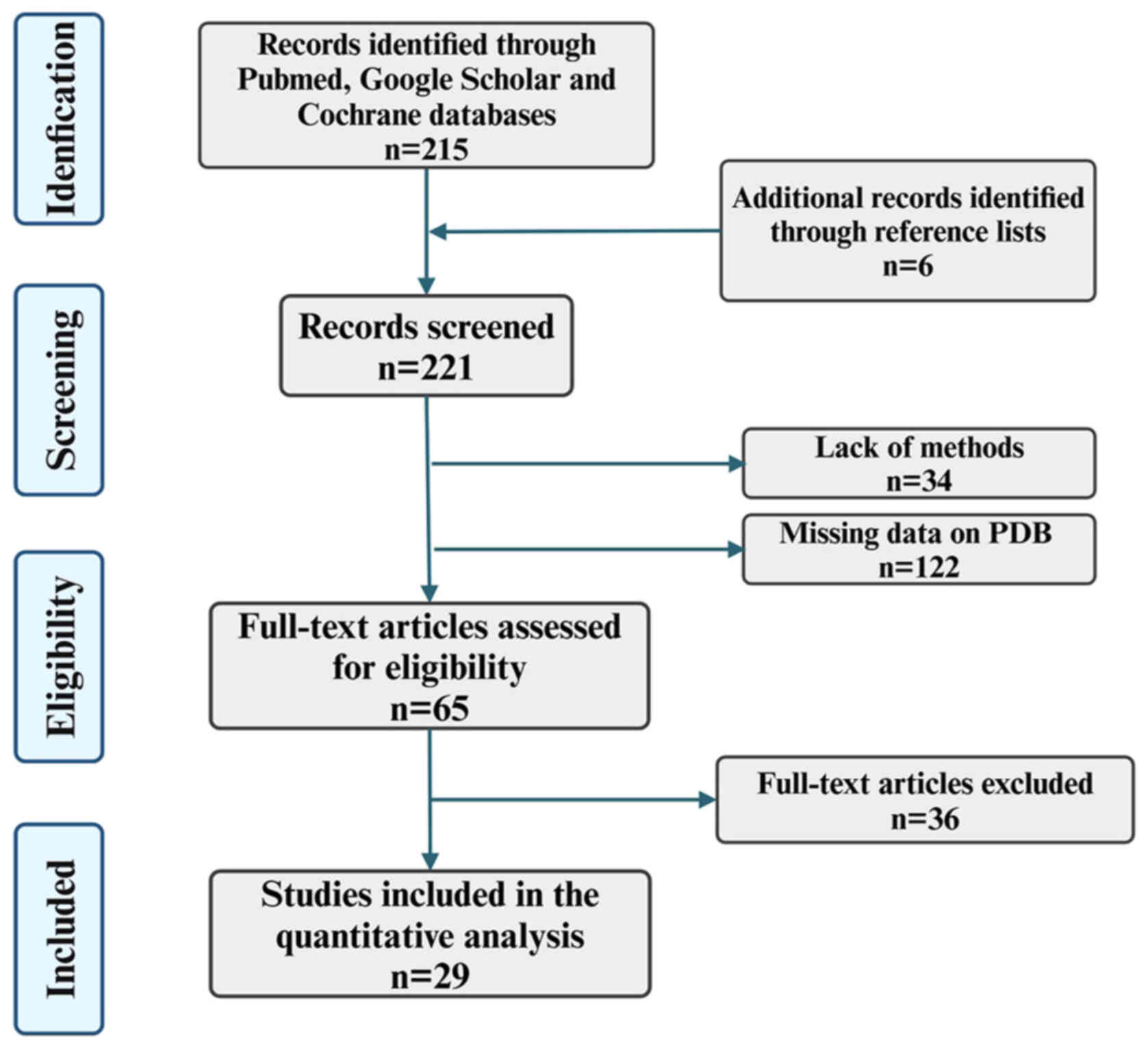
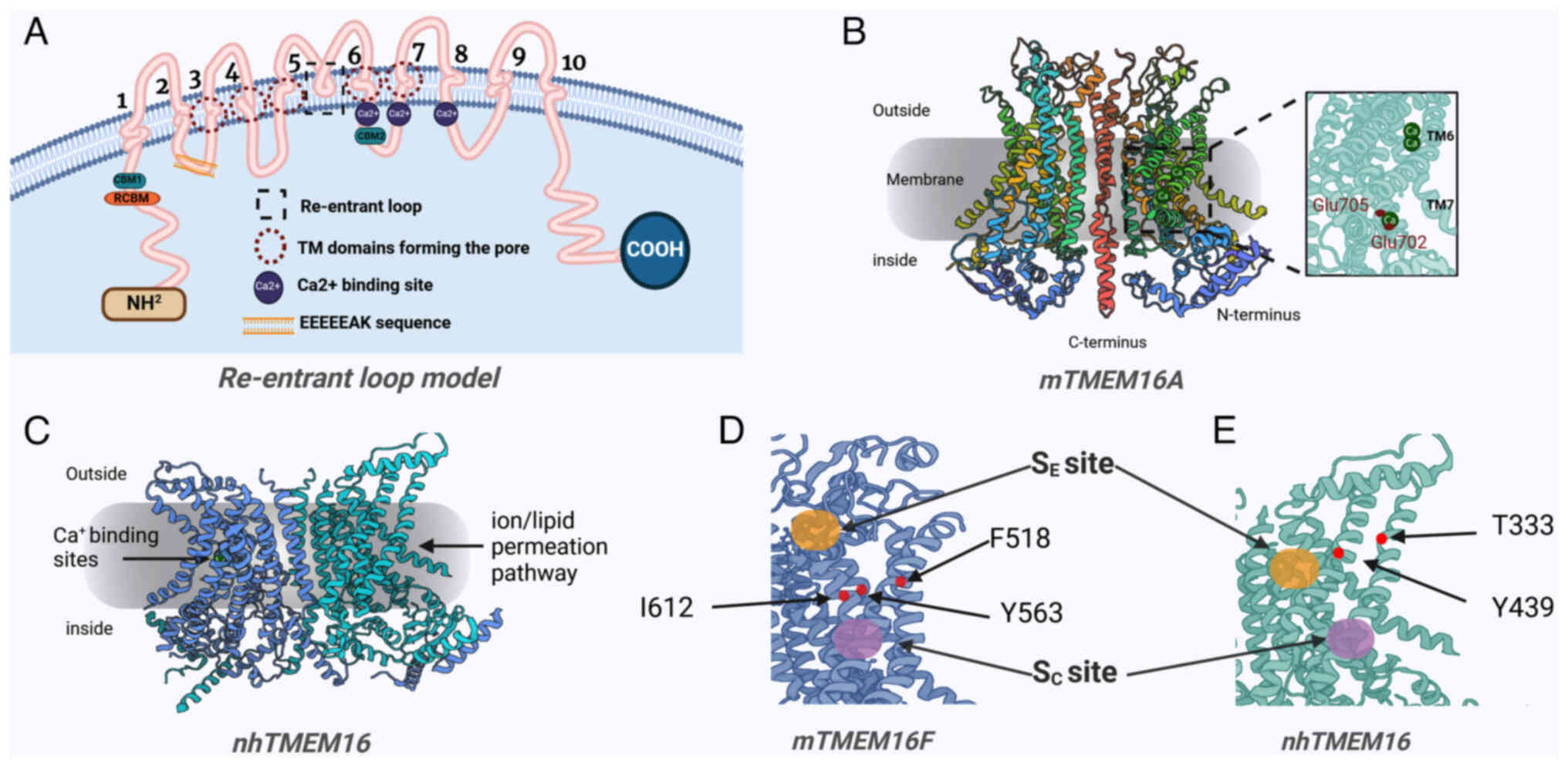
The present narrative review followed the Assessment
of Narrative Review Articles flowchart (Fig. 1) (8). The main purpose of the present
review was to summarize the evidence of potential therapeutic
targets in TMEM16 protein research and to understand the basic
characteristic structures, gene mutations and treatment strategies
for related diseases. The English terms ['TMEM16' (Mesh)] OR
['TMEM16' (Mesh)] AND ['Structure' (Mesh)] OR ['Treatment' (Mesh)]
were searched on the PubMed (https://pubmed.ncbi.nlm.nih.gov), Google Scholar
(https://scholar.google.com) and Cochrane
databases (https://www.cochranelibrary.com). The screening
results included literature published in the past 15 years.
Articles that met the following criteria were included in the
present review: i) The data reported in the study was from animals;
ii) the structure of TMEM16 protein could be found in the
corresponding data in the Protein Data Bank (PDB, https://www.rcsb.org); iii) cases of clinical diseases
were not individual case studies; iv) clearly stated the specific
methods and evidence was supported by referenced citations; and v)
the articles were not practical guidelines, guidelines,
meta-analyses, systematic reviews, narrative reviews, case series
and case reports. Articles that did not describe the methods and
those that were not strictly related to the research objectives
were excluded. The search strategy identified 221 articles, of
which 192 were excluded after evaluating the title and abstract.
Then, based on the importance of the journal, including comparison
of research designs and methods, evaluation of journal papers,
journal impact factors, academic reputation of scholars and
academic status of institutions, three independent reviewers
studied the titles and abstracts of the articles. To prioritize
analysis of the various subtypes of TMEM16 protein and their
corresponding cellular functions, and to analyze the disease and
treatment targets based on the collected literature, 29 articles
were selected for quality evaluation. The abstracts and images of
all selected articles were reviewed and the data and content from
the complete article were ultimately used to write the present
review.
Calmodulin can interact with compounds such as
1-EBIO, DCEBIO and riluzole, that induce the opening of
Ca2+-activated K+ channels with low and
medium conductivities and can also activate TMEM16A (15). The TMEM16 proteins link
Ca2+ signals with cellular electrical activity and lipid
transport and play a critical role in CF (16). The aforementioned compounds
within amino acid supplement tablets can activate the efflux of
Ca2+ from cells in CF (15). Moreover, calmodulin can interact
with and regulate the activity of Ca2+ channels
(15). Although several
structural analyses of the TMEM16 proteins, including fungal and
mouse orthologues, have been completed, the binding sites for lipid
scramblase activity are yet to be identified. The TMEM16 proteins
that function as phospholipid scramblases have an essential role in
almost all human physiological processes (2,17). Therefore, dysregulated activity
of TMEM16 as a phospholipid scramblase may lead to unfavorable
consequences. In summary, although these structural and functional
studies provide important insights into the voltage-dependent
activation mechanisms of TMEM16A as a CaCC, further studies are
needed to comprehensively understand the dual functionalities of
TMEM16 proteins as ion channels and phospholipid scramblases.
In clinical practice, prognostic markers can predict
the poor clinical outcomes of treatment methods in patients with
cancer. However, the ambiguity in the molecular functions of these
markers makes the accurate prediction of the progression of tumors
difficult. TMEM16 protein is not a prognostic marker for tumors,
but it is closely related to the occurrence and development of
tumors (18). TMEM16 proteins
are distributed throughout the human body, with different types
distributed in different tissues or organs, and are associated with
various diseases (Table I).
TMEM16A and B function as both CaCCs and phospholipid scramblases
that promote the bidirectional mobility of membrane lipids
(6). Additionally, TMEM16A and B
control the release of Ca2+ stored in the cytoplasmic
membrane, enhance intracellular Ca2+ signaling, amplify
Ca2+ signaling activated by G protein-coupled receptors
and regulate ion channel trafficking (6). TMEM16A is mainly involved in
trans-epithelial Cl− transport (1,4,19) and smooth muscle tone regulation
(20-22), and is widely expressed throughout
the body, serving as a receptor to sense injury stimuli and cell
proliferation (particularly when upregulated in cancer). In
addition, an induction of the production of angiotensin II
stimulates the contraction of cerebral vessels via the
TMEM16A-mediated Ras homolog family member A/Rho-associated protein
kinase signaling pathway (23).
Furthermore, the P38/JNK signaling pathway is also activated by
TMEM16A expression, thereby increasing the apoptosis rate of
podocytes in mice with diabetic nephropathy, which can exacerbate
the injury caused to the kidneys (24). TMEM16A regulates the
proliferation of the epithelial cells lining the bile duct via the
ATP-stimulated-Ca2+-protein kinase C signaling pathway
to induce the synthesis and secretion of bile (25). TMEM16B regulates sensory
processes such as smell and vision and can control the excitability
of neuronal and glial cells (26-28). TMEM16B mutations can cause
multiple sclerosis and schizophrenia (4,29-31). TMEM16C is typically expressed in
the central and peripheral nervous systems of humans, mice and
rats, and interacts with Na+-activated K+
channels to improve the susceptibility of Na+ and the
activity of K+ channels (32-34). TMEM16C has a role in certain
other cellular functions including the regulation of pain and heat
processing (34). Previous
studies have revealed that genetic mutations in TMEM16C can
cause craniocervical dystonia in humans (35-37). TMEM16D also functions as a
non-selective ion channel and a phospholipid scramblase (38), is mainly expressed in the brain
and endocrine glands, and it can control the mean arterial pressure
and secrete aldosterone (39). A
mutation in the gene encoding TMEM16D can lead to
neurological diseases, such as Alzheimer's disease (40). TMEM16E acts as a non-selective
ion channel and scramblase, and is mainly expressed in skeletal
muscle, participating in the repair and maintenance of
intracellular calcium stability of skeletal muscle, and activates
the janus kinase (JAK)/STAT3 signaling pathway for cell migration
and invasion (12).
TMEM16E causes gnathodiaphyseal dysplasia (GDD) in cases of
missense mutations (41) and
muscular dystrophy (MD) in cases of functional mutations (42-44). TMEM16F also acts as a
non-selective ion channel and scramblase activated by very high
concentrations of Ca2+ and promotes the translocation of
phospholipid and phosphatidylserine (PS) from the inner leaflet of
the plasma membrane to the outer leaflet (45,46). Mutations in the gene encoding
TMEM16F are associated with the development of Scott syndrome, a
hemorrhagic disease caused by phospholipid-related disorders in the
membranes of platelets (7,47,48). Moreover, TMEM16F mediates the
proliferation of myoblasts; it plays an essential role in C2C12
myoblast proliferation, likely via regulating the ERK/AKT signaling
pathway (49). The roles of
TMEM16G and H have not yet been fully elucidated (50). The levels of TMEM16G are
upregulated in cancer, particularly prostate cancer, and interact
with the other upregulated proteins such as intracellular vesicle
proteins (51). Hence, TMEM16G
may be a potential biomarker for diagnosis and a target for
prostate cancer immunotherapy (51). TMEM16G is also involved in the
perturbation of the lipid bilayer in cell lines with a deletion in
TMEM16F (7,50). TMEM16H forms junctions between
the endoplasmic reticulum (ER) and the cell membrane in
intracellular signaling and is involved in the transport of bile
salts and the manifestation of intrahepatic cholestasis of
pregnancy (52,53). The intrinsic process of TMEM16H
may involve an interaction between proteins such as the
matrix-interacting molecule 1, and receptors such as the inositol
1,4,5-trisphosphate receptor, that induce the release of
Ca2+ from the cells (52). TMEM16J is a non-selective cation
channel with scramblase activity (54,55), is activated by cAMP-dependent
protein kinase A (5) and is
associated with the development of certain types of cancer, such as
gastric cancer, pancreatic cancer and esophageal squamous cell
carcinoma (56-58). TMEM16K is mainly localized to the
membranes of intracellular compartments, is the most studied
phospholipid scramblase and demonstrates non-specific ion channel
activity that is optimally regulated by Ca2+ and
short-chain lipids (59).
TMEM16K is involved in spindle assembly (60) and affects macrophage volume
regulation (61). TMEM16K
deficiency leads to spinocerebella ataxia autosomal receiving type
10 (59). In addition, TMEM16K
also forms contact sites with endosomes and is associated with
Ca2+ signaling, cell volume regulation and apoptosis
(59-61).
CF is a genetic disease that affects multiple
organs. It is caused by abnormal CFTR transport through the
epithelial layer and is characterized by a loss of function in
various systems. This disease, caused by mutations in a single gene
encoding CFTR, can shorten the lifespan of humans (62,63). Patients with CF present with
symptoms that indicate the effects on a wide range of organs in the
body (62). These include
obstruction of the ducts of the mucinous glands and changes in
membrane composition in lung epithelium (62). However, certain non-malignant
types of CF are virtually asymptomatic and are diagnosed only in
adulthood; they affect only a single organ, as there is no systemic
involvement (63). The more
severe types of CF cause afflictions that affect multiple organs,
including male infertility, severe respiratory dysfunction
(including bronchiectasis, emphysema and pulmonary edema) and
pancreatic and intestinal complications (64). The progressive deterioration of
lung function and multiple organ failure are the main causes of
mortality in patients with CF (65). Furthermore, multiple therapies
have been clinically approved (NCT04058366) for treating the
individual conditions associated with CF (66-68). However, novel modulators,
multiple experimental approaches and advanced cellular models in
patients with CF should be developed to accurately and reliably
predict the drugs in clinical settings.
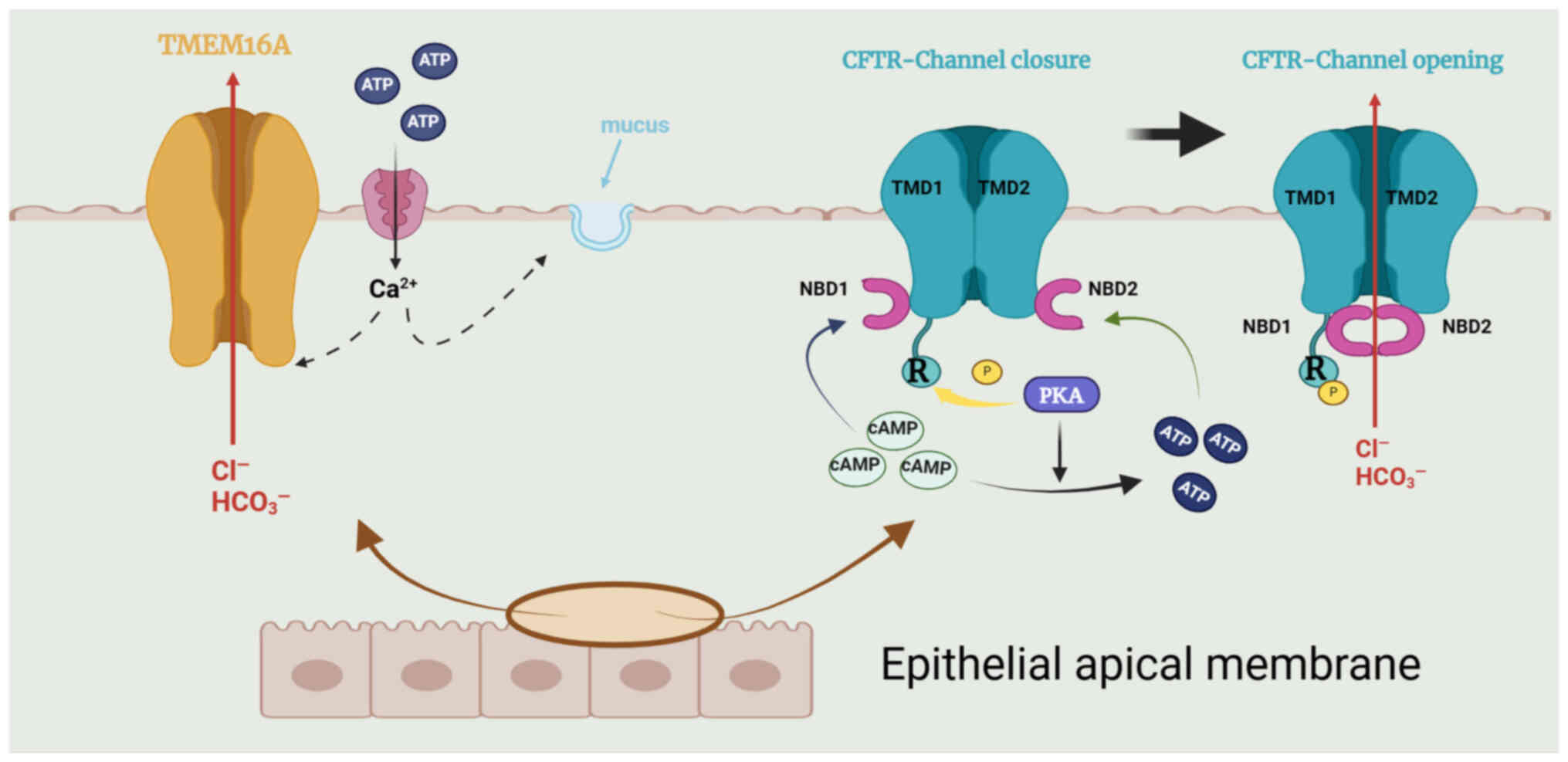
CFTR, a transmembrane conductance regulator protein
with 1,480 amino acids, is a Cl− channel driven by cAMP
(64) and is located in the
apical membrane of secretory epithelial cells. CFTR can transport
both Cl− and HCO3− into epithelial
cells. The channel is an ATP-binding transporter comprising five
domains: Two transmembrane domains that form the channel pore, a
regulatory domain (R) and two nucleotide-binding domains
(NBD1/NBD2) (69). High levels
of Cl− are necessary for the important physiological
actions of CFTR in the epithelial cells of the airways, including
moistening the mucosal surface and removing the mucosal cilia
(63,69). Patients with CF lack CFTR, which
causes mucus aggregation and tracheal blockage and leads to
susceptibility to chronic bacterial infections (63). These patients are therefore at
risk of respiratory diseases. In addition, these airway epithelial
cells also express TMEM16A, which functions as a second
Cl− channel; the cytosolic Ca2+
concentrations control its activity, and several inhibitors or
agonists have been identified (69) (Fig. 3).
In total, ~90% of patients with CF harbor a mutation
referred to as F508del, which leads to the degradation of proteases
and retention of the ER. A minimal increase in the occurrence of
F508del in the CFTR gene was observed in the plasma membrane
of apical cells (66,70,71). In total, ~50% of patients with CF
are homozygous for F508del, which not only have a processing defect
but also significantly reduces the stability and flexibility of the
cell surface in channel gating, if the mutation affects CFTR
localization to the plasma membrane (66). In such patients, administering
lumacaftor as a monotherapy may reduce the levels of Cl−
in the sweat by up to 8 mmol/l, in a dose-dependent manner.
However, lumacaftor failed to improve abnormal lung function in
phase II trials (71-73). In an in vitro preclinical
trial (NCT01225211), the addition of high concentrations of
ivacaftor as an enhancer was twice as effective as lumacaftor
alone, achieving ~25% of the normal CFTR activity (71). Thus, adding enhancers
significantly improved lung dysfunction (predicted forced
expiratory volume value of 1%) by 3-4% and reduced lung
functionality deterioration rates (71,73,74). In particular, the combined use of
two corrective agents and one enhancer
[elexacaftor-tezacaftor-ivacaftor (Trikafta) combination,
NCT03525444] was significantly effective in treating the most
commonly manifested defects in the membrane transport and gating
caused by CFTR mutations (69,70,75). The addition of tezacaftor, a
corrective agent, for treating patients homozygous for F508del
markedly improved diminished respiratory function (70). Administration of
Kalydeco®, a pharmacokinetic enhancer, can be used to
restore the damage caused to gated membrane transport by CFTR
channels due to missense mutations in CFTR (66,74,76).
Before the discovery of TMEM16A, evidence suggested
the occurrence of a second Cl−-channel expressed in the
epithelial cells of the airways of patients with and without CF
(80). This second channel,
referred to as a CaCC, is regulated by the cellular concentrations
of the Ca solute. Stimulation of the apical membranes of the
epithelial cells of the airways with the purinergic agonist ATP
in vitro and in vivo elicited a large but transient
Cl− secretory response (16,81,82). The putative physiological role of
CaCCs in the epithelial cells of the airways can involve mechanical
stimuli, such as those caused by normal tidal breathing or
coughing, that can induce the release of ATP, thereby promoting the
secretion of Cl− by the mucosal layer of the airways
through the binding of ATP to the purinergic and CaCC-associated
receptors and finally triggering the influx of Ca2+
(21,83). The secretion of mucus in the
airways involves TMEM16A (84).
By contrast, TMEM16A plays a key role in the
movement of the tracheal cilia and reducing the discharge of mucus
(85). TMEM16A simultaneously
guides chlorine gas and bicarbonate through the airway epithelium
and is expressed in the surface epithelium and submucosal glands,
removing mucosal cilia by enhancing anion influx (85). In addition, the inhibition of
TMEM16A by pharmacological compounds reduced the production of
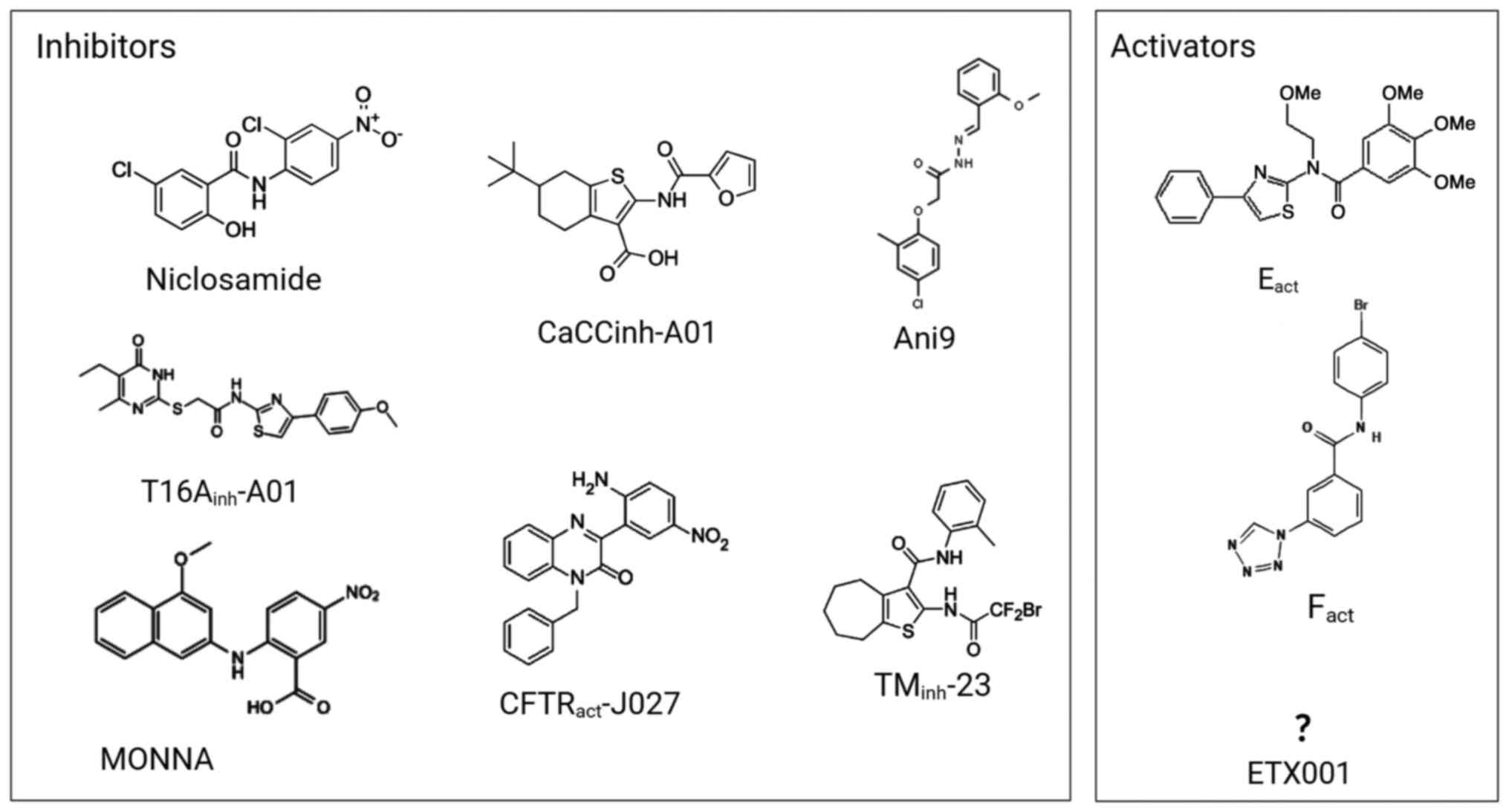
fluids on the surfaces of the airways (84). For instance, niclosamide reduced
mucus production in the airways of sensitized mice (16) and it was reported to also affect
intracellular calcium homeostasis by inhibiting the SERCA calcium
pump (86). Therefore, the
reported effects, such as the inhibition of mucus and cytokine
release, bronchodilation and antibacterial activity, make
niclosamide a potential drug suitable for the treatment of
inflammatory diseases of the airways such as CF, asthma and chronic
obstructive pulmonary disease (86). A recently identified TMEM16A
potentiator, ETX001, triggers the secretion of fluids and
accelerates the mucus clearance process without causing
bronchoconstriction (77,87).
The structure, the detailed mechanism of action, the location of
the binding site and the selectivity profile of ETX001 have not yet
been reported, although ETX001 may not interfere with
Ca2+ signaling (77).
Moreover, the functional efficiency of CFTR in epithelial cells can
be improved by blocking microRNA (miR)-based RNA silencing and
post-transcriptional regulation to increase the expression of
TMEM16A (88).
Although TMEM16A may represent a potential target
for the pharmacotherapy of CF, the widespread expression of TMEM16A
is a serious concern since systemic administration may produce a
broad range of side effects. Thus, any treatment targeting TMEM16A
requires selective treatment regimens using specific drugs.
Tumor growth is critically associated with cell
differentiation and proliferation. The regulation of the
intracellular Ca2+ levels by the TMEM16 proteins may
affect tumor development or regulate the exocytosis of the cell
membrane by controlling the intracellular concentrations of
Cl− (3,11). The activation of CaCCs by
cellular Ca2+ mainly occurs in the proliferative
potential cells and different types of cancer cells (89). The expression levels of TMEM16
proteins in diverse types of cancer, including TMEM16A-mediated
gastrointestinal stromal tumor (18), leiomyosarcoma (90), head and neck cancer (91), carcinoma of the lungs (92), pancreatic cancer (93), prostate cancer (94), breast cancer (95), colorectal cancer (96), gastric cancer (97), glioma and glioblastoma (98), esophageal cancer (99) and chondroblastoma (100), are indicated in Table II. TMEM16A participates in
cancer proliferation and migration by influencing the MAPK and
Ca2+/calmodulin-dependent protein kinase (CAMK)
signaling pathways and interacts with epidermal growth factor
receptor (EGFR) in head and neck squamous cell carcinoma (HNSCC)
(91). TMEM16E promotes the
development of colorectal (92)
and thyroid (101) cancer.
TMEM16G promotes the development of prostate (51) and breast (102) cancer, and TMEM16J promotes the
development of pancreatic cancer (57). Therefore, ascertaining the links
between TMEM16 proteins and pathways or mechanisms in tumor cells
is important for inhibiting tumor growth and proliferation and
developing therapeutic methods in clinical settings. However, to
the best of our knowledge, all commercially available TMEM16
protein detection kits are for research purposes only and not for
clinical practice. Due to a lack of availability of antibodies
against the human-derived TMEM16 proteins and significant
interspecific differences in the sequences of the TMEM16 proteins,
cross-reactivity is unlikely to occur. Thus, the development of a
TMEM16 detection kit for the diagnosis of cancer requires the
production of antibodies against the TMEM16 protein in humans.
In summary, TMEM16 can regulate the
biochemical/molecular processes in tumors, and its abnormal
expression in malignant tumors provides the possibility of
employing it as a clinical biomarker for early diagnosis and a
therapeutic target for reducing the occurrence and/or growth of
tumors.
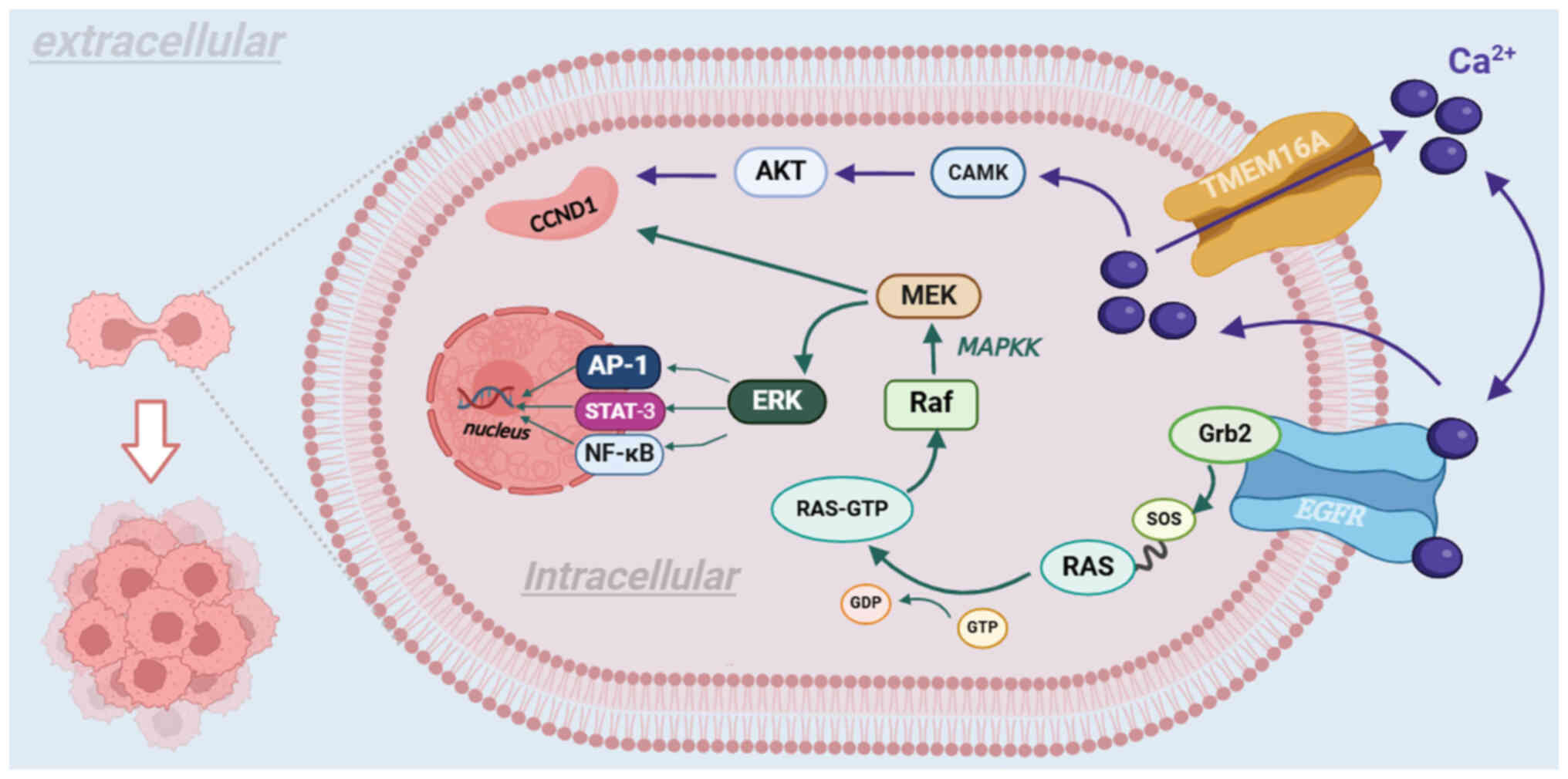
TMEM16A, in conjunction with EGFR, mediates the
growth of tumors through two routes. First, as a CaCC, TMEM16A
promotes the expression of cyclin D1 (CCND1) via the CAMK and AKT
signaling pathways in succession, thereby leading to tumor
proliferation (103). Second,
TMEM16A participates in cancer proliferation and migration by
influencing the MAPK and CAMK signaling pathways. Subsequently, the
MAPK signaling pathway activates the MAPKK signaling pathway, which
binds to the Grb2 binding site on EGFR with son of sevenless
protein on Ras protein, altering the synthesis and activation of
the Raf protein, which subsequently activates CCND1 via the
phosphorylation of MEK (104).
Furthermore, activation of the MAPKK signaling pathway promotes
angiogenesis in vascular endothelial cells (104). As EGFR regulates the expression
levels of tumor-associated genes, different signaling pathways
mediate the development of several types of cancer (105-107). For instance, the development of
glioma is mediated by activation of the NF-κB signaling pathway
(108,109) and HNSCC is mediated by the
Ras/Raf/MEK/ERK1/2 signaling pathway (110). Another set of signaling
pathways, p38 and ERK1/2, are associated with promoting
hepatocarcinogenesis (Fig. 5)
(111).
The number of chromosomal amplicons, the extent of
promoter methylation and miRs regulate the expression levels and
functions of TMEM16A. The amplicon in chromosome 11q13 consists of
TMEM16A-encoding and apoptosis-related genes, such as
FAS-associated death domain protein (112). Upon expression, the amplicon
drives the proliferation of cancer cells. Hypermethylation of the
TMEM16A gene promoter promotes the metastasis of cancer
cells but inhibits their proliferation. By contrast,
hypomethylation enhances the proliferation of cancer cells but
inhibits their metastasis (113). miR-132 (114), miR-9 (115) and miR-381 (97) directly target the mRNAs of
TMEM16A, of which miR-381 downregulates epithelial-mesenchymal
transition by suppressing the TGF-signaling pathway, thereby
inhibiting germinal center cell proliferation and metastasis
(97). In addition to the
downregulation of these miRs, the levels of TMEM16A are upregulated
by transcription of the genes associated with the
IL4/IL13/JAK/STAT3/STAT6 axis, thereby activating histone
deacetylase, enhancing the production of steroids such as
testosterone, removing cells from their physiological environment,
cellular reorganization and promoting mitosis (3,116,117).
The use of niclosamide, a potent TMEM16A inhibitor
that suppresses the expression of NF-κB and the Wnt/β-catenin,
IL-6/JAK1/STAT3 and GSK-3 signaling pathways, has been approved for
use by the US Food and Drug Administration (83,118-120). Niclosamide not only controls
the cell cycle by activating the Let-7d/CDC34 axis (121), but also by blocking the Notch
signaling pathway in addition to inhibiting goblet cell metaplasia
in asthmatic mice (121-125).
Tumorigenesis is also inhibited either via knockdown of the TMEM16A
encoding gene or the exogenous administration of low concentrations
of TMEM16A (126-128). Thus, the antiproliferative
effects of niclosamide are associated with its inhibitory effects
on TMEM16A and have been used in clinical trials in patients with
prostate and colorectal cancer (86,94,119,129). In summary, the various
anticancer effects of niclosamide may be related to inhibiting the
multiple cancer-promoting mechanisms of TMEM16A.
Bee venom is a complex mixture of natural products
such as peptides, enzymes, bioactive amines and non-peptide
components with various pharmacological properties (130,131). Bee venom peptide is a potent
activator of TMEM16; it promoted the Cl− currents in
cells overexpressing the genes encoding TMEM16A, F, J and K; these
proteins can also stimulate phospholipase A2 (PLA2) (7,11,132). Furthermore, reactive oxygen
species (ROS) and lipid peroxidation can activate the TMEM16
proteins (133). The enhanced
production of ROS and its associated lipid peroxidation causes
ferroptosis (134), which is
mainly characterized by iron-dependent lipid peroxide
damage-induced cell death occurring in the mitochondria. Lipid
peroxidation can be induced by erastin inhibition of cysteine
import through the transporter system Xc−, which leads
to the depletion of glutathione and the inactivation of glutathione
peroxidase (135). The death of
cancer cells can be induced by bee venom peptide, and
PLA2-dependent activation of metalloproteinase is essential for
this effect (132). Therefore,
the bee venom peptide promotes the iron-dependent death of cells,
including cancer cells, a process in which TMEM16A and F are
activated, in turn leading to the activation of PLA2 (132,136), which then finally induces the
death of cancer cells (133,137). A number of studies have
unraveled the underlying mechanisms and supported the potential
therapeutic applications of TMEM16 protein, but its side effects on
the human body still need further study.
TMEM16F is the most widely expressed TMEM16 protein
that functions as both an ion channel and a phospholipid scramblase
and plays a significant role in several physiological processes of
various cells (46). The PS that
arises on the surface of the activated platelets necessitates the
involvement of phospholipid scramblases, such as TMEM16F, in the
formation of the thrombin and prothrombin complex (138). Hence, TMEM16F-mediated exposure
to PS is an important process in platelet aggregation and release
to the blood (139). TMEM16F
and its closest paralog, TMEM16E, both support coagulation on
endothelial cells via PS externalization (138,140). As aforementioned, mutated
TMEM16F protein can cause Scott Syndrome, a hemorrhagic disease
with symptoms such as defects in blood coagulation, long-term
bleeding and thrombosis (141).
TMEM16F is a Ca2+-activated non-selective channel, which
plays an essential role in the exposure of PS and the repair of the
plasma membrane after pore formation (141-143). Therefore, TMEM16F may be a
target for the innovation of novel drugs that can help treat
hemostasis and thrombotic diseases (such as stroke and heart
attack) in humans. In addition, endothelial cells play a thrombotic
role in hyperuricemia via the TMEM16F-mediated exposure of PS and
the release of particulate molecules into the blood (144).
As the name suggests, COVID-19 was first identified
in 2019 and has become a serious worldwide health concern due to
the large number of fatalities. To reduce the mortality rate in
critical patients of COVID-19, targeted therapeutics are
continuously being developed based on biological and etiological
characteristics. COVID-19 causes severe respiratory conditions,
such as pulmonary edema and thrombosis, acute respiratory distress
syndrome and other diseases (145-147). The development of syncytia in
the lungs of patients is a characteristic feature of infection by
coronavirus. The host cell acts on angiotensin-converting enzyme 2
(ACE2) to activate the severe acute respiratory syndrome
coronavirus 2 (SARS-CoV-2) spike protein in a two-step hydrolytic
process (148). The first step
involves breaking the spike between the S1 and S2 subunits of the
spike protein before or after binding to the receptor. The second
step involves the hydrolysis and exposure of the S2 subunit, which
immediately binds to the cell membrane and generates a protease to
invade normal lung cells (149). Infected lung cells present with
a multinucleated and abnormal morphology; SARS-CoV-2 and Middle
East Respiratory Syndrome Coronavirus can fuse with the cells
expressing the relevant receptors to form syncytia (150).
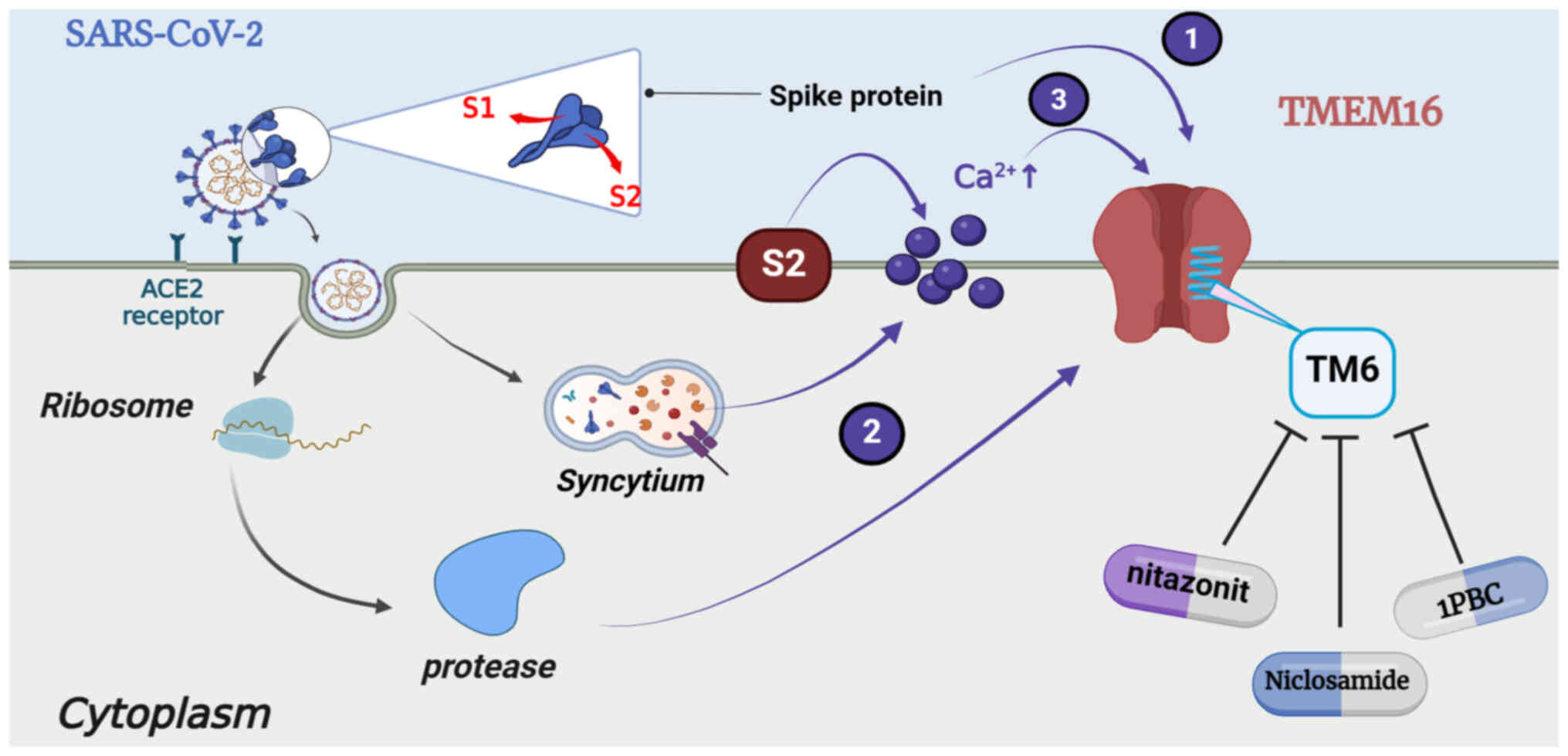
The cells that form syncytia and express the
SARS-CoV-2 spike protein on their surface demonstrate increased
concentrations and enhanced oscillations in the levels of
Ca2+ along with high expression of the
Ca2+-activated TMEM16 proteins in the cytoplasmic
membrane, resulting in the relocation of PS and secretion of
Cl− (149). The
associations between the levels of TMEM16, the levels of
Ca2+ and the activation of TMEM16 by the SARS-CoV-2
spike protein may enhance the magnitude of Ca2+-based
signaling spontaneously (150).
At least three mechanisms explaining the activation of TMEM16 have
been proposed. The first involves the direct cis-binding and
activation of spike-protein-expressing cells, the second involves
trans-binding and the initiation of protease activity via binding
to ACE2 and the third involves indirect activation by inducing the
release of Ca2+ (150). TMEM16F can significantly reduce
calcium oscillations and membrane conductivity in spike expressing
cells, used to expose PS on the cell surface (150). However, the overexpression of
TMEM16F significantly stimulated the SARS-CoV-2 spike
protein-induced formation of syncytia. Therefore, the expression of
the spike protein, which is required for the formation of syncytia,
can be reduced by the downregulation of TMEM16F. Thus, TMEM16F is
identified as the major bifunctional cell membrane-phospholipid
scramblase and Ca2+ channel these cells (Fig. 6).
In patients affected by COVID-19,
inflammation-induced damage to endothelial cells may lead to the
release of a large amount of plasmin activator, thus producing high
concentrations of D-dimers and degradation products of fibrin. A
specific cytokine storm composed of high concentrations of
proinflammatory cytokines and chemokines occurs in the body
(146). These proinflammatory
factors include TNF-α, IL-1 and IL-6. TNF-α and IL-1 are the
primary mediators driving the inhibition of the endogenous
anticoagulant pathway (147).
IL-6 can induce the expression of tissue factors on monocytes,
subsequently initiating the activation of coagulation and
generation of thrombin (150).
The PS on the surface of the activated platelets requires the
activity of the phospholipid scramblase to participate in the
formation of the thrombin and prothrombin complex, and the
TMEM16F-mediated exposure of PS is crucial for the aggregation of
platelets and release to the blood (151). The activation of TMEM16F
increases with the formation of syncytia, enhances the aggregation
of platelets and utilization of the coagulation factors, which
leads to the production of an abnormal amount of thrombin and
fibrin and finally causes disseminated intravascular coagulation
(DIC) and microangiopathy (146-151). The involvement of TMEM16F in
COVID-19-related disorders in blood coagulation results in a
combination of low-grade DIC and local, pulmonary and thrombotic
microvascular disease, which may significantly impact the
dysfunction of the organs in the most severely affected
patients.
Specific drugs should be developed to combat
COVID-19 and to provide strategies for treating similar
coronaviruses by analyzing the mechanisms behind the formation of
viral syncytia and mining for the drugs acting on the
Cl− channels. Niclosamide, a drug that suppresses the
formation of syncytium by inhibiting TMEM16F, has been identified
as a promising drug for the treatment of severe COVID-19 and has
been approved for use by the US Food and Drug Administration
(150,152). Niclosamide is highly
hydrophobic and therefore has poor solubility in aqueous solution
(153). The hydrophobic helix
formed by the TM1-6 grooves in both TMEM16A and F and the residues
in this helix are essential for drug binding (11). TM6 functions as the main gating
element of the channel and is part of the ion-conduction pore
formed by TMEM16A and F (2,154). The antagonist simultaneously
locks both of these ion-conduction pores by binding to the upper
region of the TM6 in a closed configuration. In addition to
niclosamide, nitazoxanide and 1PBC bind to the same conserved sites
(11,155).
Osteoporosis is a systemic disease resulting in a
decrease in the mineral density and quality of the bones due to
various causes, thus leading to changes in the microstructure and
increased fragility of bones, thereby leading to fractures
(158). TMEM16A is directly
regulated by the cytosolic concentrations of Ca2+ and
indirectly by interactions with calmodulin (12). Osteoporosis causes changes in the
Ca2+ concentrations, which affects the levels and
function of TMEM16A and therefore can be used as a marker for the
early diagnosis and targeted intervention of osteoporosis.
Furthermore, a deletion in the gene encoding TMEM16A caused severe
defects in the tracheal cartilage in mice, which could be fatal in
certain cases (159). In
addition, the TMEM16A blockers, benzbromarone and CaCCinh-A0, 1 can
significantly inhibit the differentiation of osteoclasts (159).
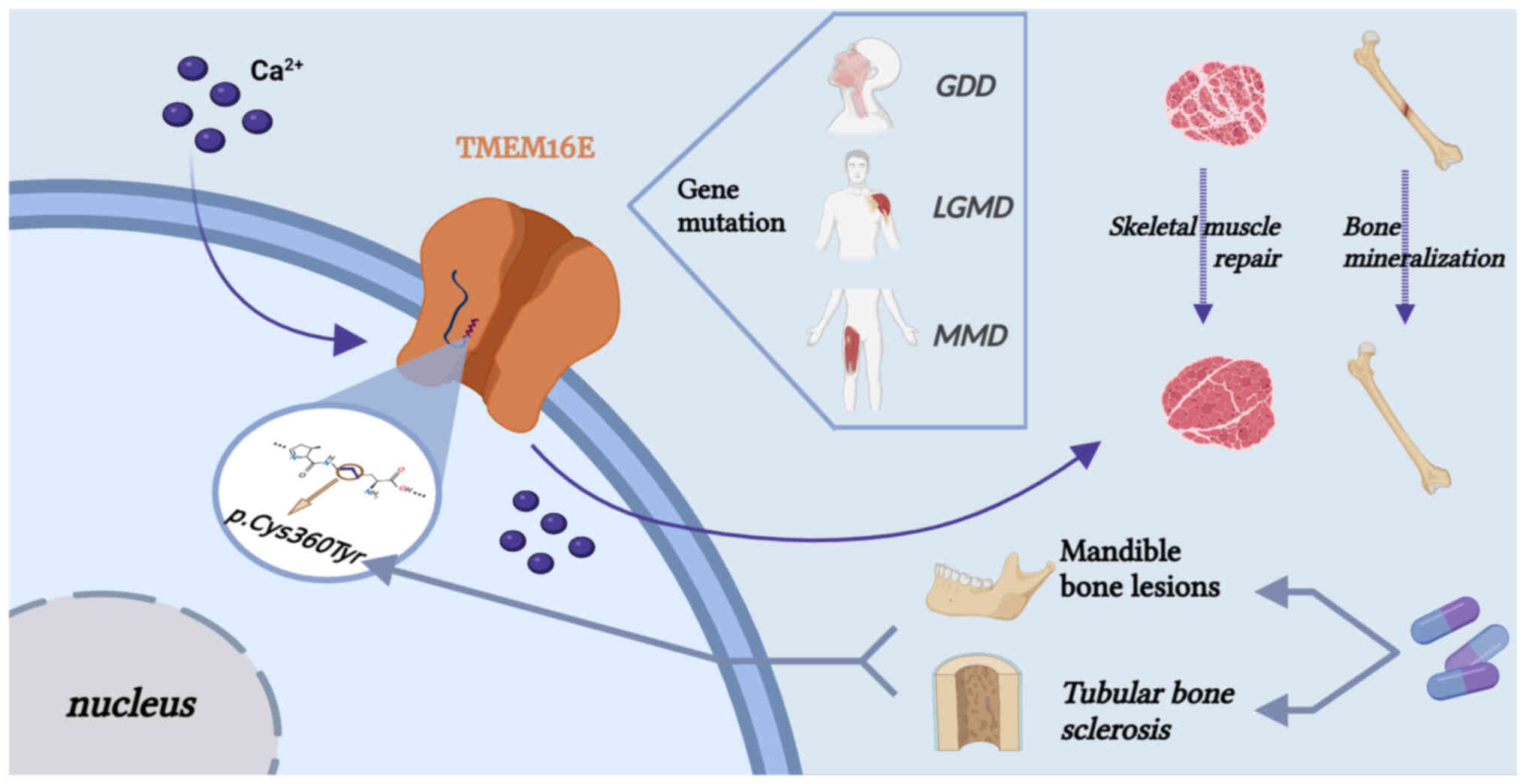
TMEM16E is highly associated with TMEM16F; both are
dual-function proteins with non-selective ion channel and
phospholipid scramblase activities. In addition to participating in
the exposure of PS and promoting different physiological processes,
TMEM16E is also involved in bone mineralization and skeletal muscle
repair (11). Expression of
TMEM16E is highest in the cardiac and skeletal muscles and in
growth plate chondrocytes and osteoblasts (43). Dysfunctional TMEM16E protein
causes bone dysplasia and fragility and suppurative osteomyelitis
of the lower jaw; in addition, LGMD-related amino acid
substitutions cause a loss of function, while GDD-related
substitutions lead to a constitutive lipid scramblase activity
without the requirement of elevated cytosolic Ca2+
levels (42). The dysregulation
of TMEM16E also causes arthritis and MD (162,163). Mutated TMEM16C protein causes
muscle-related diseases associated with involuntary muscle spasms
caused by cranial-neck dystonia (165). The discovery of the
relationship between GDD-related mutations and the functional
phenotypes confirmed the speculation that interventions based on
genetic patterns may greatly improve the treatment outcomes of
genetic diseases in humans, which is of great significance for the
health and survival of affected individuals. Therefore, it is
crucial to understand the mechanism of action of TMEM16 in
genetic-related orthopedic diseases and to decipher the molecular
mechanisms underlying the activation of gating and regulatory
functions to develop novel targeted drug-based therapies towards
the early intervention of diseases in the future.
In summary, the TMEM16 proteins have emerged as
important pharmacological targets for the treatment of several
associated diseases. Since the discovery and identification of
TMEM16 as a Cl− channel, its roles in various human
diseases have been established. The availability of a large amount
of structural information has led to significant progress in
understanding the role of TMEM16 in disease progression at the
molecular level. Thus, its role in the pathogenesis of human
diseases, research involving the therapy of such diseases based on
the targeting of TMEM16 and understanding the mechanisms of action
of the proposed drugs with an enhanced potential should be
addressed further. The studies on the structural characteristics of
TMEM16 have led to the revelation of its various functions, ranging
from ion transport to the modulation of the dynamics of the plasma
membrane phospholipids and the underlying molecular mechanisms.
However, further research is needed to fully understand the roles
played by the other anoctamins besides TMEM16A and B. This requires
efforts based on multiple methods, including gene silencing in
wild-type cells, overexpression in hematopoiesis systems and
generating conditional gene knockouts in mice.
Based on the various research models, such as the
'modular design' model explaining TMEM16 assembly and the
'clam-shell' and the 'pore-dilation' gating/permeation models
elaborating the scramblase and channel activities of TMEM16, it is
possible to address: i) The study and analyses of the synergistic
effects of the three Ca2+-binding sites on the TMEM16
protein under normal physiological environments; ii) a more
in-depth analysis of the mutated sites in the gene encoding the
TMEM16 protein that is related to the manifestation of genetic
diseases in humans to develop novel therapeutic drugs with improved
specificity and targeting; and iii) the requirement of the careful
designing of such drugs to avoid side effects as TMEM16A, F and
other anoctamins are expressed in multiple tissues of the human
body. It is thus hoped that future research on the structural
properties of TMEM16 proteins can capture the open conformations
and assist the designing of potential drugs that target
Cl− channels via utilizing the existing models more
comprehensively and promoting development and innovation in this
field.
Not applicable.
ZH, ZI, ZZ, XC, AM, JL, WL and ZD participated in
the literature review, ZH and ZD performed the figure design and ZH
wrote the manuscript. ZH, ZI and ZD revised the paper. All authors
have read and approved the final version of the manuscript. Data
authentication is not applicable.
Not applicable.
Not applicable.
The authors declare that they have no competing
interests.
Wencui Li ORCID ID: 0000-0003-2787-5360. Zhiqin
Deng ORCID ID: 0000-0002-0819-8504.
Not applicable.
This study was supported by The National Natural Science
Foundation of China (grant no. 81972085, 82172465), China
University Industry-University-Research Innovation Fund (grant no.
2021JH037), The Natural Science Foundation of Guangdong Province
(grant nos. 2021A1515010706 and 2023A1515010102), Guangdong
Provincial Key Clinical Discipline-Orthopedics (grant no. 2000005),
The Sanming Project of Shenzhen Health and Family Planning
Commission (grant no. SZSM202311008), Shenzhen Science and
Technology Planning (grant no. GJHZ20210705142007023) and The
Shenzhen Key Medical Discipline Construction Fund (grant no.
SZXK025).
|
1
|
Vocke K, Dauner K, Hahn A, Ulbrich A,
Broecker J, Keller S, Frings S and Möhrlen F: Calmodulin-dependent
activation and inactivation of anoctamin calcium-gated chloride
channels. J Gen Physiol. 142:381–404. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Whitlock JM and Hartzell HC:
Anoctamins/TMEM16 proteins: Chloride channels flirting with lipids
and extracellular vesicles. Annu Rev Physiol. 79:119–143. 2017.
View Article : Google Scholar :
|
|
3
|
Kunzelmann K, Ousingsawat J, Benedetto R,
Cabrita I and Schreiber R: Contribution of Anoctamins to cell
survival and cell death. Cancers. 11:3822019. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Scudieri P, Sondo E, Ferrera L and
Galietta LJV: The anoctamin family: TMEM16A and TMEM16B as
calcium-activated chloride channels. Exp Physiol. 97:177–183. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kim H, Kim H, Lee J, Lee B, Kim HR, Jung
J, Lee MO and Oh U: Anoctamin 9/TMEM16J is a cation channel
activated by cAMP/PKA signal. Cell Calcium. 71:75–85. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Khelashvili G, Falzone ME, Cheng X, Lee
B-C, Accardi A and Weinstein H: Dynamic modulation of the lipid
translocation groove generates a conductive ion channel in
Ca2+-bound nhTMEM16. Nat Commun. 10:49722019. View Article : Google Scholar :
|
|
7
|
Agostinelli E and Tammaro P: Polymodal
control of TMEM16x channels and Scramblases. Int J Mol Sci.
23:15802022. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Baethge C, Goldbeck-Wood S and Mertens S:
SANRA-a scale for the quality assessment of narrative review
articles. Res Integr Peer Rev. 4:52019. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Falzone ME, Malvezzi M, Lee BC and Accardi
A: Known structures and unknown mechanisms of TMEM16 scramblases
and channels. J Gen Physiol. 150:933–947. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Falzone ME, Rheinberger J, Lee BC, Peyear
T, Sasset L, Raczkowski AM, Eng ET, Di Lorenzo A, Andersen OS,
Nimigean CM and Accardi A: Structural basis of Ca2+-dependent
activation and lipid transport by a TMEM16 scramblase. ELife.
8:e432292019. View Article : Google Scholar :
|
|
11
|
Cheng Y, Feng S, Puchades C, Ko J,
Figueroa E, Chen Y, Wu H, Gu S, Han T, Li J, et al: Identification
of a conserved drug binding pocket in TMEM16 proteins. Res Sq.
View Article : Google Scholar
|
|
12
|
Pedemonte N and Galietta LJV: Structure
and function of TMEM16 proteins (Anoctamins). Physiol Rev.
94:419–459. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Yu K, Duran C, Qu Z, Cui YY and Hartzell
HC: Explaining calcium-dependent gating of Anoctamin-1 chloride
channels requires a revised topology. Circ Res. 110:990–999. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Jung J, Nam JH, Park HW, Oh U, Yoon JH and
Lee MG: Dynamic modulation of ANO1/TMEM16A HCO3−
permeability by Ca2+/calmodulin. Proc Natl Acad Sci USA.
110:360–365. 2012. View Article : Google Scholar
|
|
15
|
Tian Y, Kongsuphol P, Hug M, Ousingsawat
J, Witzgall R, Schreiber R and Kunzelmann K: Calmodulin-dependent
activation of the epithelial calcium-dependent chloride channel
TMEM16A. FASEB J. 25:1058–1068. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Hahn A, Salomon JJ, Leitz D, Feigenbutz D,
Korsch L, Lisewski I, Schrimpf K, Millar-Büchner P, Mall MA, Frings
S and Möhrlen F: Expression and function of Anoctamin 1/TMEM16A
calcium-activated chloride channels in airways of in vivo mouse
models for cystic fibrosis research. Pflugers Arch. 470:1335–1348.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Falzone ME, Feng Z, Alvarenga OE, Pan Y,
Lee B, Cheng X, Fortea E, Scheuring S and Accardi A: TMEM16
scramblases thin the membrane to enable lipid scrambling. Nat
Commun. 13:26042022. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Jansen K and Steurer S: DOG1 expression is
in common human tumors: A tissue microarray study on more than
15,000 tissue samples. Am J Clin Pathol. 156(Suppl): S108–S109.
2021. View Article : Google Scholar
|
|
19
|
Lam AK and Dutzler R: Calcium-dependent
electrostatic control of anion access to the pore of the
calcium-activated chloride channel TMEM16A. ELife. 7:e391222018.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Huang WC, Xiao S, Huang F, Harfe BD, Jan
YN and Jan L: Calcium-Activated chloride channels (CaCCs) regulate
action potential and synaptic response in hippocampal neurons.
Neuron. 74:179–192. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Davis AJ, Forrest AS, Jepps TA, Valencik
ML, Wiwchar M, Singer CA, Sones WR, Greenwood IA and Leblanc N:
Expression profile and protein translation of TMEM16A in murine
smooth muscle. Am J Physiol Cell Physiol. 299:C948–C959. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Thomas-Gatewood C, Neeb ZP, Bulley S,
Adebiyi A, Bannister JP, Leo MD and Jaggar JH: TMEM16A channels
generate Ca2+-activated Cl-currents in cerebral artery
smooth muscle cells. Am J Physiol Circ Physiol. 301:H1819–H1827.
2011. View Article : Google Scholar
|
|
23
|
Li RS, Wang Y, Chen HS, Jiang FY, Tu Q, Li
WJ and Yin RX: TMEM16A contributes to angiotensin II-induced
cerebral vasoconstriction via the RhoA/ROCK signaling pathway. Mol
Med Report. 13:3691–3699. 2016. View Article : Google Scholar
|
|
24
|
Lian H, Cheng Y and Wu X: TMEM16A
exacerbates renal injury by activating P38/JNK signaling pathway to
promote podocyte apoptosis in diabetic nephropathy mice. Biochem
Biophys Res Commun. 487:201–208. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Dutta AK, Khimji AK, Liu S, Karamysheva Z,
Fujita A, Kresge C, Rockey DC and Feranchak AP: PKCα regulates
TMEM16A-mediated Cl-secretion in human biliary cells. Am J Physiol
Liver Physiol. 310:G34–G42. 2016.
|
|
26
|
Arreola J, López-Romero AE, Pérez-Cornejo
P and Rodríguez-Menchaca AA: Phosphatidylinositol 4,5-bisphosphate
and cholesterol regulators of the calcium-activated chloride
channels TMEM16A and TMEM16B. Adv Exp Med Biol. 1422:279–304. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Lee D, Lim H, Lee J, Ha GE, No KT and
Cheong E: Intracellular loop in the brain isoforms of anoctamin 2
channels regulates calcium-dependent activation. Exp Neurobiol.
32:133–146. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Pietra G, Dibattista M, Menini A, Reisert
J and Boccaccio A: The Ca2+-activated Cl-channel TMEM16B
regulates action potential firing and axonal targeting in olfactory
sensory neurons. J Gen Physiol. 148:293–311. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Ayoglu B, Mitsios N, Kockum I, Khademi M,
Zandian A, Sjöberg R, Forsström B, Bredenberg J, Lima Bomfim I,
Holmgren E, et al: Anoctamin 2 identified as an autoimmune target
in multiple sclerosis. Proc Natl Acad Sci USA. 113:2188–2193. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Ha GE, Lee J, Kwak H, Song K, Kwon J, Jung
SY, Hong J, Chang GE, Hwang EM, Shin HS, et al: The
Ca2+-activated chloride channel anoctamin-2 mediates
spike-frequency adaptation and regulates sensory transmission in
thalamocortical neurons. Nat Commun. 7:137912016. View Article : Google Scholar
|
|
31
|
Zhang Y, Zhang Z, Xiao S, Tien J, Le S, Le
T, Jan LY and Yang H: Inferior Olivary TMEM16B mediates cerebellar
motor learning. Neuron. 95:1103–1111.e4. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Kim H, Kim E and Lee BC: Investigation of
phosphatidylserine-transporting activity of human TMEM16C isoforms.
Membranes (Basel). 12:10052022. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Wang TA, Chen C, Huang F, Feng S, Tien J,
Braz JM, Basbaum AI, Jan YN and Jan LY: TMEM16C is involved in
thermoregulation and protects rodent pups from febrile seizures.
Proc Natl Acad Sci USA. 118:e20233421182021. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Huang F, Wang X, Ostertag EM, Nuwal T,
Huang B, Jan YN, Basbaum AI and Jan LY: TMEM16C facilitates
Na(+)-activated K+ currents in rat sensory neurons and regulates
pain processing. Nat Neurosci. 16:1284–1290. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Carvalho V, Martins J, Correia F, Costa M,
Massano J and Temudo T: Another twist in the tale: Intrafamilial
phenotypic heterogeneity in ANO3-related dystonia. Mov Disord Clin
Pract. 8:758–762. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Stamelou M, Charlesworth G, Cordivari C,
Schneider SA, Kägi G, Sheerin UM, Rubio-Agusti I, Batla A, Houlden
H, Wood NW and Bhatia KP: The phenotypic spectrum of DYT24 due to
ANO3 mutations. Mov Disord. 29:928–934. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Esposito M, Trinchillo A, Piceci-Sparascio
F, D'Asdia MC, Consoli F and De Luca A: A novel ANO3 variant in two
siblings with different phenotypes. Parkinsonism Relat Disord.
111:1054132023. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Reichhart N, Milenkovic VM, Wetzel CH and
Strauß O: Prediction of functional consequences of missense
mutations in ANO4 Gene. Int J Mol Sci. 22:27322021. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Maniero C, Scudieri P, Haris Shaikh L,
Zhao W, Gurnell M, Galietta LJV and Brown MJ: ANO4 (Anoctamin 4) Is
a novel marker of zona glomerulosa that regulates stimulated
aldosterone secretion. Hypertension. 74:1152–1159. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Sherva R, Tripodis Y, Bennett DA, Chibnik
LB, Crane PK, de Jager PL, Farrer LA, Saykin AJ, Shulman JM, Naj A,
et al: Genome-wide association study of the rate of cognitive
decline in Alzheimer's disease. Alzheimers Dement. 10:45–52. 2014.
View Article : Google Scholar
|
|
41
|
Di Zanni E, Gradogna A, Scholz-Starke J
and Boccaccio A: Gain of function of TMEM16E/ANO5 scrambling
activity caused by a mutation associated with gnathodiaphyseal
dysplasia. Cell Mol Life Sci. 75:1657–1670. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Di Zanni E, Gradogna A, Picco C,
Scholz-Starke J and Boccaccio A: TMEM16E/ANO5 mutations related to
bone dysplasia or muscular dystrophy cause opposite effects on
lipid scrambling. Hum Mutat. 41:1157–1170. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Whitlock JM, Yu K, Cui YY and Hartzell HC:
Anoctamin 5/TMEM16E facilitates muscle precursor cell fusion. J Gen
Physiol. 150:1498–1509. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Foltz SJ, Cui YY, Choo HJ and Hartzell HC:
ANO5 ensures trafficking of annexins in wounded myofibers. J Cell
Biol. 220:e2020070592021. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
van Kruchten R, Mattheij NJ, Saunders C,
Feijge MA, Swieringa F, Wolfs JL, Collins PW, Heemskerk JW and
Bevers EM: Both TMEM16F-dependent and TMEM16F-independent pathways
contribute to phosphatidylserine exposure in platelet apoptosis and
platelet activation. Blood. 121:1850–1857. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Arndt M, Alvadia C, Straub MS, Clerico
Mosina V, Paulino C and Dutzler R: Structural basis for the
activation of the lipid scramblase TMEM16F. Nat Commun.
13:66922022. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Fujii T, Sakata A, Nishimura S, Eto K and
Nagata S: TMEM16F is required for phosphatidylserine exposure and
microparticle release in activated mouse platelets. Proc Natl Acad
Sci USA. 112:12800–12805. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Millington-Burgess SL and Harper MT: Gene
of the issue: ANO6 and Scott syndrome. Platelets. 31:964–967. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Li H, Xu L, Gao Y, Zuo Y, Yang Z, Zhao L,
Chen Z, Guo S and Han R: BVES is a novel interactor of ANO5 and
regulates myoblast differentiation. Cell Biosci. 11:2222021.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Guo J, Wang D, Dong Y, Gao X, Tong H, Liu
W, Zhang L and Sun M: ANO7: Insights into topology, function, and
potential applications as a biomarker and immunotherapy target.
Tissue Cell. 72:1015462021. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Kaikkonen E, Rantapero T, Zhang Q, Taimen
P, Laitinen V, Kallajoki M, Jambulingam D, Ettala O, Knaapila J,
Boström PJ, et al: ANO7 is associated with aggressive prostate
cancer. Int J Cancer. 143:2479–2487. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Jha A, Chung WY, Vachel L, Maleth J, Lake
S, Zhang G, Ahuja M and Muallem S: Anoctamin 8 tethers endoplasmic
reticulum and plasma membrane for assembly of Ca2+
signaling complexes at the ER/PM compartment. EMBO J.
38:e1014522019. View Article : Google Scholar
|
|
53
|
Liu X, Lai H, Zeng X, Xin S, Nie L, Liang
Z, Wu M, Chen Y, Zheng J and Zou Y: Whole-exome sequencing reveals
ANO8 as a genetic risk factor for intrahepatic cholestasis of
pregnancy. BMC Pregnancy Childbirth. 20:5442020. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Schreiber R, Ousingsawat J and Kunzelmann
K: Targeting of intracellular TMEM16 proteins to the plasma
membrane and activation by purinergic signaling. Int J Mol Sci.
21:40652020. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Katsurahara K, Shiozaki A, Kosuga T, Kudou
M, Shoda K, Arita T, Konishi H, Komatsu S, Kubota T, Fujiwara H, et
al: ANO9 regulated cell cycle in human esophageal squamous cell
carcinoma. Ann Surg Oncol. 27:3218–3230. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Katsurahara K, Shiozaki A, Kosuga T,
Shimizu H, Kudou M, Arita T, Konishi H, Komatsu S, Kubota T,
Fujiwara H, et al: ANO9 regulates PD-L2 expression and binding
ability to PD-1 in gastric cancer. Cancer Sci. 112:1026–1037. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Jun I, Park HS, Piao H, Han JW, An MJ, Yun
BG, Zhang X, Cha YH, Shin YK, Yook JI, et al: ANO9/TMEM16J promotes
tumourigenesis via EGFR and is a novel therapeutic target for
pancreatic cancer. Br J Cancer. 117:1798–1809. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Schreiber R, Talbi K, Ousingsawat J and
Kunzelmann K: A TMEM16J variant leads to dysregulated cytosolic
calcium which may lead to renal disease. FASEB J. 37:e226832023.
View Article : Google Scholar
|
|
59
|
Chrysanthou A, Ververis A and
Christodoulou K: ANO10 function in health and disease. Cerebellum.
22:447–467. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Wanitchakool P, Ousingsawat J, Sirianant
L, Cabrita I, Faria D, Schreiber R and Kunzelmann K: Cellular
defects by deletion of ANO10 are due to deregulated local calcium
signaling. Cell Signal. 30:41–49. 2017. View Article : Google Scholar
|
|
61
|
Hammer C, Wanitchakool P, Sirianant L,
Papiol S, Monnheimer M, Faria D, Ousingsawat J, Schramek N, Schmitt
C, Margos G, et al: A coding variant of ANO10, affecting volume
regulation of macrophages, is associated with borrelia
seropositivity. Mol Med. 21:26–37. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Gentzsch M and Mall MA: Ion channel
modulators in cystic fibrosis. Chest. 154:383–393. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Shteinberg M, Haq IJ, Polineni D and
Davies JC: Cystic fibrosis. Lancet. 397:2195–2211. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Lopes-Pacheco M, Pedemonte N and Veit G:
Discovery of CFTR modulators for the treatment of cystic fibrosis.
Expert Opin Drug Discov. 16:897–913. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Villamizar O, Waters SA, Scott T, Grepo N,
Jaffe A and Morris KV: Mesenchymal Stem Cell exosome delivered Zinc
Finger Protein activation of cystic fibrosis transmembrane
conductance regulator. J Extracell Vesicles. 10:e120532021.
View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Simon MA and Csanady L: Understanding
impact of δF508 and G551D CFTR mutations on CFTR/PKA-c interaction.
Biophys J. 122:112a2023. View Article : Google Scholar
|
|
67
|
Harrison MJ, Murphy DM and Plant BJ:
Ivacaftor in a G551D homozygote with cystic fibrosis. N Engl J Med.
369:1280–1282. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Ramsey BW, Davies J, McElvaney NG, Tullis
E, Bell SC, Dřevínek P, Griese M, McKone EF, Wainwright CE, Konstan
MW, et al: A CFTR potentiator in patients with cystic fibrosis and
theG551Dmutation. N Engl J Med. 365:1663–1672. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Fiedorczuk K and Chen J: Mechanism of CFTR
correction by type I folding correctors. Cell. 185:158–168.e11.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Veit G, Roldan A, Hancock MA, Da Fonte DF,
Xu H, Hussein M, Frenkiel S, Matouk E, Velkov T and Lukacs GL:
Allosteric folding correction of F508del and rare CFTR mutants by
Elexacaftor-Tezacaftor-Ivacaftor (Trikafta) combination. JCI
Insight. 5:e1399832020. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Rowe SM, McColley SA, Rietschel E, Li X,
Bell SC, Konstan MW, Marigowda G, Waltz D and Boyle MP;
VX09-809-102 Study Group: Lumacaftor/Ivacaftor treatment of
patients with cystic fibrosis heterozygous for F508del-CFTR. Ann Am
Thorac Soc. 14:213–219. 2017. View Article : Google Scholar :
|
|
72
|
Wainwright CE, Elborn JS, Ramsey BW,
Marigowda G, Huang X, Cipolli M, Colombo C, Davies JC, De Boeck K,
Flume PA, et al: Lumacaftor-Ivacaftor in patients with cystic
fibrosis homozygous for Phe508delCFTR. N Engl J Med. 373:220–231.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Clancy JP, Rowe SM, Accurso FJ, Aitken ML,
Amin RS, Ashlock MA, Ballmann M, Boyle MP, Bronsveld I, Campbell
PW, et al: Results of a phase IIa study of VX-809, an
investigational CFTR corrector compound, in subjects with cystic
fibrosis homozygous for theF508del-CFTRmutation. Thorax. 67:12–18.
2011. View Article : Google Scholar
|
|
74
|
Flume PA, Harris RS, Paz-Diaz H, Ahluwalia
N, Higgins M, Campbell D, Berhane I, Shih JL and Sawicki G:
Long-term tezacaftor/ivacaftor safety and efficacy in people with
cystic fibrosis and an F508del-CFTR mutation: 96-week, open-label
extension of the EXTEND trial. J Cyst Fibros. 22:464–470. 2023.
View Article : Google Scholar
|
|
75
|
Bruscia EM: The effects of
Elexacaftor/Tezacaftor/Ivacaftor beyond the epithelium: Spurring
macrophages to fight infections. Eur Respir J. 61:23002162023.
View Article : Google Scholar
|
|
76
|
Sawicki GS, Van Brunt K, Booth J, Bailey
E, Millar SJ, Konstan MW and Flume PA: Disease burden in people
with cystic fibrosis heterozygous for F508del and a minimal
function mutation. J Cyst Fibros. 21:96–103. 2022. View Article : Google Scholar :
|
|
77
|
Galietta LJV: TMEM16A (ANO1) as a
therapeutic target in cystic fibrosis. Curr Opin Pharmacol.
64:1022062022. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Simões FB, Quaresma MC, Clarke LA, Silva
IA, Pankonien I, Railean V, Kmit A and Amaral MD: TMEM16A chloride
channel does not drive mucus production. Life Sci Alliance.
2:e2019004622019. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Ruffin M, Voland M, Marie S, Bonora M,
Blanchard E, Blouquit-Laye S, Naline E, Puyo P, Le Rouzic P,
Guillot L, et al: Anoctamin 1 dysregulation alters bronchial
epithelial repair in cystic fibrosis. Biochim Biophys Acta.
1832:2340–2351. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Kirk KL and Wang W: A unified view of
cystic fibrosis transmembrane conductance regulator (CFTR) gating:
Combining the allosterism of a ligand-gated channel with the
enzymatic activity of an ATP-binding cassette (ABC) transporter. J
Biol Chem. 286:12813–12819. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Deng Z, Chen X, Lin Z, Alahdal M, Wang D,
Liu J and Li W: The homeostasis of cartilage matrix remodeling and
the regulation of volume-sensitive ion channel. Aging Dis.
13:787–800. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Talbi K, Ousingsawat J, Centeio R,
Schreiber R and Kunzelmann K: Calmodulin-dependent regulation of
overexpressed but not endogenous TMEM16A expressed in airway
epithelial cells. Membranes (Basel). 11:7232021. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Cabrita I, Benedetto R, Schreiber R and
Kunzelmann K: Niclosamide repurposed for the treatment of
inflammatory airway disease. JCI Insight. 4:e1284142019. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Danahay H, Fox R, Lilley S, Charlton H,
Adley K, Christie L, Ansari E, Ehre C, Flen A, Tuvim MJ, et al:
Potentiating TMEM16A does not stimulate airway mucus secretion or
bronchial and pulmonary arterial smooth muscle contraction. FASEB
Bioadv. 2:464–477. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Danahay HL, Lilley S, Fox R, Charlton H,
Sabater J, Button B, McCarthy C, Collingwood SP and Gosling M:
TMEM16A potentiation: A novel therapeutic approach for the
treatment of cystic fibrosis. Am J Respir Crit Care Med.
201:946–954. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Ousingsawat J, Centeio R, Cabrita I, Talbi
K, Zimmer O, Graf M, Göpferich A, Schreiber R and Kunzelmann K:
Airway delivery of hydrogel-encapsulated niclosamide for the
treatment of inflammatory airway disease. Int J Mol Sci.
23:10852022. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Centeio R, Ousingsawat J, Cabrita I,
Schreiber R, Talbi K, Benedetto R, Doušová T, Verbeken EK, De Boeck
K, Cohen I and Kunzelmann K: Mucus release and airway constriction
by TMEM16A may worsen pathology in inflammatory lung disease. Int J
Mol Sci. 22:78522021. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Sonneville F, Ruffin M, Coraux C,
Rousselet N, Le Rouzic P, Blouquit-Laye S, Corvol H and Tabary O:
MicroRNA-9 downregulates the ANO1 chloride channel and contributes
to cystic fibrosis lung pathology. Nat Commun. 8:7102017.
View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Kamaleddin MA: Molecular, biophysical, and
pharmacological properties of calcium-activated chloride channels.
J Cell Physiol. 233:787–798. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Sah SP and McCluggage WG: DOG1
immunoreactivity in uterine leiomyosarcomas. J Clin Pathol.
66:40–43. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Filippou A, Pehkonen H, Karhemo PR,
Väänänen J, Nieminen AI, Klefström J, Grénman R, Mäkitie AA,
Joensuu H and Monni O: ANO1 expression orchestrates
p27Kip1/MCL1-Mediated signaling in head and neck squamous cell
carcinoma. Cancers (Basel). 13:11702021. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Ishaque N, Abba ML, Hauser C, Patil N,
Paramasivam N, Huebschmann D, Leupold JH, Balasubramanian GP,
Kleinheinz K, Toprak UH, et al: Whole genome sequencing puts
forward hypotheses on metastasis evolution and therapy in
colorectal cancer. Nat Commun. 9:47822018. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Sauter DRP, Novak I, Pedersen SF, Larsen
EH and Hoffmann EK: ANO1 (TMEM16A) in pancreatic ductal
adenocarcinoma (PDAC). Pflugers Arch. 467:1495–1508. 2015.
View Article : Google Scholar :
|
|
94
|
Song Y, Gao J, Guan L, Chen X, Gao J and
Wang K: Inhibition of ANO1/TMEM16A induces apoptosis in human
prostate carcinoma cells by activating TNF-α signaling. Cell Death
Dis. 9:7032018. View Article : Google Scholar
|
|
95
|
Britschgi A, Bill A, Brinkhaus H, Rothwell
C, Clay I, Duss S, Rebhan M, Raman P, Guy CT, Wetzel K, et al:
Calcium-activated chloride channel ANO1 promotes breast cancer
progression by activating EGFR and CAMK signaling. Proc Natl Acad
Sci USA. 110:E1026–E1034. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Sui Y, Sun M, Wu F, Yang L, Di W, Zhang G,
Zhong L, Ma Z, Zheng J, Fang X and Ma T: Inhibition of TMEM16A
expression suppresses growth and invasion in human colorectal
cancer cells. PLoS One. 9:e1154432014. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Cao Q, Liu F, Ji K, Liu N, He Y, Zhang W
and Wang L: MicroRNA-381 inhibits the metastasis of gastric cancer
by targeting TMEM16A expression. J Exp Clin Cancer Res. 36:292017.
View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Lee YS, Lee JK, Bae Y, Lee BS, Kim E, Cho
CH, Ryoo K, Yoo J, Kim CH, Yi GS, et al: Suppression of
14-3-3γ-mediated surface expression of ANO1 inhibits cancer
progression of glioblastoma cells. Sci Rep. 6:264132016. View Article : Google Scholar
|
|
99
|
Shang L, Hao JJ, Zhao XK, He JZ, Shi ZZ,
Liu HJ, Wu LF, Jiang YY, Shi F, Yang H, et al: ANO1 protein as a
potential biomarker for esophageal cancer prognosis and
precancerous lesion development prediction. Oncotarget.
7:24374–24382. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Akpalo H, Lange C and Zustin J: Discovered
on gastrointestinal stromal tumour 1 (DOG1): A useful
immunohistochemical marker for diagnosing chondroblastoma.
Histopathology. 60:1099–1106. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Chang Z, Cai C, Han D, Gao Y, Li Q, Feng
L, Zhang W, Zheng J, Jin J, Zhang H and Wei Q: Anoctamin5 regulates
cell migration and invasion in thyroid cancer. Int J Oncol.
51:1311–1319. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Li Y, Wang X, Vural S, Mishra NK, Cowan KH
and Guda C: Exome analysis reveals differentially mutated gene
signatures of stage, grade and subtype in breast cancers. PLoS One.
10:e01193832015. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Chen W, Gu M, Gao C, Chen B, Yang J, Xie
X, Wang X, Sun J and Wang J: The prognostic value and mechanisms of
TMEM16A in human cancer. Front Mol Biosci. 8:5421562021. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Liu F, Yang X, Geng M and Huang M:
Targeting ERK, an Achilles' heel of the MAPK pathway, in cancer
therapy. Acta Pharm Sin B. 8:552–562. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Wang H, Yao F, Luo S, Ma K, Liu M, Bai L,
Chen S, Song C, Wang T, Du Q, et al: A mutual activation loop
between the Ca2+-activated chloride channel TMEM16A and
EGFR/STAT3 signaling promotes breast cancer tumorigenesis. Cancer
Lett. 455:48–59. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Bai W, Liu M and Xiao Q: The diverse roles
of TMEM16A Ca2+-activated Cl-channels in inflammation. J
Adv Res. 33:53–68. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Lin Z, Deng Z, Liu J, Lin Z, Chen S, Deng
Z and Li W: Chloride channel and inflammation-mediated pathogenesis
of osteoarthritis. J Inflamm Res. 15:953–964. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Liu J, Liu Y, Ren Y, Kang L and Zhang L:
Transmembrane protein with unknown function 16A overexpression
promotes glioma formation through the nuclear factor-κB signaling
pathway. Mol Med Report. 9:1068–1074. 2014. View Article : Google Scholar
|
|
109
|
Zhou L, Deng ZZ, Li HY, Jiang N, Wei ZS,
Hong MF, Chen XD, Wang JH, Zhang MX, Shi YH, et al: TRIM31 promotes
glioma proliferation and invasion through activating NF-κB pathway.
Onco Targets Ther. 12:2289–2297. 2019. View Article : Google Scholar :
|
|
110
|
Duvvuri U, Shiwarski DJ, Xiao D, Bertrand
C, Huang X, Edinger RS, Rock JR, Harfe BD, Henson BJ, Kunzelmann K,
et al: TMEM 16 A induces MAPK and contributes directly to
tumorigenesis and cancer progression. Cancer Res. 72:3270–3281.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Deng L, Yang J, Chen H, Ma B, Pan K, Su C,
Xu F and Zhang J: Knockdown of TMEM16A suppressed MAPK and
inhibited cell proliferation and migration in hepatocellular
carcinoma. Onco Targets Ther. 9:325–333. 2016.PubMed/NCBI
|
|
112
|
Ruiz C, Martins JR, Rudin F, Schneider S,
Dietsche T, Fischer CA, Tornillo L, Terracciano LM, Schreiber R,
Bubendorf L and Kunzelmann K: Enhanced expression of ANO1 in head
and neck squamous cell carcinoma causes cell migration and
correlates with poor prognosis. PLoS One. 7:e432652012. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Dixit R, Kemp C, Kulich S, Seethala R,
Chiosea S, Ling S, Ha PK and Duvvuri U: TMEM16A/ANO1 is
differentially expressed in HPV-negative versus HPV-positive head
and neck squamous cell carcinoma through promoter methylation. Sci
Rep. 5:166572015. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Mokutani Y, Uemura M, Munakata K, Okuzaki
D, Haraguchi N, Takahashi H, Nishimura J, Hata T, Murata K,
Takemasa I, et al: Down-regulation of microRNA-132 is associated
with poor prognosis of colorectal cancer. Ann Surg Oncol.
23:599–608. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Lin S and Gregory RI: MicroRNA biogenesis
pathways in cancer. Nat Rev Cancer. 15:321–333. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Wang H, Zou L, Ma K, Yu J, Wu H, Wei M and
Xiao Q: Cell-specific mechanisms of TMEM16A
Ca2+-activated chloride channel in cancer. Mol Cancer.
16:1522017. View Article : Google Scholar
|
|
117
|
Wanitchakool P, Wolf L, Koehl GE,
Sirianant L, Schreiber R, Kulkarni S, Duvvuri U and Kunzelmann K:
Role of anoctamins in cancer and apoptosis. Philos Trans R Soc Lond
B Biol Sci. 369:201300962014. View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Ahn SY, Yang JH, Kim NH, Lee K, Cha YH,
Yun JS, Kang HE, Lee Y, Choi J, Kim HS and Yook J: Anti-helminthic
niclosamide inhibits Ras-driven oncogenic transformation via
activation of GSK-3. Oncotarget. 8:31856–31863. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Miner K, Labitzke K, Liu B, Wang P,
Henckels K, Gaida K, Elliott R, Chen JJ, Liu L, Leith A, et al:
Drug repurposing: The anthelmintics niclosamide and nitazoxanide
are potent TMEM16A antagonists that fully bronchodilate airways.
Front Pharmacol. 10:512019. View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Jin Y, Lu Z, Ding K, Li J, Du X, Chen C,
Sun X, Wu Y, Zhou J and Pan J: Antineoplastic mechanisms of
niclosamide in acute myelogenous leukemia stem cells: Inactivation
of the NF-κB pathway and generation of reactive oxygen species.
Cancer Res. 70:2516–2527. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Han Z, Li Q, Wang Y, Wang L, Li X, Ge N,
Wang Y and Guo C: Niclosamide induces cell cycle arrest in G1 phase
in head and neck squamous cell carcinoma through Let-7d/CDC34 Axis.
Front Pharmacol. 9:15442019. View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Li Y, Li PK, Roberts MJ, Arend RC, Samant
RS and Buchsbaum DJ: Multi-targeted therapy of cancer by
niclosamide: A new application for an old drug. Cancer Lett.
349:8–14. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Arend RC, Londoño-Joshi AI, Gangrade A,
Katre AA, Kurpad C, Li Y, Samant RS, Li PK, Landen CN, Yang ES, et
al: Correction: Niclosamide and its analogs are potent inhibitors
of Wnt/β-catenin, mTOR and STAT3 signaling in ovarian cancer.
Oncotarget. 9:19459. 2018. View Article : Google Scholar
|
|
124
|
Lafkas D, Shelton A, Chiu C, de Leon
Boenig G, Chen Y, Stawicki SS, Siltanen C, Reichelt M, Zhou M, Wu
X, et al: Therapeutic antibodies reveal Notch control of
transdifferentiation in the adult lung. Nature. 528:127–131. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Danahay H, Pessotti AD, Coote J,
Montgomery BE, Xia D, Wilson A, Yang H, Wang Z, Bevan L, Thomas C,
et al: Notch2 is required for inflammatory cytokine-driven goblet
cell metaplasia in the lung. Cell Rep. 10:239–252. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Seo Y, Kim J, Chang J, Kim SS, Namkung W
and Kim I: Synthesis and biological evaluation of novel Ani9
derivatives as potent and selective ANO1 inhibitors. Eur J
Medicinal Chem. 160:245–255. 2018. View Article : Google Scholar
|
|
127
|
Burock S, Daum S, Keilholz U, Neumann K,
Walther W and Stein U: Phase II trial to investigate the safety and
efficacy of orally applied niclosamide in patients with
metachronous or sychronous metastases of a colorectal cancer
progressing after therapy: The NIKOLO trial. BMC Cancer.
18:2972018. View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Schweizer MT, Haugk K, McKiernan JS,
Gulati R, Cheng HH, Maes JL, Dumpit RF, Nelson PS, Montgomery B,
McCune JS, et al: Correction: A phase I study of niclosamide in
combination with enzalutamide in men with castration-resistant
prostate cancer. PLoS One. 13:e02027092018. View Article : Google Scholar : PubMed/NCBI
|
|
129
|
Yan Y, Ding X, Han C, Gao J, Liu Z, Liu Y
and Wang K: Involvement of TMEM16A/ANO1 upregulation in the
oncogenesis of colorectal cancer. Biochim Biophys Acta Mol Basis
Dis. 1868:1663702022. View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Khalil A, Elesawy BH, Ali TM and Ahmed OM:
Bee venom: From venom to drug. Molecules. 26:49412021. View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Badawi JK: Bee venom components as
therapeutic tools against prostate cancer. Toxins (Basel).
13:3372021. View Article : Google Scholar : PubMed/NCBI
|
|
132
|
Schreiber R, Ousingsawat J, Wanitchakool
P, Sirianant L, Benedetto R, Reiss K and Kunzelmann K: Regulation
of TMEM16A/ANO1 and TMEM16F/ANO6 ion currents and phospholipid
scrambling by Ca2+ and plasma membrane lipid. J Physiol.
596:217–229. 2017. View Article : Google Scholar
|
|
133
|
Simões F, Ousingsawat J, Wanitchakool P,
Fonseca A, Cabrita I, Benedetto R, Schreiber R and Kunzelmann K:
CFTR supports cell death through ROS-dependent activation of
TMEM16F (anoctamin 6). Pflugers Arch. 470:305–314. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
134
|
Stockwell BR, Friedmann Angeli JP, Bayir
H, Bush AI, Conrad M, Dixon SJ, Fulda S, Gascón S, Hatzios SK,
Kagan VE, et al: Ferroptosis: A regulated cell death nexus linking
metabolism, redox biology, and disease. Cell. 171:273–285. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
135
|
Xue Q, Yan D, Chen X, Li X, Kang R,
Klionsky DJ, Kroemer G, Chen X, Tang D and Liu J: Copper-dependent
autophagic degradation of GPX4 drives ferroptosis. Autophagy.
19:1982–1996. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
136
|
El-Didamony SE, Amer RI and El-Osaily GH:
Formulation, characterization and cellular toxicity assessment of a
novel bee-venom microsphere in prostate cancer treatment. Sci Rep.
12:132132022. View Article : Google Scholar : PubMed/NCBI
|
|
137
|
Schreiber R, Buchholz B, Kraus A, Schley
G, Scholz J, Ousingsawat J and Kunzelmann K: Lipid peroxidation
drives renal cyst growth in vitro through activation of TMEM16A. J
Am Soc Nephrol. 30:228–242. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
138
|
Schmaier AA, Anderson PF, Chen SM,
El-Darzi E, Aivasovsky I, Kaushik MP, Sack KD, Hartzell HC, Parikh
SM, Flaumenhaft R and Schulman S: TMEM16E regulates endothelial
cell procoagulant activity and thrombosis. J Clin Invest.
133:e1638082023. View Article : Google Scholar : PubMed/NCBI
|
|
139
|
Fujii Y, Taniguchi M, Nagaya S, Ueda Y,
Hashizume C, Watanabe K, Takeya H, Kosaka T and Okazaki T: A novel
mechanism of thrombocytopenia by PS exposure through TMEM16F in
sphingomyelin synthase 1 deficiency. Blood Adv. 5:4265–4277. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
140
|
Filep JG: Two to tango: Endothelial cell
TMEM16 scramblases drive coagulation and thrombosis. J Clin Invest.
133:e1706432023. View Article : Google Scholar : PubMed/NCBI
|
|
141
|
Wu N, Cernysiov V, Davidson D, Song H,
Tang J, Luo S, Lu Y, Qian J, Gyurova IE, Waggoner SN, et al:
Critical role of lipid scramblase TMEM16F in phosphatidylserine
exposure and repair of plasma membrane after pore formation. Cell
Rep. 30:1129–1140.e5. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
142
|
Taylor KA and Mahaut-Smith MP: A major
interspecies difference in the ionic selectivity of megakaryocyte
Ca2+-activated channels sensitive to the TMEM16F
inhibitor CaCCinh-A01. Platelets. 30:962–966. 2019. View Article : Google Scholar :
|
|
143
|
Yang H, Kim A, David T, Palmer D, Jin T,
Tien J, Huang F, Cheng T, Coughlin SR, Jan YN and Jan LY: TMEM16F
Forms a Ca2+-activated cation channel required for lipid
scrambling in platelets during blood coagulation. Cell.
151:111–122. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
144
|
Yu H, Wang Z, Li Z, An Y, Yan M, Ji S, Xu
M, Wang L, Dong W, Shi J and Gao C: Hyperuricemia enhances
procoagulant activity of vascular endothelial cells through TMEM16F
regulated phosphatidylserine exposure and microparticle release.
FASEB J. 35:e218082021. View Article : Google Scholar : PubMed/NCBI
|
|
145
|
Goyal P, Choi JJ, Pinheiro LC, Schenck EJ,
Chen R, Jabri A, Satlin MJ, Campion TR Jr, Nahid M, Ringel JB, et
al: Clinical characteristics of Covid-19 in new york city. N Engl J
Med. 382:2372–2374. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
146
|
Levi M, Thachil J, Iba T and Levy JH:
Coagulation abnormalities and thrombosis in patients with COVID-19.
Lancet Haematol. 7:e438–e440. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
147
|
Edler C, Schröder AS, Aepfelbacher M,
Fitzek A, Heinemann A, Heinrich F, Klein A, Langenwalder F,
Lütgehetmann M, Meißner K, et al: Correction to: Dying with
SARS-CoV-2 infection-an autopsy study of the first consecutive 80
cases in Hamburg, Germany. Int J Legal Med. 134:19772020.
View Article : Google Scholar : PubMed/NCBI
|
|
148
|
Hoffmann M, Kleine-Weber H, Schroeder S,
Krüger N, Herrler T, Erichsen S, Schiergens TS, Herrler G, Wu NH,
Nitsche A, et al: SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2
and is blocked by a clinically proven protease inhibitor. Cell.
181:271–280.e8. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
149
|
Hoffmann M, Hofmann-Winkler H and Pöhlmann
S: Priming time: How Cellular Proteases Arm Coronavirus Spike
Proteins. Springer International Publishing; Cham: pp. 71–98.
2018
|
|
150
|
Braga L, Ali H, Secco I, Chiavacci E,
Neves G, Goldhill D, Penn R, Jimenez-Guardeño JM, Ortega-Prieto AM,
Bussani R, et al: Drugs that inhibit TMEM16 proteins block
SARS-CoV-2 spike-induced syncytia. Nature. 594:88–93. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
151
|
Cappelletto A, Allan HE, Crescente M,
Schneider E, Bussani R, Ali H, Secco I, Vodret S, Simeone R,
Mascaretti L, et al: SARS-CoV-2 Spike protein activates
TMEM16F-mediated platelet procoagulant activity. Front Cardiovasc
Med. 9:10132622023. View Article : Google Scholar : PubMed/NCBI
|
|
152
|
Abdulamir AS, Gorial FI, Saadi SJ, Maulood
MF, Hashim HA, Alnuaimi AS and Abdulrrazaq MK: A randomised
controlled trial of effectiveness and safety of Niclosamide as add
on therapy to the standard of care measures in COVID-19 management.
Ann Med Surg (Lond). 69:1027792021.PubMed/NCBI
|
|
153
|
Chen W, Mook RA Jr, Premont RT and Wang J:
Niclosamide: Beyond an antihelminthic drug. Cell Signal. 41:89–96.
2018. View Article : Google Scholar
|
|
154
|
Kalienkova V, Clerico Mosina V and Paulino
C: The Groovy TMEM16 family: Molecular mechanisms of lipid
scrambling and ion conduction. J Mol Biol. 433:1669412021.
View Article : Google Scholar : PubMed/NCBI
|
|
155
|
Lam AKM, Rutz S and Dutzler R: Inhibition
mechanism of the chloride channel TMEM16A by the pore blocker 1PBC.
Nat Commun. 13:27982022. View Article : Google Scholar : PubMed/NCBI
|
|
156
|
Bill A, Gutierrez A, Kulkarni S, Kemp C,
Bonenfant D, Voshol H, Duvvuri U and Gaither LA: ANO1/TMEM16A
interacts with EGFR and correlates with sensitivity to
EGFR-targeting therapy in head and neck cancer. Oncotarget.
6:9173–9188. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
157
|
Zhang X, Zhang G, Zhai W, Zhao Z, Wang S
and Yi J: Inhibition of TMEM16A Ca2+-activated
Cl-channels by avermectins is essential for their anticancer
effects. Pharmacol Res. 156:1047632020. View Article : Google Scholar
|
|
158
|
Fang H, Deng Z, Liu J, Chen S, Deng Z and
Li W: The mechanism of bone remodeling after bone aging. Clin
Interv Aging. 17:405–415. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
159
|
Genovese M, Buccirossi M, Guidone D, De
Cegli R, Sarnataro S, di Bernardo D and Galietta LJV: Analysis of
inhibitors of the anoctamin-1 chloride channel (transmembrane
member 16A, TMEM16A) reveals indirect mechanisms involving
alterations in calcium signalling. Br J Pharmacol. 180:775–785.
2023. View Article : Google Scholar
|
|
160
|
Shaibani A, Khan S and Shinawi M:
Autosomal dominant ANO5-Related disorder associated with myopathy
and gnathodiaphyseal dysplasia. Neurol Genet. 7:e6122021.
View Article : Google Scholar : PubMed/NCBI
|
|
161
|
Liu X, Wang X, Ma X, Li H, Miao C, Tian Z
and Hu Y: Genetic disruption of Ano5 leads to impaired
osteoclastogenesis for gnathodiaphyseal dysplasia. Oral Dis.
30:1403–1415. 2024. View Article : Google Scholar
|
|
162
|
Chandra G, Defour A, Mamchoui K, Pandey K,
Mishra S, Mouly V, Sreetama S, Mahad Ahmad M, Mahjneh I, Morizono
H, et al: Dysregulated calcium homeostasis prevents plasma membrane
repair in Anoctamin 5/TMEM16E-deficient patient muscle cells. Cell
Death Discov. 5:1182019. View Article : Google Scholar : PubMed/NCBI
|
|
163
|
Thiruvengadam G, Sreetama SC, Charton K,
Hogarth M, Novak JS, Suel-Petat L, Chandra G, Allard B, Richard I
and Jaiswal JK: Anoctamin 5 Knockout mouse model recapitulates
LGMD2L muscle pathology and offers insight into in vivo functional
deficits. J Neuromuscul Dis. 8(Suppl): S243–S255. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
164
|
Li H, Wang X, Chen E, Liu X, Ma X, Miao C,
Tian Z, Dong R and Hu Y: Introduction of a Cys360Tyr Mutation in
ANO5 creates a mouse model for gnathodiaphyseal dysplasia. J Bone
Miner Res. 37:515–530. 2022. View Article : Google Scholar
|
|
165
|
Jiang LT, Li LX, Liu Y, Zhang XL, Pan YG,
Wang L, Wan XH and Jin LJ: The expanding clinical and genetic
spectrum of ANO3 dystonia. Neurosci Lett. 746:1355902021.
View Article : Google Scholar : PubMed/NCBI
|