Introduction
Lung cancer remains a leading cause of
cancer-related mortality worldwide, with non-small cell lung cancer
(NSCLC) accounting for ~85% of all lung cancer cases (1,2)
Despite advances in diagnostic and therapeutic strategies, the
prognosis for patients with NSCLC remains poor, primarily due to
the late-stage diagnosis and the limited efficacy of conventional
treatments (3,4). Chemotherapy, particularly with
platinum-based drugs such as cisplatin (CDDP), is a cornerstone in
the management of NSCLC (5,6).
However, the clinical use of CDDP is significantly hampered by its
systemic toxicity, poor solubility and the development of drug
resistance in tumour cells. These challenges underscore the urgent
need for innovative strategies that can enhance the delivery and
therapeutic efficacy of CDDP, while minimizing its adverse effects
(7).
Advances in nanotechnology have provided promising
avenues for the targeted delivery of chemotherapeutic agents.
Liposomes, which are spherical vesicles consisting of one or more
phospholipid bilayers, have emerged as versatile drug delivery
systems due to their biocompatibility, ability to encapsulate both
hydrophilic and hydrophobic drugs, and potential for surface
modification to achieve targeted delivery (8-10). The encapsulation of CDDP in
liposomes has been explored as a strategy to improve its
pharmacokinetic profile and reduce its toxicity (11). However, the challenge of
specifically directing these liposomal formulations to tumour
tissues remains a significant hurdle in maximizing their
therapeutic efficacy against NSCLC (12).
The concept of active targeting, which involves the
modification of liposomal surfaces with ligands that can
specifically bind to receptors upregulated on cancer cells,
represents a viable strategy for overcoming this challenge
(13-17). MUC1, a membrane-bound mucin,
exhibits a unique expression pattern on tumour cells, presenting a
targetable epitope (18-20). Peanut agglutinin (PNA), a plant
lectin with high affinity for the Thomsen-Friedenreich (TF)
antigen, has garnered attention in this context (21,22). Notably, the TF antigen is a core
antigenic site of MUC1, which is expressed on the surface of tumour
cells, providing conditions for PNA binding, thus making it a
potential target for PNA-modified liposomes (PNA-Lip) (23). The present study aimed to explore
the development and evaluation of PNA-Lip for the targeted delivery
of CDDP to NSCLC cells. By leveraging the specific binding affinity
of PNA to the TF antigen, the study aimed to enhance the
accumulation of CDDP in NSCLC tissues, thereby improving its
therapeutic efficacy and reducing systemic toxicity. Through a
comprehensive investigation encompassing liposome formulation,
characterization, in vitro cytotoxicity assays and in
vivo antitumour efficacy analyses, this research may provide a
novel and effective approach for the treatment of NSCLC with
CDDP-loaded PNA-modified liposomes (CDDP-PNA-Lip).
Materials and methods
Materials
Soy phospholipids (SPC), DPPG, cholesterol (Chol)
and DSPE-PEG2K were purchased from AVT (Shanghai) Pharmaceutical
Technology Co., Ltd. DSPE-PEG2K-NHS was purchased from Xi'an Ruixi
Biological Technology Co., Ltd. PNA was purchased from Medicago AB.
CDDP and sulfo-Cyanine7 carboxylic acid (Cy7) were purchased from
Dalian Meilun Biology Technology Co., Ltd.
The human lung cancer cell lines A549 (MUC1-high)
and H460 (MUC1-low) were purchased from The Cell Bank of Type
Culture Collection of The Chinese Academy of Sciences. A549 cells
were cultured in Dulbecco's modified Eagle's medium (Dalian Meilun
Biology Technology Co., Ltd.) supplemented with 10%
heat-inactivated foetal bovine serum (FBS; Biological Industries),
100 U/ml penicillin and 100 μg/ml streptomycin in a
humidified atmosphere containing 5% CO2 at 37°C. H460
cells were cultured in RPMI 1640 medium (Dalian Meilun Biology
Technology Co., Ltd.) supplemented with 10% heat-inactivated FBS,
100 U/ml penicillin and 100 μg/ml streptomycin at 37°C in a
humidified atmosphere containing 5% CO2. A total of 29
male BALB/C-nu mice (weight, 13-17 g; age, 4-6 weeks) were
purchased from Beijing Vital River Laboratory Animal Technology
Co., Ltd. The Ethical Review Committee of Shandong Second Medical
University (Weifang, China) approved the present study (approval
no. 2022SDL448), which complied with institutional animal care
guidelines.
The Cancer Genome Atlas (TCGA) analysis
of MUC1
The relationship between MUC1 expression and the
clinicopathological characteristics of patients with lung cancer
was statistically analysed using relevant clinical data from TCGA
database (Project Name: Lung Adenocarcinoma; portal.gdc.cancer.gov/projects/TCGA-LUAD), and
the figures were generated using GEPIA (gepia.cancer-pku.cn) and cbioportal (cbioportal.org) (24). Imaging data were obtained from
the Human Protein Atlas (proteinatlas.org). The present study complies with the
National Institutes of Health TCGA Human Guidelines for Subject
Protection and Data Access Policy, as well as the proteinatlas.org citation regulations for images.
Dialysis bag pretreatment
Due to the presence of trace amounts of sulphides,
heavy metals and some UV-absorbing impurities in the dialysis bags,
they need to be pretreated prior to use. Briefly, after the bags
were cut to size, they were boiled for 10 min in 2% sodium
bicarbonate and 1 mmol/l EDTA (pH 8.0), and 1 mmol/l EDTA (pH 8.0),
respectively. The bags were then washed on the inside and outside
surfaces with purified water and stored at 4°C.
DSPE-PEG2K-PNA synthesis
A total of 10 mg PNA was weighed, completely
dissolved in PBS (pH 7.4), reconstituted into a PNA solution with a
concentration of 5 mg/ml and set aside. Subsequently, 100 μl
DSPE-PEG2K-NHS in DMSO solution (100 mg/ml) was added to the PNA
solution, and after 4 h of incubation at room temperature, the
entire solution was transferred to treated dialysis bags. Dialysis
was performed three times using ddH2O (2 h each time),
and DSPE-PEG2K-PNA was obtained after freeze-drying at -60°C for 24
h. PNA and DSPE-PEG2K-PNA (10 μg) then underwent SDS-PAGE on
12% gels. Subsequently, the gels were stained with 0.25% Coomassie
brilliant blue staining solution. After the SDS-PAGE was
decolorized with decolorization solution (cat. no. P0017C; Beyotime
Institute of Biotechnology) until it was nearly colourless and the
bands were clear, the SDS-PAGE mixture was stained with PEG dye to
confirm the successful synthesis of DSPE-PEG2K-PNA. The infrared
absorption spectra of PNA, DSPE-PEG2K and DSPE-PEG2K-PNA were
examined using an infrared absorption spectrometer, and the
characteristic peaks of PNA, DSPE-PEG2K and DSPE-PEG2K-PNA were
compared to determine the successful synthesis of
DSPE-PEG2K-PNA.
Preparation of the pretreated CDDP
solution
CDDP pretreatment was performed according to the
methods reported by Zahednezhad et al (24). According to Le Chatelier's
principle, CDDP undergoes a hydration reaction in water to produce
Pt(NH3)2Cl2 + 2 H2O ⇆
[Pt(NH3)2(H2O)2]2+
+ 2Cl−, and the CDDP generated using this derivative
approach has a positive charge on its surface, enabling its active
loading in liposomes. The product generated by this reaction is the
form in which CDDP undergoes active intracellular action, and thus
its antitumour effect is not altered (25). A pretreated CDDP solution was
prepared by dissolving CDDP in pure water. The solution was then
placed in a magnetic stirrer and set at a speed of 150 rpm. The
mixture was incubated at 65°C for 6 h. Finally, the solution was
stored in the dark at 4°C.
Preparation of PNA-modified CDDP-loaded
liposomes (CDDP-Lip)
Film dispersion was used to create blank liposomes
(Lip) (26,27). In a clean round-bottomed flask,
the phospholipids needed to make liposomes were measured in
accordance with a mass ratio of SPC:DPPG:Chol:DSPE-PEG2K=15:4:15:4
(SPC:DPPG:Chol:DSPE-PEG2K:DSPE-PEG2K-PNA=15:4:15:4:3 for the
PNA-modified liposomes). An ultrasonic cell washer was used to
sonicate (20 kHz; 20% power at 0°C; duration, 2 sec) the
aforementioned phospholipids to thoroughly dissolve and combine
them; a chloroform-methanol ratio of 2:1 was used for this purpose.
The chloroform-methanol mixture with dissolved lipid material was
then transferred to an eggplant flask and linked to a rotary
evaporator (Yamato Scientific Shanghai Corp.). The rotary
evaporator speed, water bath temperature and pressure were adjusted
so that all the organic solvents were evaporated slowly and
uniformly until a homogeneous lipid film was visible to the naked
eye, after which the rotary evaporator pressure was adjusted to the
maximum and maintained for >30 min to ensure that all the
organic reagents were removed 5 ml ddH2O was then added
to the eggplant bottle, which was submerged in the ultrasonic cell
washer for 5 min to ensure that all the lipid films were eluted;
the liposomes were hydrated using the rotary evaporator; and after
1 h, the hydrated liposomes were sonicated (20 kHz; 20% power at
0°C) for 5 min using the probe and filtered three times using 0.45
and 0.22-μm filters. The dialysis bags were removed and
completely washed with ethanol on the surface and inside with pure
water, and all of the filtered liposomes were then transferred to
the washed bags for dialysis. The liposomes were dialyzed using
ddH2O for 6 h to eliminate any phospholipids that did
not form liposomes. The water was replaced every 2 h to ensure
thorough dialysis. Finally, the liposomes were collected from the
dialysis bag and stored at 4°C.
The method used for the preparation of CDDP-Lip was
essentially the same as that aforementioned. Briefly, the formed
lipid film was subsequently hydrated with the pretreated CDDP
solution at a ratio of 10:1 total phospholipid mass to drug mass,
and the hydration time was suitably extended to allow the
pretreated CDDP to be well encapsulated by the liposomes, followed
by sonication (20 kHz; 20% power at 0°C; duration, 2 sec) of the
CDDP-Lip for 10 min using a probe. CDDP-Lip were obtained by
dialysis with ddH2O three times (2 h each time) and were
stored at 4°C.
Liposomes were labelled with Rhodamine B (RhB) and
Cy7 fluorescent probes to study the cellular absorption and
biodistribution of the preparations in vitro (RhB) and in
vivo (Cy7) (28-30). The method used for the
preparation of Cy7-loaded liposomes (Cy7-Lip) or RhB-loaded
liposomes (RhB-Lip) was essentially the same as that
aforementioned. Notably, the total mass of phospholipids relative
to the mass of the fluorescent marker was 50:1 during hydration and
when the fluorescent marker was added and hydrated.
Characterization of liposomes
Several types of liposomes (CDDP-PNA-Lip for
electron microscopy; Lip, CDDP-Lip, PNA-Lip and CDDP-PNA-Lip for
characterisation measurements) were diluted 10 times with
ddH2O. Subsequently, a transmission electron microscope
(TEM; JEOL, Ltd.) was used to examine the samples. Samples were
prepared by negative staining with phosphotungstic acid. Briefly,
the sample was added dropwise directly onto a copper grid and left
to stand at room temperature for 3 min before excess liquid was
aspirated. After waiting for the sample to dry completely, 2%
phosphotungstic acid was added dropwise, and after 2 min of
staining at room temperature, the excess liquid was aspirated, and
the sample was left to dry naturally. Using a Malvern Zetasizer
Nano ZS 90 (Malvern Panalytical, Ltd.), the ζ potentials and
particle size distributions were measured.
High-performance liquid chromatography (HPLC) was
used to quantify drug loading (DL) and entrapment efficiency (EE).
Briefly, the HPLC was performed using an Agilent 1260 Infinity II
system (Agilent Technologies, Inc.), with a column size of 4.6×200
mm packed with Hypersil ODS2 (filler particle size, 5 μm;
Dalian Elite Analytical Instruments Co., Ltd.) on a sample size of
20 μl. The mobile phase consisted of a mixture of methanol
and water (v/v=75:25) at a flow rate of 1 ml/min, and the column
temperature was maintained at 25°C. The standard was purchased from
Shanghai Yuanye Bio-Technology Co., Ltd. (cat. no. B24462), and the
peak areas of 100, 75 and 50 μg/ml were measured to
determine the standard curve (31). Triton X-100 (0.1%) was applied to
quickly release the internal CDDP solution and destroy the
CDDP-Lip. After mixing sodium diethyldithiocarbamate (DDTC) and the
sample, the mixture was subsequently incubated in a water bath at
37°C. Afterward, the sample was thoroughly mixed with chloroform,
and the complex formed by DDTC and CDDP was extracted and detected.
The EE and DL of the CDDP-Lip were calculated using the following
formulas:
Stability analysis and drug release from
CDDP-Lip
The stabilities of the liposomes, PNA-liposomes,
CDDP-Lip and CDDP-PNA-Lip were investigated by removing liposomes
stored in ddH2O at 4°C every 2 days for 14 days, and
measuring their particle sizes and ζ potentials.
The drug release profiles of CDDP, CDDP-Lip and
CDDP-PNA-Lip were measured using in vitro dialysis. The CDDP
concentrations of the different groups were adjusted to the same
level using PBS and then added to the dialysis bags, which were
subsequently placed into conical flasks filled with 100 ml PBS. The
samples were incubated at 37°C in a constant temperature shaker at
100 rpm, and the PBS in the conical flask was removed at different
time points and replenished to 100 ml with fresh PBS. The
cumulative in vitro release curves of CDDP were plotted
using HPLC, as aforementioned, to determine the CDDP concentration
in the samples at different time points, with time as the
horizontal coordinate and the cumulative percentage release of CDDP
as the vertical coordinate.
Detection of MUC1-positive cells
MUC1-positive cells were detected via western
blotting. Briefly, the A549 and H460 cell lines were inoculated
into culture dishes and cultured for 48 h in complete medium
supplemented with 10% FBS. After the cells were washed with PBS,
the proteins were extracted from the remaining cells with protein
extraction buffer [50 mM Tris (pH 7.4), 150 mM NaCl, 10 mM
MgCl2, 1% Triton X-100 (v/v), 1% sodium deoxycholate
(v/v), 0.1% SDS (v/v), 5 mg/ml sodium orthovanadate]. Protein
quantification was performed using a BCA assay. Subsequently,
proteins (10 μg) were separated by SDS-PAGE (10% resolving
gel and 4% stacking gel). The proteins were then transferred to
BioTrace nitrocellulose membranes, which were blocked with 5%
non-fat dry milk at 37°C for 1 h. The membranes were then incubated
with the MUC1 (D9O8K) XP® Rabbit mAb (1:1,000; cat. no.
14161; Cell Signaling Technology, Inc.) and β-actin antibody as a
loading control (1:1,000; cat. no. sc-47778; Santa Cruz
Biotechnology, Inc.) overnight at 4°C, followed by incubation with
anti-rabbit IgG and anti-mouse IgG, HRP-linked secondary antibody
(1:2,000; cat. nos. 7074S and 7076S; Cell Signaling Technology,
Inc.) for 1 h at room temperature. The blots were visualized using
WesternBright ECL, (cat. no. 230329-20; Advantsa Inc.) and
underwent densitometric analysis using ImageJ (version 2.9 011.53t;
National Institutes of Health). Subsequently, MUC1 expression in
the cell lines was detected by western blotting to distinguish
between MUC1-positive cell lines and MUC1-negative cell lines.
In vitro cellular liposome uptake
assay
The cellular uptake of CDDP-PNA-Lip was simulated by
observing the in vitro uptake of RhB fluorescent liposomes
by A549 and H460 cells through laser confocal microscopy. The
targeting efficiency of CDDP-PNA-Lip was evaluated. A549 and H460
cells were seeded at a density of 5×105 cells/dish in
Petri dishes suitable for laser confocal microscopy using complete
medium supplemented with 10% FBS. The cells were incubated at 37°C
for 24 h. Subsequently, different concentrations of RhB, RhB-Lip
and RhB-loaded PNA-modified liposomes (RhB-PNA-Lip) were added to
the medium (at a final RhB concentration of 50 μg/ml), and
the mixture was incubated for 6 h at 37°C. Afterwards, the medium
was removed, and the cell surface was cleaned using precooled PBS
at 4°C. Subsequently, the cells were fixed with 4% paraformaldehyde
for 10 min at room temperature and washed 3-5 times with PBS to
remove residual paraformaldehyde and unfixed cells. Next, the cells
were treated with 0.1% Triton X-100 for 10 min at room temperature
to permeabilize the cell and nuclear membranes, and were washed 3-5
times with PBS until the PBS was clear and foam free. The nuclei
were then stained with DAPI staining solution for 10 min at room
temperature and washed 3-5 times with PBS to remove any residual
DAPI and the samples were finally observed using a laser confocal
microscope.
Antitumour efficacy of CDDP and hydrated
CDDP in vitro
The cytotoxicity of various drugs was assessed using
the MTS assay, and the reaction product was dissolved in DMEM).
Briefly, A549 and H460 cells were inoculated into 96-well plates at
a density of 5,000 cells/well and were cultured in complete medium
for 24 h. Once cell adhesion and extension were observed, various
concentrations of CDDP and hydrated CDDP (concentration gradient,
50.00, 25.00, 12.50, 6.25, 3.12, 1.56, 0.78 and 0.39 μg/ml)
were added. After incubation for 48 h, the optical density (OD)
values were measured using a microplate reader (PerkinElmer, Inc.)
at 490 nm. The rate of cell viability was calculated using the
following equation: Cell viability
(%)=(ODsample-ODblank)/(ODcontrol-OD-blank) ×100.
Antitumour efficacy of CDDP-PNA-Lip in
vitro
The cytotoxicity of CDDP, CDDP-Lip and CDDP-PNA-Lip
was assessed using the MTS assay, and the reaction product was
dissolved in DMEM. Briefly, A549 and H460 cells were inoculated
into 96-well plates at a density of 5,000 cells/well and were
cultured in complete medium for 24 h. Once cell adhesion and
extension were observed, various concentrations of drugs
(concentration gradient, 50.00, 25.00, 12.50, 6.25, 3.12, 1.56,
0.78 and 0.39 μg/ml) were added. After incubation for 48 and
24 h, the OD values were measured using a microplate reader
(PerkinElmer, Inc.) at 490 nm. The rate of cell viability was
calculated using the following equation: Cell viability
(%)=(ODsample-ODblank)/(ODcontrol-OD-blank) ×100.
Detection of apoptosis-inducing effects
of CDDP-PNA-Lip in vitro
The percentage of NSCLC cells that underwent
apoptosis after various drug treatments was analysed using the FITC
Annexin V Apoptosis Assay kit (cat. no. 556547; BD Biosciences).
A549 and H460 cells were inoculated into 6-well plates at a density
of 3×105 cells/well, cultured in complete medium for 24
h, and were treated with 2.5 μg/ml CDDP, CDDP-Lip or
CDDP-PNA-Lip for 48 h at 37°C. Subsequently, all of the cells were
collected and washed twice with precooled PBS at 4°C. The cells
were diluted to 1×106 cells/ml using 1X binding buffer,
and were then co-incubated with FITC-conjugated Annexin V and PI
(binding buffer:FITC-conjugated Annexin V/PI=20:1, v/v) staining
solution for 15 min at room temperature in the dark. Subsequently,
the incubation was terminated by the addition of 1X binding buffer,
and the cells were analysed using the BD Accuri C6 Plus Flow
Cytometer (BD Biosciences) and BD Accuri C6 Plus software (version
1.0.34.1; BD Biosciences).
Detection of antimigratory efficacy of
CDDP-PNA-Lip in vitro
The effect of CDDP, CDDP-Lip and CDDP-PNA-Lip
treatments on the lateral migration ability of NSCLC cells was
examined using a cell scratch wound healing assay. A549 and H460
cells in the logarithmic growth phase were inoculated into separate
6-well plates. Once the cells had grown to cover the entire well,
various drugs were added to each well (2.5 μg/ml, with
normal saline used as a control) and the medium was replaced with
medium containing 1% FBS. The cell surface (100% confluence) was
then scratched using a sterile 200-μl pipette tip and the
cells were incubated at 37°C for 24 h. Each scratch was imaged at 0
and 24 h using an inverted light microscope (Nikon Corporation).
The rate of cell migration was measured using ImageJ Plus (version
2.9 011.53t; National Institutes of Health).
Scratch wound healing rate (%)=(initial scratch
width-scratch width at observation)/initial scratch width ×100.
Transwell assays (6.5 mm Transwell® with
0.4 μm pore polycarbonate membrane insert; cat. no. 3413;
Corning, Inc.) were used to measure the longitudinal migration
capacity of NSCLC cells treated with different formulations. A
total of 2,000 A549 and H460 cells were separately inoculated into
Transwell chambers containing medium supplemented with 1% FBS.
Complete medium containing 10% FBS was added to the lower chamber.
After 24 h incubation at 37°C, the dead cells were washed and
removed from the inside of the Transwell chambers. Subsequently,
DAPI staining was performed as aforementioned. The cells were then
observed and images were captured using an inverted fluorescence
microscope (Motic Incorporation, Ltd.). The number of cells was
calculated using ImageJ Plus.
Targeted action of CDDP-PNA-Lip in
vivo
In the present study, the construction of a
xenograft mouse model was performed by subcutaneous injection. A
total of 29 male nude mice (age, 4-6 weeks; weight, 15±2 g) were
used in the present study; 9 mice underwent this experiment and 20
mice underwent the subsequent experiment. The mice were maintained
in a specific pathogen-free environment at a temperature of 20-26°C
and humidity of 40-70%. The mice were provided free access to
sterile food and water and were maintained under a 12-h light-dark
cycle. The humane endpoints were as follows: Tumour volume reaching
10% of body weight of the mouse; the tumour severely affected the
mobility or quality of life of the mouse; or if the mouse showed
signs of significant pain and distress. Each nude mouse was
subcutaneously injected with 5×106 A549 cells diluted
with 0.2 ml PBS in the left hind limb. After the tumours were
considered palpable, the tumour volume was measured every 2 days.
The subcutaneous tumour volume was close to 200 mm3 when
mice with comparable tumour volumes and body weights were selected
and randomly divided into three groups [Cy7, Cy7-Lip, Cy7-loaded
PNA-modified liposomes (Cy7-PNA-Lip); n=3/group]. Cy7 instead of
CDDP was encapsulated into liposomes and the position of Cy7 was
reflected by small animal imaging. Cy7-Lip and Cy7-PNA-Lip were
prepared to mimic the distribution of CDDP in tumour-bearing nude
mice. The mice were injected with 10 mg/kg Cy7, Cy7-Lip or
Cy7-PNA-Lip via tail vein within 15 min. Fluorescence images of Cy7
in tumour-bearing nude mice were acquired and analysed using the
IVIS Spectrum (PerkinElmer, Inc.) after mice were anaesthetized by
inhalation using isoflurane at 2, 4, 8, 12, 24 and 48 h
post-injection. During the induction phase, the isoflurane
concentration was set at 4%, and during maintenance anaesthesia,
the isoflurane concentration was reduced to 2% to maintain stable
anaesthesia while reducing the effects on the physiological
functions of the mice. The anaesthetic gas was dispersed through
oxygen to ensure that the mice received an adequate supply of
oxygen throughout the process. All experimental animals were
euthanized by CO2 inhalation 48 h after injection
(volumetric emission rate of 4 l/min; ~40% container volume/min) to
detect the distribution of Cy7 in major organs and tumour sites in
nude mice. Fluorescence images of Cy7 were then acquired and
analysed using the IVIS Spectrum Small Animal Live Imaging System
and the accompanying software Living Image (version 4.7.2.20319;
PerkinElmer, Inc.).
Antitumour efficacy of CDDP-PNA-Lip in
vivo
The present study investigated the in vivo
antitumour activity of CDDP-PNA-Lip by treating nude mice with
subcutaneous tumours. An animal model of subcutaneous
tumour-bearing nude mice was established using male BALB/C-nu nude
mice (age, 4-6 weeks; weight, 15±2 g). The nude mice were housed
for 1 week in a specific pathogen-free grade environment at a
temperature of 20-26°C and, humidity of 40-70%. The mice were
provided free access to sterile food and water and were maintained
under a 12-h light-dark cycle. Each nude mouse was injected
subcutaneously with 5×106 A549 cells diluted with 0.2 ml
PBS in the left hind limb, and the tumour volume and body weight of
the mice were measured every 2 days. A total of 14 days after the
subcutaneous injection, the subcutaneous tumour volume in the mice
approached 100 mm3. The mice were then randomly divided
into four groups (n=5/group) and administered the following
solutions: Saline, CDDP saline solution, CDDP-Lip saline solution
or CDDP-PNA-Lip saline solution via intraperitoneal injection. All
four solutions were administered at a dose of 2 mg/kg every 4 days
for a total of 24 days. Tumour volume and body weight were measured
every 2 days. After the length and width of the tumour were
measured using digital Vernier callipers, the tumour volume was
estimated using the following formula: Tumour volume=(tumour
length) × (tumour width)2 ×0.5. Tumour growth inhibition
was measured using the following formula: Tumour growth inhibition
value (%)=[1-RTV (experimental group)/RTV (control group)] ×100,
where RTV indicates relative tumour volume.
After 24 days, the animals were euthanized by
CO2 inhalation, blood was collected from the heart, and
the tumours, liver, kidney, heart, spleen and lungs were also
collected. The tissues were weighed, fixed in 4% paraformaldehyde
at room temperature for 6 h and processed. The blood was treated
with sodium heparin and centrifuged at 500 × g and 4°C for 10 min
to extract the serum. All organs and tumour tissues were embedded
in paraffin for haematoxylin and eosin (H&E) staining. Sections
(6 μm) were stained using haematoxylin for 7 min at room
temperature and eosin for 1 min at room temperature and were
observed under a light microscope, while the serum was analysed for
liver and kidney function indicators. Alanine transaminase (ALT;
cat. no. H001), alkaline phosphatase (ALP; cat. no. H004), blood
urea nitrogen (BUN; cat. no. H044) and creatinine (Cr; cat. no.
H047) test kits were obtained from Weifang Kanghua Biotechnology
Co., Ltd.
Statistical analysis
The experiments were independently conducted three
times. After the data were entered into GraphPad Prism (version
8.0; Dotmatics), statistical analysis was performed using an
independent sample t-test for two groups of data or one-way ANOVA
followed by Dunnett's multiple comparisons test for three or more
groups of data. Data are presented as the mean ± SD. P<0.05 was
considered to indicate a statistically significant difference.
Results
Bioinformatics analysis of MUC1 in
clinical specimens
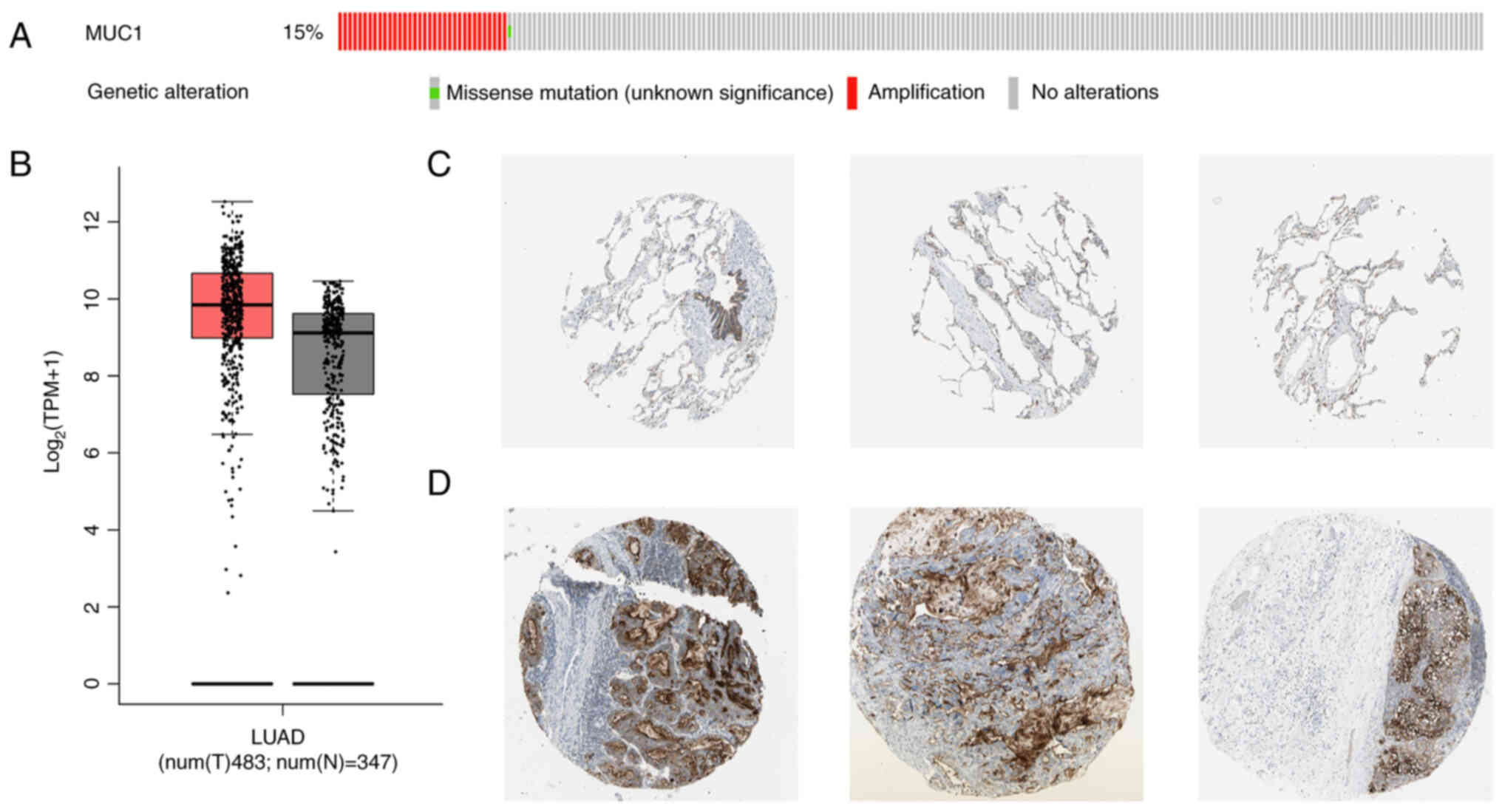
The bioinformatics analysis of NSCLC tissues
revealed that, compared with in normal tissues from patients with
lung adenocarcinoma, there was a trend towards elevated expression
levels of MUC1 in NSCLC tissues; however, this difference was not
significant (Fig. 1B). In
addition, >15% of patients with NSCLC exhibited upregulated MUC1
expression (Fig. 1A). These
findings suggested that the development of therapeutic regimens
targeting MUC1 may be important for patients with NSCLC and has
significant clinical implications. Moreover, immunohistochemical
staining results from the Human Protein Atlas database revealed
that MUC1 was widely and intensively expressed in malignant tumours
(Fig. 1C and D), which was
consistent with the results of the bioinformatics analysis, further
indicating that the present study has good clinical application
potential. The results revealed that cells with abnormally elevated
tumour MUC1 expression were mainly distributed on the tumour
surface, which facilitated the targeting of liposomes for more
precise localization at the tumour site.
Synthesis of PNA-modified DSPE
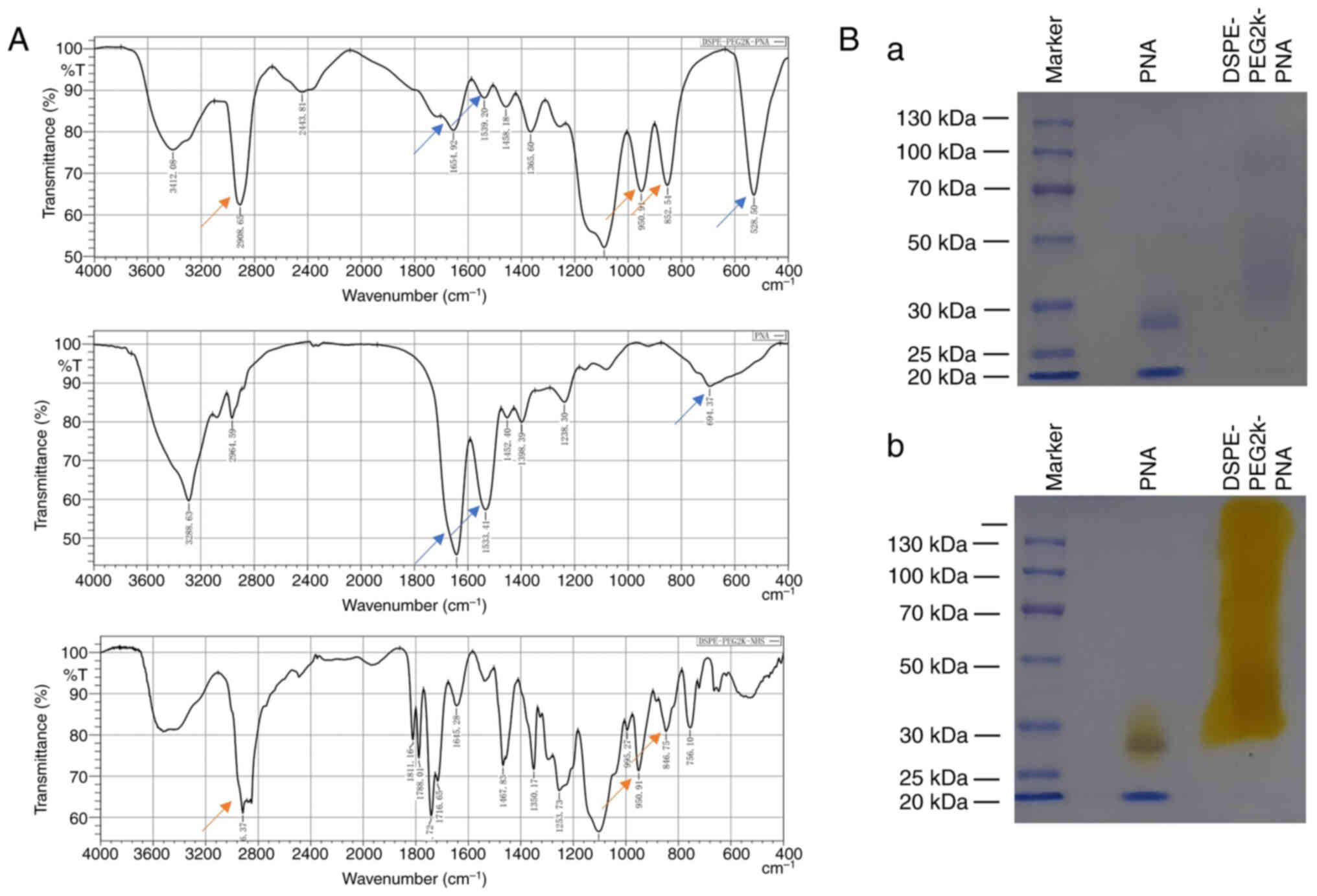
PNA-modified DSPE-PEG2K was synthesized by reacting
the active ester (-NHS) of DSPE-PEG2K-NHS with an amino group
(-NH2) on PNA to form an amide bond (-CO-NH-). The
successful synthesis of DSPE-PEG2K-PNA was verified by the
characteristic peaks of DSPE-PEG2K-PNA detected using infrared
spectroscopy and SDS-PAGE (Fig. 2A
and B). Images of PEG distribution using PEG dye (Fig. 2Ba) and Coomassie brilliant blue
staining of SDS-PAGE gels (Fig.
2Bb) are shown.
Preparation of stable liposomes and
CDDP-Lip
As shown in Table
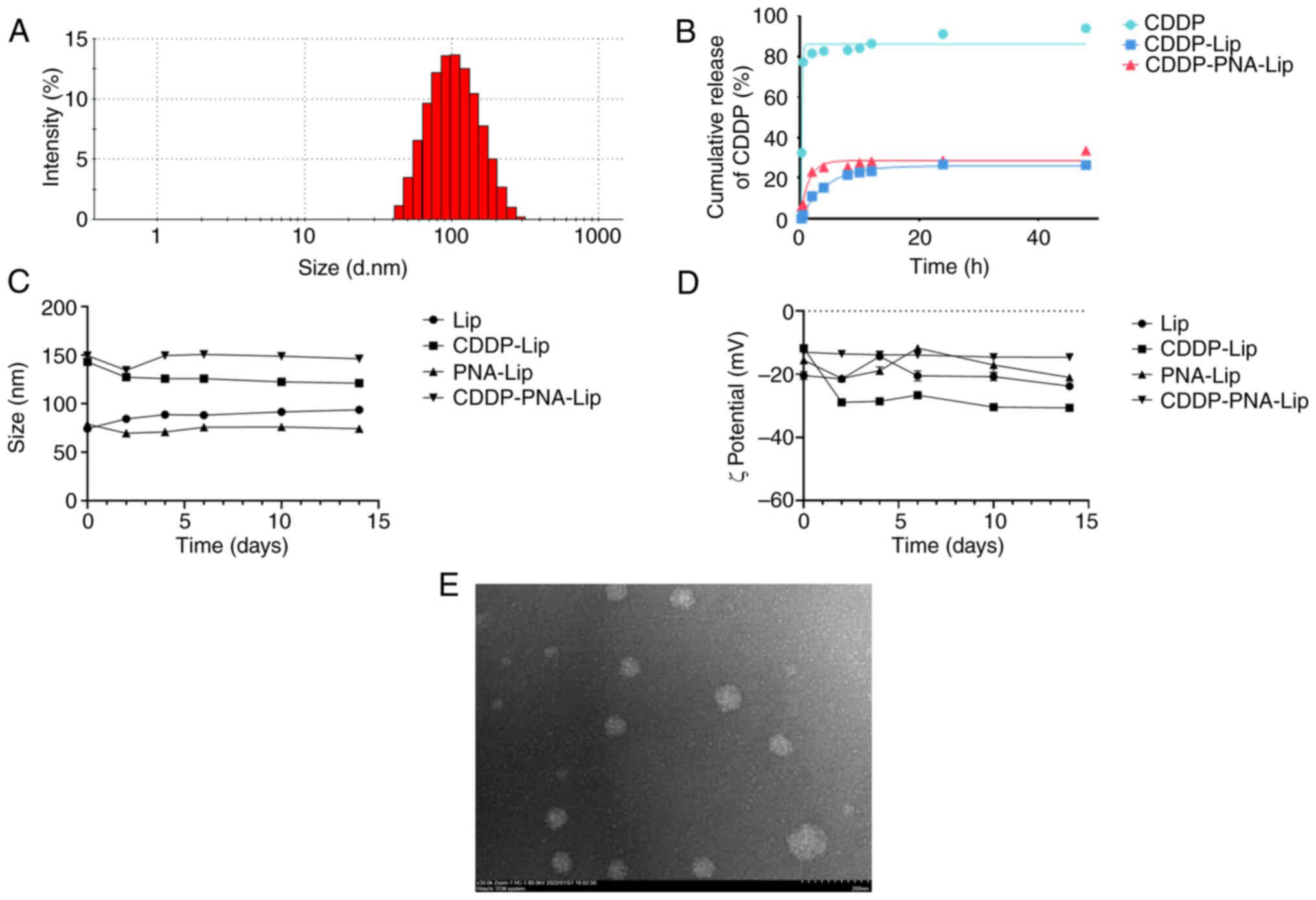
I and Fig. 3A, the prepared
CDDP-PNA-Lip were heterogeneous and stable, and could effectively
encapsulate CDDP. The particle sizes of Lip and PNA-Lip were ~80
nm, while the particle size of the liposomes increased to ~120 nm
after encapsulating CDDP. This increase in size is likely due to
the electrostatic force of some of the CDDP molecules on the
surface of the liposomes. The polydispersity index of each liposome
was ~0.2, which represented good lipid homogeneity. The ζ potential
of CDDP-Lip was ~−30 mV, and the encapsulation of CDDP did not
result in a change in the surface charge. This finding is
consistent with the findings of other protocols for DL that rely on
electrostatic forces of adsorption (24). The EE was >35%, and the DL
capacity was greater than 3%. This DL capacity was greater than
that of reported CDDP-Lip (32),
which achieved only a 1% DL capacity. Since the DL of CDDP was
achieved through electrostatic adsorption of liposomes, it led to
an increase in the particle size of liposomes encapsulating CDDP
compared to those that were not encapsulated. The morphology of the
CDDP-PNA-Lip was observed using a TEM. The results indicated that
the PNA-CDDP-Lip were uniform three-dimensional spheres with smooth
surfaces and particle sizes (Fig.
3E).
 | Table ICharacterization of different types
of liposomes. |
Table I
Characterization of different types
of liposomes.
| Liposome type | Size, nm | PdI | ζ, mV | EE, % | DL, % |
|---|
| Lip | 81.4±0.77 | 0.258±0.01 | -33.7±1.15 | - | - |
| PNA-Lip | 83.5±1.92 | 0.227±0.01 | -30.1±1.19 | - | - |
| CDDP-Lip | 114.1±3.04 | 0.214±0.02 | -31.4±0.72 | 35.4±0.24 | 3.13±0.02 |
| CDDP-PNA-Lip | 114.2±1.03 | 0.203±0.03 | -30.7±0.74 | 35.9±0.14 | 2.91±0.01 |
The in vitro dialysis experiments were
performed using dialysis bags, and the in vitro release
profile of CDDP-Lip is shown in Fig.
3B. Depending on the release rate, the cumulative release of
CDDP from unmodified liposomes exhibited two distinct phases, i.e.,
a rapid release phase and a sustained release phase. The rapid
release phase lasted for ~2 h, during which ~20% of the CDDP was
released from the liposomes; this phase was followed by a sustained
release phase, during which CDDP continued to be slowly released;
the cumulative release of the drug was ~35% after 48 h of the
experiment. Although the cumulative release of CDDP from the
modified liposomes also showed two phases, its release profile was
smoother than that of unmodified liposomes, and it had a better
slow-release effect than that of unmodified liposomes within 12 h.
Notably, ~20% of CDDP was released from CDDP-PNA-Lip in the first 4
h, followed by a sustained release phase at a slower rate; the
first 20% seemed to be more quickly released from CDDP-PNA-Lip than
from CDDP-Lip within 4 h. It was hypothesized that this result may
be due to the addition of phospholipids containing PEG components
within the liposomes, which allowed CDDP-Lip and CDDP-PNA-Lip to
exhibit a sustained and stable release during the experiment. The
results suggested that encapsulating CDDP with liposomes may
prolong the release time of the drug, which is beneficial for
achieving sustained release at the tumour target (Fig. 3B).
All the liposomes were diluted 10-fold with
ddH2O and stored at 4°C in the dark. The particle size
and potential of the liposomes were monitored every 2 days, and the
corresponding change curves are presented in Fig. 3C and D. The particle size of all
the liposomes fluctuated within a range of ±10 nm and did not
change significantly across 15 days. Similarly, the potential did
not vary significantly, indicating that the liposomes prepared in
the present study were stable.
Determination of MUC1 expression in
cells
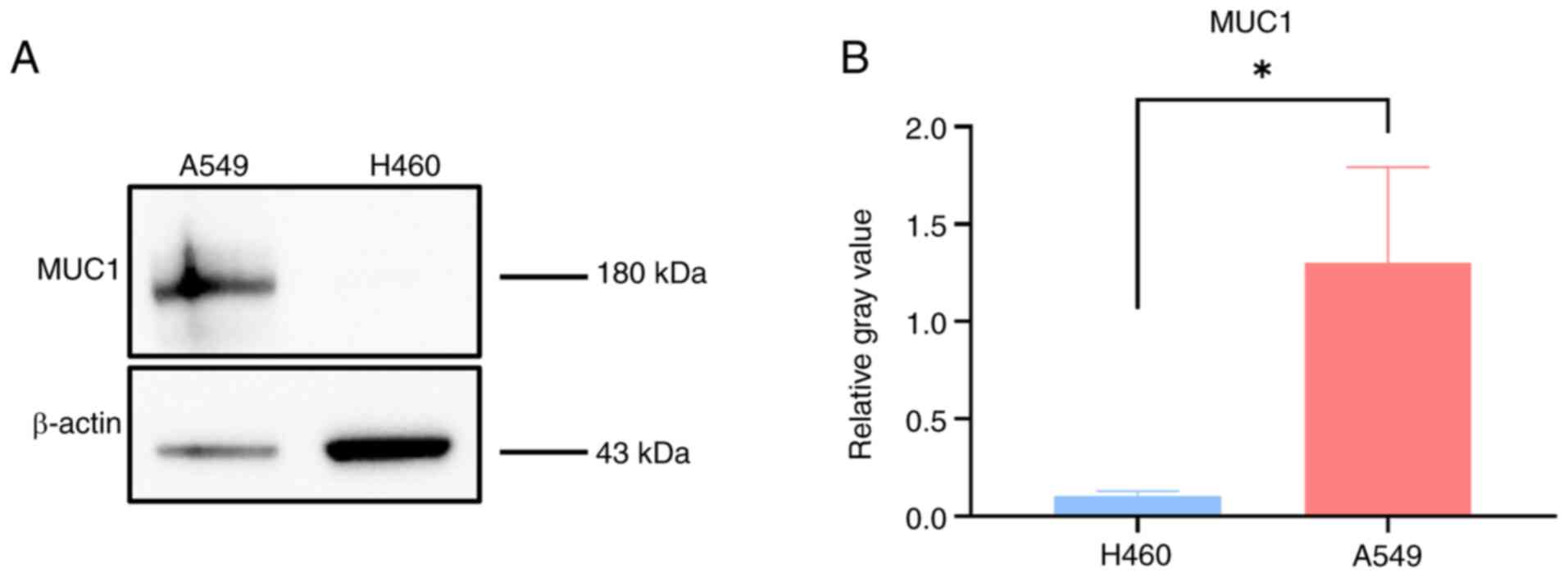
Western blotting was used to detect the expression
levels of MUC1 in the A549 and H460 cell lines. The grayscale
analysis revealed that MUC1 was expressed at significantly higher
levels in A549 cells than in H460 cells (Fig. 4A and B).
PNA-Lip enhance the delivery efficiency
to MUC1-positive NSCLC cells
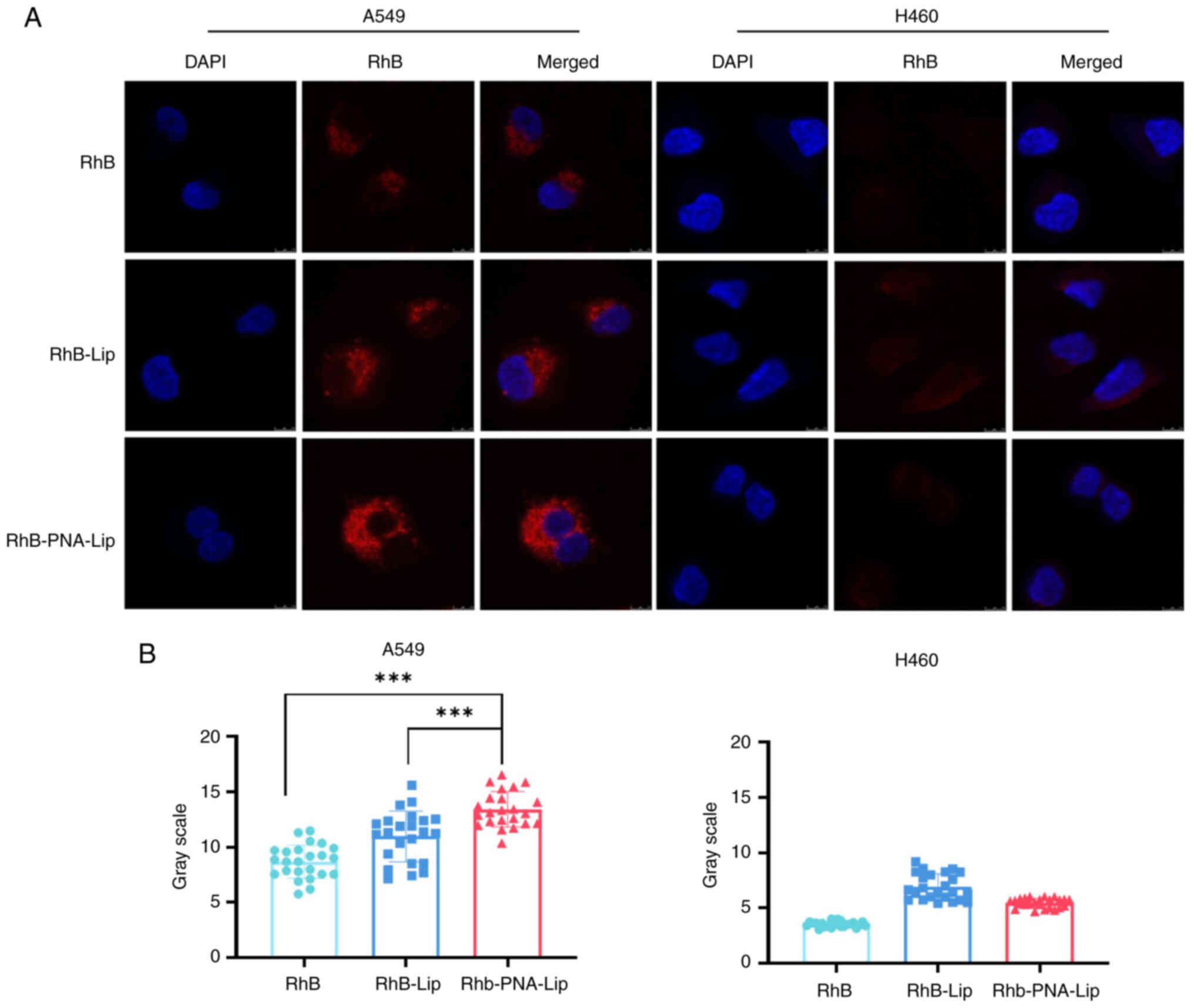
The effect of ligand modification on improving the
liposomal delivery efficiency was explored by performing in
vitro cellular uptake assays (Fig. 5). The MUC1-negative NSCLC cell
line used in the experiment was H460, and the MUC1-positive NSCLC
cell line was A549. The impact of the ligand modification on
enhancing the efficiency of liposomal drug delivery was studied by
examining the cellular uptake of each liposome type using confocal
laser scanning microscopy (CLSM). The uptake of each type of RhB
liposome was assessed in A549 and H460 cells after 6 h. As shown in
Fig. 5A and B, CLSM revealed the
strongest red fluorescence in the A549 cell line treated with
RhB-PNA-Lip. The quantitative analysis showed that the intensities
of red fluorescence in the RhB-PNA-Lip group were 1.53 and
1.24-fold greater than those in the RhB and RhB-Lip groups,
respectively. These results indicated that the PNA modification
enhanced the uptake of liposomes by A549 cells. By contrast,
RhB-PNA-Lip did not exhibit a stronger fluorescence intensity in
the H460 cell line. These findings suggested that the modification
of liposomes with PNA significantly improved their targeting effect
on MUC1-positive NSCLC cells, such as A549 cells.
PNA-Lip exhibit enhanced antitumour
efficacy in MUC1-positive NSCLC cells in vitro
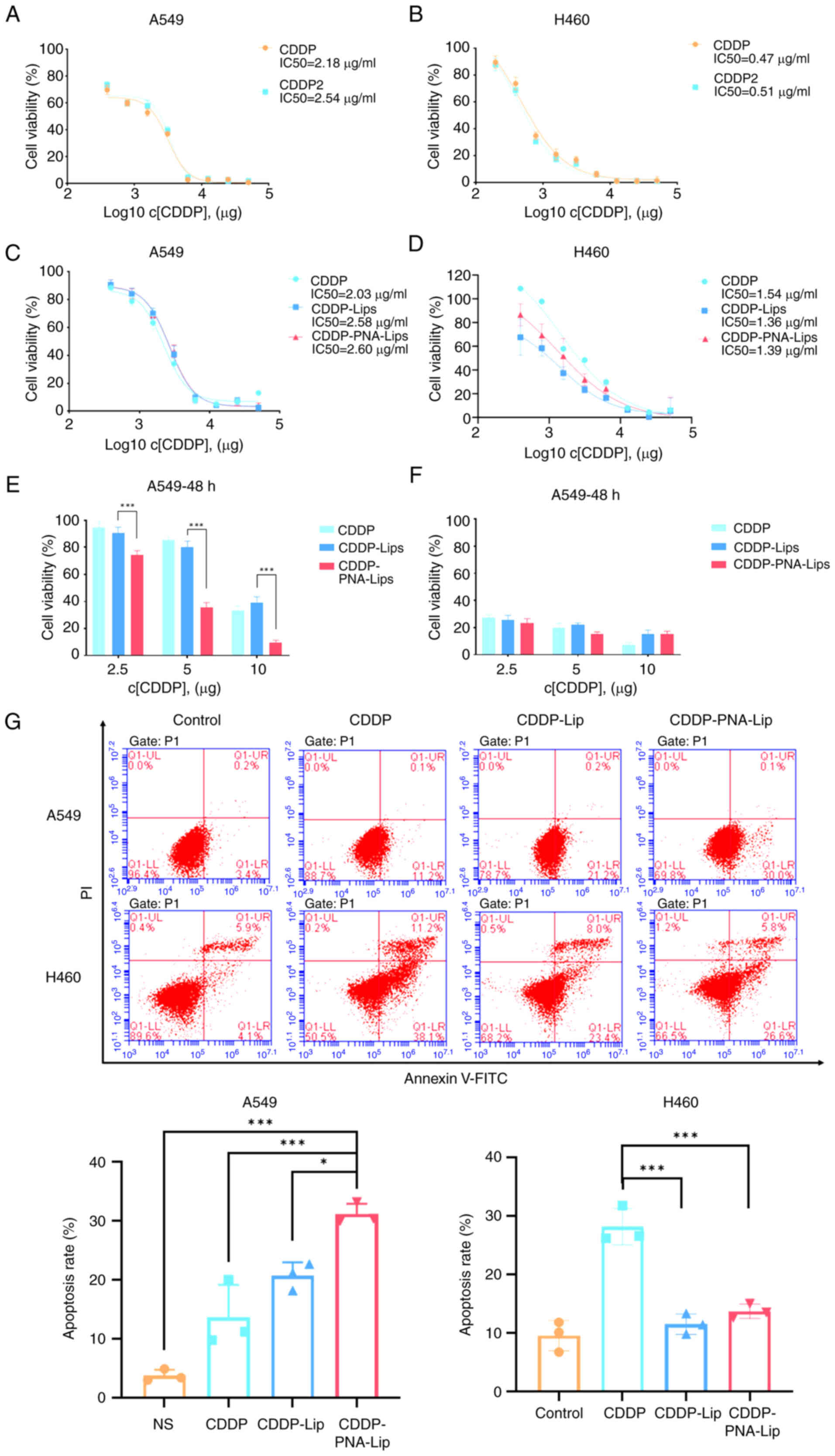
Investigations into the cytotoxic efficiency of
ligand-modified liposomes were conducted using the MTS assay to
determine the viability of A549 and H460 cells treated with
CDDP-Lip. The MTS assay results showed no significant difference in
cytotoxicity between CDDP and hydrated CDDP in the two cell lines
(Fig. 6A and B), which was
previously described by Lippard (31), where hydrated CDDP regenerated
CDDP in the presence of Cl−, which did not affect its
antitumour activity. After treating A549 and H460 cells with CDDP,
CDDP-Lip and CDDP-PNA-Lip for different durations, it was revealed
that CDDP had a dose-dependent antitumour effect. After 48 h of
drug treatment, the A549 cells in the CDDP-PNA-Lip treatment group
exhibited the lowest cellular viability, which was not observed in
the three treatment groups of H460 cells (Fig. 6C-F). This finding suggested that
the ligand-modified liposomes may have greater delivery efficiency
and stronger cytotoxicity against MUC1-positive NSCLC cell
lines.
The results of the MTS experiments prompted the
evaluation of the apoptosis of A549 and H460 cells treated for 48 h
with 2.5 μg/ml CDDP and liposomes. The levels of Annexin V
on the cell surface were detected using flow cytometry. The results
revealed no significant difference in the effects of CDDP-Lip or
CDDP-PNA-Lip on early apoptosis in MUC1-negative H460 cells
(Fig. 6G). For MUC1-positive
A549 cells, the percentage of apoptotic cells in the CDDP-PNA-Lip
group was 1.5 times higher than that in the CDDP-Lip group
(Fig. 6G). These findings
suggested that modification of CDDP-Lip with PNA may induce the
apoptosis of MUC1-positive NSCLC cells.
Notably, the utilization of liposomes to encapsulate
CDDP seems to diminish its antitumour efficacy on H460 cells. It
may be hypothesized that this is because liposomes exhibit a
sustained-release effect on the drug, and cells uptake liposomes
with varying efficiency. This phenomenon was also observed by Diao
et al (33).
PNA-Lip enhance the antimigratory
efficacy of MUC1-positive NSCLC cells in vitro
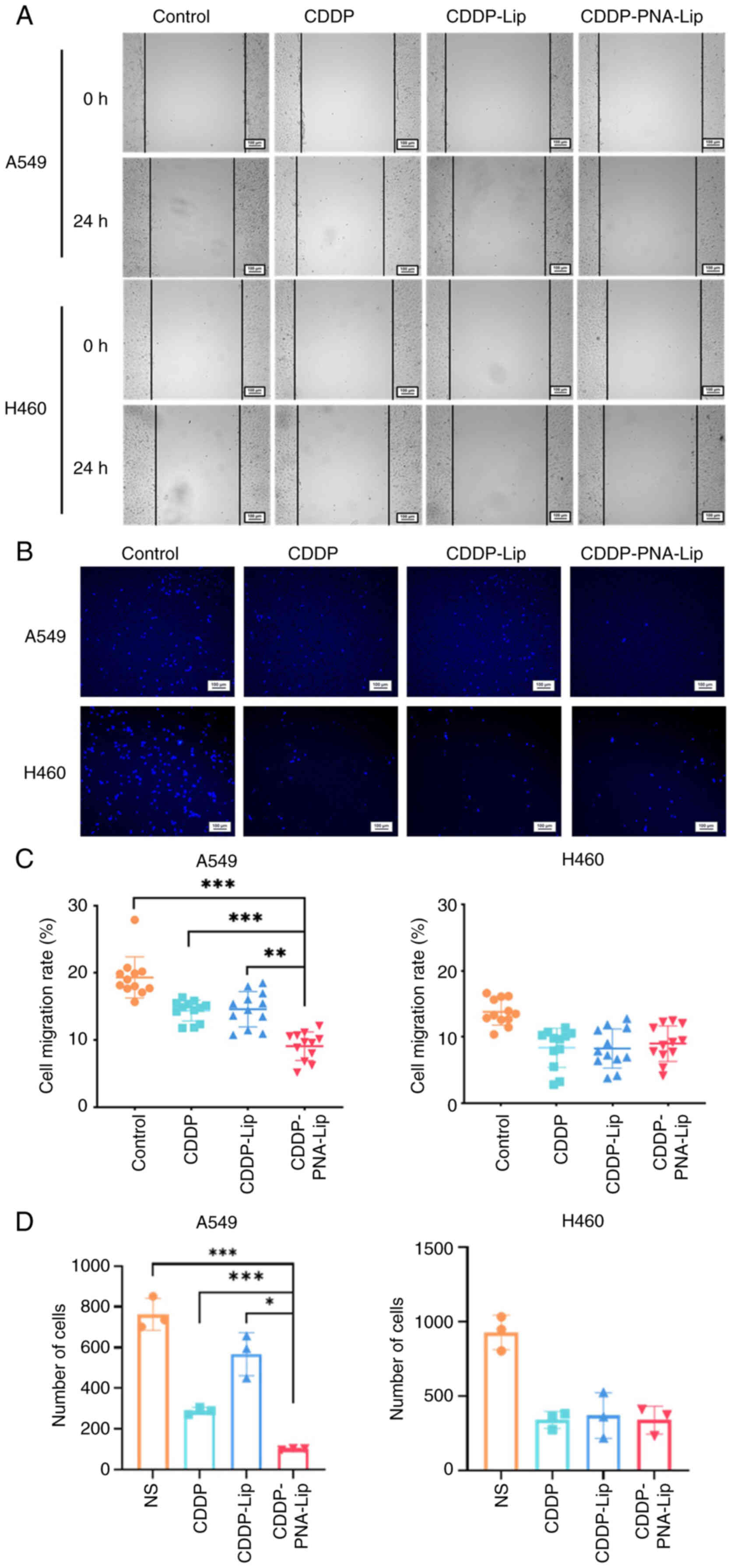
Cell scratch wound assays and Transwell assays were
used to verify the lateral and longitudinal migration abilities of
the different PNA-modified liposomes. For the cell scratch wound
assay, normal saline, free CDDP, CDDP-Lip or CDDP-PNA-Lip were
added to the scratched cells in 6-well plates, which were
subsequently cocultured with the cells for 24 h. Images of the
cells were captured at 0 and 24 h. The scratch wound healing rate
of the A549 cells in the normal saline group was 19.32±2.93%,
whereas that in the CDDP-PNA-Lip group was the smallest, with a
scratch wound healing rate of 9.08±2.03% (Fig. 7A and C). These rates were
significantly lower than those in the CDDP group and CDDP-Lip group
(14.38±1.48 and 14.59±2.51%, respectively). The healing rate of the
saline group (13.77±1.89%) was notably higher than that of the free
CDDP, CDDP-Lip and CDDP-PNA-Lip groups (8.36±2.81, 8.26±2.83 and
9.00±2.58%, respectively) in A549 cells; however, no significant
differences were observed among the drug treatment groups in H460
cells (Fig. 7A and C).
In the Transwell assay, the cells were cultured for
24 h, stained with DAPI and images were captured using an inverted
fluorescence microscope. The number of A549 cells treated with
CDDP-PNA-Lip was the lowest in the field of view and was
significantly lower than that in the other groups; however, no
significant differences were observed in H460 cells (Fig. 7B and D). These results indicated
that the ability of liposomes to inhibit tumour cell migration was
enhanced when they were modified with PNA.
PNA-Lip exhibit enhanced tumour targeting
in a xenograft mouse model
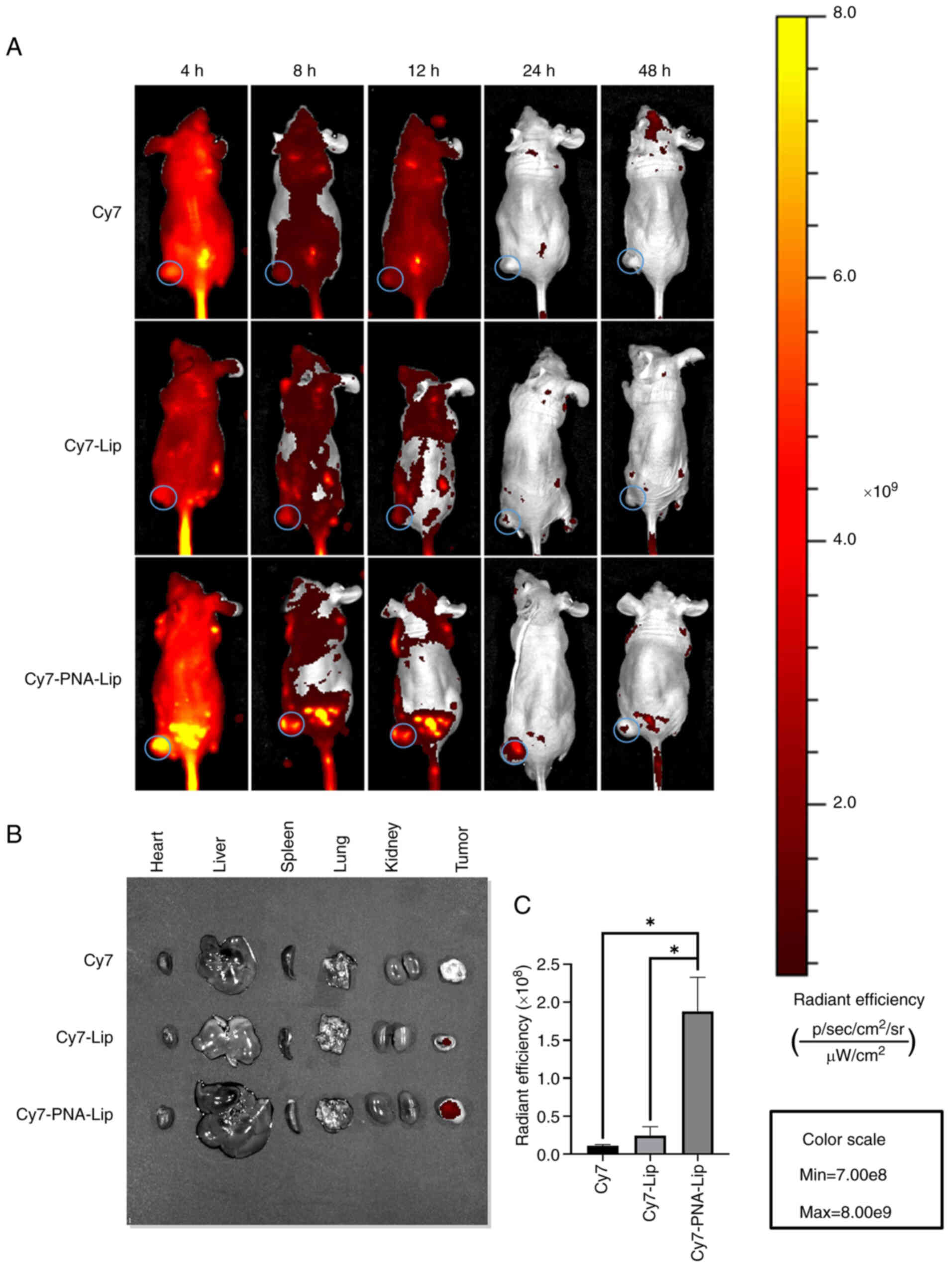
The effects of the ligand modification on enhancing
target acquisition in vivo were investigated by monitoring
animals with near-infrared fluorophore imaging. Cy7 was
encapsulated in liposomes to mimic the circulation of CDDP in nude
mice. Cy7, Cy7-Lip and Cy7-PNA-Lip were then injected into the nude
mice via the tail vein. A total of 4 h after injection, Cy7-PNA-Lip
began to distribute predominantly to the tumour site in nude mice
and was most evident at 8 h. Over time, Cy7 was metabolized and
excreted from the nude mice. The fluorescent signal in the bodies
of the mice gradually weakened, and all residual fluorescence
disappeared completely after 24 h (Fig. 8A). Observations of mice injected
with Cy7-PNA-Lip revealed that the fluorescent signal at the tumour
site was markedly stronger than that in mice injected with Cy7 or
Cy7-Lip. This high signal intensity persisted for 48 h. Before the
nude mice were euthanized, the mice injected with Cy7-PNA-Lip still
exhibited notably greater fluorescent signals at the tumour site
than the other two groups (Fig.
8A). Subsequently, the mice were euthanized, fluorescence
imaging was conducted, and an analysis of their major organs and
tumour sites was performed. The results revealed that the tumours
of nude mice injected with Cy7-PNA-Lip still exhibited strong
fluorescent signals even after 48 h (Fig. 8B and C). Furthermore, due to the
absence of an epidermal barrier, the fluorescence intensity at the
tumour sites was significantly higher in the Cy7-PNA-Lip-injected
nude mice than in the other two groups. These results indicated
that the in vivo targeting ability of PNA-Lip aligns with
the results of cellular uptake. Furthermore, the PNA modification
of liposomes may enable them to actively target tumours. In
addition, based on the change in the fluorescence intensity of Cy7
within the tumours of Balb/c-nu mice, the modification of liposomes
with PNA extends the duration of drug presence in tumours and
enhances drug accumulation at the tumour site. It was thus
hypothesized that the utilization of PEG and PNA may enhance the
active targeting ability of a drug and prolong its presence at the
tumour site. These characteristics, in turn, could reduce the
required drug dosage and frequency of administration.
PNA-Lip exhibit enhanced antitumour
efficacy in a xenograft mouse model
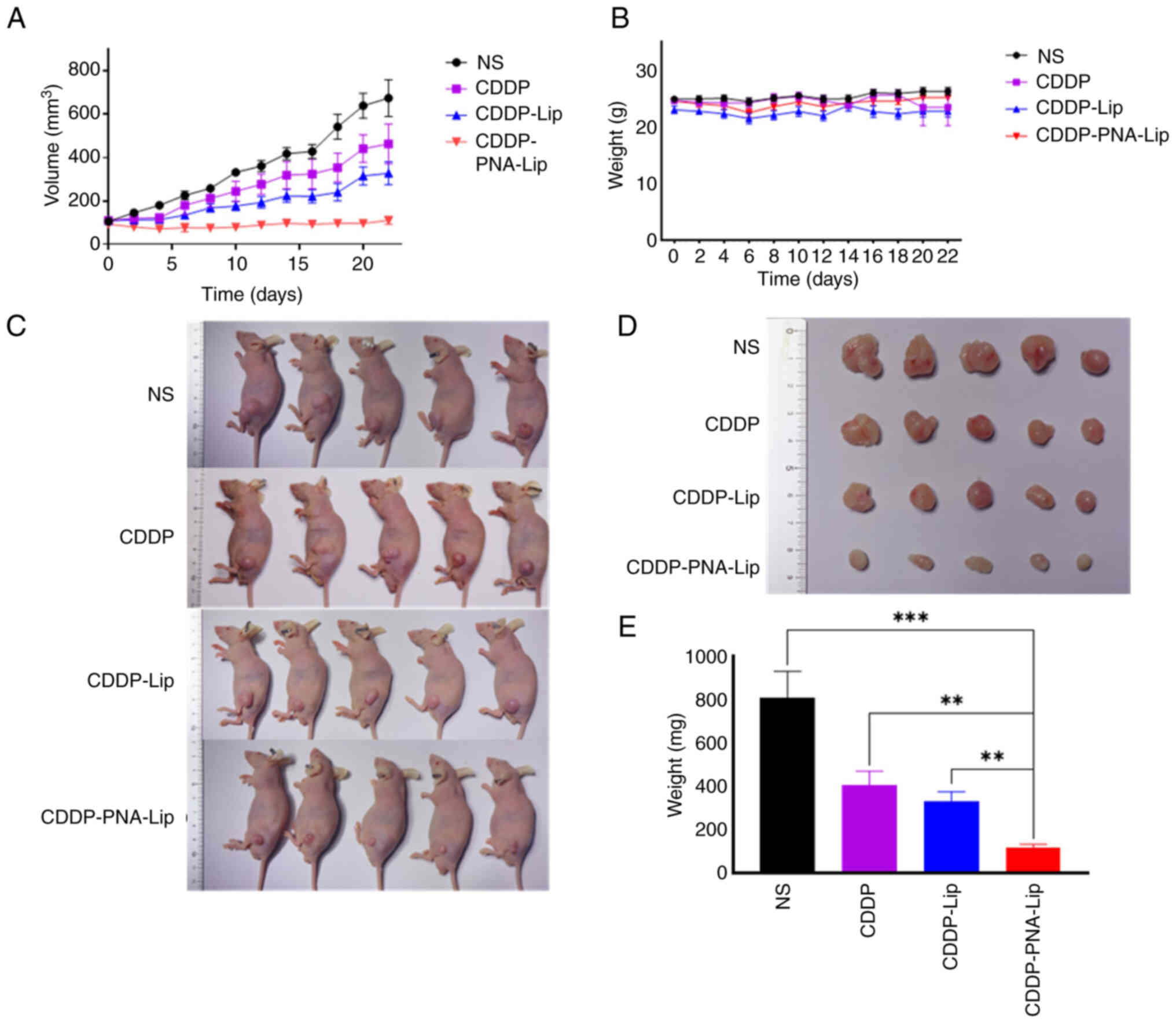
The in vivo antitumour efficacy of
CDDP-PNA-Lip was evaluated by treating nude mice bearing
subcutaneous tumours with saline, CDDP, CDDP-Lip or CDDP-PNA-Lip.
By monitoring and analysing the body weights of the animals, no
significant weight loss was observed in any of the four groups
after drug administration (Fig.
9B). This finding suggested that the drug treatment had few
toxic side effects. After the initial treatment (day 0; Fig. 9A), the growth of the tumour
tissue stopped in all groups of subcutaneous tumour-bearing nude
mice. However, as the number of doses increased (from day 4;
Fig. 9A), the tumour tissue in
the subcutaneous tumour-bearing nude mice treated with CDDP and
CDDP-Lip continued to grow, whereas that in the mice treated with
CDDP-PNA-Lip showed sustained tumour suppression. The results
indicated that CDDP-PNA-Lip exerted the strongest antitumour
effect, with a tumour inhibition rate of 83.1%. The tumour volume
of the mice treated with CDDP was 2.65 times larger than that of
the mice treated with CDDP-PNA-Lip, while the tumour volume of the
mice treated with CDDP-Lip was 1.63 times larger than that of the
mice treated with CDDP-PNA-Lip. At the end of the treatment, all
mice were sacrificed and the tumours were weighed, the results
showed that mice treated with CDDP-PNA-Lip had the smallest tumour
weights, which were significantly different compared with the other
groups (Fig. 9C-E). H&E
staining revealed that the tumour tissues in the saline control
group were densely packed, whereas those in the CDDP-PNA-Lip group
exhibited the highest levels of apoptosis and necrosis. A large
number of vacuoles appeared between the tissues, indicating the
significant antitumour efficacy of CDDP-PNA-Lip (Fig. 10A). The modification of
liposomes with PNA significantly enhanced the antitumour efficacy
of the liposomes against NSCLC tissues with high MUC1
expression.
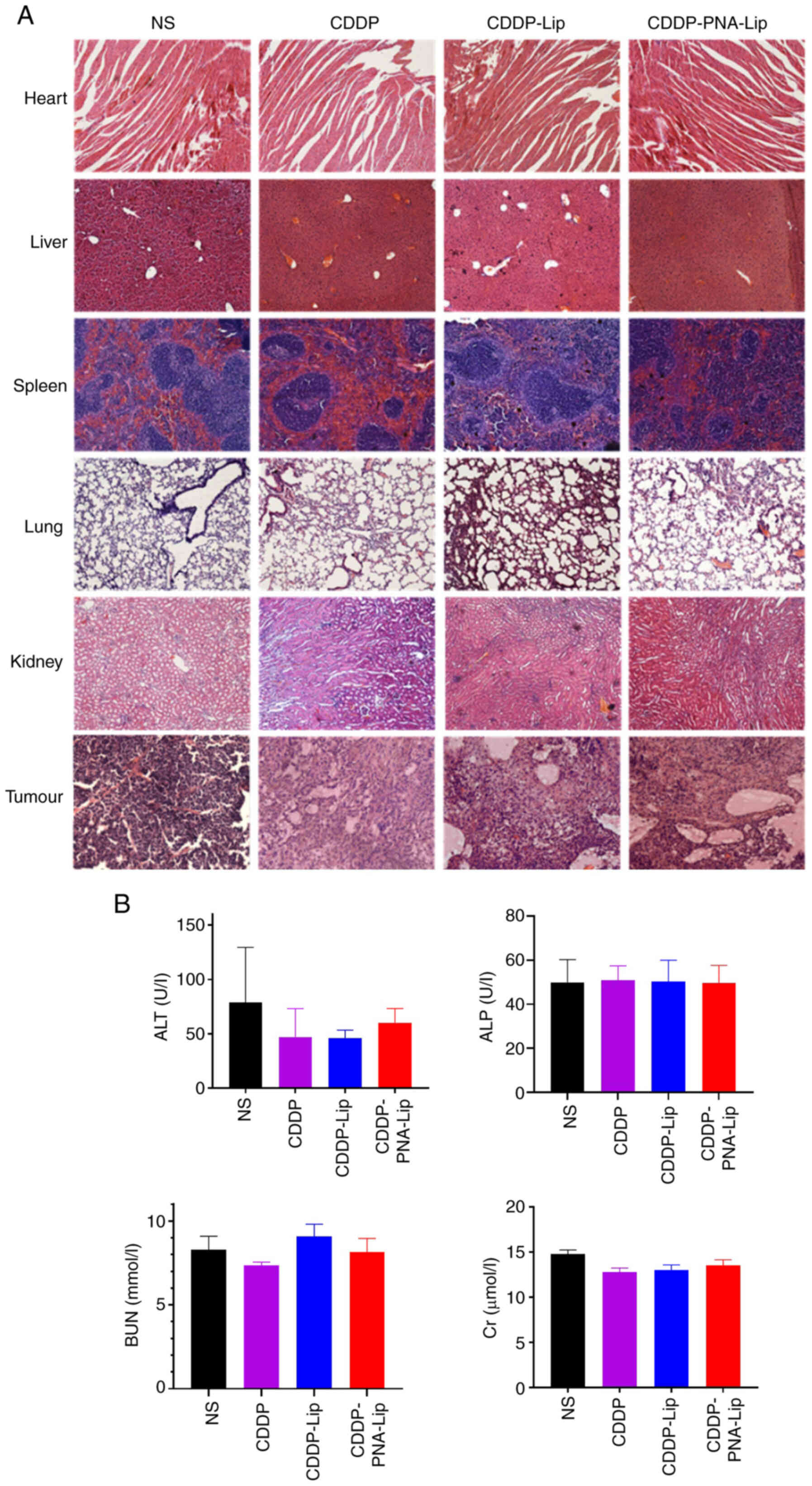 | Figure 10A xenograft mouse model shows that
PNA-modified liposomes have no systemic toxicity in vivo.
(A) Haematoxylin and eosin staining of vital organs and tumours
(magnification, ×20). (B) Analysis of physiological indices of the
liver and kidney. ALP, alkaline phosphatase; ALT, alanine
transaminase; BUN, blood urea nitrogen; CDDP, cisplatin; Cr,
creatinine; NS, normal saline; PNA, peanut agglutinin; CDDP-Lip,
CDDP-loaded liposomes; CDDP-PNA-Lip, CDDP-loaded PNA-modified
liposomes. |
PNA-Lip improve the toxicity and side
effects of CDDP in vivo
The biosafety and antitumour effects of CDDP-PNA-Lip
were evaluated using H&E staining. After histological
sectioning and staining of the heart, liver, spleen, lungs and
kidneys, no histopathological abnormalities were found in any of
the four groups of animals (Fig.
10A). The effect of CDDP-PNA-Lip on liver and kidney function
in nude mice was evaluated by measuring physiological indicators of
liver and kidney function in serum (Fig. 10B). The results showed that
biochemical indices, such as Cr, ALT, ALP and BUN, were not
significantly altered in any of the groups. These results indicated
that liposomes modified with PNA and containing CDDP colloids do
not cause significant systemic toxicity in experimental mice.
Discussion
Lung cancer ranks second in global cancer incidence
and first in mortality, and is therefore considered one of the
greatest threats to human health (34). In addition, primary treatments
for lung cancer, such as surgery, radiotherapy or chemotherapy,
carry a significant risk of recurrence. Based on the
histopathological classification, lung cancer can be divided into
two groups: Small cell lung cancer and NSCLC (34,35). Among lung cancer types, NSCLC has
a high incidence rate, accounting for 80-85% of all lung cancer
cases, and is particularly difficult to cure (36). Among the therapeutic options for
NSCLC, CDDP, a broad-spectrum anticancer drug, has shown efficacy
as a first-line treatment for lung cancer (37,38). However, due to the evident
nephrotoxicity of CDDP, its clinical application has been somewhat
restricted (7). It has been
shown that the use of liposome-encapsulated drugs not only
decreases their toxic side effects but also extends the duration of
drug circulation in the body. Due to the ease of modifying the
liposome surface, the binding of ligands to the liposome surface
can enable the liposome to acquire an active targeting ability.
This property, in turn, reduces nonspecific drug release from
liposomes (39).
Based on previous research, it was hypothesized that
using liposomes to encapsulate CDDP and modification of their
surface is a feasible option for NSCLC treatment. Modification of
liposomes with PNA enables specific active targeting of tumour
tissues in NSCLC with high expression of MUC1 (23,33). The surface of MUC1 expressed in
normal tissues is highly glycosylated due to the properties of
MUC1, which makes it untargeted. As observed in the present animal
experiments, fluorescence did not accumulate outside the tumour
site in the Cy7-PNA-Lip group, and no marked damage was observed in
the major organs of the animals in the CDDP-PNA-Lip group. This
finding suggested that targeted therapy against tumour MUC1 may
have promising applications. Notably, in a study by Lozano et
al (40), the utilization of
the MUC1 monoclonal antibody hCTM01 to modify PGE-conjugated
liposomes for the smooth encapsulation of antitumour drugs resulted
in marked antitumour effects both in vivo and ex
vivo. Furthermore, the efficacy of antitumour therapy can be
enhanced by combining the targeting of MUC1 with other targeting
sites. This combination was reported in a study by Kim et al
(41), where co-targeting of
CD44 and MUC1 was achieved through dual aptamer modification of
liposomes, leading to targeted killing effects on tumour stem cells
and tumour cells. In addition, targeting MUC1 via antibody-drug
conjugates (ADCs) has been suggested as another effective method of
antibody therapy. In a study by Panchamoorthy et al
(42), a monomethyl auristatin
E-conjugated MUC1-C monoclonal antibody ADC was constructed and was
shown to achieve targeted killing of MUC1-C-overexpressing tumours
in vitro and in vivo. Ranjbar-Navazi et al
(43) constructed a nanosystem
consisting of a MUC1 aptamer and nanohydrogels and quantum dots,
and achieved effective loading of paclitaxel and sodium oxalate. In
in vitro studies, this nanosystem was able to target MCF-7
breast cancer cells and significantly induced mitochondria-mediated
apoptosis.
For liposomal encapsulation, the solubility of the
drug is one of the key factors that affects the encapsulation
efficiency. The present study used the hydration reaction of CDDP
to pretreat the drug and create a positively charged surface.
Moreover, DPPG, a commonly functionalized phospholipid, was added
to the liposomes to create a negatively charged surface. Hydrated
CDDP can be efficiently carried through electrostatic adsorption.
By combining the aforementioned techniques with our research on
liposome preparation, the stable encapsulation and targeted
delivery of CDDP was successfully achieved. Compared with the DL
capacity of SPI-077 (a class of liposomes coated with CDDP), the
liposomes created in the present study achieved a significant
increase in the DL capacity of CDDP (3%), while also enabling
active targeting to tumour sites in animals (11,44). These results were validated in
both in vivo and in vitro experiments. Although the
apoptosis-inducing efficiency of CDDP decreased after encapsulation
with PNA-Lip in in vitro experimental studies on H460 cells,
this phenomenon may be attributed to the variance in cellular
uptake of liposomes and CDDP. The cellular uptake of the free drug
is primarily influenced by drug concentration; however, in the
present in vivo experiments, small-animal imaging showed
that liposome-encapsulated drugs accumulated more effectively at
the tumour site. Consequently, the drug concentration at the tumour
site was notably higher than that of the free drug until the drug
was fully metabolized, thereby not affecting the application of
CDDP. Moreover, the liposomes may be further optimized by using
alternative lipid compositions, surface modifications or
encapsulation techniques, and these could be investigated in the
future to enhance the DL capacity, stability and controlled release
properties.
In addition, the present cytological studies
revealed that CDDP-PNA-Lip had significant antitumour effects after
only 48 h of treatment, even at low concentrations. It was
speculated that this phenomenon may be related to the antitumour
mechanism of CDDP, which is activated by replacing one of the
chlorine ligands with a water ligand once the agent enters the cell
because water is a better leaving group than chlorine (the core of
CDDP prefers to bind to chlorine), and because the concentration of
chlorine ions in the cytoplasm is relatively low (4-20 mM). The
pretreatment approach adopted in the present study resulted in the
activation of liposome-encapsulated CDDP, which could exert its
antitumour effect faster and more efficiently compared to direct
CDDP use. Moreover, after an intravenous injection of CDDP in
sterile saline via the conventional mode of administration, 65-95%
of CDDP can bind to plasma proteins and lead to its inactivation
within 24 h of administration due to the relatively high
concentration of chloride (100 nM) in the blood (45). The present study was able to
protect CDDP from high chloride concentrations and plasma proteins,
and improve drug utilization after its encapsulation in liposomes.
In the experiments using mice, the CDDP-PNA-Lip combination had a
tumour inhibition rate of 83.1% when only 2.5 mg/kg CDDP was used
(~50% of the standard dose), which was 2.65 times higher than that
of the CDDP group and 1.63 times greater than that of the CDDP-Lip
group. Compared with other studies, a stronger tumour-suppressive
effect was achieved using only 50% of the CDDP dose in the same
time period.
In the in vitro experiments, A549 cells
showed an increase in apoptotic rate in response to CDDP-PNA-Lip
compared with CDDP and CDDP-Lip; however, H460 cells showed a
decrease in apoptotic rate in response to CDDP-Lip and CDDP-PNA-Lip
compared with CDDP. This difference may be attributed to the
variances in cellular uptake mechanisms of free and liposomal
drugs. Free CDDP is primarily absorbed by cells through passive
diffusion, which is greatly influenced by the drug concentration.
By contrast, as shown in our previous study (23), the liposomes were internalized
via receptor-mediated endocytosis. However, it is important to note
that different cells may demonstrate alternate uptake efficiencies.
The varying uptake efficiencies of cells for liposomal drugs may
have contributed to this situation. Consequently, the effectiveness
of the drug may be reduced, leading to this phenomenon. However, it
was demonstrated through small animal imaging that the use of
liposome-encapsulated drugs may effectively concentrate the drug at
the tumour site compared with the free drug. The drug concentration
at the tumour site was shown to remain significantly higher than
that of the free drug until the drug was fully metabolized. This
process does not affect the application of CDDP. Notably, the use
of only A549 and H460 cell lines is a limitation of the present
study and further NSCLC cell lines, as well as MUC1-positive and
MUC1-negative cells, should be used in the future to investigate
the specificity of the targeting mechanism.
In addition, the CDDP-Lip targeting MUC1
constructed in the present study hold promise for application in
other types of cancer or disease. Platinum-based compounds are
currently the most commonly used chemotherapeutic agents in
clinical practice. They are employed as first-line treatment for a
wide range of malignant tumours or as alternative therapies
following the development of resistance to the initial
chemotherapeutic agent. For example, in addition to NSCLC, CDDP is
widely used in treating solid tumours, including melanoma, breast
cancer, colon cancer and liver cancer (46). In addition, the target site of
choice, MUC1, is a mucin widely expressed in a variety of malignant
tumours. The MUC1 antigenic epitopes expressed on the surface of
malignant tumours are exposed due to alterations in their
glycosylation levels. This makes the targeting of MUC1 even more
conducive to reducing non-tumour site-specific release. For
example, in the study by Li et al (47), the expression of MUC1 was
revealed to be significantly upregulated in breast cancer. As CDDP
is a commonly used drug for breast cancer, developing CDDP
liposomes that can target MUC1 is expected to enable drug-targeted
delivery to patients with breast cancer and high MUC1 expression.
Furthermore, our previous studies have demonstrated the targeting
of the generated liposomes to hepatocellular carcinoma and
colorectal cancer (23,33). It is expected that targeted CDDP
can be delivered to patients with these types of cancer using our
specialized nanodelivery system. In addition, CDDP shows good
clinical synergy with gemcitabine and pemetrexed; the
gemcitabine/CDDP regimen has been extensively evaluated in Phase II
and Phase III randomized trials in advanced NSCLC, with response
rates ranging from 21 to 40%, and a median survival of ~9 months
(48). Additionally, in a study
by Cui et al (49), the
combination of farnesyltamol and CDDP successfully inhibited the
progression of NSCLC by targeting the DUSP26-mediated signalling
pathway, suggesting potential clinical applications of NSCLC.
CDDP is widely used to treat various types of
malignant tumours. For example, it has shown synergistic antitumour
effects with temozolomide (TMZ) in several clinical studies
(50,51), such as in the treatment of
advanced malignant melanoma (50). TMZ creates methyl adducts at the
0.6 position of guanine, while CDDP can reduce the activity of the
DNA repair enzyme AGAT, which inhibits the formation of methyl
adducts, thereby boosting the antitumour effects of TMZ (52). The chosen target, MUC1, is a
tumour-specific protein found extensively on the surface of various
malignant tumours. Targeting MUC1 has also been proven to be
effective in studies of other types of malignant tumours.
HuMNC2-CAR44 CAR-T cells are being utilized in a clinical trial
(NCT04020575; classic.clinicaltrials.gov/ct2/show/NCT04020575) to
treat metastatic breast cancer, which is currently enrolling
participants. In another clinical study (NCT02544880;
classic.clinicaltrials. gov/ct2/show/NCT02544880), researchers
assessed the safety and immunological effectiveness of tadalafil
combined with an antitumour vaccine containing MUC1 and poly(ICLC);
this study revealed that tadalafil had a beneficial
immunomodulatory effect on patients with recurrent primary squamous
cell carcinoma of the head and neck. In addition, the antitumour
vaccine MUC1/polyICLC can be used as an adjuvant to tadalafil
therapy in primary head and neck squamous cell carcinoma (53).
In conjunction with our previous studies, it may be
hypothesized that in addition to pre-preparation of CDDP, which
enables it to better exert its effects on organisms, the enhanced
antitumour effect may also be due to liposomes assisting in the
intracellular delivery of CDDP. In addition to passive diffusion,
the transport efficiency of CDDP is influenced by copper
transporter protein 1 (CTR1), which is crucial for the
internalization of CDDP in tumour cells (54). Several in vitro and in
vivo studies (55,56) have shown that CDDP resistance in
cancer is associated with changes in the levels of CTR1 (55). Differences in the efficacy of
CDDP therapy and the development of resistance between different
types of cancer or individuals may also be associated with the
activity of CTR1. We previously reported that PNA-Lip can be
generated through lattice protein-mediated endocytosis, this
explains the mechanism by which liposomes were endocytosed by cells
in this study (57,58). Furthermore, we revealed that
tumour cells express a significant amount of MUC1 on their surface
(23). This property allows for
unrestricted transport of CDDP into the cell without having to
receive the rate limitation of CRT1 transport as is the case with
direct CDDP, allowing for more effective targeting of tumours.
The CDDP-PNA-Lip prepared in the present study were
revealed to have a significant anti-NSCLC effect, and by adding
DPPG to the liposome formulation, the stability and DL capacity of
the liposomes were better than those of traditional liposomes.
However, the present study has several limitations. First, the
cytotoxicity of CDDP after pretreatment was tested, and the
cytotoxicity of CDDP did not differ significantly before and after
treatment, but other characteristics were not detected, such as its
integration capacity with DDTC. Another limitation of this study is
that CDDP is mainly administered intravenously in clinical
practice, whereas the present study used intraperitoneal injection.
Although studies have shown that small molecules with a particle
size <300 nm can produce therapeutic effects comparable to those
of intravenous injection when administered intraperitoneally, some
variability still exists that hinders the complete replication of
the therapeutic effects of intravenous injection. At present, this
process must be considered when advancing clinical trials to
determine whether pretreatment with CDDP may result in unforeseen
toxic side effects, necessitating further toxicological analysis.
The in vivo clearance of liposomes is also an issue that
requires further attention. According to Ishida et al
(59), the complement system
serves a key role in enhancing the clearance of liposomes when they
are negatively charged and relatively large in size. Therefore,
constructing smaller liposomes may offer an effective solution to
this issue. In addition, there may be some variability between the
two cell lines used in the present study. Although the ability of
PNA to target MUC1 was shown in our previous study (33), a more systematic approach may be
warranted in future studies. This could involve isolating primary
cells from patients or constructing MUC1-overexpressing cell lines
using a lentivirus. This step is necessary to clarify the
specificity of the targeting mechanism and to assess the potential
toxicity of the formulation to normal lung cell lines.
In conclusion, in the present study, CDDP-loaded
ligand-modified liposomes were successfully prepared; these
materials were well characterized and stable, suggesting that they
could be used for the delivery of CDDP in vivo. This
approach effectively improved the time of CDDP in animals and
increased the local drug concentration and residence time at the
tumour site through targeting. In vitro experiments proved
that CDDP-PNA-Lip had a significant inhibitory effect on tumour
cells. Therefore, the present study is highly important for
evaluating the clinical application of CDDP, improving the
antitumour efficacy of drugs and reducing the administration dose,
and indicated that CDDP-loaded ligand-modified liposomes may be
used as a potential treatment for NSCLC.
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
ZG performed conceptual design. WY conducted
methodology and performed experiments. The data were quantified and
analysed by RK, FW and MS. LW, SZ and JW performed bioinformatics
analyses. FY and MQ examined the physiological indicators of the
animals. SS, AW and HW conducted animal experiments. YiW and YuW
supervised and participated in the cell experiments. YiW, YuW and
BY wrote the original manuscript. ZG and BY reviewed and edited the
article, supervised by ZG. ZG was responsible for project
management. ZG, WY and MQ obtained funds. ZG and WY confirming the
authenticity of all the raw data. All authors read and approved the
final version of the manuscript.
Ethics approval and consent to
participate
The animal experimental protocol used in the
present study was approved by the Ethical Review Committee of
Shandong Second Medical University.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Use of artificial intelligence tools
During the preparation of this work, AI tools were
used to improve the readability and language of the manuscript or
to generate images, and subsequently, the authors revised and
edited the content produced by the AI tools as necessary, taking
full responsibility for the ultimate content of the present
manuscript.
Acknowledgements
Not applicable.
Funding
This research was funded by The Natural Science Foundation of
Shandong Province, (grant no. ZR20180709017), the National Natural
Science Foundation of China, (grant nos. 82070856, 81274093 and
81871892) and the Weifang City Science and Technology Project Plan
(grant no. 2022YX038).
References
|
1
|
Nooreldeen R and Bach H: Current and
future development in lung cancer diagnosis. Int J Mol Sci.
22:86612021. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Wu F, Wang L and Zhou C: Lung cancer in
China: Current and prospect. Curr Opin Oncol. 33:40–46. 2021.
View Article : Google Scholar
|
|
3
|
Teramoto K, Ozaki Y, Hanaoka J, Sawai S,
Tezuka N, Fujino S, Daigo Y and Kontani K: Predictive biomarkers
and effectiveness of MUC1-targeted dendritic-cell-based vaccine in
patients with refractory non-small cell lung cancer. Ther Adv Med
Oncol. 9:147–157. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Sun M, Shi Y, Dang UJ and Di Pasqua AJ:
Phenethyl isothiocyanate and cisplatin co-encapsulated in a
liposomal nanoparticle for treatment of non-small cell lung cancer.
Molecules. 24:8012019. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Wang Y, Qian J, Yang M, Xu W, Wang J, Hou
G, Ji L and Suo A: Doxorubicin/cisplatin co-loaded hyaluronic
acid/chitosan-based nanoparticles for in vitro synergistic
combination chemotherapy of breast cancer. Carbohydr Polym.
225:1152062019. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Nasrollahi F, Koh YR, Chen P, Varshosaz J,
Khodadadi AA and Lim S: Targeting graphene quantum dots to
epidermal growth factor receptor for delivery of cisplatin and
cellular imaging. Mater Sci Eng C Mater Biol Appl. 94:247–257.
2019. View Article : Google Scholar
|
|
7
|
Gualdani R, de Clippele M, Ratbi I, Gailly
P and Tajeddine N: Store-operated calcium entry contributes to
cisplatin-induced cell death in non-small cell lung carcinoma.
Cancers (Basel). 11:4302019. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ashique S, Sandhu NK, Chawla V and Chawla
PA: Targeted drug delivery: Trends and perspectives. Curr Drug
Deliv. 18:1435–1455. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Tavakkoli Yaraki M, Daqiqeh Rezaei S and
Tan YN: Simulation guided design of silver nanostructures for
plasmon-enhanced fluorescence, singlet oxygen generation and SERS
applications. Phys Chem Chem Phys. 22:5673–5687. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Sarfaraz N and Khan I: Plasmonic gold
nanoparticles (AuNPs): Properties, synthesis and their advanced
energy, environmental and biomedical applications. Chem Asian J.
16:720–742. 2021. View Article : Google Scholar
|
|
11
|
Mittal D, Singh A, Kohli K and Verma AK:
Engineering biosafe cisplatin loaded nanostructured lipid carrier:
Optimisation, synthesis, pharmacokinetics and biodistribution. J
Microencapsul. 39:522–538. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Herrera-Juarez M, Serrano-Gomez C,
Bote-de-Cabo H and Paz-Ares L: Targeted therapy for lung cancer:
Beyond EGFR and ALK. Cancer. 129:1803–1820. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Moosavian SA, Abnous K, Akhtari J, Arabi
L, Gholamzade Dewin A and Jafari M: 5TR1 aptamer-PEGylated
liposomal doxorubicin enhances cellular uptake and suppresses
tumour growth by targeting MUC1 on the surface of cancer cells.
Artif Cells Nanomed Biotechnol. 46:2054–2065. 2018.
|
|
14
|
Noble GT, Stefanick JF, Ashley JD,
Kiziltepe T and Bilgicer B: Ligand-targeted liposome design:
Challenges and fundamental considerations. Trends Biotechnol.
32:32–45. 2014. View Article : Google Scholar
|
|
15
|
Filipczak N, Pan J, Yalamarty SSK and
Torchilin VP: Recent advancements in liposome technology. Adv Drug
Deliv Rev. 156:4–22. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Saw PE, Park J, Lee E, Ahn S, Lee J, Kim
H, Kim J, Choi M, Farokhzad OC and Jon S: Effect of PEG pairing on
the efficiency of cancer-targeting liposomes. Theranostics.
5:746–754. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Underwood C, van Eps AW, Ross MW, Laverman
P, van Bloois L, Storm G and Schaer TP: Intravenous technetium-99m
labelled PEG-liposomes in horses: A safety and biodistribution
study. Equine Vet J. 44:196–202. 2012. View Article : Google Scholar
|
|
18
|
Namba M, Hattori N, Hamada H, Yamaguchi K,
Okamoto Y, Nakashima T, Masuda T, Sakamoto S, Horimasu Y, Miyamoto
S, et al: Anti-KL-6/MUC1 monoclonal antibody reverses resistance to
trastuzumab-mediated antibody-dependent cell-mediated cytotoxicity
by capping MUC1. Cancer Lett. 442:31–39. 2019. View Article : Google Scholar
|
|
19
|
Bouillez A, Adeegbe D, Jin C, Hu X, Tagde
A, Alam M, Rajabi H, Wong KK and Kufe D: MUC1-C promotes the
suppressive immune microenvironment in non-small cell lung cancer.
Oncoimmunology. 6:e13389982017. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kumar P, Lindberg L, Thirkill TL, Ji JW,
Martsching L and Douglas GC: The MUC1 extracellular domain subunit
is found in nuclear speckles and associates with spliceosomes. PLoS
One. 7:e427122012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Beack S, Cho M, Kim YE, Ahn GO and Hahn
SK: Hyaluronate-peanut agglutinin conjugates for target-specific
bioimaging of colon cancer. Bioconjug Chem. 28:1434–1442. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wu X, McFall-Boegeman H, Rashidijahanabad
Z, Liu K, Pett C, Yu J, Schorlemer M, Ramadan S, Behren S,
Westerlind U and Huang X: Synthesis and immunological evaluation of
the unnatural β-linked mucin-1 Thomsen-Friedenreich conjugate. Org
Biomol Chem. 19:2448–2455. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Li X, Diao W, Xue H, Wu F, Wang W, Jiang
B, Bai J, Lian B, Feng W, Sun T, et al: Improved efficacy of
doxorubicin delivery by a novel dual-ligand-modified liposome in
hepatocellular carcinoma. Cancer Lett. 489:163–173. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zahednezhad F, Zakeri-Milani P, Shahbazi
Mojarrad J and Valizadeh H: The latest advances of cisplatin
liposomal formulations: essentials for preparation and analysis.
Expert Opin Drug Deliv. 17:523–541. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Long DF and Repta AJ: Cisplatin:
Chemistry, distribution and biotransformation. Biopharm Drug
Dispos. 2:1–16. 1981. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Cheng Y, Zhao P, Wu S, Yang T, Chen Y,
Zhang X, He C, Zheng C, Li K, Ma X and Xiang G: Cisplatin and
curcumin co-loaded nano-liposomes for the treatment of
hepatocellular carcinoma. Int J Pharm. 545:261–273. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Saracchini S, Foltran L, Tuccia F, Bassini
A, Sulfaro S, Micheli E, Del Conte A, Bertola M, Gion M, Lorenzon M
and Tumolo S: Phase II study of liposome-encapsulated doxorubicin
plus cyclophosphamide, followed by sequential trastuzumab plus
docetaxel as primary systemic therapy for breast cancer patients
with HER2 overexpression or amplification. Breast. 22:1101–1107.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Wang H, Wu H, Shen H, Geng S, Wang B, Wang
Y, Ma X, Li G and Tan M: A bimodal MRI and NIR liposome nanoprobe
for tumor targeted molecular imaging. J Mater Chem B. 3:8832–8841.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Theodosiou M, Sakellis E, Boukos N,
Kusigerski V, Kalska-Szostko B and Efthimiadou E: Iron oxide
nanoflowers encapsulated in thermosensitive fluorescent liposomes
for hyperthermia treatment of lung adenocarcinoma. Sci Rep.
12:86972022. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Zhang X, Lü S, Han J, Sun S, Wang L and Li
Y: Preparation, characterization and in vivo distribution of solid
lipid nanoparticles loaded with syringopicroside. Pharmazie.
66:404–407. 2011.PubMed/NCBI
|
|
31
|
Lippard SJ: New chemistry of an old
molecule: Cis-[Pt(NH3)2Cl2]. Science. 218:1075–1082. 1982.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Newman MS, Colbern GT, Working PK, Engbers
C and Amantea MA: Comparative pharmacokinetics, tissue
distribution, and therapeutic effectiveness of cisplatin
encapsulated in long-circulating, pegylated liposomes (SPI-077) in
tumor-bearing mice. Cancer Chemother Pharmacol. 43:1–7. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Diao W, Yang B, Sun S, Wang A, Kou R, Ge
Q, Shi M, Lian B, Sun T, Wu J, et al: PNA-modified liposomes
improve the delivery efficacy of CAPIRI for the synergistic
treatment of colorectal cancer. Front Pharmacol. 13:8931512022.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Bar J, Ofek E, Barshack I, Gottfried T,
Zadok O, Kamer I, Urban D, Perelman M and Onn A: Transformation to
small cell lung cancer as a mechanism of resistance to
immunotherapy in non-small cell lung cancer. Lung Cancer.
138:109–115. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Ettinger DS, Wood DE, Aisner DL, Akerley
W, Bauman JR, Bharat A, Bruno DS, Chang JY, Chirieac LR, D'Amico
TA, et al: Non-small cell lung cancer, version 3.2022, NCCN
clinical practice guidelines in oncology. J Natl Compr Canc Netw.
20:497–530. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Riaz SP, Lüchtenborg M, Coupland VH,
Spicer J, Peake MD and Møller H: Trends in incidence of small cell
lung cancer and all lung cancer. Lung Cancer. 75:280–284. 2012.
View Article : Google Scholar
|
|
37
|
Zhong WZ, Yan HH, Chen KN, Chen C, Gu CD,
Wang J, Yang XN, Mao WM, Wang Q, Qiao GB, et al: Erlotinib versus
gemcitabine plus cisplatin as neoadjuvant treatment of stage
IIIA-N2 EGFR-mutant non-small-cell lung cancer: Final overall
survival analysis of the EMERGING-CTONG 1103 randomised phase II
trial. Signal Transduct Target Ther. 8:762023. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Stinchcombe TE: Flashback foreword:
Cisplatin/pemetrexed in non-small-cell lung cancer. J Clin Oncol.
41:2455–2456. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Makwana V, Karanjia J, Haselhorst T,
Anoopkumar-Dukie S and Rudrawar S: Liposomal doxorubicin as
targeted delivery platform: Current trends in surface
functionalization. Int J Pharm. 593:1201172021. View Article : Google Scholar
|
|
40
|
Lozano N, Al-Ahmady ZS, Beziere NS,
Ntziachristos V and Kostarelos K: Monoclonal antibody-targeted
PEGylated liposome-ICG encapsulating doxorubicin as a potential
theranostic agent. Int J Pharm. 482:2–10. 2015. View Article : Google Scholar
|
|
41
|
Kim DM, Kim M, Park HB, Kim KS and Kim DE:
Anti-MUC1/CD44 dual-aptamer-conjugated liposomes for cotargeting
breast cancer cells and cancer stem cells. ACS Appl Bio Mater.
2:4622–4633. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Panchamoorthy G, Jin C, Raina D, Bharti A,
Yamamoto M, Adeebge D, Zhao Q, Bronson R, Jiang S, Li L, et al:
Targeting the human MUC1-C oncoprotein with an antibody-drug
conjugate. JCI Insight. 3:e998802018. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Ranjbar-Navazi Z, Fathi M, Abdolahinia ED,
Omidi Y and Davaran S: MUC-1 aptamer conjugated InP/ZnS quantum
dots/nanohydrogel fluorescent composite for mitochondria-mediated
apoptosis in MCF-7 cells. Mater Sci Eng C Mater Biol Appl.
118:1114692021. View Article : Google Scholar
|
|
44
|
Marzban E, Alavizadeh SH, Ghiadi M,
Khoshangosht M, Khashayarmanesh Z, Abbasi A and Jaafari MR:
Optimizing the therapeutic efficacy of cisplatin PEGylated
liposomes via incorporation of different DPPG ratios: In vitro and
in vivo studies. Colloids Surf B Biointerfaces. 136:885–891. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Zia MK, Siddiqui T, Ali SS, Rehman AA,
Ahsan H and Khan FH: Chemotherapeutic drugs and plasma proteins:
Exploring new dimensions. Curr Protein Pept Sci. 19:937–947. 2018.
View Article : Google Scholar
|
|
46
|
Romani AMP: Cisplatin in cancer treatment.
Biochem Pharmacol. 206:1153232022. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Li Z, Yang D, Guo T and Lin M: Advances in
MUC1-mediated breast cancer immunotherapy. Biomolecules.
12:9522022. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Crinò L, Calandri C, Maestri A and
Marrocolo F: Gemcitabine and cisplatin combination in early-stage
non-small-cell lung cancer. Oncology (Williston Park). 15(3 Suppl
6): S40–S42. 2001.
|
|
49
|
Cui Z, Li D, Zhao J and Chen K: Falnidamol
and cisplatin combinational treatment inhibits non-small cell lung
cancer (NSCLC) by targeting DUSP26-mediated signal pathways. Free
Radic Biol Med. 183:106–124. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Daponte A, Ascierto PA, Gravina A, Melucci
M, Scala S, Ottaiano A, Simeone E, Palmieris G and Comella G:
Temozolomide and cisplatin in avdanced malignant melanoma.
Anticancer Res. 25:1441–1447. 2005.PubMed/NCBI
|
|
51
|
Christodoulou C, Bafaloukos D, Linardou H,
Aravantinos G, Bamias A, Carina M, Klouvas G and Skarlos D;
Hellenic Cooperative Oncology Group: Temozolomide (TMZ) combined
with cisplatin (CDDP) in patients with brain metastases from solid
tumors: A hellenic cooperative oncology group (HeCOG) phase II
study. J Neurooncol. 71:61–65. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Bafaloukos D, Tsoutsos D, Kalofonos H,
Chalkidou S, Panagiotou P, Linardou E, Briassoulis E, Efstathiou E,
Polyzos A and Fountzilas G: Temozolomide and cisplatin versus
temozolomide in patients with advanced melanoma: A randomized phase
II study of the Hellenic cooperative oncology group. Ann Oncol.
16:950–957. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Weed DT, Zilio S, Reis IM, Sargi Z,
Abouyared M, Gomez-Fernandez CR, Civantos FJ, Rodriguez CP and
Serafini P: The reversal of immune exclusion mediated by tadalafil
and an anti-tumor vaccine also induces PDL1 Upregulation in
recurrent head and neck squamous cell carcinoma: interim analysis
of a phase I clinical trial. Front Immunol. 10:12062019. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Ishida S, Lee J, Thiele DJ and Herskowitz
I: Uptake of the anticancer drug cisplatin mediated by the copper
transporter Ctr1 in yeast and mammals. Proc Natl Acad Sci USA.
99:14298–14302. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Akhter J, Goswami P, Ali Beg MM, Ahmad S,
Najmi AK and Raisuddin S: Protective effect of rosmarinic acid on
the transmembrane transporter Ctr1 expression in cisplatin-treated
mice. J Cancer Res Ther. 19:1753–1759. 2023. View Article : Google Scholar
|
|
56
|
Huang CP, Fofana M, Chan J, Chang CJ and
Howell SB: Copper transporter 2 regulates intracellular copper and
sensitivity to cisplatin. Metallomics. 6:654–661. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Akerfeldt MC, Tran CMN, Shen C, Hambley TW
and New EJ: Interactions of cisplatin and the copper transporter
CTR1 in human colon cancer cells. J Biol Inorg Chem. 22:765–774.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Zhang P, Li B, Chen Q, Wang H and Feng Q:
Glucose restriction induces ROS-AMPK-mediated CTR1 expression and
increases cisplatin efficiency in NSCLC. Cancer Lett.
543:2157932022. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Ishida T, Harashima H and Kiwada H:
Liposome clearance. Biosci Rep. 22:197–224. 2002. View Article : Google Scholar : PubMed/NCBI
|