketoglutarate).
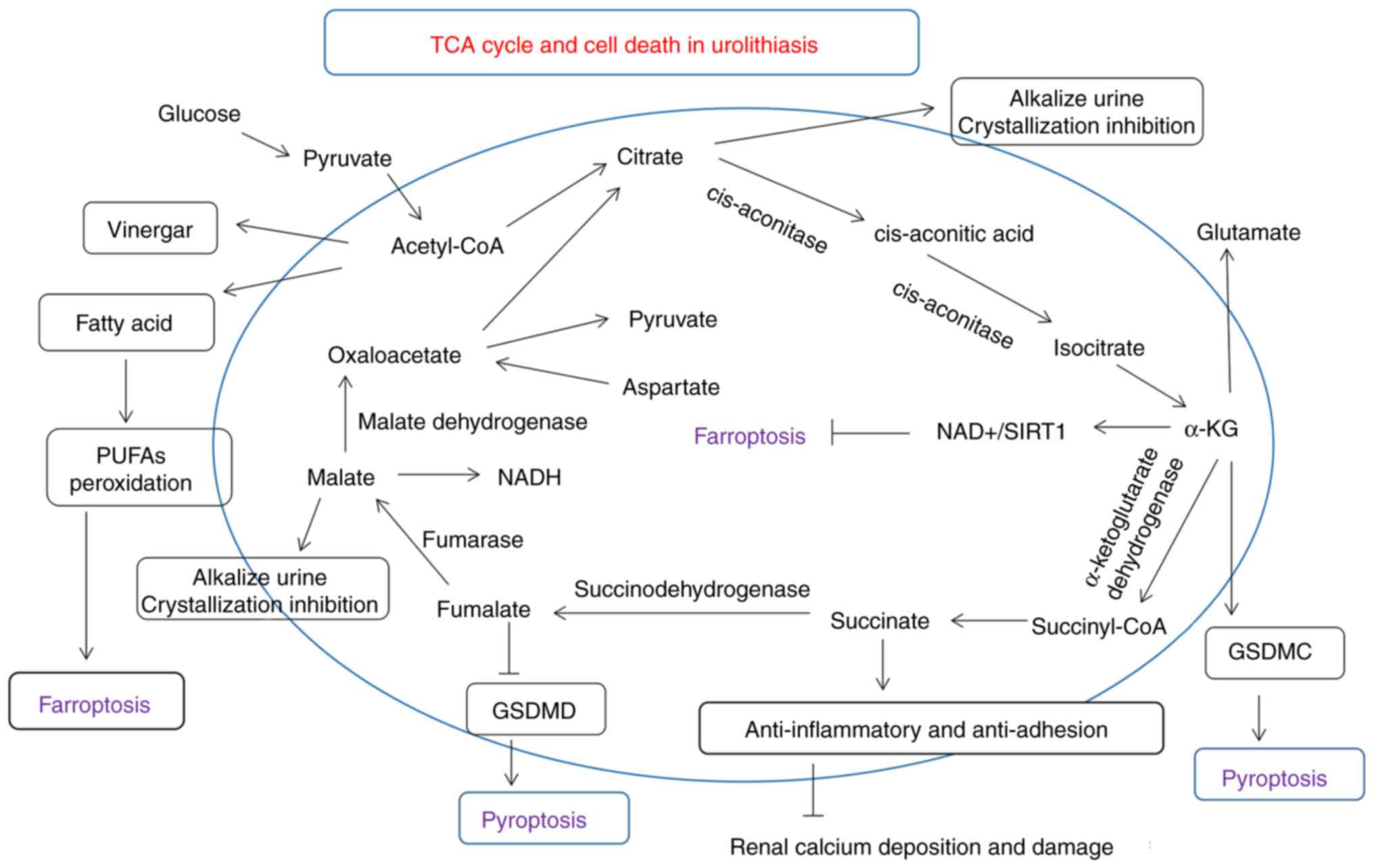 | Figure 2Roles of small organic acids, TCA
cycle-related molecules (i.e., citric acid, malate, fumarate and
succinate), and cell death in urolithiasis are described in detail.
Various molecules involved in the TCA cycle, particularly citrate,
which is considered to be a stone-inhibiting substance, may
regulate molecules associated with kidney cell damage and cell
death. GSDMD, gasdermin D; GSDMC, gasdermin C; PUFAs,
polyunsaturated fatty acids; TCA, tricarboxylic acid; α-KG,
α-ketoglutaric acid. |
5. Conclusion and future directions
Numerous types of urinary calculus are related to
cell death, whose progression commonly causes renal tubular cell
damage, often mediated by apoptosis, pyroptosis, necroptosis and
ferroptosis, are related to oxidative stress, inflammation and
lipid peroxidation molecular signals caused by high oxalate and
calcium crystal aggregation and precipitation. In addition, the
roles of cell death-related metabolic disorders and non-unique cell
death (e.g., PANoptosis) in urolithiasis processes remain to be
explored. However, there are still limitations as the changes in
in vitro experiments and animal models of CaOx-induced
kidney injury cannot always represent the actual situation of the
real stone formation in human.
The understanding of cell death was initially
limited to apoptosis, and various types of cell death (i.e.,
necroptosis, pyroptosis and ferroptosis) and their mechanisms and
evolution are increasingly being explored. Specific biomarkers are
required to identify cell death types, and the exploration of cell
death physiology and pathology and their roles in urolithiasis is
essential. Elucidation of the regulatory mechanisms of renal
inflammatory injury, renal cell death, and related crosstalk is
important for the prevention and treatment of kidney stone-related
diseases. Bioinformatic technology, metabolomics, intestinal flora,
spatial transcription analysis, artificial intelligence and other
methods are developing rapidly and will be increasingly used to
study the urine composition and environment in patients with kidney
stones, which can open new avenues for the exploration of
urolithiasis (15). In addition,
the application of organoids and other new models, which can
simulate the pathological conditions of human urinary calculi is
anticipated to be used to explore the corresponding mechanisms of
the disease in future.
Availability of data and materials
Not applicable.
Authors' contributions
All authors (LW, XX, CH, YL and LT) contributed to
the conception and design of the study. The first draft of the
manuscript was written by LW, XX and LT. YL and CH contributed to
the literature search and the preparation of the figures. All
authors commented and critically revised on previous versions of
the manuscript. All authors have read and agreed to the final
version of manuscript. Data authentication is not applicable.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Use of artificial intelligence tools
During the preparation of this work, AI tools were
used to improve the readability and language of the manuscript, and
subsequently, the authors revised and edited the content produced
by the AI tools as necessary, taking full responsibility for the
ultimate content of the present manuscript.
Abbreviations:
|
CaOx
|
calcium oxalate
|
|
COM
|
calcium oxalate monohydrate
|
|
AKI
|
acute kidney injury
|
|
ROS
|
reactive oxygen species
|
|
ER
|
endoplasmic reticulum
|
|
Nrf2
|
nuclear factor erythroid 2-related
factor
|
|
GPX4
|
glutathione peroxidase 4
|
|
TNF
|
tumor necrosis factor
|
|
UPEC
|
uropathogenic Escherichia
coli
|
|
TNFR
|
tumor necrosis factor receptor
|
|
ERS
|
endoplasmic reticulum stress
|
|
SOCE
|
store-operated Ca2+
entry
|
|
Mp
|
macrophage polarization
|
|
lncRNA
|
long non-coding RNA
|
|
SIRT
|
sirtuin
|
|
GSDM
|
gasdermin
|
|
GSH
|
glutathione
|
|
PUFAs
|
polyunsaturated fatty acids
|
|
ACSL4
|
acyl coenzyme A synthase long-chain
family member 4
|
|
DHA
|
dihydroxyadenine
|
|
TFEB
|
transcription factor EB
|
|
TCA
|
tricarboxylic acid
|
|
α-KG
|
α-ketoglutaric acid
|
|
NF-κB
|
nuclear factor κB
|
|
HO-1
|
heme oxygenase-1
|
Acknowledgments
Not applicable.
Funding
The present work was supported by the Futian Healthcare Research
Project (grant no. FTWS2023081).
References
|
1
|
Wigner P, Grębowski R, Bijak M, Szemraj J
and Saluk-Bijak J: The molecular aspect of nephrolithiasis
development. Cells. 10:19262021. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Liu Y, Chen Y, Liao B, Luo D, Wang K, Li H
and Zeng G: Epidemiology of urolithiasis in Asia. Asian J Urol.
5:205–214. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Lai Y, Zheng H, Sun X, Lin J, Li Q, Huang
H, Hou Y, Zhong H, Zhang D, Fucai T and He Z: The advances of
calcium oxalate calculi associated drugs and targets. Eur J
Pharmacol. 935:1753242022. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Tang D, Kang R, Berghe TV, Vandenabeele P
and Kroemer G: The molecular machinery of regulated cell death.
Cell Res. 29:347–364. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Vitale I, Pietrocola F, Guilbaud E,
Aaronson SA, Abrams JM, Adam D, Agostini M, Agostinis P, Alnemri
ES, Altucci L, et al: Apoptotic cell death in disease-current
understanding of the NCCD 2023. Cell Death Differ. 30:1097–1154.
2023. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Tsvetkov P, Coy S, Petrova B, Dreishpoon
M, Verma A, Abdusamad M, Rossen J, Joesch-Cohen L, Humeidi R,
Spangler RD, et al: Copper induces cell death by targeting
lipoylated TCA cycle proteins. Science. 375:1254–1261. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Liu X, Nie L, Zhang Y, Yan Y, Wang C,
Colic M, Olszewski K, Horbath A, Chen X, Lei G, et al: Actin
cytoskeleton vulnerability to disulfide stress mediates
disulfidptosis. Nat Cell Biol. 25:404–414. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Martin-Sanchez D, Fontecha-Barriuso M,
Sanchez-Niño MD, Ramos AM, Cabello R, Gonzalez-Enguita C,
Linkermann A, Sanz AB and Ortiz A: Cell death-based approaches in
treatment of the urinary tract-associated diseases: A fight for
survival in the killing fields. Cell Death Dis. 9:1182018.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Yang L, Liu Y, Zhou S, Feng Q, Lu Y, Liu D
and Liu Z: Novel insight into ferroptosis in kidney diseases. Am J
Nephrol. 54:184–199. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Bayir H, Dixon SJ, Tyurina YY, Kellum JA
and Kagan VE: Ferroptotic mechanisms and therapeutic targeting of
iron metabolism and lipid peroxidation in the kidney. Nat Rev
Nephrol. 19:315–336. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Sun XY and Ouyang JM: New view in cell
death mode: Effect of crystal size in renal epithelial cells. Cell
Death Dis. 6:e20132015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Gan QZ, Sun XY, Bhadja P, Yao XQ and
Ouyang JM: Reinjury risk of nano-calcium oxalate monohydrate and
calcium oxalate dihydrate crystals on injured renal epithelial
cells: Aggravation of crystal adhesion and aggregation. Int J
Nanomedicine. 11:2839–2854. 2016.PubMed/NCBI
|
|
13
|
Sanz AB, Sanchez-Niño MD, Ramos AM and
Ortiz A: Regulated cell death pathways in kidney disease. Nat Rev
Nephrol. 19:281–299. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Abhishek A, Benita S, Kumari M, Ganesan D,
Paul E, Sasikumar P, Mahesh A, Yuvaraj S, Ramprasath T and Selvam
GS: Molecular analysis of oxalate-induced endoplasmic reticulum
stress mediated apoptosis in the pathogenesis of kidney stone
disease. J Physiol Biochem. 73:561–573. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Canela VH, Bowen WS, Ferreira RM, Syed F,
Lingeman JE, Sabo AR, Barwinska D, Winfree S, Lake BB, Cheng YH, et
al: A spatially anchored transcriptomic atlas of the human kidney
papilla identifies significant immune injury in patients with stone
disease. Nat Commun. 14:41402023. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Liu J, Huang J, Gong B, Cheng S, Liu Y,
Chen Y, Feng Q, Li J, Qiu M, Yu G and Liao Y: Polydatin protects
against calcium oxalate crystal-induced renal injury through the
cytoplasmic/mitochondrial reactive oxygen species-NLRP3
inflammasome pathway. Biomed Pharmacother. 167:1156212023.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Singh P, Harris PC, Sas DJ and Lieske JC:
The genetics of kidney stone disease and nephrocalcinosis. Nat Rev
Nephrol. 18:224–240. 2022. View Article : Google Scholar
|
|
18
|
Shastri S, Patel J, Sambandam KK and
Lederer ED: Kidney stone pathophysiology, evaluation and
management: Core curriculum 2023. Am J Kidney Dis. 82:617–634.
2023. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Grases F, Rodriguez A and Costa-Bauza A:
Efficacy of mixtures of magnesium, citrate and phytate as calcium
oxalate crystallization inhibitors in urine. J Urol. 194:812–819.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wang Z, Zhang Y, Zhang J, Deng Q and Liang
H: Recent advances on the mechanisms of kidney stone formation
(review). Int J Mol Med. 48:1492021. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Letavernier E, Bouderlique E, Zaworski J,
Martin L and Daudon M: Pseudoxanthoma elasticum, kidney stones and
pyrophosphate: From a rare disease to urolithiasis and vascular
calcifications. Int J Mol Sci. 20:63532019. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Dedinszki D, Szeri F, Kozák E, Pomozi V,
Tőkési N, Mezei TR, Merczel K, Letavernier E, Tang E, Le Saux O, et
al: Oral administration of pyrophosphate inhibits connective tissue
calcification. EMBO Mol Med. 9:1463–1470. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Robinson TE, Hughes EAB, Wiseman OJ,
Stapley SA, Cox SC and Grover LM: Hexametaphosphate as a potential
therapy for the dissolution and prevention of kidney stones. J
Mater Chem B. 8:5215–5224. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zeng G, Zhu W, Robertson WG, Penniston KL,
Smith D, Pozdzik A, Tefik T, Prezioso D, Pearle MS, Chew BH, et al:
International alliance of urolithiasis (IAU) guidelines on the
metabolic evaluation and medical management of urolithiasis.
Urolithiasis. 51:42022. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Liu BC, Tang TT, Lv LL and Lan HY: Renal
tubule injury: A driving force toward chronic kidney disease.
Kidney Int. 93:568–579. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Honarpisheh M, Foresto-Neto O, Desai J,
Steiger S, Gómez LA, Popper B, Boor P, Anders HJ and Mulay SR:
Phagocytosis of environmental or metabolic crystalline particles
induces cytotoxicity by triggering necroptosis across a broad range
of particle size and shape. Sci Rep. 7:155232017. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Guo H, Wang M, Shang Y, Zhang B, Zhang S,
Liu X, Cao P, Fan Y and Tan K: Apoptosis-related prognostic
biomarkers and potential targets for acute kidney injury based on
machine learning algorithm and in vivo experiments. Apoptosis.
29:303–320. 2024. View Article : Google Scholar
|
|
28
|
Klinkhammer BM, Buchtler S, Djudjaj S,
Bouteldja N, Palsson R, Edvardsson VO, Thorsteinsdottir M, Floege
J, Mack M and Boor P: Current kidney function parameters
overestimate kidney tissue repair in reversible experimental kidney
disease. Kidney Int. 102:307–320. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kumar R, Soni H, Afolabi JM, Kanthakumar
P, Mankuzhy PD, Iwhiwhu SA and Adebiyi A: Induction of reactive
oxygen species by mechanical stretch drives endothelin production
in neonatal pig renal epithelial cells. Redox Biol. 55:1023942022.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Li J, Lin Q, Shao X, Li S, Zhu X, Wu J,
Mou S, Gu L, Wang Q, Zhang M, et al: HIF1α-BNIP3-mediated mitophagy
protects against renal fibrosis by decreasing ROS and inhibiting
activation of the NLRP3 inflammasome. Cell Death Dis. 14:2002023.
View Article : Google Scholar
|
|
31
|
Li Y, Yuan Y, Huang ZX, Chen H, Lan R,
Wang Z, Lai K, Chen H, Chen Z, Zou Z, et al: GSDME-mediated
pyroptosis promotes inflammation and fibrosis in obstructive
nephropathy. Cell Death Differ. 28:2333–2350. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Zhang B, Chen X, Ru F, Gan Y, Li B, Xia W,
Dai G, He Y and Chen Z: Liproxstatin-1 attenuates unilateral
ureteral obstruction-induced renal fibrosis by inhibiting renal
tubular epithelial cells ferroptosis. Cell Death Dis. 12:8432021.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Jung HD, Cho S and Lee JY: Update on the
effect of the urinary microbiome on urolithiasis. Diagnostics
(Basel). 13:9512023. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
An L, Wu W, Li S, Lai Y, Chen D, He Z,
Chang Z, Xu P, Huang Y, Lei M, et al: Escherichia coli aggravates
calcium oxalate stone formation via PPK1/flagellin-mediated renal
oxidative injury and inflammation. Oxid Med Cell Longev.
2021:99496972021. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Wang M, Lin X, Yang X and Yang Y: Research
progress on related mechanisms of uric acid activating NLRP3
inflammasome in chronic kidney disease. Ren Fail. 44:615–624. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Yifan Z, Luming S, Wei C, Luwei X, Zheng X
and Ruipeng J: Cystine crystal-induced reactive oxygen species
associated with NLRP3 inflammasome activation: Implications for the
pathogenesis of cystine calculi. Int Urol Nephrol. 54:3097–3106.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Mayayo-Vallverdú C, López de Heredia M,
Prat E, González L, Espino Guarch M, Vilches C, Muñoz L, Asensi MA,
Serra C, Llebaria A, et al: The antioxidant l-Ergothioneine
prevents cystine lithiasis in the Slc7a9-/- mouse model
of cystinuria. Redox Biol. 64:1028012023. View Article : Google Scholar
|
|
38
|
Rao CY, Sun XY and Ouyang JM: Effects of
physical properties of nano-sized hydroxyapatite crystals on
cellular toxicity in renal epithelial cells. Mater Sci Eng C Mater
Biol Appl. 103:1098072019. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Yuan J and Ofengeim D: A guide to cell
death pathways. Nat Rev Mol Cell Bio. Dec;–18. 2023.Epub ahead of
print.
|
|
40
|
Ai Y, Meng Y, Yan B, Zhou Q and Wang X:
The biochemical pathways of apoptotic, necroptotic, pyroptotic, and
ferroptotic cell death. Mol Cell. 84:170–179. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Sun Y, Kang J, Guan X, Xu H, Wang X and
Deng Y: Regulation of endoplasmic reticulum stress on the damage
and apoptosis of renal tubular epithelial cells induced by calcium
oxalate crystals. Urolithiasis. 49:291–299. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Howles SA and Thakker RV: Genetics of
kidney stone disease. Nat Rev Urol. 17:407–421. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Cil O, Chu T, Lee S, Haggie PM and Verkman
AS: Small-molecule inhibitor of intestinal anion exchanger SLC26A3
for treatment of hyperoxaluria and nephrolithiasis. JCI Insight.
7:e1533592022. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Ming S, Tian J, Ma K, Pei C, Li L, Wang Z,
Fang Z, Liu M, Dong H, Li W, et al: Oxalate-induced apoptosis
through ERS-ROS-NF-κB signalling pathway in renal tubular
epithelial cell. Mol Med. 28:882022. View Article : Google Scholar
|
|
45
|
Wu D, Huang LF, Chen XC, Huang XR, Li HY,
An N, Tang JX, Liu HF and Yang C: Research progress on endoplasmic
reticulum homeostasis in kidney diseases. Cell Death Dis.
14:4732023. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Sharma M, Naura AS and Singla SK: A
deleterious interplay between endoplasmic reticulum stress and its
functional linkage to mitochondria in nephrolithiasis. Free Radical
Bio Med. 168:70–80. 2021. View Article : Google Scholar
|
|
47
|
Wu Y, Zhang J, Li C, Hu H, Qin B, Wang T,
Lu Y and Wang S: The activation of ROS/NF-κB/MMP-9 pathway promotes
calcium-induced kidney crystal deposition. Oxid Med Cell Longev.
2021:88363552021. View Article : Google Scholar
|
|
48
|
Yiu AJ, Ibeh CL, Roy SK and Bandyopadhyay
BC: Melamine induces Ca2+-sensing receptor activation
and elicits apoptosis in proximal tubular cells. Am J Physiol Cell
Physiol. 313:C27–C41. 2017. View Article : Google Scholar
|
|
49
|
Wu CF, Liu CC, Tsai YC, Chen CC, Wu MT and
Hsieh TJ: Diminishment of Nrf2 antioxidative defense aggravates
nephrotoxicity of melamine and oxalate coexposure. Antioxidants
(Basel). 10:14642021. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Peng Y, Fang Z, Liu M, Wang Z, Li L, Ming
S, Lu C, Dong H, Zhang W, Wang Q, et al: Testosterone induces renal
tubular epithelial cell death through the HIF-1alpha/BNIP3 pathway.
J Transl Med. 17:622019. View Article : Google Scholar
|
|
51
|
Gombedza FC, Shin S, Kanaras YL and
Bandyopadhyay BC: Abrogation of store-operated Ca2+
entry protects against crystal-induced ER stress in human proximal
tubular cells. Cell Death Discov. 5:1242019. View Article : Google Scholar
|
|
52
|
Yan L, Chen J and Fang W: Exosomes derived
from calcium oxalate-treated macrophages promote apoptosis of HK-2
cells by promoting autophagy. Bioengineered. 13:2442–2450. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Khan SR, Canales BK and
Dominguez-Gutierrez PR: Randall's plaque and calcium oxalate stone
formation: Role for immunity and inflammation. Nat Rev Nephrol.
17:417–433. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
He J, Cao Y, Zhu Q, Wang X, Cheng G, Wang
Q, He R, Lu H, Weng Y, Mao G, et al: Renal macrophages monitor and
remove particles from urine to prevent tubule obstruction.
Immunity. 57:106–123.e7. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Liu Q, Liu Y, Guan X, Wu J, He Z, Kang J,
Tao Z and Deng Y: Effect of M2 macrophages on injury and apoptosis
of renal tubular epithelial cells induced by calcium oxalate
crystals. Kidney Blood Press Res. 44:777–791. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Lu H, Sun X, Jia M, Sun F, Zhu J, Chen X,
Chen K and Jiang K: Rosiglitazone suppresses renal crystal
deposition by ameliorating tubular injury resulted from oxidative
stress and inflammatory response via promoting the Nrf2/HO-1
pathway and shifting macrophage polarization. Oxid Med Cell Longev.
2021:55271372021. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Xi J, Chen Y, Jing J, Qi W and Zhang Y:
LncRNA LINC01197 inhibited the formation of calcium oxalate-induced
kidney stones by regulating miR-516b-5p/SIRT3/FOXO1 signaling
pathway. Cell Tissue Res. 392:553–563. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Xi J, Jing J, Zhang Y, Liang C, Hao Z,
Zhang L and Chen Y: SIRT3 inhibited the formation of calcium
oxalate-induced kidney stones through regulating NRF2/HO-1
signaling pathway. J Cell Biochem. 120:8259–8271. 2019. View Article : Google Scholar
|
|
59
|
Li Y, Ding T, Hu H, Zhao T, Zhu C, Ding J,
Yuan J and Guo Z: LncRNA-ATB participates in the regulation of
calcium oxalate crystal-induced renal injury by sponging the
miR-200 family. Mol Med. 27:1432021. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Su B, Han H, Ji C, Hu W, Yao J, Yang J,
Fan Y and Li J: MiR-21 promotes calcium oxalate-induced renal
tubular cell injury by targeting PPARA. Am J Physiol Renal Physiol.
319:F202–F214. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Cabuzu D, Ramakrishnan SK, Moor MB,
Harmacek D, Auberson M, Durussel F and Bonny O: Loss of Ecrg4
improves calcium oxalate nephropathy. PLoS One. 17:e2759722022.
View Article : Google Scholar
|
|
62
|
Gao X, Peng Y, Fang Z, Li L, Ming S, Dong
H, Li R, Zhu Y, Zhang W, Zhu B, et al: Inhibition of EZH2
ameliorates hyperoxaluria-induced kidney injury through the
JNK/FoxO3a pathway. Life Sci. 291:1202582022. View Article : Google Scholar
|
|
63
|
Zhou Z, Zhou X, Zhang Y, Yang Y, Wang L
and Wu Z: Butyric acid inhibits oxidative stress and inflammation
injury in calcium oxalate nephrolithiasis by targeting CYP2C9. Food
Chem Toxicol. 178:1139252023. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Song Q, Song C, Chen X, Xiong Y, Li L,
Liao W, Xue L and Yang S: FKBP5 deficiency attenuates calcium
oxalate kidney stone formation by suppressing cell-crystal
adhesion, apoptosis and macrophage M1 polarization via inhibition
of NF-κB signaling. Cell Mol Life Sci. 80:3012023. View Article : Google Scholar
|
|
65
|
Xun Y, Zhou P, Yang Y, Li C, Zhang J, Hu
H, Qin B, Zhang Z, Wang Q, Lu Y and Wang S: Role of Nox4 in high
calcium-induced renal oxidative stress damage and crystal
deposition. Antioxid Redox Sign. 36:15–38. 2022. View Article : Google Scholar
|
|
66
|
Thomas K, Zondler L, Ludwig N, Kardell M,
Lüneburg C, Henke K, Mersmann S, Margraf A, Spieker T, Tekath T, et
al: Glutamine prevents acute kidney injury by modulating oxidative
stress and apoptosis in tubular epithelial cells. JCI Insight.
7:e1631612022. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Li Y, Lu X, Yu Z, Wang H and Gao B:
Meta-data analysis of kidney stone disease highlights ATP1A1
involvement in renal crystal formation. Redox Biol. 61:1026482023.
View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Ye QL, Wang DM, Wang X, Zhang ZQ, Tian QX,
Feng SY, Zhang ZH, Yu DX, Ding DM and Xie DD: Sirt1 inhibits kidney
stones formation by attenuating calcium oxalate-induced cell
injury. Chem Biol Interact. 347:1096052021. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Ji N, Qi Z, Wang Y, Yang X, Yan Z, Li M,
Ge Q and Zhang J: Pyroptosis: A new regulating mechanism in
cardiovascular disease. J Inflamm Res. 14:2647–2666. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Vande WL and Lamkanfi M: Drugging the
NLRP3 inflammasome: From signalling mechanisms to therapeutic
targets. Nat Rev Drug Discov. 23:43–66. 2024. View Article : Google Scholar
|
|
71
|
Que X, Zheng S, Song Q, Pei H and Zhang P:
Fantastic voyage: The journey of NLRP3 inflammasome activation.
Genes Dis. 11:819–829. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Darisipudi MN and Knauf F: An update on
the role of the inflammasomes in the pathogenesis of kidney
diseases. Pediatr Nephrol. 31:535–544. 2016. View Article : Google Scholar
|
|
73
|
Chen Y, Yang S, Kong H, Wang Q, Chen S,
Wang X, Chen L and Qi S: Oxalate-induced renal pyroptotic injury
and crystal formation mediated by NLRP3-GSDMD signaling in vitro
and in vivo. Mol Med Rep. 28:2092023. View Article : Google Scholar
|
|
74
|
Gu Y, Shen Y, Chen W, He H, Ma Y, Mei X,
Ju D and Liu H: Protective effects of interleukin-22 on
oxalate-induced crystalline renal injury via alleviating
mitochondrial damage and inflammatory response. Appl Microbiol
Biot. 106:2637–2649. 2022. View Article : Google Scholar
|
|
75
|
Zhang Y, Wang S, Dai X, Liu T, Liu Y, Shi
H, Yin J, Xu T, Zhang Y, Zhao D, et al: Simiao San alleviates
hyperuricemia and kidney inflammation by inhibiting NLRP3
inflammasome and JAK2/STAT3 signaling in hyperuricemia mice. J
Ethnopharmacol. 312:1165302023. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Gan XG, Wang ZH and Xu HT: Mechanism of
miRNA-141-3p in calcium oxalate-induced renal tubular epithelial
cell injury via NLRP3-mediated pyroptosis. Kidney Blood Press Res.
47:300–308. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Ding T, Zhao T, Li Y, Liu Z, Ding J, Ji B,
Wang Y and Guo Z: Vitexin exerts protective effects against calcium
oxalate crystal-induced kidney pyroptosis in vivo and in vitro.
Phytomedicine. 86:1535622021. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Song Z, Zhang Y, Gong B, Xu H, Hao Z and
Liang C: Long noncoding RNA LINC00339 promotes renal tubular
epithelial pyroptosis by regulating the miR-22-3p/NLRP3 axis in
calcium oxalate-induced kidney stone. J Cell Biochem.
120:10452–10462. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Liu J, Yang K, Jin Y, Liu Y, Chen Y, Zhang
X, Yu S, Song E, Chen S, Zhang J, et al: H3 relaxin protects
against calcium oxalate crystal-induced renal inflammatory
pyroptosis. Cell Prolif. 53:e129022020. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Yifan Z, Benxiang N, Zheng X, Luwei X,
Liuhua Z, Yuzheng G and Ruipeng J: Ceftriaxone Calcium crystals
induce acute kidney injury by NLRP3-mediated inflammation and
oxidative stress injury. Oxid Med Cell Longev. 2020:64284982020.
View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Sundaram B, Pandian N, Mall R, Wang Y,
Sarkar R, Kim HJ, Malireddi RKS, Karki R, Janke LJ, Vogel P and
Kanneganti TD: NLRP12-PANoptosome activates PANoptosis and
pathology in response to heme and PAMPs. Cell. 186:2783–2801. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Mulay SR, Shi C, Ma X and Anders HJ: Novel
insights into crystal-induced kidney injury. Kidney Dis (Basel).
4:49–57. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Hou B, Liu M, Chen Y, Ni W, Suo X, Xu Y,
He Q, Meng X and Hao Z: Cpd-42 protects against calcium oxalate
nephrocalcinosis-induced renal injury and inflammation by targeting
RIPK3-mediated necroptosis. Front Pharmacol. 13:10411172022.
View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Sedmaki K, Karnam K, Sharma P, Mahale A,
Routholla G, Ghosh B and Prakash Kulkarni O: HDAC6 inhibition
attenuates renal injury by reducing IL-1β secretion and RIP kinase
mediated necroptosis in acute oxalate nephropathy. Int
Immunopharmacol. 110:1089192022. View Article : Google Scholar
|
|
85
|
Prajapati S, Tomar B, Srivastava A,
Narkhede YB, Gaikwad AN, Lahiri A and Mulay SR:
6,7-Dihydroxycoumarin ameliorates crystal-induced necroptosis
during crystal nephropathies by inhibiting MLKL phosphorylation.
Life Sci. 271:1191932021. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Mulay SR, Eberhard JN, Desai J, Marschner
JA, Kumar SV, Weidenbusch M, Grigorescu M, Lech M, Eltrich N,
Müller L, et al: Hyperoxaluria requires TNF receptors to initiate
crystal adhesion and kidney stone disease. J Am Soc Nephrol.
28:761–768. 2017. View Article : Google Scholar :
|
|
87
|
Sun S, Shen J, Jiang J, Wang F and Min J:
Targeting ferroptosis opens new avenues for the development of
novel therapeutics. Signal Transduct Target Ther. 8:3722023.
View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Chen X, Kang R, Kroemer G and Tang D:
Broadening horizons: The role of ferroptosis in cancer. Nat Rev
Clin Oncol. 18:280–296. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
He Z, Liao W, Song Q, Li B, Liu J, Xiong
Y, Song C and Yang S: Role of ferroptosis induced by a high
concentration of calcium oxalate in the formation and development
of urolithiasis. Int J Mol Med. 47:289–301. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Ye Z, Xia Y, Li L, Li B, Chen L, Yu W,
Ruan Y, Rao T, Zhou X and Cheng F: p53 deacetylation alleviates
calcium oxalate deposition-induced renal fibrosis by inhibiting
ferroptosis. Biomed Pharmacother. 164:1149252023. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Song Q, Liao W, Chen X, He Z, Li D, Li B,
Liu J, Liu L, Xiong Y, Song C and Yang S: Oxalate activates
autophagy to induce ferroptosis of renal tubular epithelial cells
and participates in the formation of kidney stones. Oxid Med Cell
Longev. 2021:66303432021. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Xie J, Ye Z, Li L, Xia Y, Yuan R, Ruan Y
and Zhou X: Ferrostatin-1 alleviates oxalate-induced renal tubular
epithelial cell injury, fibrosis and calcium oxalate stone
formation by inhibiting ferroptosis. Mol Med Rep. 26:2562022.
View Article : Google Scholar :
|
|
93
|
Martin-Saiz L, Guerrero-Mauvecin J,
Martin-Sanchez D, Fresnedo O, Gómez MJ, Carrasco S, Cannata-Ortiz
P, Ortiz A, Fernandez JA and Sanz AB: Ferrostatin-1 modulates
dysregulated kidney lipids in acute kidney injury. J Pathol.
257:285–299. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Xia C, Xing X, Zhang W, Wang Y, Jin X,
Wang Y, Tian M, Ba X and Hao F: Cysteine and homocysteine can be
exploited by GPX4 in ferroptosis inhibition independent of GSH
synthesis. Redox Biol. 69:1029992024. View Article : Google Scholar :
|
|
95
|
Ide S, Ide K, Abe K, Kobayashi Y, Kitai H,
McKey J, Strausser SA, O'Brien LL, Tata A, Tata PR and Souma T: Sex
differences in resilience to ferroptosis underlie sexual dimorphism
in kidney injury and repair. Cell Rep. 41:1116102022. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Chu LK, Cao X, Wan L, Diao Q, Zhu Y, Kan
Y, Ye LL, Mao YM, Dong XQ, Xiong QW, et al: Autophagy of OTUD5
destabilizes GPX4 to confer ferroptosis-dependent kidney injury.
Nat Commun. 14:83932023. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Song J, Wang H, Sheng J, Zhang W, Lei J,
Gan W, Cai F and Yang Y: Vitexin attenuates chronic kidney disease
by inhibiting renal tubular epithelial cell ferroptosis via NRF2
activation. Mol Med. 29:1472023. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Lee J and Roh JL: SLC7A11 as a gateway of
metabolic perturbation and ferroptosis vulnerability in cancer.
Antioxidants (Basel). 11:24442022. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Zhao J, Wu Y, Zhou K, Huang M, Sun Y, Kang
J, Su Q, Zhao Y, Liu Q and Li C: Ferroptosis in calcium oxalate
kidney stone formation and the possible regulatory mechanism of
ANKRD1. Biochim Biophys Acta Mol Cell Res. 1870:1194522023.
View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Hao W, Zhang H, Hong P, Zhang X, Zhao X,
Ma L, Qiu X, Ping H, Lu D and Yin Y: Critical role of
VHL/BICD2/STAT1 axis in crystal-associated kidney disease. Cell
Death Dis. 14:6802023. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Chen X, Li J, Kang R, Klionsky DJ and Tang
D: Ferroptosis: Machinery and regulation. Autophagy. 17:2054–2081.
2021. View Article : Google Scholar :
|
|
102
|
Liu Q, Tang J, Chen Z, Wei L, Chen J and
Xie Z: Polyunsaturated fatty acids ameliorate renal stone-induced
renal tubular damage via miR-93-5p/Pknox1 axis. Nutrition.
105:1118632023. View Article : Google Scholar
|
|
103
|
Li L, Ye Z, Xia Y, Li B, Chen L, Yan X,
Yuan T, Song B, Yu W, Rao T, et al: YAP/ACSL4 pathway-mediated
ferroptosis promotes renal fibrosis in the presence of kidney
stones. Biomedicines. 11:26922023. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Mishima E, Ito J, Wu Z, Nakamura T, Wahida
A, Doll S, Tonnus W, Nepachalovich P, Eggenhofer E, Aldrovandi M,
et al: A non-canonical vitamin K cycle is a potent ferroptosis
suppressor. Nature. 608:778–783. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Nishizawa H, Yamanaka M and Igarashi K:
Ferroptosis: Regulation by competition between NRF2 and BACH1 and
propagation of the death signal. FEBS J. 290:1688–1704. 2023.
View Article : Google Scholar
|
|
106
|
Dong C, Song C, He Z, Song Q, Song T, Liu
J, Xiong Y, Su X, Zhou J, Yang S and Liao W: Protective efficacy of
Schizandrin B on ameliorating nephrolithiasis via regulating
GSK3β/Nrf2 signaling-mediated ferroptosis in vivo and in vitro. Int
Immunopharmacol. 117:1100422023. View Article : Google Scholar
|
|
107
|
Zhou D, Wu Y, Yan H, Shen T, Li S, Gong J,
Li G, Mai H, Wang D and Tan X: Gallic acid ameliorates calcium
oxalate crystal-induced renal injury via upregulation of Nrf2/HO-1
in the mouse model of stone formation. Phytomedicine.
106:1544292022. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Li Y, Zhang J, Liu H, Yuan J, Yin Y, Wang
T, Cheng B, Sun S and Guo Z: Curcumin ameliorates
glyoxylate-induced calcium oxalate deposition and renal injuries in
mice. Phytomedicine. 61:1528612019. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Zhu J, Wang Q, Li C, Lu Y, Hu H, Qin B,
Xun Y, Zhu Y, Wu Y, Zhang J and Wang S: Inhibiting inflammation and
modulating oxidative stress in oxalate-induced nephrolithiasis with
the Nrf2 activator dimethyl fumarate. Free Radical Bio Med.
134:9–22. 2019. View Article : Google Scholar
|
|
110
|
Ushimoto C, Sugiki S, Kunii K, Inoue S,
Kuroda E, Akai R, Iwawaki T and Miyazawa K: Dynamic change and
preventive role of stress response via Keap1-Nrf2 during renal
crystal formation. Free Radic Bio Med. 207:120–132. 2023.
View Article : Google Scholar
|
|
111
|
Song Q, He Z, Li B, Liu J, Liu L, Liao W,
Xiong Y, Song C, Yang S and Liu Y: Melatonin inhibits
oxalate-induced endoplasmic reticulum stress and apoptosis in HK-2
cells by activating the AMPK pathway. Cell Cycle. 19:2600–2610.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Zhou J, Meng L, He Z, Song Q, Liu J, Su X,
Wang C, Ke H, Dong C, Liao W and Yang S: Melatonin exerts a
protective effect in ameliorating nephrolithiasis via targeting
AMPK/PINK1-Parkin mediated mitophagy and inhibiting ferroptosis in
vivo and in vitro. Int Immunopharmacol. 124:1108012023. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Su X, Song C, He Z, Song Q, Meng L, Dong
C, Zhou J, Ke H, Xiong Y, Liu J, et al: Ambra1 in exosomes secreted
by HK-2 cells damaged by supersaturated oxalate induce mitophagy
and autophagy-ferroptosis in normal HK-2 cells to participate in
the occurrence of kidney stones. Biochim Biophys Acta Mol Cell Res.
1871:1196042024. View Article : Google Scholar
|
|
114
|
Khan MA, Nag P, Grivei A, Giuliani KTK,
Wang X, Diwan V, Hoy W, Healy H, Gobe G and Kassianos AJ: Adenine
overload induces ferroptosis in human primary proximal tubular
epithelial cells. Cell Death Dis. 13:1042022. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Liu S, Yao S, Yang H, Liu S and Wang Y:
Autophagy: Regulator of cell death. Cell Death Dis. 14:6482023.
View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Lee S, Hwang N, Seok BG, Lee S, Lee SJ and
Chung SW: Autophagy mediates an amplification loop during
ferroptosis. Cell Death Dis. 14:4642023. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Bhatia D and Choi ME: Autophagy and
mitophagy: Physiological implications in kidney inflammation and
diseases. Am J Physiol Renal Physiol. 325:F1–F21. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Duan X, Kong Z, Mai X, Lan Y, Liu Y, Yang
Z, Zhao Z, Deng T, Zeng T, Cai C, et al: Autophagy inhibition
attenuates hyperoxaluria-induced renal tubular oxidative injury and
calcium oxalate crystal depositions in the rat kidney. Redox Biol.
16:414–425. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Sun Y, Kang J, Tao Z, Wang X, Liu Q, Li D,
Guan X, Xu H, Liu Y and Deng Y: Effect of endoplasmic reticulum
stress-mediated excessive autophagy on apoptosis and formation of
kidney stones. Life Sci. 244:1172322020. View Article : Google Scholar
|
|
120
|
Kang J, Sun Y, Deng Y, Liu Q, Li D, Liu Y,
Guan X, Tao Z and Wang X: Autophagy-endoplasmic reticulum stress
inhibition mechanism of superoxide dismutase in the formation of
calcium oxalate kidney stones. Biomed Pharmacother. 121:1096492020.
View Article : Google Scholar
|
|
121
|
Kumar P, Laurence E, Crossman DK, Assimos
DG, Murphy MP and Mitchell T: Oxalate disrupts monocyte and
macrophage cellular function via Interleukin-10 and mitochondrial
reactive oxygen species (ROS) signaling. Redox Biol. 67:1029192023.
View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Nakamura S, Shigeyama S, Minami S, Shima
T, Akayama S, Matsuda T, Esposito A, Napolitano G, Kuma A,
Namba-Hamano T, et al: LC3 lipidation is essential for TFEB
activation during the lysosomal damage response to kidney injury.
Nat Cell Biol. 22:1252–1263. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Wu Y, Xun Y, Zhang J, Hu H, Qin B, Wang T,
Wang S, Li C and Lu Y: Resveratrol attenuates oxalate-induced renal
oxidative injury and calcium oxalate crystal deposition by
regulating TFEB-induced autophagy pathway. Front Cell Dev Biol.
9:6387592021. View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Dong F, Jiang S, Tang C, Wang X, Ren X,
Wei Q, Tian J, Hu W, Guo J, Fu X, et al: Trimethylamine N-oxide
promotes hyperoxaluria-induced calcium oxalate deposition and
kidney injury by activating autophagy. Free Radic Bio Med.
179:288–300. 2022. View Article : Google Scholar
|
|
125
|
Alaygut D, Ozturk I, Ulu S and Gungor O:
NETosis and kidney disease: What do we know? Int Urol Nephrol.
55:1985–1994. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Makki MS, Winfree S, Lingeman JE, Witzmann
FA, Worcester EM, Krambeck AE, Coe FL, Evan AP, Bledsoe S,
Bergsland KJ, et al: A precision medicine approach uncovers a
unique signature of neutrophils in patients with brushite kidney
stones. Kidney Int Rep. 5:663–677. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Cai Z, Wu X, Song Z, Sun S, Su Y, Wang T,
Cheng X, Yu Y, Yu C, Chen E, et al: Metformin potentiates
nephrotoxicity by promoting NETosis in response to renal
ferroptosis. Cell Discov. 9:1042023. View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Malireddi RKS, Kesavardhana S and
Kanneganti TD: ZBP1 and TAK1: Master regulators of NLRP3
inflammasome/pyroptosis, apoptosis, and necroptosis (PAN-optosis).
Front Cell Infect Microbiol. 9:4062019. View Article : Google Scholar : PubMed/NCBI
|
|
129
|
Hadian K and Stockwell BR: The therapeutic
potential of targeting regulated non-apoptotic cell death. Nat Rev
Drug Discov. 22:723–742. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Peerapen P and Thongboonkerd V: Kidney
stone prevention. Adv Nutr. 14:555–569. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Baltazar P, de Melo Junior AF, Fonseca NM,
Lança MB, Faria A, Sequeira CO, Teixeira-Santos L, Monteiro EC,
Campos Pinheiro L, Calado J, et al: Oxalate (dys)metabolism:
Person-to-person variability, kidney and cardiometabolic toxicity.
Genes (Basel). 14:17192023. View Article : Google Scholar : PubMed/NCBI
|
|
132
|
Marengo SR and Romani AMP: Oxalate in
renal stone disease: The terminal metabolite that just won't go
away. Nat Clin Pract Nephrol. 4:368–377. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
133
|
Grocholski C, Derain Dubourg L,
Guebre-Egziabher F, Acquaviva-Bourdain C, Abid N, Bacchetta J,
Chambrier C and Lemoine S: Oxalate: From physiology to pathology.
Nephrol Ther. 19:201–214. 2023.In French. View Article : Google Scholar : PubMed/NCBI
|
|
134
|
Kim D, Rimer JD and Asplin JR:
Hydroxycitrate: A potential new therapy for calcium urolithiasis.
Urolithiasis. 47:311–320. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
135
|
Lunyera J, Diamantidis CJ, Bosworth HB,
Patel UD, Bain J, Muehlbauer MJ, Ilkayeva O, Nguyen M, Sharma B, Ma
JZ, et al: Urine tricarboxylic acid cycle signatures of early-stage
diabetic kidney disease. Metabolomics. 18:52021. View Article : Google Scholar : PubMed/NCBI
|
|
136
|
Chen L, Min J and Wang F: Copper
homeostasis and cuproptosis in health and disease. Signal Transduct
Target Ther. 7:3782022. View Article : Google Scholar : PubMed/NCBI
|
|
137
|
Humphries F, Shmuel-Galia L,
Ketelut-Carneiro N, Li S, Wang B, Nemmara VV, Wilson R, Jiang Z,
Khalighinejad F, Muneeruddin K, et al: Succination inactivates
gasdermin D and blocks pyroptosis. Science. 369:1633–1637. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
138
|
Zhang XZ, Lei XX, Jiang YL, Zhao LM, Zou
CY, Bai YJ, Li YX, Wang R, Li QJ, Chen QZ, et al: Application of
metabolomics in urolithiasis: The discovery and usage of succinate.
Signal Transduct Target Ther. 8:412023. View Article : Google Scholar : PubMed/NCBI
|
|
139
|
Sun K, Zhi Y, Ren W, Li S, Zhou X, Gao L
and Zhi K: The mitochondrial regulation in ferroptosis signaling
pathway and its potential strategies for cancer. Biomed
Pharmacother. 169:1158922023. View Article : Google Scholar : PubMed/NCBI
|
|
140
|
Duan X, Zhang T, Ou L, Kong Z, Wu W and
Zeng G: 1H NMR-based metabolomic study of metabolic
profiling for the urine of kidney stone patients. Urolithiasis.
48:27–35. 2020. View Article : Google Scholar
|
|
141
|
Hernandez Y, Costa-Bauza A, Calvó P,
Benejam J, Sanchis P and Grases F: Comparison of two dietary
supplements for treatment of uric acid renal lithiasis: Citrate vs
Citrate + theobromine. Nutrients. 12:20122020. View Article : Google Scholar
|
|
142
|
Eisner BH, Asplin JR, Goldfarb DS, Ahmad A
and Stoller ML: Citrate, malate and alkali content in commonly
consumed diet sodas: Implications for nephrolithiasis treatment. J
Urol. 183:2419–2423. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
143
|
Zhu W, Liu Y, Lan Y, Li X, Luo L, Duan X,
Lei M, Liu G, Yang Z, Mai X, et al: Dietary vinegar prevents kidney
stone recurrence via epigenetic regulations. EBioMedicine.
45:231–250. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
144
|
Zhang JY, Zhou B, Sun RY, Ai YL, Cheng K,
Li FN, Wang BR, Liu FJ, Jiang ZH, Wang WJ, et al: The metabolite
α-KG induces GSDMC-dependent pyroptosis through death receptor
6-activated caspase-8. Cell Res. 31:980–997. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
145
|
Kumar P, Saini K, Saini V and Mitchell T:
Oxalate alters cellular bioenergetics, redox homeostasis,
antibacterial response, and immune response in macrophages. Front
Immunol. 12:6948652021. View Article : Google Scholar : PubMed/NCBI
|
|
146
|
Cai W, Wannemuehler Y, Dell'anna G,
Nicholson B, Barbieri NL, Kariyawasam S, Feng Y, Logue CM, Nolan LK
and Li G: A novel two-component signaling system facilitates
uropathogenic Escherichia coli's ability to exploit abundant host
metabolites. PLoS Pathog. 9:e10034282013. View Article : Google Scholar : PubMed/NCBI
|
|
147
|
Yu H, Gan D, Luo Z, Yang Q, An D, Zhang H,
Hu Y, Ma Z, Zeng Q, Xu D and Ren H: α-Ketoglutarate improves
cardiac insufficiency through NAD+-SIRT1
signaling-mediated mitophagy and ferroptosis in pressure
overload-induced mice. Mol Med. 30:152024. View Article : Google Scholar
|














