Introduction
Osteoarthritis (OA) is the most prevalent
degenerative joint disease globally and the primary cause of joint
pain, discomfort and limited activity among middle-aged and elderly
individuals. As of 2019, ~250 million people worldwide were
affected (1,2). During OA progression, new blood
vessels form in the subchondral bone, extend to the tidemark and
infiltrate the avascular cartilage. Notably, an increase in the
permeability of these new blood vessels promotes infiltration of
various inflammatory factors into the cartilage, which exacerbates
cartilage degeneration and OA severity (3,4).
Additionally, studies have indicated that inhibiting formation of
new blood vessels and reducing permeability in subchondral bone can
mitigate progression of OA (5,6).
However, the specific mechanism underlying increased vascular
permeability in the subchondral bone during OA remains elusive.
Vascular permeability primarily depends on adhesive
connections and tight junctions between endothelial cells (ECs)
(7,8). RhoA is a member of the Rho GTP
family and plays a key role in governing the rearrangement of the
cytoskeleton and intercellular junctions of ECs (9,10). RhoA enhances retinal vascular
permeability by inhibiting the junctions between ECs (11). Additionally, RhoA induces EC
apoptosis and amplifies EC permeability in rats with chronic renal
failure (12). However, it
remains uncertain whether RhoA influences the vascular permeability
of the subchondral bone in OA. Comprehensive understanding of the
mechanism of RhoA in the pathogenesis of OA may facilitate the
development of prevention and treatment strategies.
Ferroptosis is a form of cell death. During
ferroptosis, the antioxidant axis of solute carrier family 7 member
11/glutathione peroxidase 4 (SLC7A11/GPX4) is deactivated,
resulting in intracellular oxidative stress. An increase in
oxidative stress upregulates lipid peroxidation and induces
programmed cell death, with acyl-CoA synthase long-chain family
member 4 (ACSL4) serving a key role (13-15). Ferroptosis influences vascular
permeability (16). For example,
inhibiting ferroptosis in ECs preserves the integrity of the
blood-spinal cord barrier and enhances spinal cord injury recovery
(17). Additionally,
upregulation of lipid peroxidation and inflammatory responses in
retinal vascular ECs prompt EC dysfunction and increase
permeability (18). However, the
role and molecular mechanisms of RhoA in OA are yet to be
elucidated. Therefore, the present study aimed to investigate the
role and molecular mechanisms of RhoA in OA, EC ferroptosis and
vascular permeability.
Materials and methods
Patients and specimens
The present study was approved by the Ethics
Committee of General Hospital of Ningxia Medical University
(Yinchuan, China; approval no. KYLL-2021-269) and all experimental
procedures adhered to the Declaration of Helsinki. Human
subchondral bone samples were obtained from individuals undergoing
total knee arthroplasty. Written informed consent was provided by
the patients.
Tibial plateau samples were collected from 60
patients (male:female ratio, 1:3; age range, 58-82) diagnosed with
knee OA and undergoing total knee arthroplasty at the General
Hospital of Ningxia Medical University, China, between February
2022 and February 2023. The subchondral bone was categorized into
damaged area (OA sample) and corresponding undamaged area (control
sample). OA diagnosis was based on diagnostic criteria of the
American Rheumatology Association (19). Patients with secondary OA, such
as those with trauma or connective tissue disease, were
excluded.
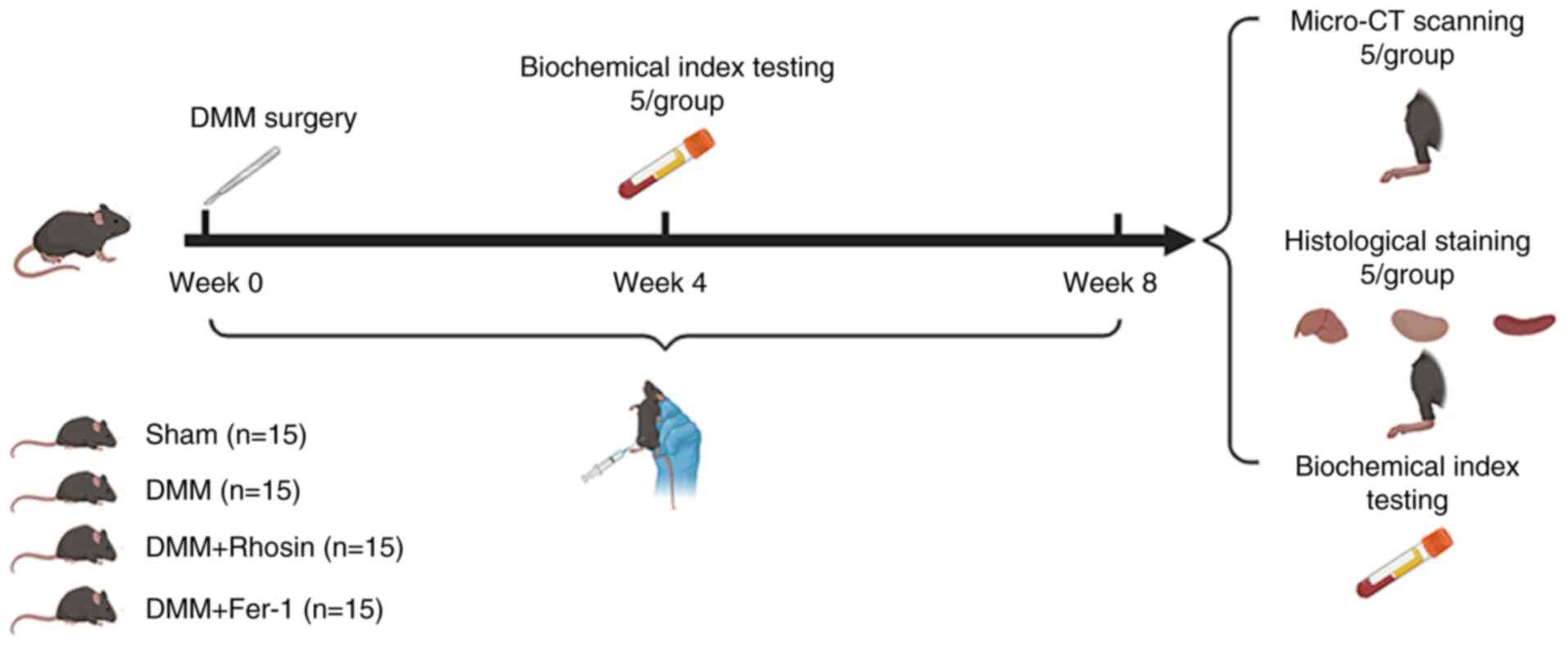
Animal model
In total, 60 healthy adult female C57BL/6 mice (age:
6 weeks; weight: 16g) were acquired from the Experimental Animal
Center of Ningxia Medical University, China. Mice were maintained
in a specific pathogen-free environment under standard conditions
(22±1°C, 55% humidity and 12/12-h light/dark cycle) with free
access to food and water. Destabilization of the medial meniscus
(DMM) was induced in the right knee joint to establish the OA
model. All mice were randomly divided into four groups (n=15
mice/group): Control (sham-operated), OA (DMM), ferrostatin-1 (DMM
+ ferrostatin-1) and rhosin (DMM + rhosin). After anesthetizing
mice with an intraperitoneal injection of pentobarbital (40 mg/kg),
incisions were made in the skin and joint cavity. The meniscus
ligament of the right knee joint was transversely cut to release
the meniscus anterior segment. In the control group, the meniscus
ligament was left intact (20).
Mice in the DMM + ferroastatin-1 and DMM + rhosin groups received
intraperitoneal ferroastatin-1 (15 mg/kg) and rhosin (40 mg/kg),
respectively, twice/week (21,22), while mice in the control and DMM
groups were administered an equivalent volume of normal saline on
the same schedule. At 4 and 8 weeks after surgery, mice were
anesthetized via intraperitoneal injection of pentobarbital (40
mg/kg) and blood samples (volume, 0.4 ml) were collected using the
cardiac puncture method for biochemical analysis. Subsequently,
mice were sacrificed via cervical dislocation. The knee joint,
liver, kidney and spleen tissues were harvested for further
experiments at 8 weeks after surgery. The animal experimental
procedure is illustrated in Fig.
1.
Isolation and culturing of ECs from
bone
Briefly, six healthy adult female C57BL/6 mice (age,
6 weeks; weight, 16 g) were acquired from the Experimental Animal
Center of Ningxia Medical University, China. Mice were maintained
in a specific pathogen-free environment under standard conditions
(22±1°C, 55% humidity and 12/12-h light/dark cycle) with free
access to water and food. Mice were anesthetized via
intraperitoneal injection of pentobarbital (40 mg/kg), followed by
sacrifice via cervical dislocation. Femur and tibia bones were
collected from mice and preserved in sterile PBS devoid of
Ca2+ and Mg2+. The bones were ground using a
mortar and pestle and digested with collagenase (Sigma-Aldrich;
Merck KGaA) to achieve a single-cell suspension. ECs were isolated
from the cell suspension using Magnetic-Activated Cell Sorting
(MACS) with CD31 antibody (50 μg/5×107 cells;
cat. no. ab7388; Abcam) for 15 min at 4°C. The sorted ECs were
seeded (1×106) in a culture dish pre-coated with
fibronectin (cat. no. 354403; Corning, Inc.), followed by the
addition of endothelial cell growth medium (EBM-2) supplemented
with EGM-2 SingleQuots (CC-4176; both Clonetics; Lonza), cells were
maintained in a humidified environment at 37°C under a 5%
CO2 atmosphere for 48 h. During the first passage, the
cells were sorted with CD31 antibody using MACS sorting kit (cat.
no. 11061D; Thermo Fisher Scientific, Inc.) according to the
manufacturer's instructions. Cells were maintained in a humidified
environment at 37°C under a 5% CO2 atmosphere for 48 h.
ECs between the second and fifth passages were used for subsequent
experiments. ECs were stimulated with interleukin-1β (100 ng/ml)
for 24 h with or without 50 μM ferroptosis inhibitor
ferrostatin-1 or 10 μM ferroptosis activator erastin at 37°C
(all MedChemExpress).
Transfection
Lentiviral vectors (psi-LVRU6P) were purchased from
GeneCopoeia to stably knock down RhoA in ECs. The lentiviral
vectors include RhoA (MSH031463-LVRU6P) and scrambled short hairpin
(sh)RNA as the negative control (CSHCTR001-LVRU6P). Briefly, 293T
cells (cat. no. CL-0005; Pricella) were transfected with 10
μg lentivirus vector using calcium phosphate method
(23). The concentration of the
plasmid is 166 ng/μl. We used a transfection reagent
(Lipofectamine 3000; cat. no. L3000001; Thermo Fisher Scientific
Inc.). Following 48 h incubation at 37°C, the viral supernatant was
collected and filtered. ECs were incubated overnight with the
supernatant at 37°C, followed by the addition of 10 μg/ml
polybrene. RhoA knockdown cells were selected using 2 μg/ml
puromycin. Subsequently, cells were continuously screened with
medium containing 2 μg/ml puromycin and passaged for three
generations to maintain stable expression, which were then
preserved. Subsequent experiments will be conducted after 1 week.
shRNA and the negative control sequence are listed in Table SI.
Reverse transcription-quantitative (RT-q)
PCR
Total RNA was extracted from ECs using TRIzol
(Thermo Fisher Scientific, Inc.) according to the manufacturer's
instructions. RNA (1 μg) was reverse-transcribed to cDNA
using a PrimeScript RT reagent kit (cat. no. RR037A; Takara Bio,
Inc.) according to the manufacturer's instructions. qPCR was
performed on a MiniOpticon real-time PCR system (Bio-Rad
Laboratories, Inc.) using the TB Green Detection kit (Takara Bio,
Inc.) and specific primers (Table
SII) with β-actin as the internal control. Thermocycling
conditions include a Holding Stage (reps: 1; 95°C; 30 sec) and a
Cycling Stage (Number of Cycles: 40; 95°C 3 sec; 60°C 12 sec to 15
sec * 2). The relative expression of the target genes was
calculated using the 2−ΔΔCq method and normalized to
that of β-actin (24). All
reactions were performed in quintuplicate.
Western blotting
Briefly, ECs were seeded in 6-well plates at
a density of 5×105/well and incubated with IL-1β (100
ng/ml) for 24 h in the presence or absence of 50 μM
ferroptosis inhibitor ferrostatin-1 or 10 μM ferroptosis
activator erastin at 37°C. Following three washes with PBS, protein
was extracted from the cells on ice using 80 μl Mammalian
Protein Extraction Reagent (Thermo Fisher Scientific, Inc.). The
protein concentration was determined using the BCA) method.
Thereafter, proteins (30 μg/lane) from each sample were
separated using 10/15% SDS-PAGE and transferred to PVDF membranes.
After blocking with 5% skimmed milk for 1 h at room temperature,
membranes were incubated with specific primary antibodies overnight
at 4°C. After five washes with TBST (0.05% Tween), the membranes
were incubated with the secondary antibody for 1 h at room
temperature. Protein bands were visualized using a enhanced
chemiluminescent (ECL) kit (cat. no. RM0021; ABclonal) and images
were captured using ChemiDoc™ Imaging Systems (Bio-Rad
Laboratories, Inc.). The expression levels of the proteins were
quantified using ImageJ v1.8.0 software (National Institutes of
Health). The following primary antibodies were used: Ras homolog
family member A (RhoA; 1:2,000; cat. no. ab187027; Abcam), zona
occludens-1 (ZO-1; 1:1,000; cat. no. ab96587; Abcam), connexin 43
(1:1,000; cat. no. ab235282; Abcam), intercellular adhesion
Molecule-1(ICAM-1; 1:500; cat. no. ab171123; Abcam), glutathione
peroxidase 4 (GPX4; 1:1,000; cat. no. ab125066; Abcam), solute
carrier family 7 member 11 (SLC7A11; 1:1,000; cat. no. ab307601;
Cell Signaling Technology, Inc.), acyl-CoA synthase long-chain
family member 4 (ACSL4; cat. no. 22401-1-AP; Proteintech Group,
Inc.), tumor necrosis factor-alpha (TNF-α; cat. no. 11948; Cell
Signaling Technology, Inc.) and β-actin (1:1,000; cat. no.
66009-1-Ig; Proteintech Group, Inc.). The following secondary
antibodies were used: Horseradish peroxidase-conjugated goat
anti-rabbit (1:10,000; cat. no. ab205718; Abcam) and anti-mouse IgG
(1:10,000; cat. no. ab205719; Abcam).
Cell viability assay
Briefly, the viability of ECs was assessed using a
Cell Counting Kit-8 (CCK-8) assay (Dojindo Laboratories, Inc.). ECs
were seeded in 96-well plates at a density of 3,000 cells/well (six
replicate wells for each condition), followed by treatment with
IL-1β (0, 1, 10, 100, 200, 500 and 1,000 ng/ml), ferrostatin-1 (0,
1, 10, 50, 100 and 200 μM) or erastin (0, 1, 5, 10, 50, 100
μM) alone for 24, 48, 72 h at 37°C. After discarding the
medium, 100 μl 10% CCK-8 solution was added to each well,
followed by incubation at 37°C for 1 h. The absorbance was measured
at 460 nm using an enzyme-linked instrument (Infinite®
200 PRO; Tecan Group. Ltd.).
Wound healing assay
ECs were evenly seeded
(5×105/well) into 6-well plates and incubated for 12 h
at 37°C. Thereafter, scratches were formed on the EC monolayer and
cultured using serum-free medium, followed by treatment with
interleukin-1β (100 ng/ml) with or without 50 μM ferroptosis
inhibitor ferrostatin-1 or 10 μM ferroptosis activator
erastin for 24 h at 37°C in 5% CO2. The confluence on
either side of the wound is 1×106/well. Images were
captured using a fluorescence microscope (cat. no. BX53; Olympus
Corporation) at 0 and 24 h. The extent of cell migration was
quantified by comparing the remaining wound area with the area at 0
h post-scratch.
Fluorescence analysis
ECs were treated with IL-1β (100 ng/ml) for 24 h
with or without 50 μM ferroptosis inhibitor ferrostatin-1 or
10 μM ferroptosis activator erastin at 37°C, rinsed with
PBS, fixed with 4% paraformaldehyde for 10 min and permeabilized
with 0.1% Triton X-100 (Beyotime Institute of Biotechnology) for 5
min at 25°C. After three washes with PBS, the cells were blocked
with 1% BSA (Beijing Solarbio Science & Technology Co., Ltd.)
for 30 min at 25°C. For immunofluorescence assay, ECs were
incubated with primary antibodies overnight at 4°C, followed by
incubation with secondary antibodies in the dark at room
temperature for 2 h. For phalloidin staining, ECs were incubated
with 200 μl Tetramethylrhodamine Isothiocyanat Phalloidin
working solution (100 nM; Beijing Solarbio Science & Technology
Co., Ltd.) in the dark for 30 min at 25°C. To assess lipid
peroxidation and mitochondrial membrane potential, cells were
incubated in the dark with 200 μl Liperfluo working solution
(1 μM; Invitrogen; Thermo Fisher Scientific, Inc.) for 30
min at 37°C, followed by incubation in the dark with Rhodamine 123
working solution (2 μM; Beyotime Institute of Biotechnology)
for 15 min at 37°C. Following thorough washing with PBS, nuclei
were stained with DAPI (5 μM; Invitrogen; Thermo Fisher
Scientific, Inc.) for 30 min at 25°C and sealed. The primary
antibodies were as follows: RhoA (1:200; cat. no. ab187027; Abcam),
ZO-1 (1:500; cat. no. ab96587; Abcam), connexin 43 (1:200; cat. no.
ab235282; Abcam), GPX4 (1:200; cat. no. ab125066; Abcam), ACSL4
(1:300; cat. no. 22401-1-AP; Proteintech) and VE-cadherin (1:400;
cat. no. 2500T; Cell Signaling Technology, Inc.). Fluorescence
signals were visualized using a fluorescence microscope (cat. no.
BX53; Olympus Corporation) and analyzed using ImageJ v1.8.0
software (National Institutes of Health). Brightness and contrast
adjustments were performed during image processing. For consistent
comparison, all images were captured using identical collection
settings.
Histology and immunohistochemical (IHC)
staining
The knee joint, liver, kidney and spleen tissue was
fixed in 4% paraformaldehyde buffer for 24 h at 4°C and decalcified
in 10% EDTA for 4 weeks at 25°C. After dehydration with graded
ethanol, tissues were embedded in paraffin, sectioned (thickness, 4
μm) and subjected to hematoxylin and eosin (H&E),
safranin-O-fast green and Masson staining. According to the
instructions of the H&E staining kit (cat. no. G1076;
Servicebio Technology), the sections were dewaxed and dehydrated,
then stained with hematoxylin followed by eosin staining (both 5
min at 20°C). For safranin O-fast green staining (cat. no. G1053;
Servicebio Technology), the deparaffinization of slides was
consistent with the description above. Subsequently, they were
stained with Fast Green for 6 min, washed at 20°C, dehydrated, and
then stained with Safranin O at 20°C for 3 min. The Osteoarthritis
Research Society International (OARSI) scoring system was used to
evaluate the degeneration of articular cartilage (25). The distance from the tidemark to
the surface of the articular cartilage was measured and recorded as
the thickness of the hyaline cartilage (HC), while the distance
from the tidemark to the subchondral bone plate was recorded as the
thickness of the calcified cartilage (CC). For Masson staining
(cat. no. G1006; Servicebio Technology), sections were soaked in
2.5% potassium dichromate staining solution overnight at 25°C and
incubated for 30 min at 65°C. Next, the sections were immersed in
Weigert's iron hematoxylin staining solution for 1 min, followed by
staining in Van Gieson's acid fuchsin for 6 min at 25°C.
Subsequently, the sections were immersed in 1% phosphomolybdic acid
solution for 1 min, and then stained in 2.5% aniline blue solution
for 30 sec at 25°C. For IHC, tibial plateau samples were prepared
as aforementioned. Following dewaxing and rehydration, 0.1% trypsin
was used for antigen retrieval at 37°C for 30 min. Subsequently,
endogenous peroxidase activity was quenched with 3% hydrogen
peroxide for 10 min at 25°C. After blocking with 5% normal goat
serum (cat. no. G1208; Servicebio Technology) at 37°C for 30 min,
the sections were incubated overnight at 4°C with the following
primary antibodies: RhoA (1:300; cat. no. ab54835; Abcam), ZO-1
(1:500; cat. no. ab221547; Abcam), GPX4 (1:250; cat. no. ab125066;
Abcam) and ACSL4 (1:500; cat. no. 22401-1-AP; Proteintech Group,
Inc.). The secondary antibodies were used: Horseradish
peroxidase-conjugated goat anti-rabbit (1:10,000; cat. no.
ab205718; Abcam) and anti-mouse IgG (1:10,000; cat. no. ab205719;
Abcam) for 30 min at 25°C. The sections were stained using DAB
(cat. no. G1212; Servicebio Technology) and counterstained with
hematoxylin for 5 min at 25°C. Images were captured using an
optical microscope (cat. no. CX43; Olympus Corporation) and
analyzed using ImageJ v1.8.0 software (National Institutes of
Health).
Micro-computed tomography (Micro-CT)
analysis
After removing the surrounding skin and muscles, the
right knee joint of the mice was fixed with 4% paraformaldehyde at
room temperature for 48 h. Scans were performed using a micro-CT
(SkyScan 1176; Bruker Belgium S.A./N.V.) with a resolution of 9
μm/pixel. Analyzed indices included bone volume fraction
(BV/TV), trabecular separation (Tb.Sp), trabecular thickness
(Tb.Th) and tissue mineral density (TMD).
Determination of serum biochemical
indexes
The acquired mouse whole blood sample was
centrifuged at 3,000 rpm for 15 min, and the supernatant was
collected as the serum sample. This was stored at -80°C for
subsequent use. Working solutions were prepared according to the
guidelines of the reagent manufacturer (cat. no. BC2, BC4, BC5,
BC7; LWPOCT). Biochemical indices, including Albumin (ALB), Direct
bilirubin (DBIL), Total bile acids (TBA), Triglycerides (TG), Total
cholesterol (TC), High-density lipoprotein cholesterol (HDL-C),
Low-density lipoprotein cholesterol (LDL-C), Glucose (GLU),
Glycated serum protein (GSP), Creatine kinase (CK), Creatine
kinase-MB (CK-MB), Lactate dehydrogenase (LDH), Hydroxybutyrate
dehydrogenase (HBDH), Superoxide dismutase (SOD), UREA, Creatinine
(CRE), and Uric acid (UA) were assessed using an automatic
biochemical analyzer, and the results were documented. Serum
indices for Malondialdehyde (MDA), glutathione (GSH), and Total
antioxidant capacity (T-AOC) in mice were determined based on the
kit's instructions (cat. no. A003-1, A006-2-1, A015-2-1;
Jiancheng), with the final data obtained using the microplate
reader (cat. no. Epoch; Bio TEK).
Liquid chromatography-tandem mass
spectrometry (LC-MS/MS) and label-free quantification of tissue
proteomes
Paired OA and control tissue samples from 30
patients were combined at a ratio of 10:1, after which the tissue
proteins were subjected to LC-MS/MS for label-free quantification.
LC-MS/MS analysis was performed on a Q Exactive mass spectrometer
coupled to Easy nLC (Thermo Fisher Scientific, Inc.). The mass
spectrometer was operated in positive ion mode. The nitrogen gas
temperature is 180°C with a flow rate of 3 l/min. Parallel reaction
monitoring (PRM) transitions assessed are within the 300-1,800 scan
range. LC-MS/MS was used for data collection, followed by a
database search (uniprot.org) to identify proteins.
Adjusted P<0.05 and Log (Fold Change) >1 or Log (Fold Change)
<-1" were defined as the threshold for the differential
expression of protein. Gene Ontology (GO; https://www.geneontology.org) functional annotation
and Kyoto Encyclopedia of Genes and Genomes (KEGG; http://geneontology.org/) pathway enrichment analysis
were performed on the DEPs. The results were plotted by R (version:
4.3.3) software (https://www.r-project.org).
Statistical analysis
All data are presented as the means ± SD. All
independent experiments were repeated at least three times. All
statistical analyses were performed using GraphPad Prism 9.0
(GraphPad Software, Inc.; Dotmatics). Statistical comparisons
between two groups were performed using unpaired Student's t test.
For ≥3 groups, a variance homogeneity test was first conducted,
followed by one-way ANOVA and Tukey's multiple comparison post hoc
test. P<0.05 was considered to indicate a statistically
significant difference.
Results
RhoA is highly expressed in the OA
clinical samples
The present study analyzed age, BMI, Visual Analog
Scale (VAS)score, Lysholm score, WOMAC (Western Ontario and
McMaster Universities Osteoarthritis Index) score and
Kellgren-Lawrence (K-L) grades of the 60 clinical samples (Fig. S1A-F) from 30 patients with OA
who had not received any drugs. Pre- and postoperative
anteroposterior and lateral radiographs and preoperative MRI scans
were performed (Fig. S1G).
Overall, the average age was 68.72±7.941 years. The BMI was
27.22±1.480. The VAS score was 7.15±1.424, the Lysholm score was
76.43±8.001, and the WOMAC score was 46±7.105. All patients had a
K-L grade of 3-4, consistent with characteristics of OA.
Bioinformatics analysis of the proteome data showed
292 DEPs in the OA vs. control group, among which RhoA was
significantly upregulated (Fig. 2A
and B). KEGG analysis indicated that most DEPs were enriched in
processes such as 'cell migration' (Fig. S2B). GO analysis showed that the
DEPs were primarily enriched in biological processes associated
with 'PPAR signaling pathway', 'ferroptosis' and 'pentose phosphate
pathway' (Fig. S2A). Overall,
these results suggested that RhoA may be associated with these
biological processes.
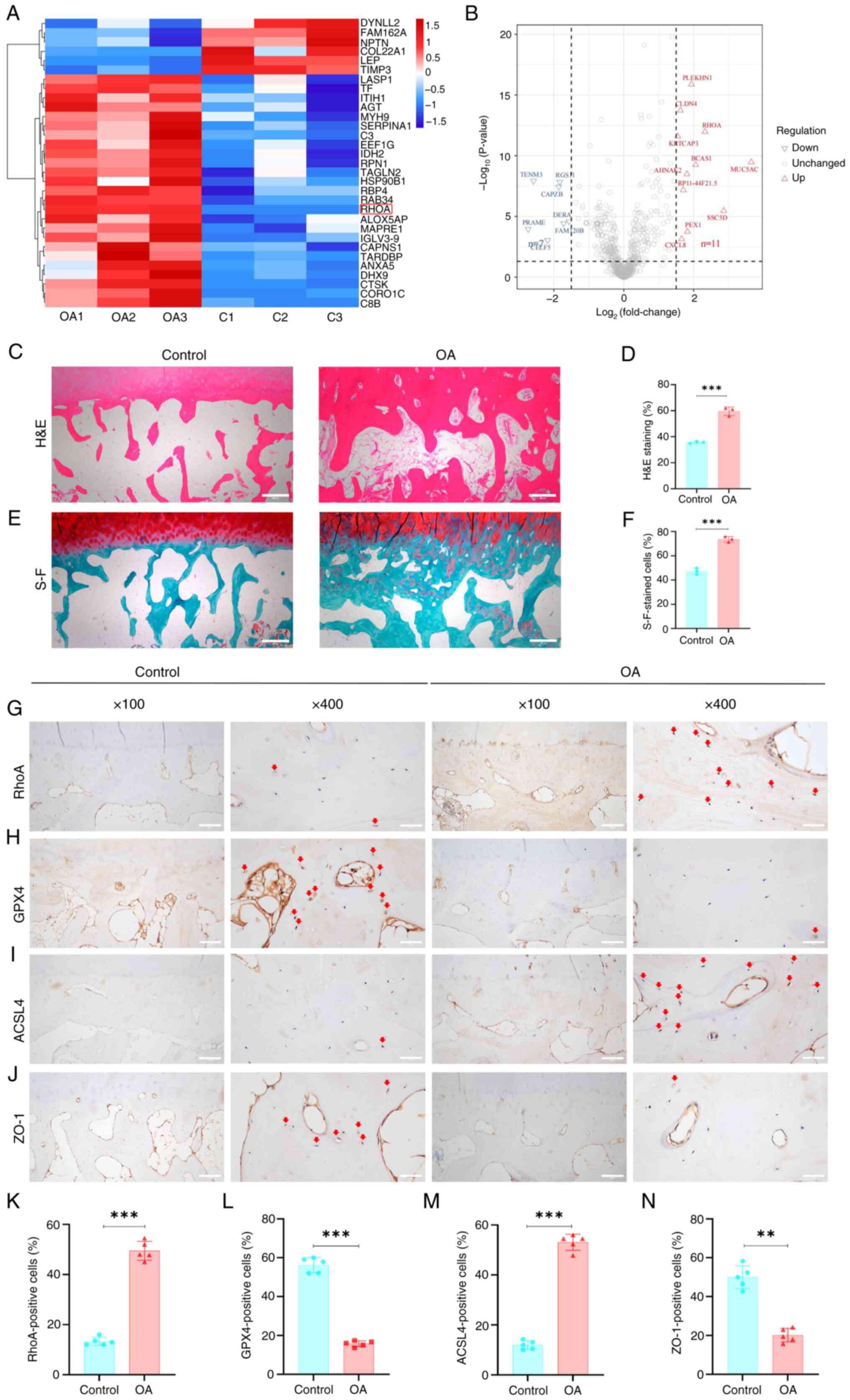 | Figure 2RhoA expression, GPX4 and vascular
adhesion are significantly downregulated in clinical samples from
patients with OA, while ferroptosis core factor-ACSL4 significantly
upregulated. (A) Heatmap and (B) volcano plot of differentially
expressed proteins. (C) H&E-stained clinical samples. (D)
Subchondral bone relative to the total area in H&E-stained
samples. (E) S-F green-stained clinical samples. Scale bar, 500
μm. (F) Ratio of subchondral bone area to the total area in
S-F green-stained samples. n=3. Immunohistochemical staining for
(G) RhoA, key ferroptosis proteins (H) GPX4 and (I) ACSL4 and (J)
ZO-1 in clinical samples. Red arrows indicate positive cells. Scale
bar, 200 and 50 μm. Percentage of immunoreactive positive
cells for (K) RhoA, (L) GPX4, (M) ACSL4 and (N) ZO-1. n= 5.
**P<0.01 and ***P<0.001. RhoA, Ras
homolog family member A; OA, osteoarthritis; H&E, hematoxylin
and eosin; S-F, Safranin-O-fast green; GPX4, Glutathione peroxidase
4; ASCL4, Acyl-CoA synthase long-chain family member 4; ZO-1, Zona
occludens-1. |
Furthermore, H&E and safranin-O-fast green
staining indicated noticeable subchondral bone remodeling and
sclerosis in clinical samples of OA (Fig. 2C-F). IHC showed a significantly
higher expression of RhoA in OA than in undamaged subchondral bone
(Fig. 2G and K). Collectively,
these results indicated that RhoA was upregulated in the
subchondral bone remodeling phase of OA.
GPX4 and vascular adhesion are
significantly downregulated in OA samples, while ACSL4
significantly upregulated
To investigate ferroptosis in OA, expression of GPX4
and ACSL4, key regulators of ferroptosis (26), was assessed in OA and undamaged
tissue. There was a significant decrease in GPX4 expression and an
increase in ACSL4 expression in OA-damaged tissue (Fig. 2H, I, L and M). To determine
whether there was an alteration in vascular permeability during OA
development, expression of the cell adhesion molecule ZO-1 was
assessed (27,28). ZO-1 expression was significantly
downregulated in OA compared with controls (Fig. 2J and N). Overall, these results
indicated GPX4 and vascular adhesion are significantly
downregulated during OA progression, while ferroptosis core
factor-ACSL4 significantly upregulated.
RhoA enhanced the permeability of
ECs
Ferrostatin-1 and erastin are inhibitors and
activators of ferroptosis, respectively (29). Cell viability assay was performed
to examine cytotoxic effects of IL-1β, ferrostatin-1 and erastin on
ECs. IL-1β, ferrostatin-1 and erastin were not cytotoxic to ECs at
concentrations <100 ng/ml and 50 and 10 μM, respectively.
Notably, there was no significant difference in cell viability at
24, 48 and 72 h of treatment (Fig.
S3A-I).
To investigate changes in RhoA expression in
IL-1β-treated ECs, EC lentiviral transduction was performed.
Successful lentiviral transduction of ECs inhibited expression of
RhoA at the protein and mRNA levels (Fig. S4A-C). RhoA knockdown
significantly inhibited IL-1β-induced increase in RhoA expression
in ECs, evidenced by a decrease in the fluorescence intensity of
RhoA following RhoA knockdown (Fig.
S4D and E). Additionally, western blotting indicated that RhoA
knockdown significantly reversed IL-1β-induced increase in RhoA
protein expression in ECs (Fig. S4F
and G), which was confirmed by RT-qPCR (Fig. S4H). Collectively, these results
suggested that IL-1β increased RhoA expression in ECs but this
effect was reversed by RhoA knockdown.
To elucidate the effect of RhoA on inflammatory
responses in ECs, mRNA expression of inflammatory genes in ECs was
assessed. RhoA knockdown significantly attenuated IL-1β (100
ng/ml)-induced increase in mRNA expression of ICAM1, VCAM1, MCP-1,
TNF-α, IL-6 and IFN-γ (Fig.
S5A-F). Additionally, RhoA knockdown suppressed IL-1β-induced
increase in protein expression of TNF-α and ICAM1 in ECs (Fig. S5G-I). Overall, these results
suggested that RhoA knockdown ameliorated inflammatory response in
ECs.
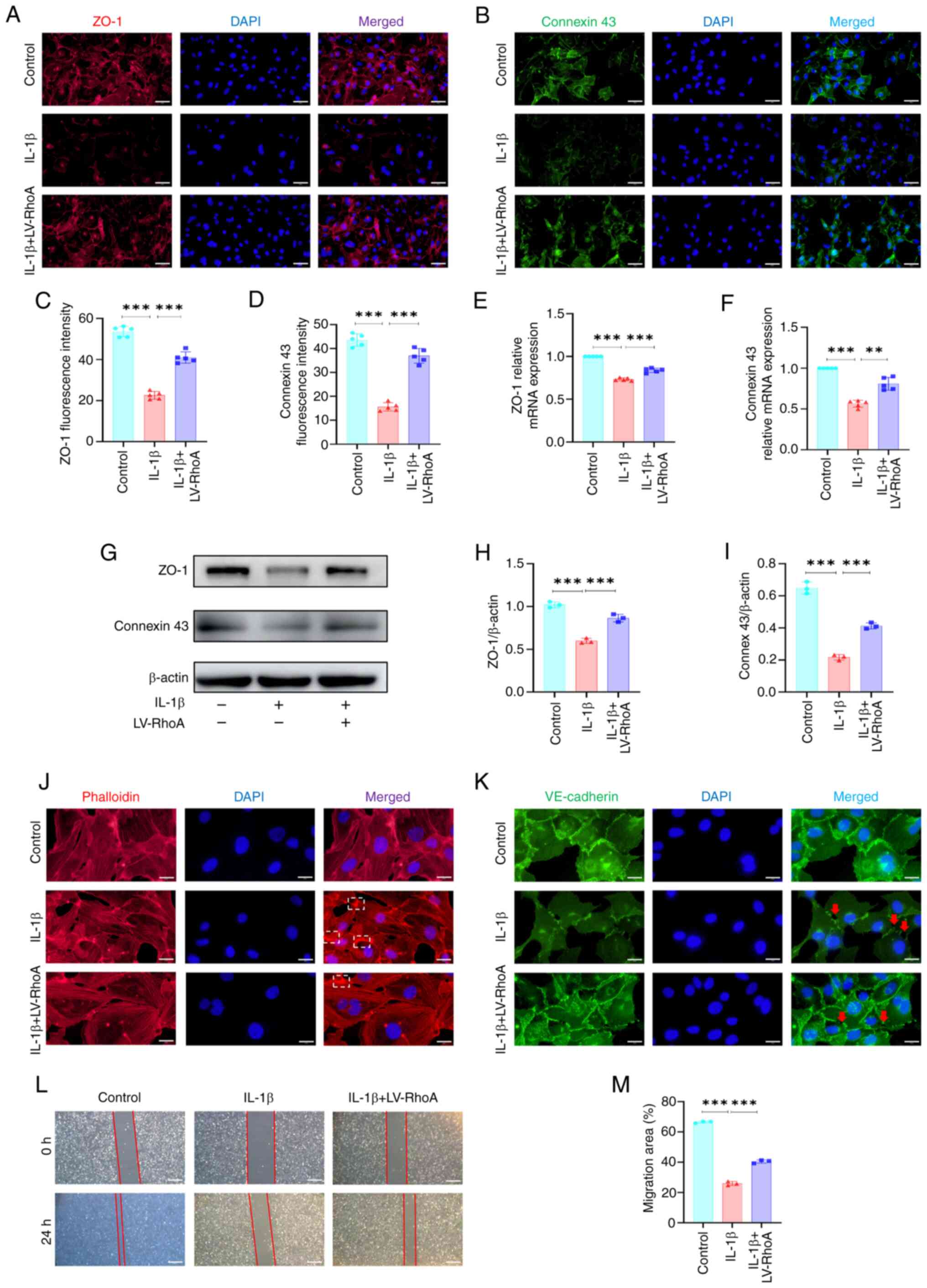
The role of RhoA in the expression of vascular
permeability-related markers was assessed. ZO-1 and connexin 43,
which are key factors for adhesion and connection between ECs,
inhibit vascular permeability (30,31). Immunofluorescence staining
revealed RhoA knockdown significantly reversed IL-1β-induced
decrease in the mean fluorescence intensity of ZO-1 and connexin 43
(Fig. 3A-D). RT-qPCR showed that
RhoA knockdown enhanced IL-1β-induced decrease of ZO-1 and connexin
43 mRNA levels in ECs (Fig. 3E and
F), which was confirmed by western blot analysis (Fig. 3G-I).
To elucidate the influence of RhoA on vascular
permeability, actin cytoskeleton and VE-cadherin staining was
performed. The actin cytoskeleton interacts with VE-cadherin to
regulate integrity of intercellular connections (32). IL-1β treatment significantly
decreased stress fibers within ECs but increased branched actin
filaments. However, RhoA knockdown ameliorated these IL-1β-induced
changes in the cytoskeleton (Fig.
3J). RhoA knockdown reversed IL-1β-induced alterations in
VE-cadherin distribution along EC junctions (Fig. 3K). Moreover, cell scratch assay
indicated that RhoA knockdown attenuated IL-1β-induced inhibition
of cell migration (Fig. 3L and
M). Collectively, these results suggested that RhoA suppression
may ameliorate IL-1β-induced increase in vascular permeability in
ECs.
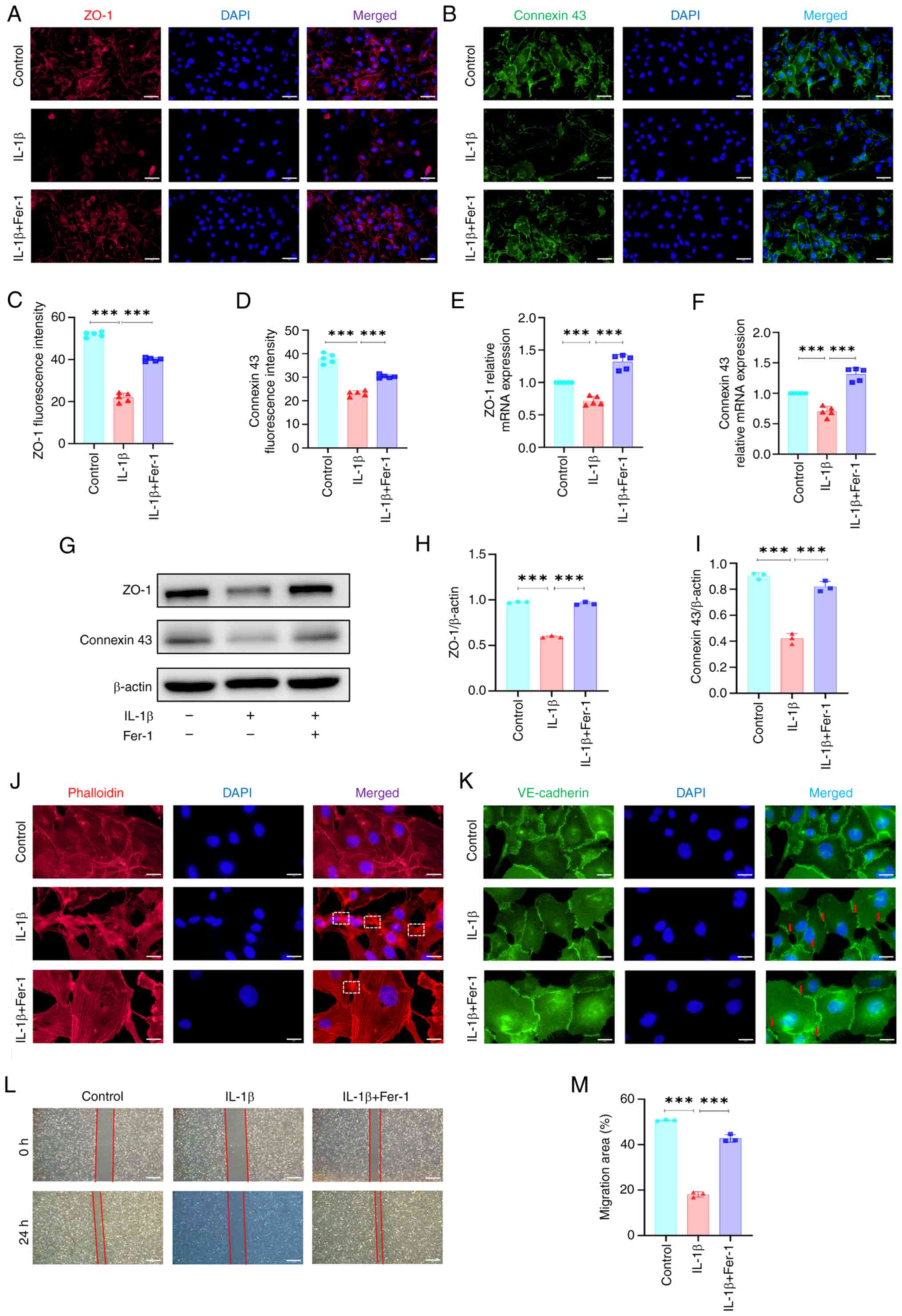
Ferroptosis enhances vascular
permeability in ECs
The present study confirmed the inhibitory effect of
ferrostatin-1 on ferroptosis in IL-1β-treated ECs.
Immunofluorescence staining showed that ferrostatin-1-induced
inhibition of ferroptosis significantly reversed IL-1β-induced
decrease in the average fluorescence intensity of GPX4 (Fig. S6A and C). By contrast,
ferrostatin-1-induced inhibition of ferroptosis decreased the
average fluorescence intensity of ACSL4 in ECs (Fig. S6B and D). Additionally, RT-qPCR
indicated that ferrostatin-1-induced inhibition of ferroptosis
significantly reversed IL-1β-induced decrease in mRNA expression of
SLC7A11 and GPX4 in ECs and decreased mRNA expression of ACSL4
(Fig. S6E-G), which was
confirmed by western blot analysis (Fig. S6H-K). ferroptosis inhibition
effectively reversed IL-1β-induced increases in ROS levels and
increased mitochondrial membrane potential (Fig. S6L-N). The present study
investigated the effects of ferrostatin-1-induced inhibition of
ferroptosis on vascular permeability in IL-1β-treated ECs.
Immunofluorescence staining showed that, compared with
IL-1β-treated ECs, ferrostatin-1-induced inhibition of ferroptosis
restored ZO-1 and connexin 43 fluorescence intensity in
IL-1β-treated ECs (Fig. 4A-D).
RT-qPCR indicated that ferroptosis inhibition enhanced
IL-1β-induced downregulation of ZO-1 and connexin 43 mRNA
expression in ECs (Fig. 4E and
F), which was confirmed by western blotting (Fig. 4G-I). Moreover, ferroptosis
inhibition ameliorated IL-1β-induced decrease in stress fibers and
decreased branched actin filaments in ECs (Fig. 4J). Furthermore, ferroptosis
inhibition reversed IL-1β-induced alteration in VE-cadherin
distribution along the EC junction (Fig. 4K). The scratch assay showed that
ferroptosis inhibition reversed the IL-1β-induced decrease in cell
migration (Fig. 4L and M).
Collectively, these results suggested that ferroptosis inhibition
may mitigate IL-1β-induced increase in vascular permeability in
ECs.
RhoA regulates ferroptosis in ECs
To investigate the potential mechanism by which RhoA
influences vascular permeability, the present study examined the
effects of RhoA inhibition on ferroptosis in IL-1β-treated ECs. The
SLC7A11/GPX4 antioxidant system and ACSL4 are key in the regulation
of ferroptosis (33,34). Immunofluorescence staining showed
that RhoA knockdown significantly reversed IL-1β-induced decrease
in the mean fluorescence intensity of GPX4 (Fig. 5A and C). By contrast, RhoA
knockdown decreased the mean fluorescence intensity of ACSL4 in ECs
(Fig. 5B and D). Additionally,
RT-qPCR indicated that RhoA knockdown significantly reversed
IL-1β-induced decrease in mRNA expression of SLC7A11 and GPX4 in
ECs and decreased the mRNA expression of ACSL4 (Fig. 5E-G), which was confirmed by
western blot analysis (Fig.
5H-K).
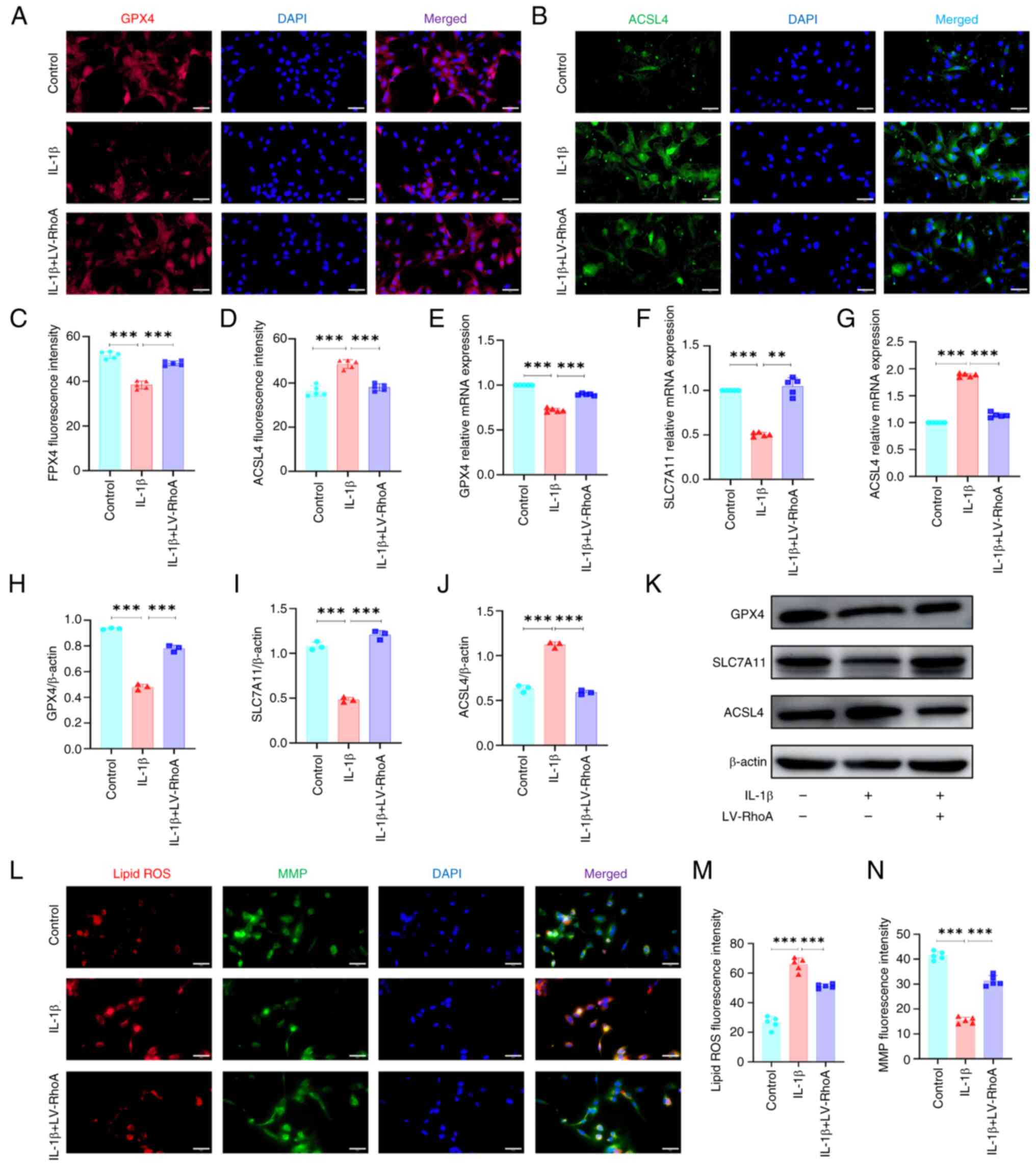 | Figure 5RhoA inhibition reverses
IL-1β-induced increase ferroptosis in ECs. ECs were transfected
with LV-RhoA and treated with or without IL-1β for 24 h.
Representative immunofluorescence staining for (A) GPX4 and (B)
ACSL4. Scale bar, 50 μm. Quantitative analysis of (C) GPX4
and (D) ACSL4 fluorescence intensity. Reverse
transcription-quantitative PCR to detect (E) GPX4, (F) SLC7A11 and
(G) ACSL4 mRNA expression. n=5. Quantitative analysis of (H) GPX4,
(I) SLC7A11 and (J) ACSL4 protein expression. n= 3. (K) Western
blotting for GPX4, SLC7A11 and ACSL4 protein expression. (L)
Assessment of (M) ROS and (N) MMP in ECs. Scale bar, 50 μm.
n=5. **P<0.01 and ***P<0.001. RhoA, Ras
homolog family member A; EC, Endothelial cell; LV-RhoA, Lentiviral
vectors-RhoA; GPX4, Glutathione peroxidase 4; ACSL4, Acyl-CoA
synthase long-chain family member 4; SLC7A11, Solute carrier family
7 member 11; ROS, reactive oxygen species; MMP, mitochondrial
membrane potential. |
To ascertain the role of RhoA in ferroptosis, the
present study assessed lipid ROS levels and mitochondrial membrane
potential in ECs. IL-1β treatment significantly increased ROS
levels in ECs and markedly decreased mitochondrial membrane
potential, as indicated by immunofluorescence staining. However,
RhoA inhibition effectively reversed IL-1β-induced increases in ROS
levels and increased mitochondrial membrane potential (Fig. 5L-N). Overall, these results
indicated that RhoA regulated ferroptosis by affecting
mitochondrial function and oxidative stress.
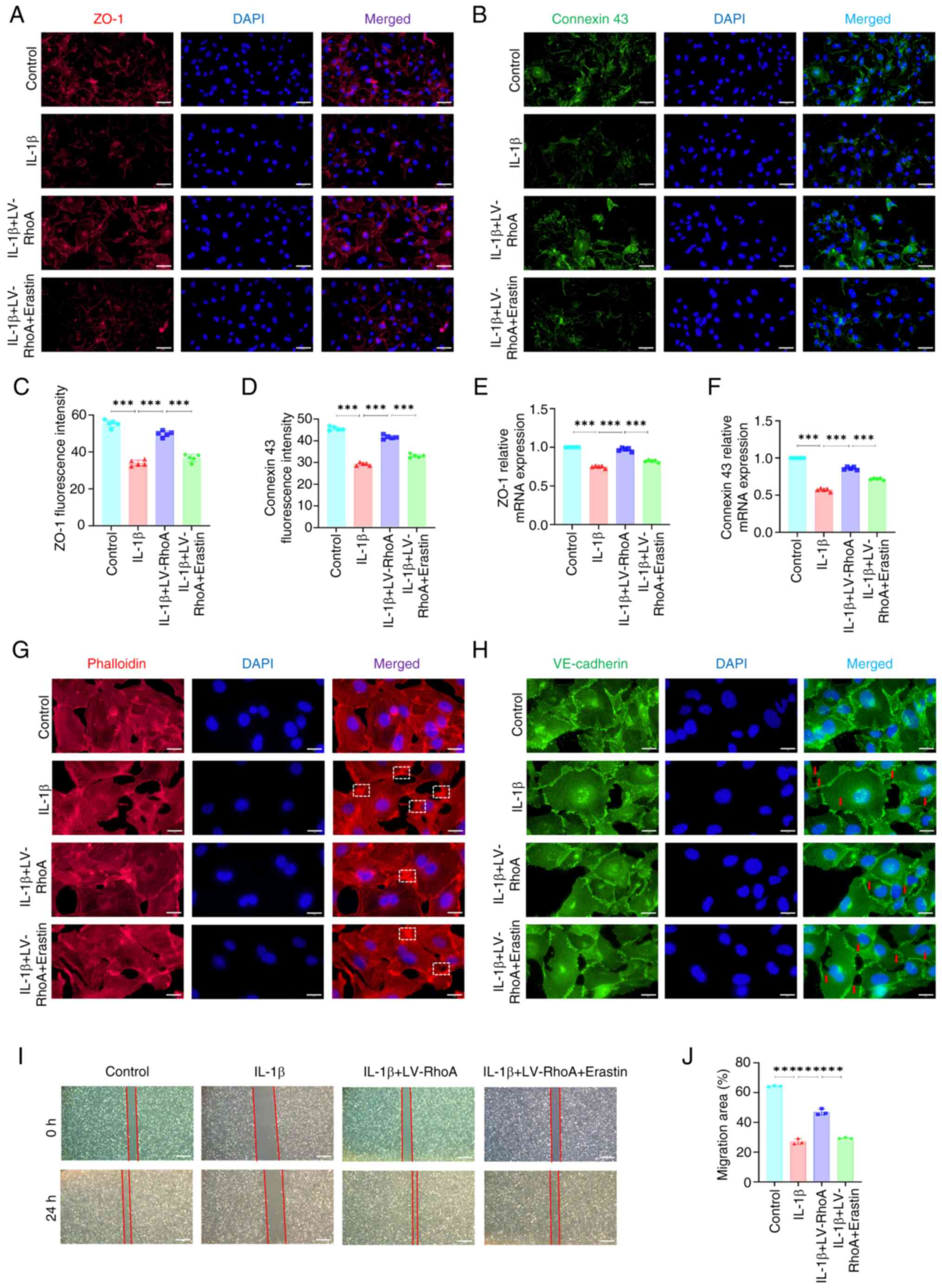
RhoA affects vascular permeability via
ferroptosis in ECs
To confirm the role of ferroptosis in RhoA-mediated
regulation of permeability in IL-1β-treated ECs, the present study
simultaneously inhibited RhoA and activated ferroptosis in ECs.
Immunofluorescence staining showed that RhoA inhibition notably
enhanced fluorescence intensity of ZO-1 and connexin 43 in
IL-1β-treated ECs; however, ferroptosis activation counteracted
these effects (Fig. 6A-D).
RT-qPCR indicated that ferroptosis activation nullified RhoA
inhibition-induced increase in ZO-1 and connexin 43 mRNA expression
in IL-1β-treated ECs (Fig. 6E and
F). Additionally, immunofluorescence staining indicated that
RhoA knockdown rescued IL-1β-induced decrease in stress fibers and
decreased branched actin filaments in ECs; however, these changes
were reversed by ferroptosis activation (Fig. 6G). Moreover, ferroptosis
activation nullified RhoA knockdown-induced amelioration of
alterations in VE-cadherin distribution along junctions in
IL-1β-treated ECs (Fig. 6H).
Wound healing assay showed that ferroptosis counteracted RhoA
knockdown-induced decrease in migration in IL-1β-treated ECs
(Fig. 6I and J). Overall, these
results indicated that RhoA influenced vascular permeability in ECs
via ferroptosis.
RhoA enhances ferroptosis in DMM
mice
To assess the role of RhoA and ferroptosis in OA,
the present study established a murine model of OA via DMM and
evaluated the toxicity of rhosin and ferrostatin-1 8 weeks
post-DMM. Rhosin and ferrostatin-1 had minimal effect on the body
weight, liver, kidney and spleen of mice at 8 weeks post-DMM
(Fig. S7A-D). Hepatic lobules
exhibited intact and well-organized tissue structure, the spleen
structure appeared largely normal, and there were no evident
pathological alterations in the glomerular and tubular structures
of the kidneys. Additionally, parameters, including biochemical
indices at 4 and 8 weeks post-DMM, were evaluated to assess the
potential toxicity of ferrostatin-1 and rhosin. Notably, there were
no significant differences in blood sugar levels (GSP and GLU),
liver function (ALB, DBIL and TBA), renal function (CRE, UA, and
UREA), and myocardial enzyme activity (CK, CK-MB, LDH and HBDH)
between groups (Figs. S8A-H and
S9A-D). Collectively, these results suggested that RhoA and
ferroptosis inhibition did not induce toxic effects in mice.
Lipid peroxidation and mitochondrial dysfunction are
key components of ferroptosis (35). Therefore, the present study
investigated the effects of RhoA and ferroptosis on lipid
metabolism and antioxidant capacity in mice at 4 and 8 weeks
post-DMM. Triglyceride (TG), total cholesterol (TC), low-density
lipoprotein cholesterol (LDL-C), high-density lipoprotein (HDL-C),
malondialdehyde (MDA) and nitric oxide (NO) levels, superoxide
dismutase (SOD) and glutathione (GSH) activity and total
antioxidant capacity (T-AOC) were assessed. Compared with the sham
group, there was a significant increase in TG, TC and LDL-C levels
and a decrease in HDL-C levels in the DMM group. However,
inhibition of ferroptosis and RhoA effectively mitigated these
alterations, suggesting that RhoA and ferroptosis inhibition may
modulate blood lipid metabolism in DMM mice (Fig. S9E-H). Additionally, there was an
increase in MDA levels and a decrease in SOD, GSH and T-AOC in the
DMM group compared with the sham group. However, ferrostatin-1 and
rhosin effectively reversed DMM-associated changes in MDA levels,
SOD and GSH activity and T-AOC (Fig. S9I-L). Overall, these results
indicated that the inhibition of RhoA and ferroptosis enhanced
antioxidant capacity in DMM mice.
Inhibition of RhoA ameliorates progress
of OA in DMM mice
To determine the effect of RhoA on OA in
vivo, OA progression in mice 8 weeks post-DMM was assessed
using H&E, safranin-O-fast green and Masson staining. The MM of
mice in the sham group was characterized by intact and smooth
articular cartilage surface, moderate cartilage thickness, distinct
tidemark, a substantial number of chondrocytes and a normal
chondrocyte structure and size (Fig.
7A-C). However, articular cartilage damage, decreased cartilage
thickness, reduced chondrocyte count, abnormal chondrocyte size and
structure, disappearance of hyaline cartilage (HC), and a
significant increase in calcified cartilage (CC) were observed 8
weeks after DMM. Notably, these changes were largely reversed in
the rhosin-treated group (Fig. 7A, D
and E). Additionally, OARSI histological score was used to
evaluate sections stained with safranin-O-fast green. Compared with
the sham group, the OARSI score was significantly higher in the DMM
group. However, rhosin treatment inhibited the increase in the
OARSI score observed in DMM mice (Fig. 7B and F). Overall, these results
suggested that RhoA inhibition ameliorated OA progression in DMM
mice.
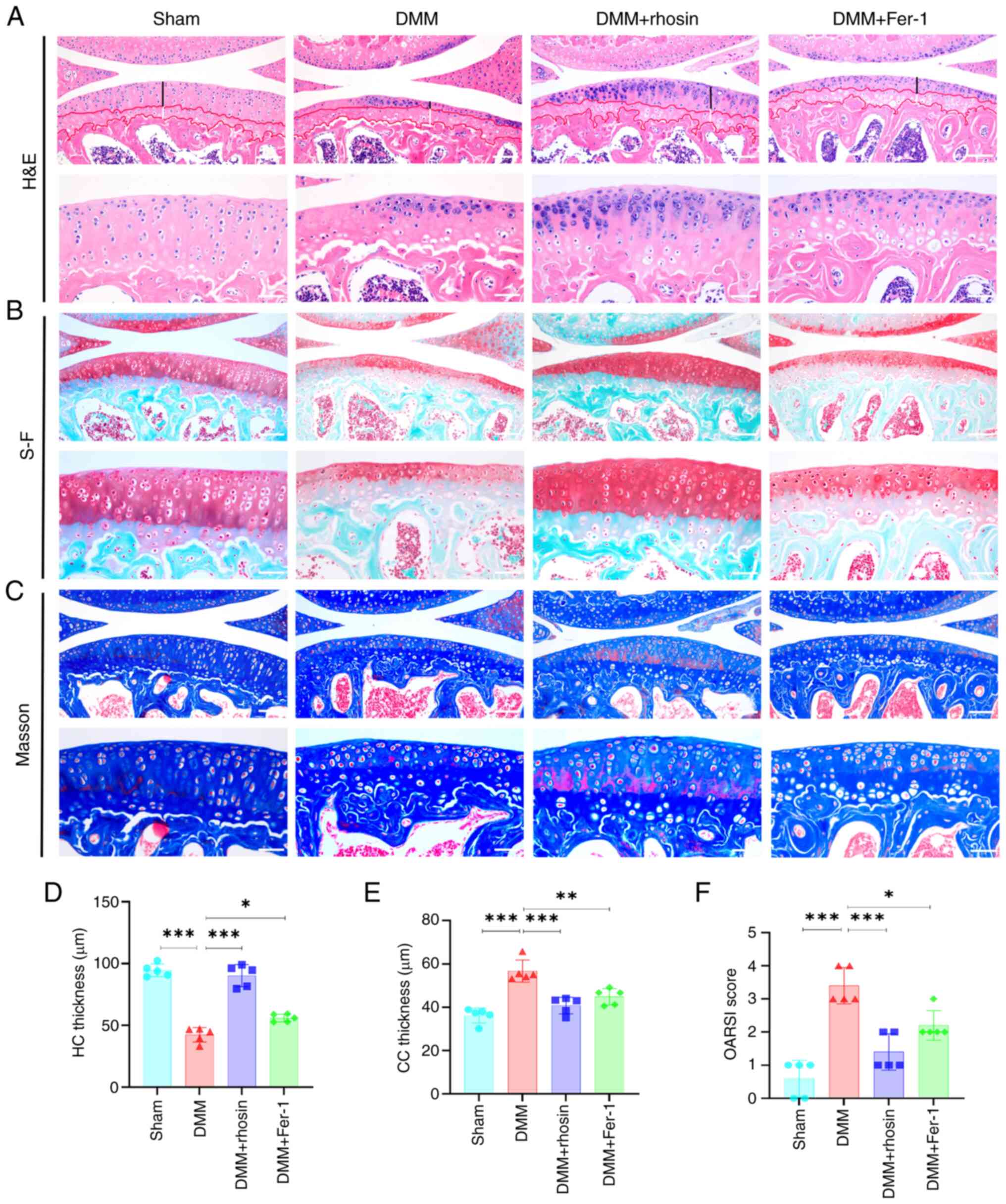 | Figure 7RhoA or ferroptosis inhibition
alleviates cartilage degeneration in mice with DMM. (A) H&E,
(B) S-F and (C) Masson's staining of knee joints in DMM mice after
RhoA or ferroptosis inhibition. Black line, HC thickness; white,
CC. Scale bar, 100 and 50 μm. Quantitative analysis of (D)
HC and (E) CC thickness in H&E-stained samples and (F) OARSI
scores of knee joint cartilage using S-F staining. n=5.
*P<0.05, **P<0.01 and
***P<0.001. RhoA, Ras homolog family member A; DMM,
destabilization of the medial meniscus; H&E, hematoxylin and
eosin; S-F, Safranin-O-fast green; HC, Hyaline cartilag; CC,
Calcified cartilage; OARSI, Osteoarthritis Research Society
International; Fer-1, Ferrostatin-1. |
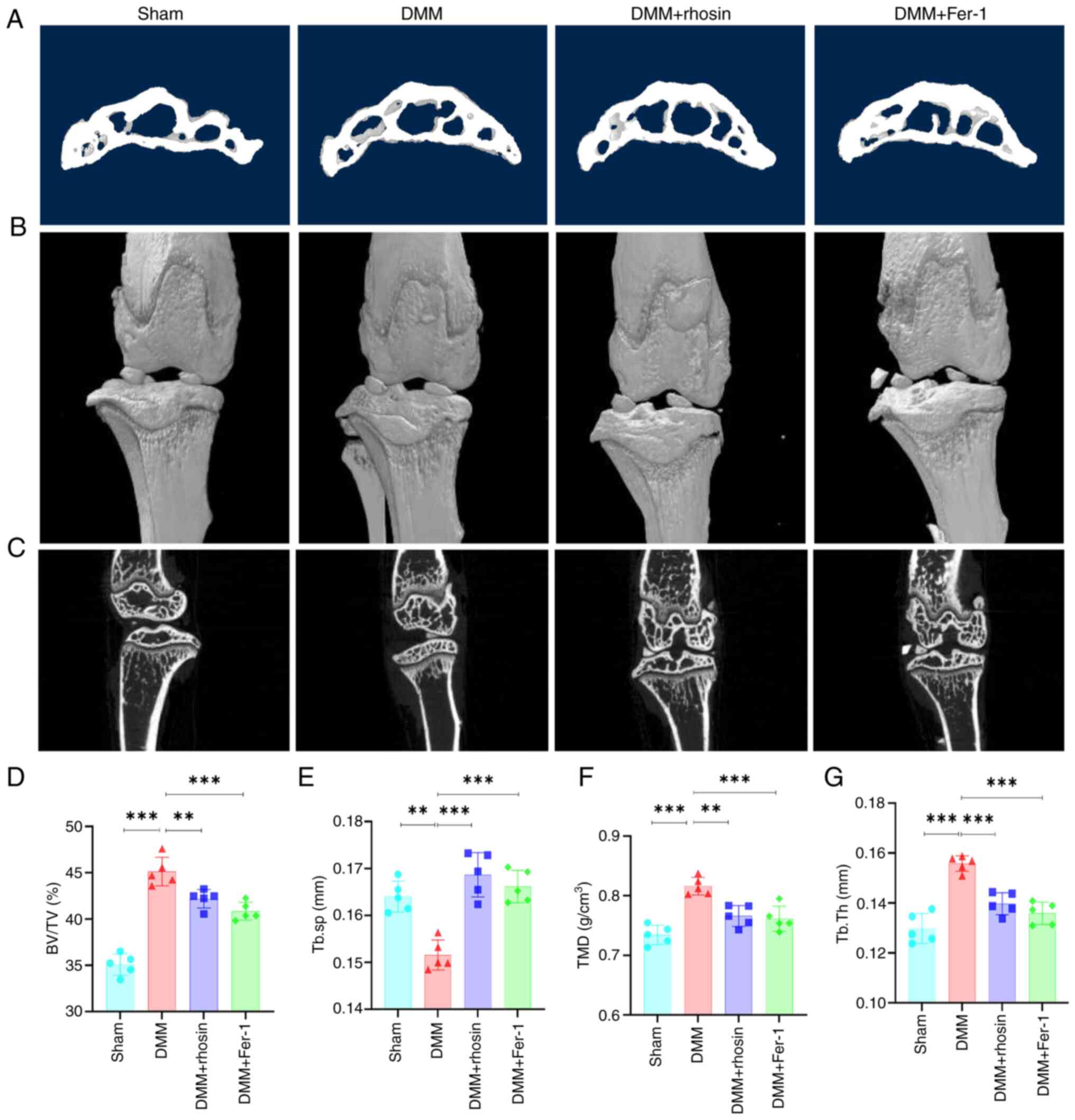
Micro-CT scan was performed on subchondral bone of
DMM mice to elucidate the role of RhoA in OA. Notable osteophyte
formation was observed in the DMM group, which was notably
ameliorated by RhoA inhibition (Fig.
8A-C). Additionally, subchondral sclerosis was indicated by
increased BV/TV, Tb.Th and TMD and reduced Tb.Sp in DMM mice.
However, RhoA inhibition partially mitigated these alterations
(Fig. 8D-G). Collectively, these
results indicated that RhoA inhibition improved subchondral bone
remodeling in mice after DMM surgery.
Inhibition of ferroptosis improves OA
progression in DMM mice
Ferroptosis inhibition mitigated cartilage damage
and enhanced OARSI score in DMM mice (Fig. 7A-F). Micro-CT scan indicated
osteophyte formation 8 weeks post-DMM surgery; however, ferroptosis
inhibition significantly reversed osteophyte development.
Additionally, ferroptosis inhibition significantly reversed
OA-associated subchondral sclerosis in DMM mice, evidenced by a
decrease in BV/TV, Tb.Th and TMD and an increase in Tb.Sp (Fig. 8D-G). Overall, these results
suggested that ferroptosis inhibition ameliorated the progression
of OA.
Discussion
During onset and progression of OA, subchondral bone
exhibits abnormal neovascularization and enhanced vascular
permeability (36). However, the
molecular mechanisms underlying increased vascular permeability in
OA remain unclear. Considering the pathogenesis of OA, identifying
and developing effective strategies to mitigate vascular
permeability may be effective for OA prevention and treatment. In
the present study, proteomic analysis indicated that RhoA was
significantly upregulated in subchondral bone of patients with OA.
RhoA is a Rho GTPase involved in biological activities, including
cell proliferation, apoptosis, migration and invasion (37). Research indicates that RhoA can
suppress the expression of several adhesion molecules such as ZO-1,
Connexin 43 in ECs, thereby increasing vascular permeability
(38). Additionally, RhoA
mediates OA progression by affects cartilage degeneration. Notably,
chondrocyte cytoskeleton remodeling is directly related to the
progression of OA and is characterized by the RhoA/Rock pathway
activation-induced chondrocyte cytoskeleton reorganization, changes
in cell shape, and stress fiber formation (39). Additionally, RhoA mediates
Wnt/β-catenin regulation of chondrocyte catabolism, hypertrophy,
and cartilage degradation (40).
However, the role of RhoA in the vascular permeability of the
subchondral bone in OA is poorly understood. However, the role of
RhoA in the vascular permeability of the subchondral bone in OA is
poorly understood. In the present study, high RhoA expression was
associated with increased vascular permeability of the subchondral
bone in patients with OA. Therefore, it is crucial to investigate
the association between RhoA and vascular permeability of the
subchondral bone in OA.
RhoA can affect blood-brain barrier permeability by
regulating tight junction proteins (41). Moreover, RhoA adjusts EC
permeability by enhancing actin filaments (42). The contraction force generated by
branched actin filaments widens the gap between cells, thereby
increasing permeability. Here, proteomic and bioinformatics
analyses indicated an association between RhoA, ferroptosis and
vascular permeability in OA. Additionally, in vitro
experiments showed that RhoA inhibition reversed IL-1β-induced
increase in vascular permeability in ECs and DMM mice. Moreover,
RhoA inhibition suppressed lipid metabolism and activated the
antioxidant system in DMM mice. Overall, these findings suggested
that RhoA may regulate vascular permeability by mediating
ferroptosis. RhoA inhibition suppressed ferroptosis by activating
the antioxidant system and suppressing lipid peroxidation in
vitro, which was confirmed by the results of biochemical
analysis of mouse serum. Additionally, RhoA inhibition ameliorated
IL-1β-induced decrease in mitochondrial membrane potential in ECs.
Therefore, it was hypothesized that RhoA may decrease in
mitochondrial membrane potential, thereby elevating ROS production
in the cytoplasm, promoting lipid peroxidation, inhibiting the
antioxidant system and inducing ferroptosis.
Many studies have shown that ferroptosis is involved
in the damage and degeneration of chondrocytes, thereby inducing
the progression of osteoarthritis (43,44). Various drugs, including
astaxanthin and biochanin A, alleviate cartilage degeneration in
osteoarthritis by inhibiting ferroptosis (45,46). Additionally, ferroptosis is
involved in the death of endothelial cells and changes in vascular
permeability. However, the exact role of ferroptosis in the
vascular permeability of subchondral bone in osteoarthritis remains
unclear. To elucidate the effect of ferroptosis on vascular
permeability of subchondral bone in OA, in vitro and in
vivo experiments were performed using a ferroptosis
inhibitor/activator. Ferroptosis inhibition effectively maintained
the permeability of ECs, whereas ferroptosis activation reversed
the protective effects of RhoA inhibition on EC permeability.
Similarly, in vivo experiments showed that ferroptosis
inhibition ameliorated subchondral bone sclerosis and remodeling in
DMM mice. Mechanistically, RhoA inhibition-induced decrease in
ferroptosis and improvement in vascular permeability may be
primarily attributed to suppression of oxidative stress and
enhancement of mitochondrial function. IL-1β-induced increase in
ROS generation and mitochondrial dysfunction in ECs may increase
permeability. By contrast, inhibiting ROS generation may regulate
cell permeability and inflammatory response (47). Lipid metabolism may also be
involved in oxidative stress- and mitochondrial dysfunction-induced
increase in vascular permeability. Systemic inflammation is
characterized by an increase in ROS production, which can
upregulate oxidized HDL and nullify the protective effects of
non-oxidized HDL on blood vessels. Additionally, ROS can promote
endothelial fibrosis, resulting in downregulation of adhesion
proteins, thereby compromising endothelial integrity and increasing
permeability (48).
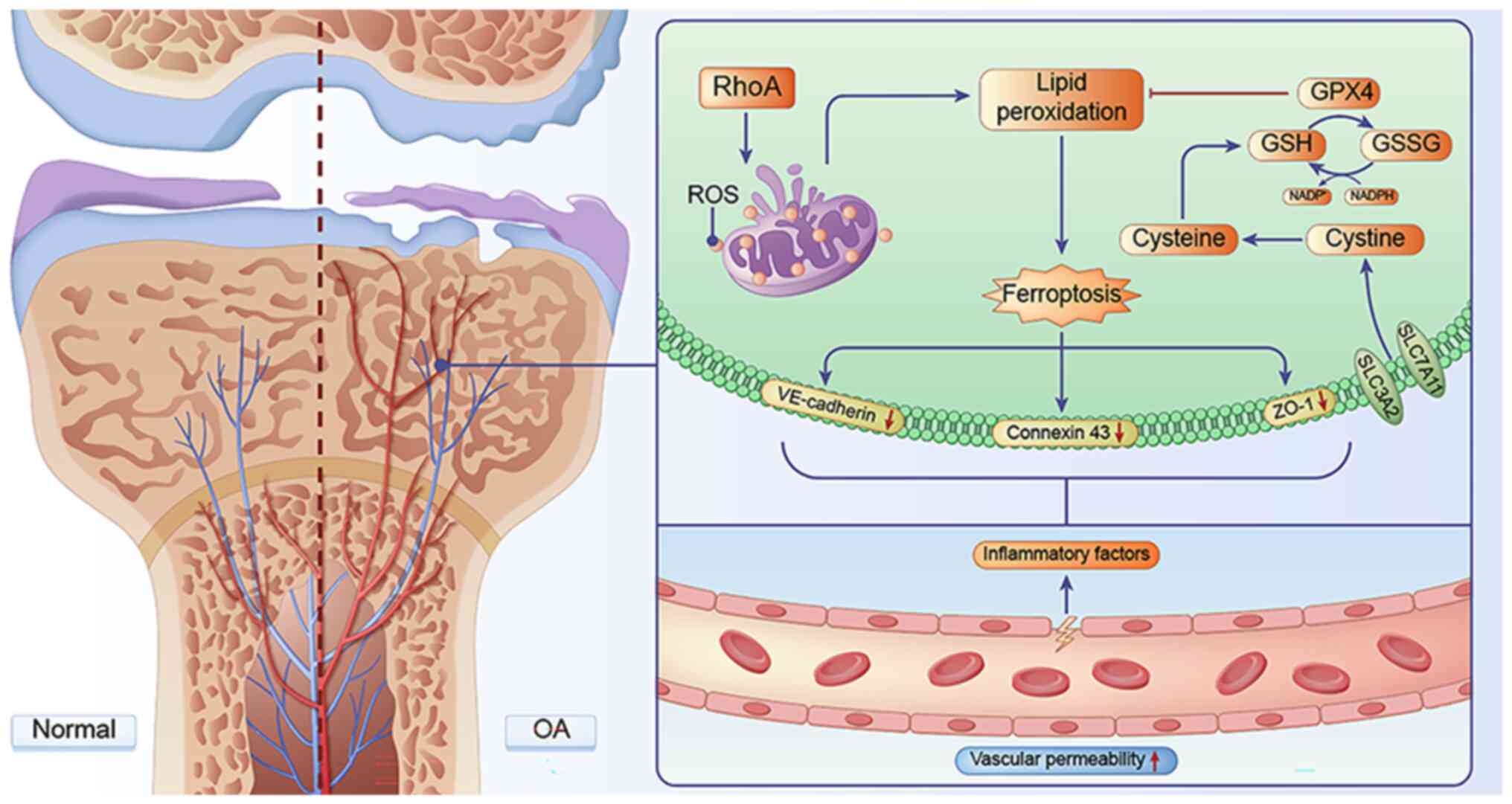
In conclusion, the present study demonstrated that
RhoA can effectively induce OA progression by modulating
ferroptosis and vascular permeability. Specifically, RhoA disrupted
mitochondrial function in ECs, leading to oxidative stress.
Excessive ROS production can induce ferroptosis in ECs and inhibit
expression of cell adhesion proteins, such as ZO-1, connexin 43 and
VE-cadherin, thereby damaging inter-EC connections and increasing
vascular permeability. Excessive release of inflammatory factors
from newly formed blood vessels contributes to the progression of
OA (Fig. 9). Moreover, RhoA may
be a potential key target for treating OA in future, thus
presenting a novel therapeutic approach for OA treatment. Although
inhibiting RhoA and ferroptosis effectively alleviated cartilage
degeneration and subchondral bone remodeling, thereby improving the
progression of OA in vivo, further study is required to
elucidate the mechanism.
Supplementary Data
Availability of data and materials
The data generated in the present study may be found
in the ProteomeXchange Consortium via the iProX partner repository
under accession number PXD051627 or at the following URL:
proteomexchange.org.
Authors' contributions
QJ, DX and XH designed the study. XH, KT, XL, XC, YS
and LM performed experiments. ZLu, ZC, LZ and PL analyzed data. GF,
XZ, ZLa and CZ analyzed and interpreted data and wrote the
manuscript. XH and KT confirm the authenticity of all the raw data.
All authors have read and approved the final manuscript.
Ethics approval and consent to
participate
The present study was approved by the Ethics
Committee of the General Hospital of Ningxia Medical University,
China (approval no. KYLL-2021-269). The study adhered to the
Helsinki Declaration. Written informed consent provided was by the
patients. All animal experiment procedures were executed in
compliance with the Guidelines for the Care and Use of Laboratory
Animals by the National Institutes of Health and were approved by
the Animal Ethics Committee of Ningxia Medical University, China
(approval no. IACUC-NY LAC-2023-100)
Patient consent for publication
The patients provided written informed consent
regarding the publication of the case details and associated
images.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgements
Not applicable.
Funding
The present study was supported by National Natural Science
Foundation of China (grant nos. U22A20285, 82160433 and 82360319),
Key R&D Project of Autonomous Region (grant nos. 2023BEG02018,
2021BEG02037 and 2022BEG03126), Scientific Research Project of
Ningxia University (grant no. NYG-2022033) and Ningxia Medical
University General Hospital 'Medical Engineering Special' (grant
no. NYZYYG-001).
References
|
1
|
Glyn-Jones S, Palmer AJ, Agricola R, Price
AJ, Vincent TL, Weinans H and Carr AJ: Osteoarthritis. Lancet.
386:376–387. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Hunter DJ and Bierma-Zeinstra S:
Osteoarthritis. Lancet. 393:1745–1759. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Mapp PI and Walsh DA: Mechanisms and
targets of angiogenesis and nerve growth in osteoarthritis. Nat Rev
Rheumatol. 8:390–398. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Walsh DA, McWilliams DF, Turley MJ, Dixon
MR, Fransès RE, Mapp PI and Wilson D: Angiogenesis and nerve growth
factor at the osteochondral junction in rheumatoid arthritis and
osteoarthritis. Rheumatology (Oxford). 49:1852–1861. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ni R, Guo XE, Yan C and Wen C: Hemodynamic
stress shapes subchondral bone in osteoarthritis: An emerging
hypothesis. J Orthop Translat. 32:85–90. 2021. View Article : Google Scholar
|
|
6
|
Peng Y, Wu S, Li Y and Crane JL: Type H
blood vessels in bone modeling and remodeling. Theranostics.
10:426–436. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Adzraku SY, Wang G, Cao C, Bao Y, Wang Y,
Smith AO, Du Y, Wang H, Li Y, Xu K, et al: Robo4 inhibits gamma
radiation-induced permeability of a murine microvascular
endothelial cell by regulating the junctions. Cell Mol Biol Lett.
28:22023. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Qian T, Qi B, Fei Y, Li J, Luo L, Lv B,
Song Y, Sheng S, Xiao W, Huang X and Wang X: PLD2 deletion
alleviates disruption of tight junctions in sepsis-induced ALI by
regulating PA/STAT3 phosphorylation pathway. Int Immunopharmacol.
114:1095612023. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Arnold TR, Stephenson RE and Miller AL:
Rho GTPases and actomyosin: Partners in regulating epithelial
cell-cell junction structure and function. Exp Cell Res. 358:20–30.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ridley AJ: Rho family proteins:
Coordinating cell responses. Trends Cell Biol. 11:471–477. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zhao H, Kong H, Wang W, Chen T, Zhang Y,
Zhu J, Feng D and Cui Y: High glucose aggravates retinal
endothelial cell dysfunction by activating the
RhoA/ROCK1/pMLC/Connexin43 signaling pathway. Invest Ophthalmol Vis
Sci. 63:222022. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Chen J, Shi W, Xu Y, Zhang H and Chen B:
Hirudin prevents vascular endothelial cell apoptosis and
permeability enhancement induced by the serum from rat with chronic
renal failure through inhibiting RhoA/ROCK signaling pathway. Drug
Dev Res. 82:553–561. 2021. View Article : Google Scholar
|
|
13
|
Dixon SJ, Lemberg KM, Lamprecht MR, Skouta
R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS,
et al: Ferroptosis: An iron-dependent form of nonapoptotic cell
death. Cell. 149:1060–1072. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Miao Y, Chen Y, Xue F, Liu K, Zhu B, Gao
J, Yin J, Zhang C and Li G: Contribution of ferroptosis and GPX4's
dual functions to osteoarthritis progression. EBioMedicine.
76:1038472022. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zhang S, Xu J, Si H, Wu Y, Zhou S and Shen
B: The role played by ferroptosis in osteoarthritis: Evidence based
on iron dyshomeostasis and lipid peroxidation. Antioxidants
(Basel). 11:16682022. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Fei Y and Huang X, Ning F, Qian T, Cui J,
Wang X and Huang X: NETs induce ferroptosis of endothelial cells in
LPS-ALI through SDC-1/HS and downstream pathways. Biomed
Pharmacother. 175:1166212024. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Li W, Zhao X, Zhang R, Liu X, Qi Z, Zhang
Y, Yang W, Pang Y, Zhao C, Fan B, et al: Ferroptosis inhibition
protects vascular endothelial cells and maintains integrity of the
blood-spinal cord barrier after spinal cord injury. Neural Regen
Res. 18:2474–2481. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Gu Y, Hao S, Liu K, Gao M, Lu B, Sheng F,
Zhang L, Xu Y, Wu D, Han Y, et al: Airborne fine particulate matter
(PM2.5) damages the inner blood-retinal barrier by
inducing inflammation and ferroptosis in retinal vascular
endothelial cells. Sci Total Environ. 838:1565632022. View Article : Google Scholar
|
|
19
|
Altman R, Asch E, Bloch D, Bole G,
Borenstein D, Brandt K, Christy W, Cooke TD, Greenwald R, Hochberg
M, et al: Development of criteria for the classification and
reporting of osteoarthritis. Classification of osteoarthritis of
the knee. Diagnostic and therapeutic criteria committee of the
American Rheumatism Association. Arthritis Rheum. 29:1039–1049.
1986. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Yan J, Ding D, Feng G, Yang Y, Zhou Y, Ma
L, Guo H, Lu Z and Jin Q: Metformin reduces chondrocyte pyroptosis
in an osteoarthritis mouse model by inhibiting NLRP3 inflammasome
activation. Exp Ther Med. 23:2222022. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Francis TC, Gaynor A, Chandra R, Fox ME
and Lobo MK: The selective RhoA inhibitor rhosin promotes stress
resiliency through enhancing D1-medium spiny neuron plasticity and
reducing hyperexcitability. Biol Psychiatry. 85:1001–1010. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Guerrero-Hue M, García-Caballero C,
Palomino-Antolín A, Rubio-Navarro A, Vázquez-Carballo C, Herencia
C, Martín-Sanchez D, Farré-Alins V, Egea J, Cannata P, et al:
Curcumin reduces renal damage associated with rhabdomyolysis by
decreasing ferroptosis-mediated cell death. FASEB J. 33:8961–8975.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhao YY, Yang YQ, Sheng HH, Tang Q, Han L,
Wang SM and Wu WY: GPX4 plays a crucial role in Fuzheng Kang'ai
decoction-induced non-small cell lung cancer cell ferroptosis.
Front Pharmacol. 13:8516802022. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Jiang A, Xu P, Yang Z, Zhao Z, Tan Q, Li
W, Song C, Dai H and Leng H: Increased Sparc release from
subchondral osteoblasts promotes articular chondrocyte degeneration
under estrogen withdrawal. Osteoarthritis Cartilage. 31:26–38.
2023. View Article : Google Scholar
|
|
25
|
Glasson SS, Chambers MG, Van Den Berg WB
and Little CB: The OARSI histopathology initiative-recommendations
for histological assessments of osteoarthritis in the mouse.
Osteoarthritis Cartilage. 18(Suppl 3): S17–S23. 2010. View Article : Google Scholar
|
|
26
|
Ahola S and Langer T: Ferroptosis in
mitochondrial cardiomyopathy. Trends Cell Biol. 34:150–160. 2024.
View Article : Google Scholar
|
|
27
|
Cao L, Yang T, Huang S, Yun X, Hou H, Wang
T, Shi D and Li X: Expression patterns of ZO-1/2 and their effects
on porcine oocyte in vitro maturation and early embryonic
development. Theriogenology. 161:262–270. 2021. View Article : Google Scholar
|
|
28
|
Schwayer C, Shamipour S, Pranjic-Ferscha
K, Schauer A, Balda M, Tada M, Matter K and Heisenberg CP:
Mechanosensation of tight junctions depends on ZO-1 phase
separation and flow. Cell. 179:937–952.e18. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Liu X, Wang T, Wang W, Liang X, Mu Y, Xu
Y, Bai J and Geng D: Emerging potential therapeutic targets of
ferroptosis in skeletal diseases. Oxid Med Cell Longev.
2022:31123882022.PubMed/NCBI
|
|
30
|
Zeng H, Hou Y, Zhou X, Lang L, Luo H, Sun
Y, Wan X, Yuan T, Wang R, Liu Y, et al: Cancer-associated
fibroblasts facilitate premetastatic niche formation through lncRNA
SNHG5-mediated angiogenesis and vascular permeability in breast
cancer. Theranostics. 12:7351–7370. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Zhu Y: Gap junction-dependent and
-independent functions of connexin43 in biology. Biology (Basel).
11:2832022.PubMed/NCBI
|
|
32
|
Liu J, Rickel A, Smith S, Hong Z and Wang
C: 'Non-cytotoxic' doses of metal-organic framework nanoparticles
increase endothelial permeability by inducing actin reorganization.
J Colloid Interface Sci. 634:323–335. 2023. View Article : Google Scholar
|
|
33
|
Doll S, Proneth B, Tyurina YY, Panzilius
E, Kobayashi S, Ingold I, Irmler M, Beckers J, Aichler M, Walch A,
et al: ACSL4 dictates ferroptosis sensitivity by shaping cellular
lipid composition. Nat Chem Biol. 13:91–98. 2017. View Article : Google Scholar :
|
|
34
|
Yang WS, SriRamaratnam R, Welsch ME,
Shimada K, Skouta R, Viswanathan VS, Cheah JH, Clemons PA, Shamji
AF, Clish CB, et al: Regulation of ferroptotic cancer cell death by
GPX4. Cell. 156:317–331. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Jiang X, Stockwell BR and Conrad M:
Ferroptosis: Mechanisms, biology and role in disease. Nat Rev Mol
Cell Biol. 22:266–282. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Liu Y, Xie HQ and Shen B: Type H vessels-a
bridge connecting subchondral bone remodelling and articular
cartilage degeneration in osteoarthritis development. Rheumatology
(Oxford). 62:1436–1444. 2023. View Article : Google Scholar
|
|
37
|
Kempers L, Driessen AJM, van Rijssel J,
Nolte MA and van Buul JD: The RhoGEF trio: A protein with a wide
range of functions in the vascular endothelium. Int J Mol Sci.
22:101682021. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Li L, Xin J, Wang H, Wang Y, Peng W, Sun
N, Huang H, Zhou Y, Liu X, Lin Y, et al: Fluoride disrupts
intestinal epithelial tight junction integrity through
intracellular calcium-mediated RhoA/ROCK signaling and myosin light
chain kinase. Ecotoxicol Environ Saf. 257:1149402023. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Yang J, Wang L, Zhang Z, Sun Q and Zhang
Y: Downregulation of HAS-2 regulates the chondrocyte cytoskeleton
and induces cartilage degeneration by activating the RhoA/ROCK
signaling pathway. Int J Mol Med. 52:572023. View Article : Google Scholar :
|
|
40
|
Liang J, Feng J, Wu WK, Xiao J, Wu Z, Han
D, Zhu Y and Qiu G: Leptin-mediated cytoskeletal remodeling in
chondrocytes occurs via the RhoA/ROCK pathway. J Orthop Res.
29:369–374. 2011. View Article : Google Scholar
|
|
41
|
Feng S, Zou L, Wang H, He R, Liu K and Zhu
H: RhoA/ROCK-2 pathway inhibition and tight junction protein
upregulation by catalpol suppresses lipopolysaccaride-induced
disruption of blood-brain barrier permeability. Molecules.
23:23712018. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Dasgupta SK, Le A, Vijayan KV and
Thiagarajan P: Dasatinib inhibits actin fiber reorganization and
promotes endothelial cell permeability through RhoA-ROCK pathway.
Cancer Med. 6:809–818. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Gao L, Hua W, Tian L, Zhou X, Wang D, Yang
Y and Ni G: Molecular mechanism of ferroptosis in orthopedic
diseases. Cells. 11:29792022. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Yao X, Sun K, Yu S, Luo J, Guo J, Lin J,
Wang G, Guo Z, Ye Y and Guo F: Chondrocyte ferroptosis contribute
to the progression of osteoarthritis. J Orthop Translat. 27:33–43.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Wang X, Liu Z, Peng P, Gong Z, Huang J and
Peng H: Astaxanthin attenuates osteoarthritis progression via
inhibiting ferroptosis and regulating mitochondrial function in
chondrocytes. Chem Biol Interact. 366:1101482022. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
He Q, Yang J, Pan Z, Zhang G, Chen B, Li
S, Xiao J, Tan F, Wang Z, Chen P and Wang H: Biochanin A protects
against iron overload associated knee osteoarthritis via regulating
iron levels and NRF2/System xc-/GPX4 axis. Biomed Pharmacother.
157:1139152023. View Article : Google Scholar
|
|
47
|
Qin X, Zhu L, Zhong Y, Wang Y, Wu G, Qiu
J, Wang G, Qu K, Zhang K and Wu W: Spontaneously
right-side-out-orientated coupling-driven ROS-sensitive
nanoparticles on cell membrane inner leaflet for efficient
renovation in vascular endothelial injury. Adv Sci (Weinh).
10:e22050932023. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Rojas M, Prado Y, Tapia P, Carreño LJ,
Cabello-Verrugio C and Simon F: Oxidized high-density lipoprotein
induces endothelial fibrosis promoting hyperpermeability,
hypotension, and increased mortality. Antioxidants (Basel).
11:24692022. View Article : Google Scholar : PubMed/NCBI
|