Diabetic kidney disease (DKD) is a prevalent
microvascular complication, and the leading cause of mortality in
individuals with diabetes (1).
Approximately 30-40% of patients with diabetes develop DKD
(2). It typically begins with
microalbuminuria and, if not managed effectively, can progress to
end-stage renal disease (3,4).
Disease progression results in kidney failure, cardiovascular
complications and premature mortality (5). Angiotensin-converting enzyme
inhibitors, angiotensin receptor blockers, sodium-glucose
cotransporter 2 inhibitors and mineralocorticoid receptor
antagonists have been used to delay DKD progression (6-8).
However, these treatments are not universally effective (9,10). A 2021 study indicated that ~537
million adults worldwide were living with diabetes, and this number
is projected to rise to 738 million by 2045. Of these individuals,
nearly 90-95% have type 2 diabetes and approximately half are
anticipated to develop DKD (11). Therefore, there is an urgent need
to develop improved strategies for the prevention and treatment of
DKD.
The pathogenesis of DKD is influenced by genetic and
environmental factors. Specific genetic variants or predispositions
can affect the susceptibility of an individual to DKD, and risk
factors such as a high-sugar diet, high salt intake, obesity and
physical inactivity contribute to the development of characteristic
histological changes in the kidneys (12-14). These changes include podocyte
depletion, tubular epithelial-mesenchymal transition (EMT),
fibroblast activation, mesangial cell dysplasia and extracellular
matrix (ECM) accumulation. Additionally, glomerular endothelial
cells may undergo endothelial-mesenchymal transformation (EndMT),
ultimately leading to irreversible renal fibrosis (15,16). Histone methylation of specific
lysine or arginine residues is a critical post-translational
modification (PTM) involved in these processes (17). The synergistic action of histone
methylases and demethylases results in the addition or removal of
methyl groups from specific sites on histones, leading to
monomethylated (me1), dimethylated (me2) and trimethylated (me3)
modifications (18,19). Although these changes only subtly
alter the primary structure of histones, they can trigger chromatin
remodeling, modify the accessibility of the underlying DNA
sequences and regulate gene activation or silencing (20). Advanced glycation end products
(AGEs) and various injury mediators produced by poor long-term
glycemic control in patients with DKD have persistent effects on
renal function by altering the distribution pattern of histone
methylation in the kidneys (21). This highlights the pivotal role
of histone methylation in mediating interactions between genes and
environmental factors. By targeting these methylation
modifications, it is possible to effectively improve the renal
histological manifestations and prevent or reverse the progression
of renal fibrosis and proteinuria in DKD, thereby offering a
promising novel therapeutic strategy for prevention and treatment.
Numerous studies have focused on the unique role of histone
methylation modifications in DKD, emphasizing the importance of an
improved understanding of the mechanisms underlying DKD and
identifying novel treatment options (21-23).
The present review elucidates the mechanisms of
histone methylation modifications and provides a comprehensive
overview of the current understanding of histone methylation in
patients with DKD. Furthermore, the present review systematically
summarizes the alterations and impacts of histone methylases in
various intrinsic renal cells of patients with DKD, including
podocytes, renal tubule cells, fibroblasts, mesangial cells and
glomerular endothelial cells, and discusses the therapeutic
potential of inhibitors targeting histone methylation modifications
as well as their prospective mechanistic implications.
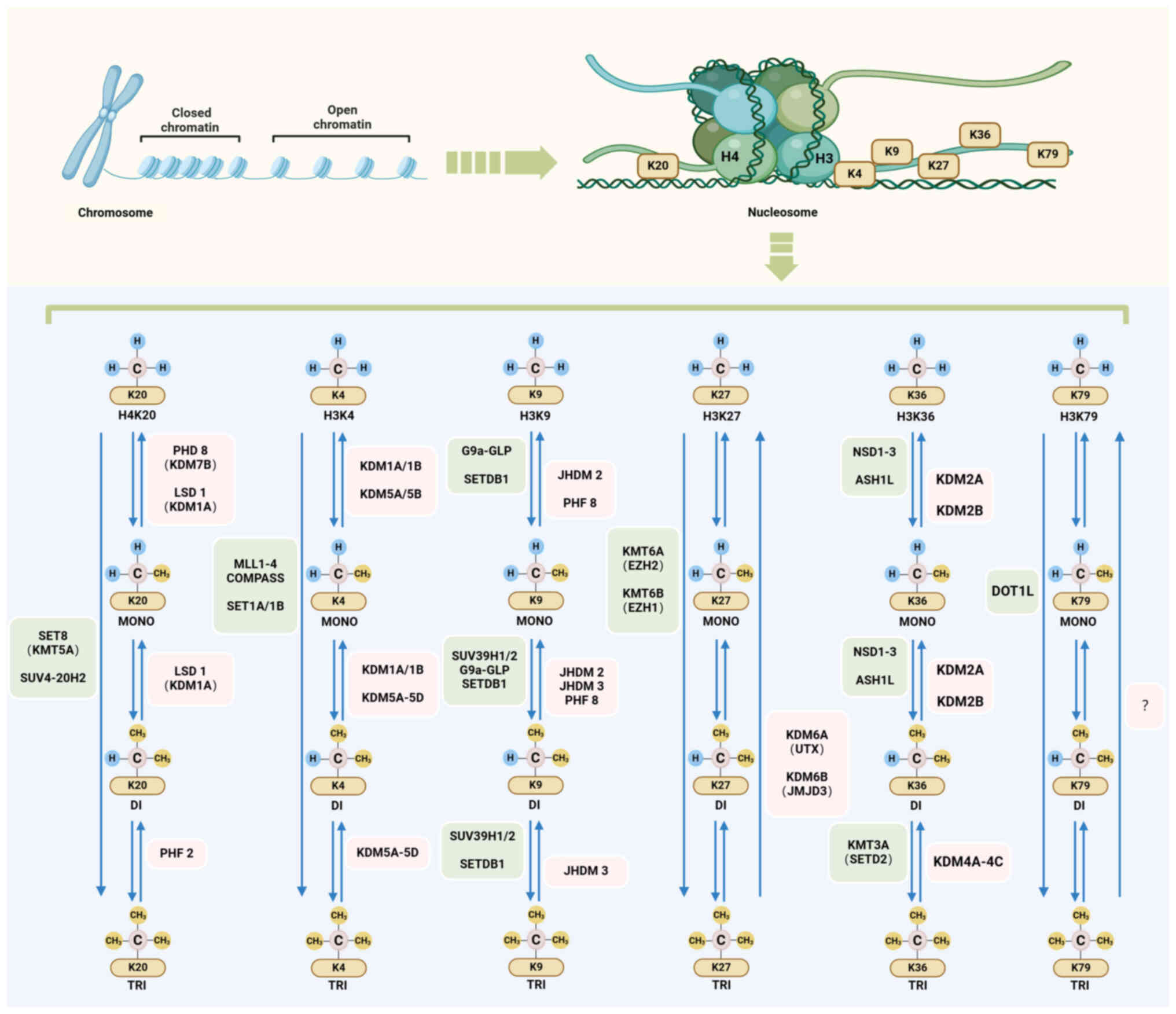
Histone methylation is a key epigenetic modification
that influences various cellular processes, including gene
expression, DNA replication and repair, chromatin structure, and
cell cycle control (24,25). Several histone residues are prone
to methylation, notably at well-known sites, such as histone H3
lysine (H3K)4, H3K9, H3K27, H3K36, H3K79 and histone H4 lysine
(H4K)20. This methylation process is dynamic and reversible,
facilitated by the interplay between histone lysine
methyltransferases (HKMTs or 'writers') and demethylases (HKDMs or
'erasers') (26). The
methylation balance at these sites is maintained using
S-adenosylmethionine (SAM) as the methyl donor (27,28).
Various types of HKMTs exist. All HKMTs except the
DOT1 family possess conserved su(var)3-9, enhancer-of-zeste and
trithorax (SET) domains, making them broadly similar from
single-cell organisms to complex multicellular organisms (29). Each HKMT has a unique substrate
specificity and catalytic function (Fig. 1). In humans, these classical
HKMTs are categorized based on their catalytic sites: H3K4
methyltransferases, including mixed lineage leukemia (MLL) family
complexes; H3K9 methyltransferases, including suppressor of
variegation 3-9 homolog 1 and 2, SET domain bifurcated 1 and
euchromatic HKMT2 (G9a); H3K27 methyltransferases, including
enhancer of zeste homolog 2 (EZH2); H3K36 methyltransferases,
including nuclear receptor binding SET domain protein 1-3; H3K79
methyltransferases, including disruptor of telomeric silencing
1-like; and H4K20 methyltransferases, including SET
domain-containing protein 8 and suppressor of variegation 4-20
homolog 2 (26). Despite the
complex recognition and binding mechanisms of HKMTs, HKMTs can
transfer one, two or three methyl groups from the donor SAM to the
ε-nitrogen of specific lysine side chains. While this enzymatic
reaction induces only subtle changes in the primary structure of
the modified polypeptide, it substantially affects the chromatin
structure and DNA sequence accessibility, thereby influencing gene
expression (27). Specifically,
methylation of H3K4, H3K36 and H3K79 serves a pivotal role in
transcriptional activation, whereas methylation of H3K9, H3K27 and
H4K20 is typically associated with transcriptional inhibition
(30). In addition, there are
several non-classical lysine methylation sites on core histones
(such as H3K23, H3K37 and H4K5) whose biological significance and
regulatory pathways remain incompletely understood (31-33).
Histone lysine residues are not exclusive substrates
of HKMTs. While numerous known histone methyltransferases are
primarily involved in promoting lysine methylation on histones,
they can also modify non-histone substrates (34). It is well-established that
methylation of non-histone substrates is prevalent in cells.
Various proteins, including transcription factors, cyclins,
metabolic enzymes and DNA repair proteins, can be methylated at
lysine residues. These changes impact the function, stability,
interaction and intracellular localization of proteins, and serve a
crucial role in the development of cancer, neurodegenerative
diseases, metabolic disorders and numerous other diseases (35,36). Disease-specific methylation of
non-histone substrates by a number of known histone methylases
serves as a biomarker for these diseases and offers potential
therapeutic targets (37,38).
Historically, histone methylation has been
considered a permanent and stable modification (39). However, the identification of
lysine-specific demethylase (LSD)1 (also known as KDM1A) from the
LSD family has revolutionized this perspective (40). HKDMs, including members of the
LSD and jumonji C domain (JmjC) domain-containing families, can
reverse changes in histone methylation (18). Specifically, the LSD family,
which includes LSD1 and LSD2, removes mono- and dimethyl groups
from histones H3K4 and H4K20 (41). Additionally, JmjC
domain-containing families, which encompass subfamilies, such as
lysine demethylase 2-6 (KDM2-6), serve substantial roles in histone
demethylation (42). Although no
H3K79 demethylases have been identified, studies have suggested
that H3K79 methylation is reversible. Observations have indicated
that H3K79 methylation levels changed dynamically during the cell
cycle, with a notable decrease after S phase, specifically in
mammalian cell lines such as HeLa cells and mouse embryonic
fibroblasts (43,44). These findings suggested the
presence of histone demethylases in these cells.
The balance of histone methylation within cells is
particularly vulnerable to external disruption. This can affect the
function of essential proteins that add or remove methylation
markers and can also change the levels of SAM, which influences the
overall pattern of methylation modifications (18). SAM availability is critical as it
is a vital methyl donor and a key intermediate in several metabolic
pathways (45). Disturbances in
metabolic processes can impair SAM synthesis and availability,
substantially affecting the expression and maintenance of histone
methylation markers (46,47).
Several proteins, known as co-factors, influence histone
methylation by interacting with HKMTs or HKDMs. These interactions
affect the localization, stability and enzymatic functions of HKMTs
and HKDMs (48,49). For example, WD repeat domain 5
and ASH2 like histone lysine methyltransferase complex subunit
(ASH2L), key components of the MLL complex, enhance H3K4
methylation by facilitating the recruitment of other MLL complex
proteins to target sites, thereby affecting the progression of
breast cancer and glioblastoma (50,51). In addition to proteins,
non-coding RNAs serve a vital role in controlling histone
methylation and substantially affect gene transcription by
recruiting histone methyltransferases (52). The interplay among histone
modifications has been the focus of epigenetic research. Histone
modification crosstalk refers to the phenomenon in which the
recognition or deposition of one epigenetic marker on a histone
influences the distribution of another marker. This intricate
crosstalk enhances the complexity and specificity of histone
modification combinations, thereby enabling precise regulation of
various biological processes, such as cell fate determination,
development and disease states (53). A prime example of this cross-talk
is the interaction between histone methylation and ubiquitination.
Specifically, ubiquitination of lysine 14 on histone H3 is crucial
for the initiation of H3K9 methylation (54). Similarly, existing histone
acetylation and phosphorylation in gene promoter regions subtly
influence the further development of histone methylation. For
example, the dynamic interchange of acetylation and methylation of
H3K27 regulates gene expression, with acetylation promoting
transcriptional activation and methylation, leading to silencing
(55). In cancer cells,
deacetylated H3K27 can be specifically targeted for methylation by
KDM6A (also known as ubiquitously transcribed tetratricopeptide
repeat, X chromosome) after treatment with inhibitory drugs,
resulting in the transcriptional suppression of MYC, BCL2,
CCND1 and other oncogenes (56). Additionally, a study showed that
phosphorylation at threonine 11 on histone H3 hinders
DOT1-catalyzed methylation at H3K79me3, thereby affecting the
chromatin state and gene transcription activities vital for
autophagy regulation and telomere silencing (57).
Substantial advancements in molecular biology and
genomics have transformed medical research, moving beyond
macroscopic phenotypic observations to explore changes in key
epigenetic markers, such as histone methylation (58,59). These changes serve a critical
role in disease pathogenesis and exert intricate molecular effects.
Dysregulation of histone methylation has been linked to various
diseases, including cancer, neurodegenerative disorders and
developmental anomalies (60,61). Rapid and dynamic changes in
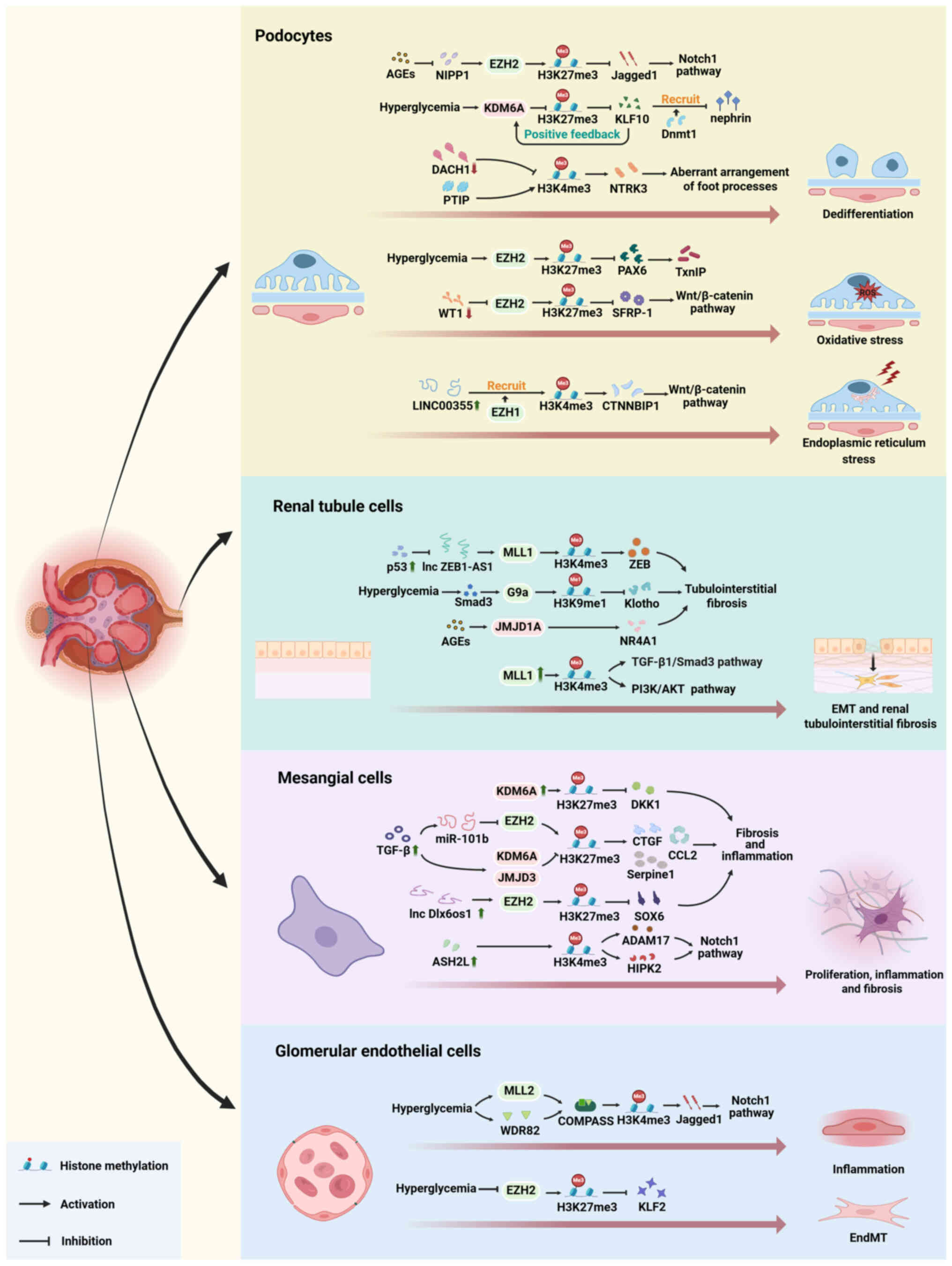
histone methylation have been observed in patients with DKD.
Research has shown an association between DKD severity and the
levels of the active marker H3K4me2 and the inhibitory marker
H3K27me3 (23,62). Further investigations have
indicated that these methylation changes can trigger pathological
processes in DKD, such as cell dedifferentiation, inflammation,
oxidative stress and fibrosis (63-66). Additionally, the response of
different kidney cell types to histone methylation varies, leading
to nonuniform pathological changes. This variability results from
the diverse functions and microenvironmental differences between
these cells (Fig. 2). In this
section, the altered distribution patterns of histone methylation
(Table I) and functional
disparities in histone methylation in specific intrinsic renal
cells under diabetic conditions are discussed.
Podocytes are specialized epithelial cells that form
intricate networks of foot processes, essential for the glomerular
filtration barrier. However, their proliferative capacity is
limited, and once damaged, they cannot be repaired, making them
particularly susceptible to DKD (67). These cells face multiple
stressors, including mechanical, oxidative and immune challenges.
Although normally adaptive to stress to maintain homeostasis,
excessive stress may lead to substantial biological changes, such
as structural disintegration and metabolic dysfunction, ultimately
causing podocyte loss (68). In
patients with DKD, podocytes disappear in response to
hyperglycemia. Subsequently, surviving podocytes undergo changes in
size and shape to compensate for the exposed basement membrane
caused by podocyte loss. This morphological transition is
accompanied by the activation of signaling pathways associated with
podocyte dedifferentiation, leading to the transformation of the
cell phenotype, such as diminished expression of cell-specific
markers, aberrant organization of foot processes and impairment of
cell-specific functionalities. Although these compensatory changes
may temporarily delay the progression of proteinuria, they
ultimately compromise podocyte damage resistance and structural
integrity, thereby exacerbating the progression of DKD (69,70).
Podocyte dedifferentiation is regulated by a complex
array of molecular mechanisms, primarily involving signaling
pathways, such as the Wnt/β-catenin, Notch and TGF-β/Smad signaling
pathways (71). These pathways
collectively govern cell fate (72). Histone methylation changes the
transcriptional status of key factors in these signaling pathways,
affecting podocyte differentiation and the occurrence of glomerular
disease. In the promoter region of the podocyte-specific marker
Wilms tumor 1 (WT1), jumonji domain-containing protein (JMDJ)3
(KDM6B), a demethylase from the JmjC family, mediates a reduction
in transcriptional inhibitory marker H3K27me3 to maintain the
normal expression of podocyte-specific markers (73). WT1 has an antagonistic effect on
histone methyltransferase EZH2, which ameliorates podocytic
injuries mediated by β-catenin in diabetic models, such as
apoptosis and oxidative stress. This gene transcriptional
de-inhibition occurs through reducing the enrichment of H3K27me3 in
the promoter of the Wnt antagonist, secreted frizzled related
protein 1, suggesting that podocytes possess compensatory
mechanisms to adapt to epigenetic modifications caused by
environmental changes (74).
Elevated levels of AGEs, which are key mediators of metabolic
memory, are associated with the epigenetic reactivation of the
long-silenced Notch pathway in DKD (75). AGEs decrease the expression of
nuclear inhibitor of protein phosphatase 1 (NIPP1), which is an
upstream regulator of EZH2, disrupting the interaction between
NIPP1 and EZH2, and reducing H3K27me3 levels in podocytes (76). Majumder et al (64) observed that decreased H3K27me3,
due to downregulation of methylase EZH2 or upregulation of
demethylase KDM6A, leads to the de-repression of Jagged1, thereby
activating the Notch1 pathway. This promotes podocyte
dedifferentiation and susceptibility to damage, and contributes to
renal function deterioration (64). However, RNA sequencing analysis
in another study revealed no substantial alterations in
Jagged1 and other Notch receptor mRNA levels in
podocytes overexpressing KDM6A. KDM6A expression was upregulated
under high-glucose conditions, but not under hypertensive
conditions. Loss of H3K27me3 led to transcriptional de-repression
of Kruppel like factor 10 (KLF10), enhancing the feedback loop
between KDM6A and KLF10, and maintaining high expression levels of
KLF10 under diabetic conditions. Increased KLF10 expression
recruited DNMT1 to the nephrin promoter, inhibited nephrin
expression and contributed to podocyte dysfunction. Upregulation of
KLF10 expression also suppressed the expression of other
podocyte-specific genes, such as WT1, Podocin and
Synaptopodin (77).
In patients with diabetic nephropathy, alterations
in H3K4 methylation in podocytes are also observed. Paired box
transactivation domain interacting protein (PTIP)-dependent H3K4
methylation is crucial for maintaining differentiation and cell
type-specific transcriptional programs in mature podocytes. In
normal podocytes, PTIP is recruited to target gene promoters by PAX
or dachshund family transcription factor 1 (DACH1), with each
binding mode exerting opposing effects on methylase activity.
Specifically, when PTIP is recruited by PAX, it promotes methylase
activity, while recruitment by DACH1 inhibits methylase activity
(78-80). This balance shapes the H3K4
methylation landscape in the podocytes. In DKD mice, reduced DACH1
expression leads to transcriptional de-repression of multiple
downstream target genes (80).
Abnormal expression of the H3K4me3 downstream gene NTRK3
results in an aberrant foot process arrangement and impaired
podocyte function (22).
In addition to inducing cellular dedifferentiation,
histone methylation also induces oxidative stress in podocytes. Low
EZH2 expression decreases H3K27me3 levels and triggers PAX6
expression in podocytes with high glucose levels. The enrichment of
PAX6 in the gene promoter promotes the expression of the
antioxidant inhibitor, thioredoxin interacting protein, thereby
enhancing oxidative stress in podocytes (66). Additionally, non-coding RNA
LINC00355 elevates H3K4me3 levels by recruiting histone methylase
EZH1 to the catenin β interacting protein 1 promoter, activating
the β-catenin signaling pathway, and promoting endoplasmic
reticulum stress-induced podocellular injury (81).
Tubular epithelial cells and the tubular
interstitium constitute >90% of the renal cortex and serve
pivotal roles in maintaining fluid and electrolyte balance,
excretion of metabolic waste and regulation of blood pressure
(82). Given their high energy
demand and reliance on aerobic metabolism, tubular epithelial cells
are particularly susceptible to diabetes-related metabolic
disorders (83,84). Structural alterations in the
diabetic renal tubules, including atrophy, interstitial fibrosis
and peritubular capillary thinning, are closely linked to decreased
renal function (85). Tubular
atrophy and interstitial fibrosis form the basis of DKD renal
fibrosis and contribute substantially to the decline in renal
function (86). TGF-β1 is a
well-established pathogenic factor in fibrotic diseases affecting
multiple organs and serves a pivotal role in the onset and
progression of DKD (87-90). In progressive renal fibrosis
associated with DKD, there is substantial upregulation of TGF-β1
synthesis and secretion, along with Smad3 phosphorylation. This
triggers a cascade of core responses, including EMT, fibroblast
activation and abnormal ECM deposition (37,91). Modification of histone
methylation serves as a key epigenetic regulatory mechanism in this
process by modulating the expression of genes related to fibrosis
(92).
H3K4 methylation and gene transcription activation
mediated by histone methylase MLL1 are essential for the activation
of TGF-β/Smad3 and AKT signaling pathways, upregulation of Snail
and downregulation of E-cadherin (93). Tao et al (63) reported that epigenetic inhibitors
targeting methylase EZH2 effectively attenuated kidney injury and
fibrosis by modulating the TGF-β/Smad3 signaling pathway via
regulation of Smad7. Smad7 is an essential inhibitory factor for
fibrosis, which suppresses the activation of the TGF-β1/Smad3
pathway by inhibiting Smad3 phosphorylation (94). In diabetic nephropathy, elevated
p53 levels inhibit the expression of the zinc finger E-box binding
homeobox 1 antisense RNA 1 (ZEB1-AS1) long non-coding RNA
(lncRNA), thereby regulating the binding of methylase MLL1 to the
ZEB promoter. This leads to the downregulation of H3K4me3 and the
inhibition of ZEB expression, contributing to the development of
renal fibrosis (95).
Furthermore, various methylation and demethylation events in renal
tubules can serve as downstream targets of fibrosis-related
signaling pathways. Irifuku et al (96) demonstrated that TGF-β1/Smad3
upregulates G9a, leading to H3K9 methylation, which facilitates the
induction of fibrosis markers and suppresses Klotho expression.
Another study on chronic kidney disease revealed that
hypoxia-inducible factor (HIF)1α/HIF1β-induced activation of
JMJD1A, a histone demethylase opposing G9a, exerts a negative
regulatory effect on the expression of renal fibrosis-related
factors (97). However, whether
the upregulation of HIF1α/HIF1β in DKD results in similar
methylation changes remains unclear. Additionally, elevated levels
of AGEs in a mouse of diabetic nephropathy can upregulate the
expression of JMJD1A, leading to an increase in nuclear receptor
subfamily 4 group A member 1 levels and ultimately inhibiting the
fibrotic process of renal tubular epithelial cells (98). Notably, H3K9 has garnered
attention as a mediator linking the TGF-β1/Smad3 signaling pathway
to Klotho (96). However,
another study indicated that the age-related decreased expression
of Klotho results from the transcriptional suppression of key
factors in the serum/glucocorticoid regulated kinase 1/FOXO3
signaling pathway driven by H3K27me3 (99). Further exploration is warranted
to determine whether H3K9 methylation upstream of Klotho
specifically promotes fibrosis in DKD.
Mesangial cells exert multiple functions in
maintaining glomerular function and structure (107). Within the glomeruli, mesangial
cells, along with their associated mesangial matrix, form a stalk
that holds together multiple capillary loops. Mesangial cells serve
a crucial role in regulating the contraction and expansion of the
capillaries to ensure a normal glomerular filtration rate. Outside
the glomeruli, mesangial cells are integral to regulating blood
pressure and fluid volume through their secretion of renin
(108,109). Additionally, these cells serve
as the primary producers of glomerular matrix (110). In DKD, mesangial cells are
primarily responsible for glomerular hypertrophy and sclerosis.
Heightened activation of mesangial cells is observed in the kidneys
of patients with DKD, which is characterized by increased
proliferative activity and excessive production of type IV collagen
and fibronectin, both of which are pivotal components that
contribute to fibrosis and sclerosis (111-113). In addition, mesangial cells
exhibit reduced responsiveness to angiotensin II (114). At the molecular level,
mesangial cells undergo epigenetic and transcriptional changes,
leading to the expression of various growth factors, fibrotic
elements, complement components and other pro-inflammatory
mediators, which collectively drive the progression of glomerular
fibrosis and sclerosis (115).
Similar to podocytes, abnormal histone methylation
has been demonstrated to mediate aberrant activation of the Notch1
signaling pathway in mesangial cells in a diabetic nephrotic model.
Zhong et al (116,117) identified ASH2L, a crucial
component of the MLL methyltransferase complex, as a pivotal
'transcriptional igniter'. The enrichment of ASH2L-mediated H3K4me3
in the promoter region of the gene substantially enhanced the
expression of a disintegrin and metalloproteinase 17 (ADAM17) and
homeodomain interacting protein kinase 2 (HIPK2) as co-factors.
Subsequently, ADAM17 and HIPK2 facilitate the expression and
nuclear translocation of Notch intracellular domain 1 in the
mesangial cells of diabetic mice, ultimately leading to the
activation of the Notch1 signaling pathway, which is implicated in
the pathogenesis of DKD fibrosis and inflammation (116,117). Furthermore, high expression
levels of the Dlx6 opposite strand transcript 1 (Dlx6os1) lncRNA
are observed in diabetic mesangial cells. Dlx6os1 recruits
methylase EZH2 to inhibit gene expression via EZH2-mediated H3K27
methylation within the SOX6 promoter region, thereby accelerating
hyperglycemic mesangial cell proliferation, fibrosis and the
release of inflammatory cytokines (118). KDM6A, a histone H3K27
demethylase, serves a crucial role in regulating inflammation in
cultured mesangial cells and db/db mouse kidneys. Dickkopf WNT
signaling pathway inhibitor 1 expression is modulated by H3K27me3,
and its upregulation effectively mitigates TGF-β1-induced fibrosis
(119). Furthermore, KDM6A
interacts with p53, leading to DNA damage (120). Notably, H3K27me3 is also
influenced by TGF-β, and the increased TGF-β levels in diabetic
conditions result in decreased methylase EZH2 expression, and
increased demethylase KDM6A and demethylase JMJD3 expression. This
promotes profibrotic and inflammatory gene activation in rat
mesangial cells through H3K27me3 depletion of genes such as
connective tissue growth factor, Serpine1 and C-C motif chemokine
ligand 2 (121).
Glomerular endothelial cells are specialized
vascular cells that form the glomerular filtration membrane and act
as primary barriers to circulating substances in the blood
(122). Glomerular endothelial
cells are particularly susceptible to hyperglycemia-induced damage,
leading to endothelial cell dysfunction in DKD, which is a critical
pathological change in the early stages (123). This dysfunction results in
increased permeability, apoptosis and loss of glomerular
endothelial cell fenestration, resulting in substantial plasma
protein wastage (124).
Although the mechanisms underlying glomerular
endothelial cell dysfunction are not fully understood, histone
methylation modifications are involved in processes such as
cellular inflammation and EndMT. For instance, elevated blood
glucose levels induce increased H3K27me3 levels in endothelial
cells by promoting the nuclear localization of methylase EZH2 and
assembly of the H3K27 methylase complex 2, leading to endothelial
inflammation through the downregulation of Kruppel like factor 2
(125). Furthermore, high
glucose-induced H3K4me3 triggers the transcriptional activity of
Serpine1, a downstream NF-κB gene, resulting in increased
expression of plasminogen activator inhibitor-1, a crucial protein
encoded by Serpine1, ultimately leading to endothelial dysfunction
(126). Huang et al
(127) revealed that methylase
lysine methyltransferase (KMT)5A interacts with its co-factor, cAMP
response element-binding protein, to cooperatively regulate protein
tyrosine phosphatase 1B expression, influencing P65 phosphorylation
and inflammatory factor levels, thereby contributing to the
development of DKD. These findings underscore the substantial role
of high glucose-driven histone methylation in DKD. Additionally,
various histone methylases have been identified as crucial
epigenetic regulators of EndMT in endothelial cells (128). For example, inflammatory
stimuli and hypoxia induce endothelial identity and functional
disorders, which are tightly controlled by the histone demethylase
JMJD2B through the modulation of H3K9me3 levels (129). Further studies are required to
determine whether similar effects occur in renal endothelial cells
under high-glucose conditions.
Histone methylation serves a role in regulating the
differentiation and plasticity of endothelial cells in DKD. Similar
to that in podocytes, histone methylation contributes to the
reactivation of the Notch1 signaling pathway in endothelial cells.
This regulatory mechanism involves the deposition of H3K4me3,
rather than the ablation of H3K27me3 markers. High glucose exposure
results in the accumulation of H3K4me3 in endothelial cells, driven
by increased expression of MLL2 and WD repeat domain 82, two
crucial protein subunits of H3K4 methyltransferase complex.
Aberrant elevation of H3K4me3 in the promoters of Jagged1 and
Jagged2 leads to the acquisition of mesenchymal properties in
endothelial cells through hyperactivation of the Notch signaling
pathway (130). Studies have
elucidated the unique role of bivalent chromosomes composed of
H3K27 and H3K4 linkages in ontogeny and cell phenotype maintenance,
suggesting that the phenotype of terminally differentiated cells is
partially sustained through interactions between active H3K4me3 and
inhibitory H3K27me3 markers (131,132). H3K27 methylation is a
well-established marker of transcriptional silencing and is crucial
for maintaining stable differentiated states (133,134). The abundant deposition of H3K4
in gene promoters often leads to the activation of gene
transcription. H3K4me3 histone methylation co-mediated by the
methylase MLL3/4 and its co-factor PTIP is necessary for the
maintenance or re-establishment of cell epithelial phenotypes
(135). H3K4me3, a marker of
gene activation, and inhibitory H3K27me3, a marker of gene
repression, together form bivalent chromatin in the kidney
(136). However, their
influence on the occurrence and progression of DKD via crosstalk
remains debatable. Therefore, it is prudent to consider the roles
of H3K27, H3K4 and bivalent chromatin when studying histone
methylation during phenotypic changes and functional
transformations, particularly those associated with the Notch1
signaling pathway.
Histone methylation affects gene expression, which
is critical for the progression of DKD, marking a key event in the
pathological timeline (38).
Most of these modifications are reversible and regulated by
intricate networks, indicating the potential of histone methylation
as a reliable biomarker and effective therapeutic target for the
treatment of DKD (137,138). However, compared with that of
classical PTMs such as acetylation, drug development targeting
histone methylation is still in its early stages, particularly for
DKD treatment (139,140). Therefore, the present review
provides a comprehensive overview of existing histone methylase
inhibitors and the potential therapeutic approaches investigated in
experimental studies (Table
II).
A series of small-molecule inhibitors targeting
histone methylation have emerged, and show promise for managing
various pathological processes in DKD. Specific HKMTs and HKDMs,
including EZH2, KDM6A and G9a, are abnormally expressed in several
intrinsic renal cells in DKD (138,141). The strategic use of targeted
therapies, particularly histone methyltransferase inhibitors, has
effectively reduced sugar-induced renal cell damage in animal
models and in vitro, substantially slowing DKD progression
(141). In 2020, The Food and
Drug Administration first approved the KMT inhibitor tazemetostat
for clinical use, although its application is currently limited to
the treatment of hematological malignancies and solid tumors
(142). This approval prompted
researchers to explore the potential of histone methyltransferase
inhibitors in the treatment of other diseases, including DKD,
malignant rhabdoid tumors and ovarian cancer, highlighting their
promise as novel therapeutic strategies (64,143,144). 3-Deazaneplanocin A (DZNep) is a
well-known inhibitor of histone methyltransferase, initially
identified for its potential in antiviral research. Its primary
mechanism involves the reduction of H3K27me3 modification by
inhibiting methylase EZH2 activity, leading to the activation of
gene expression (66). DZNep has
been widely utilized to explore its potential as an antitumor
agent, as EZH2 expression is upregulated in a variety of cancer
types, including lung cancer, breast cancer and glioblastoma, and
is closely associated with the development and progression of
tumors (145-148). However, due to the indirect
inhibition of EZH2, primarily by increasing
S-adenosyl-L-homocysteine levels, DZNep lacks specificity as a drug
candidate (149).
EPZ-6438/tazemetostat is a potent and highly selective inhibitor of
EZH2 methyltransferase, which is currently undergoing advanced
stage testing (150). In a
study on DKD, EPZ-6438 effectively inhibited podocyte
dedifferentiation and mitigated podocyte damage under adverse
conditions (64). GSK-J4, a
leading H3K27 demethylase inhibitor, exerts a potent dual
inhibitory effect on JMJD3 and KDM6A (151). GSK-J4 effectively mitigates the
pathological changes in various intrinsic renal cells associated
with DKD, thereby attenuating the progression of glomerular
disease, mesangial matrix accumulation, kidney inflammation and
fibrosis (64,106,119). A range of inhibitors targeting
the histone H3K9 methylase G9a have been developed, with BIX-01294
being a particularly potent and well-tolerated inhibitor of G9a.
This compound has been widely used in relevant research because of
its superior efficacy and reduced toxicity compared with earlier
small-molecule inhibitors (152-154). During the process of renal
fibrosis, BIX-01294 effectively suppresses the EMT of tubule cells
and attenuates the TGF-β1-induced downregulation of Klotho
(96). The aforementioned series
of small molecule drugs primarily function as inhibitors by
suppressing the activity of key enzymes involved in histone
methylation. Disruption of the interactions between core enzymes
and other co-factors within the histone methylase complex also
affects the distribution and extent of histone methylation in cells
(18). Menin serves as a crucial
scaffold protein in the methylase MLL1 complex of proteins
associated with Set1, and the interactions between MLL and menin
are essential for maintaining H3K4 methylation levels and the
expression of MLL target genes under specific pathological
conditions (155,156). MI-503 exerts a substantial
anti-fibrotic effect through targeted inhibition of the MLL-menin
interaction (93).
Compared with small-molecule inhibitors, natural
products exhibit greater selectivity, fewer side effects and
enhanced biological activity in drug development, making them a
preferred source for the identification and development of novel
therapeutic agents (157). A
limited number of natural products are available for treating
histone methylation in DKD, with a primary focus on addressing
renal fibrosis (158).
Gambogenic acid, an active constituent derived from the traditional
medicinal plant garcinia, has been extensively investigated for its
antitumor, anti-inflammatory and anti-fibrotic properties (159-161). Tao et al (63) demonstrated that gambogenic acid
modulates the TGF-β/Smad 3 signaling pathway through Smad 7
mediation, leading to amelioration of renal fibrosis. Sinefungin, a
naturally occurring nucleoside analog, was initially recognized for
its antiparasitic efficacy (63). Subsequent research has revealed
its potential as a methylase SET7/9 inhibitor, leading to improved
renal fibrosis through the inhibition of H3K4me1 (158).
The application of histone methylation inhibitors
has shown promising results in experimental studies. However, their
therapeutic efficacies have certain limitations: i) Histone
methylation is prevalent in vivo (27), and the lack of precise
therapeutic targets may affect normal cell function during
treatment; ii) the regulatory mechanism of histone methylation is
not entirely independent, and its establishment process is
influenced by metabolic mediators, co-factors and existing
epigenetic modifications (18),
which implies that targeting a histone methyl residue or related
enzyme may lead to unpredictable effects, and the aggressive use of
histone methylation modification inhibitors could potentially pose
unintended risks because of a lack of understanding of the protein;
and iii) discrepancies in histone methylation in patients with DKD
and experimental models may be attributed to variations in genome
structure and gene expression patterns across different species
(62). This implies that animal
models inadequately replicate the histone methylation process in
patients with DKD. Therefore, it is crucial to expand the currently
limited histone methylation database of human kidney samples to
develop effective therapeutic strategies for DKD.
The present review provides a comprehensive overview
of the pathological effects of histone methylation modification
disorders in various intrinsic renal cells during the onset and
progression of DKD, as well as potential therapeutic strategies
involving related inhibitors. Aberrant histone methylation,
particularly altered H3K27 and H3K4 methylation levels in
podocytes, renal tubular epithelial cells, interstitial cells and
glomerular cells in patients with DKD, is closely associated with
proteinuria and decreased glomerular filtration rate. Conversely,
research on H3K36, H3K79 and H4K20 has been limited, highlighting a
substantial gap in the epigenetic theory of DKD. Studies have
demonstrated the efficacy of histone methylation modification
inhibitors in ameliorating glomerular injury and mitigating renal
fibrosis in the management of DKD, while reducing proteinuria and
preserving renal function. These findings have attracted
considerable attention from the scientific community. However,
further research is needed to determine how to effectively utilize
these drugs while minimizing their adverse effects. In conclusion,
additional mechanistic studies are required to refine the profile
of histone methylation in specific intrinsic renal cells in DKD and
develop inhibitors with higher specificity and favorable
pharmacokinetics.
Not applicable.
PQ, LL and QJ contributed to acquisition, analysis
and interpretation of data, and drafted the manuscript. DL, YQ and
YZ revised the manuscript. QS, SR and ZL performed editing and
proofreading. LP and TL contributed to conception and design, and
critically revised the manuscript. Data authentication is not
applicable. All authors have read and approved the final version of
the manuscript.
Not applicable.
Not applicable.
The authors declare that they have no competing
interests.
Not applicable.
The present study was supported by Beijing Municipal Natural
Science Foundation of China (grant no. 7222160), the National
Natural Science Foundation of China (grant nos. 82170817 and
81970713), National High Level Hospital Clinical Research Funding
(grant no. 2023-NHLHCRF-DJZD-01), Beijing Research Ward
Construction Clinical Research Project (grant no. 2022-YJXBF-04-02)
and Elite Medical Professionals Project of China-Japan Friendship
Hospital (grant no. ZRJY2024-BJ03).
|
1
|
Thomas MC, Brownlee M, Susztak K, Sharma
K, Jandeleit-Dahm KA, Zoungas S, Rossing P, Groop PH and Cooper ME:
Diabetic kidney disease. Nat Rev Dis Primers. 1:150182015.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Johansen KL, Chertow GM, Foley RN,
Gilbertson DT, Herzog CA, Ishani A, Israni AK, Ku E, Kurella Tamura
M, Li S, et al: US renal data system 2020 annual data report:
Epidemiology of kidney disease in the United States. Am J Kidney
Dis. 77(4 Suppl 1): A7–A8. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Anders HJ, Huber TB, Isermann B and
Schiffer M: CKD in diabetes: Diabetic kidney disease versus
nondiabetic kidney disease. Nat Rev Nephrol. 14:361–377. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Thomas MC, Weekes AJ, Broadley OJ, Cooper
ME and Mathew TH: The burden of chronic kidney disease in
Australian patients with type 2 diabetes (the NEFRON study). Med J
Aust. 185:140–144. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Scilletta S, Di Marco M, Miano N,
Filippello A, Di Mauro S, Scamporrino A, Musmeci M, Coppolino G, Di
Giacomo Barbagallo F, Bosco G, et al: Update on diabetic kidney
disease (DKD): Focus on Non-Albuminuric DKD and cardiovascular
risk. Biomolecules. 13:7522023. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Parving HH, Hommel E, Jensen BR and Hansen
HP: Long-term beneficial effect of ACE inhibition on diabetic
nephropathy in normotensive type 1 diabetic patients. Kidney Int.
60:228–234. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zou H, Zhou B and Xu G: SGLT2 inhibitors:
A novel choice for the combination therapy in diabetic kidney
disease. Cardiovasc Diabetol. 16:652017. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Barrera-Chimal J, Lima-Posada I, Bakris GL
and Jaisser F: Mineralocorticoid receptor antagonists in diabetic
kidney disease-mechanistic and therapeutic effects. Nat Rev
Nephrol. 18:56–70. 2022. View Article : Google Scholar
|
|
9
|
Zhang R, Wang Q, Li Y, Li Q, Zhou X, Chen
X and Dong Z: A new perspective on proteinuria and drug therapy for
diabetic kidney disease. Front Pharmacol. 15:13490222024.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wang N and Zhang C: Recent advances in the
management of diabetic kidney disease: Slowing progression. Int J
Mol Sci. 25:30862024. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Forst T, Mathieu C, Giorgino F, Wheeler
DC, Papanas N, Schmieder RE, Halabi A, Schnell O, Streckbein M and
Tuttle KR: New strategies to improve clinical outcomes for diabetic
kidney disease. BMC Med. 20:3372022. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Regele F, Jelencsics K, Shiffman D, Paré
G, McQueen MJ, Mann JF and Oberbauer R: Genome-wide studies to
identify risk factors for kidney disease with a focus on patients
with diabetes. Nephrol Dial Transplant. 30(Suppl 4): iv26–iv34.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Cowie CC, Port FK, Wolfe RA, Savage PJ,
Moll PP and Hawthorne VM: Disparities in incidence of diabetic
end-stage renal disease according to race and type of diabetes. N
Engl J Med. 321:1074–1079. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Cefalu WT, Buse JB, Tuomilehto J, Fleming
GA, Ferrannini E, Gerstein HC, Bennett PH, Ramachandran A, Raz I,
Rosenstock J and Kahn SE: Update and next steps for real-world
translation of interventions for type 2 diabetes prevention:
Reflections from a diabetes care editors' expert forum. Diabetes
Care. 39:1186–1201. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Tervaert TW, Mooyaart AL, Amann K, Cohen
AH, Cook HT, Drachenberg CB, Ferrario F, Fogo AB, Haas M, de Heer
E, et al: Pathologic classification of diabetic nephropathy. J Am
Soc Nephrol. 21:556–563. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Caramori ML, Parks A and Mauer M: Renal
lesions predict progression of diabetic nephropathy in type 1
diabetes. J Am Soc Nephrol. 24:1175–1181. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Mohandes S, Doke T, Hu H, Mukhi D, Dhillon
P and Susztak K: Molecular pathways that drive diabetic kidney
disease. J Clin Invest. 133:e1656542023. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Li Y, Ge K, Li T, Cai R and Chen Y: The
engagement of histone lysine methyltransferases with nucleosomes:
Structural basis, regulatory mechanisms, and therapeutic
implications. Protein Cell. 14:165–179. 2023.PubMed/NCBI
|
|
19
|
Greer EL and Shi Y: Histone methylation: A
dynamic mark in health, disease and inheritance. Nat Rev Genet.
13:343–357. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Millán-Zambrano G, Burton A, Bannister AJ
and Schneider R: Histone post-translational modifications-cause and
consequence of genome function. Nat Rev Genet. 23:563–580. 2022.
View Article : Google Scholar
|
|
21
|
Keating ST, van Diepen JA, Riksen NP and
El-Osta A: Epigenetics in diabetic nephropathy immunity and
metabolism. Diabetologia. 61:6–20. 2018. View Article : Google Scholar
|
|
22
|
Lefevre GM, Patel SR, Kim D, Tessarollo L
and Dressler GR: Altering a histone H3K4 methylation pathway in
glomerular podocytes promotes a chronic disease phenotype. PLoS
Genet. 6:e10011422010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Sayyed SG, Gaikwad AB, Lichtnekert J,
Kulkarni O, Eulberg D, Klussmann S, Tikoo K and Anders HJ:
Progressive glomerulosclerosis in type 2 diabetes is associated
with renal histone H3K9 and H3K23 acetylation, H3K4 dimethylation
and phosphorylation at serine 10. Nephrol Dial Transplant.
25:1811–1817. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Husmann D and Gozani O: Histone lysine
methyltransferases in biology and disease. Nat Struct Mol Biol.
26:880–889. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Martin C and Zhang Y: The diverse
functions of histone lysine methylation. Nat Rev Mol Cell Biol.
6:838–849. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Hyun K, Jeon J, Park K and Kim J: Writing,
erasing and reading histone lysine methylations. Exp Mol Med.
49:e3242017. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Gong F and Miller KM: Histone methylation
and the DNA damage response. Mutat Res Rev Mutat Res. 780:37–47.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Black JC, Van Rechem C and Whetstine JR:
Histone lysine methylation dynamics: Establishment, regulation, and
biological impact. Mol Cell. 48:491–507. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Mohan M, Herz HM and Shilatifard A:
SnapShot: Histone lysine methylase complexes. Cell. 149:498–498.e1.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Højfeldt JW, Agger K and Helin K: Histone
lysine demethylases as targets for anticancer therapy. Nat Rev Drug
Discov. 12:917–930. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Schwartz-Orbach L, Zhang C, Sidoli S, Amin
R, Kaur D, Zhebrun A, Ni J and Gu SG: Caenorhabditis elegans
nuclear RNAi factor SET-32 deposits the transgenerational histone
modification, H3K23me3. Elife. 9:e543092020. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Shen Y, Mevius DEHF, Caliandro R,
Carrozzini B, Roh Y, Kim J, Kim S, Ha SC, Morishita M and di Luccio
E: Set7 Is a H3K37 methyltransferase in schizosaccharomyces pombe
and is required for proper gametogenesis. Structure. 27:631–638.e8.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zong Y, Weiss N, Wang K, Pagano AE,
Heissel S, Perveen S and Huang J: Development of complementary
photo-arginine/lysine to promote discovery of Arg/Lys hPTMs
Interactomes. Adv Sci (Weinh). 11:e23075262024. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Feng X, Wang AH, Juan AH, Ko KD, Jiang K,
Riparini G, Ciuffoli V, Kaba A, Lopez C, Naz F, et al: Polycomb
Ezh1 maintains murine muscle stem cell quiescence through
non-canonical regulation of Notch signaling. Dev Cell.
58:1052–1070.e10. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Wang Z and Liu H: Roles of lysine
methylation in glucose and lipid metabolism: Functions, regulatory
mechanisms, and therapeutic implications. Biomolecules. 14:8622024.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Aziz N, Hong YH, Kim HG, Kim JH and Cho
JY: Tumor-suppressive functions of protein lysine
methyltransferases. Exp Mol Med. 55:2475–2497. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Liu BC, Tang TT, Lv LL and Lan HY: Renal
tubule injury: A driving force toward chronic kidney disease.
Kidney Int. 93:568–579. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Cheng Y, Chen Y, Wang G, Liu P, Xie G,
Jing H, Chen H, Fan Y, Wang M and Zhou J: Protein methylation in
diabetic kidney disease. Front Med (Lausanne). 9:7360062022.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Allis CD, Bowen JK, Abraham GN, Glover CV
and Gorovsky MA: Proteolytic processing of histone H3 in chromatin:
A physiologically regulated event in Tetrahymena micronuclei. Cell.
20:55–64. 1980. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Shi Y, Lan F, Matson C, Mulligan P,
Whetstine JR, Cole PA, Casero RA and Shi Y: Histone demethylation
mediated by the nuclear amine oxidase homolog LSD1. Cell.
119:941–953. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Di Nisio E, Manzini V, Licursi V and Negri
R: To Erase or not to erase: non-canonical catalytic functions and
non-catalytic functions of members of histone lysine demethylase
families. Int J Mol Sci. 25:69002024. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Yang J, Hu Y, Zhang B, Liang X and Li X:
The JMJD family histone demethylases in crosstalk between
inflammation and cancer. Front Immunol. 13:8813962022. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Kim W, Kim R, Park G, Park JW and Kim JE:
Deficiency of H3K79 histone methyltransferase Dot1-like protein
(DOT1L) inhibits cell proliferation. J Biol Chem. 287:5588–5599.
2012. View Article : Google Scholar :
|
|
44
|
Feng Q, Wang H, Ng HH, Erdjument-Bromage
H, Tempst P, Struhl K and Zhang Y: Methylation of H3-lysine 79 is
mediated by a new family of HMTases without a SET domain. Curr
Biol. 12:1052–1058. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Lee YH, Ren D, Jeon B and Liu HW:
S-Adenosylmethionine: More than just a methyl donor. Nat Prod Rep.
40:1521–1549. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Gou D, Liu R, Shan X, Deng H, Chen C,
Xiang J, Liu Y, Gao Q, Li Z, Huang A, et al: Gluconeogenic enzyme
PCK1 supports S-adenosylmethionine biosynthesis and promotes
H3K9me3 modification to suppress hepatocellular carcinoma
progression. J Clin Invest. 133:e1617132023. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Lim CY, Lin HT, Kumsta C, Lu TC, Wang FY,
Kang YH, Hansen M, Ching TT and Hsu AL: SAMS-1 coordinates
HLH-30/TFEB and PHA-4/FOXA activities through histone methylation
to mediate dietary restriction-induced autophagy and longevity.
Autophagy. 19:224–240. 2023. View Article : Google Scholar :
|
|
48
|
Cenik BK and Shilatifard A: COMPASS and
SWI/SNF complexes in development and disease. Nat Rev Genet.
22:38–58. 2021. View Article : Google Scholar
|
|
49
|
Xue H, Yao T, Cao M, Zhu G, Li Y, Yuan G,
Chen Y, Lei M and Huang J: Structural basis of nucleosome
recognition and modification by MLL methyltransferases. Nature.
573:445–449. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Mitchell K, Sprowls SA, Arora S, Shakya S,
Silver DJ, Goins CM, Wallace L, Roversi G, Schafer RE, Kay K, et
al: WDR5 represents a therapeutically exploitable target for cancer
stem cells in glioblastoma. Genes Dev. 37:86–102. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Zhao Z, Rendleman EJ, Szczepanski AP,
Morgan MA, Wang L and Shilatifard A: CARM1-mediated methylation of
ASXL2 impairs tumor-suppressive function of MLL3/COMPASS. Sci Adv.
8:eadd33392022. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Lu J, Huang Y, Zhang X, Xu Y and Nie S:
Noncoding RNAs involved in DNA methylation and histone methylation,
and acetylation in diabetic vascular complications. Pharmacol Res.
170:1055202021. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Lee JS, Smith E and Shilatifard A: The
language of histone crosstalk. Cell. 142:682–685. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Stirpe A, Guidotti N, Northall SJ, Kilic
S, Hainard A, Vadas O, Fierz B and Schalch T: SUV39 SET domains
mediate crosstalk of heterochromatic histone marks. Elife.
10:e626822021. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Segelle A, Núñez-Álvarez Y, Oldfield AJ,
Webb KM, Voigt P and Luco RF: Histone marks regulate the
epithelial-to-mesenchymal transition via alternative splicing. Cell
Rep. 38:1103572022. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Hogg SJ, Motorna O, Cluse LA, Johanson TM,
Coughlan HD, Raviram R, Myers RM, Costacurta M, Todorovski I,
Pijpers L, et al: Targeting histone acetylation dynamics and
oncogenic transcription by catalytic P300/CBP inhibition. Mol Cell.
81:2183–2200.e13. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
He F, Yu Q, Wang M, Wang R, Gong X, Ge F,
Yu X and Li S: SESAME-catalyzed H3T11 phosphorylation inhibits
Dot1-catalyzed H3K79me3 to regulate autophagy and telomere
silencing. Nat Commun. 13:75262022. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Metzker ML: Sequencing technologies-the
next generation. Nat Rev Genet. 11:31–46. 2010. View Article : Google Scholar
|
|
59
|
Pulecio J, Verma N, Mejía-Ramírez E,
Huangfu D and Raya A: CRISPR/Cas9-Based engineering of the
epigenome. Cell Stem Cell. 21:431–447. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Peng X, Peng Q and Zhong L: Targeting
H3K36 methyltransferases NSDs: A promising strategy for tumor
targeted therapy. Signal Transduct Target Ther. 6:2202021.
View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Basavarajappa BS and Subbanna S: Histone
methylation regulation in neurodegenerative disorders. Int J Mol
Sci. 22:46542021. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Komers R, Mar D, Denisenko O, Xu B, Oyama
TT and Bomsztyk K: Epigenetic changes in renal genes dysregulated
in mouse and rat models of type 1 diabetes. Lab Invest. 93:543–552.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Tao S, Yang L, Wu C, Hu Y, Guo F, Ren Q,
Ma L and Fu P: Gambogenic acid alleviates kidney fibrosis via
epigenetic inhibition of EZH2 to regulate Smad7-dependent
mechanism. Phytomedicine. 106:1543902022. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Majumder S, Thieme K, Batchu SN, Alghamdi
TA, Bowskill BB, Kabir MG, Liu Y, Advani SL, White KE, Geldenhuys
L, et al: Shifts in podocyte histone H3K27me3 regulate mouse and
human glomerular disease. J Clin Invest. 128:483–499. 2018.
View Article : Google Scholar :
|
|
65
|
Paneni F, Costantino S, Battista R,
Castello L, Capretti G, Chiandotto S, Scavone G, Villano A, Pitocco
D, Lanza G, et al: Adverse epigenetic signatures by histone
methyltransferase Set7 contribute to vascular dysfunction in
patients with type 2 diabetes mellitus. Circ Cardiovasc Genet.
8:150–158. 2015. View Article : Google Scholar
|
|
66
|
Siddiqi FS, Majumder S, Thai K, Abdalla M,
Hu P, Advani SL, White KE, Bowskill BB, Guarna G, Dos Santos CC, et
al: The histone methyltransferase enzyme enhancer of zeste homolog
2 protects against podocyte oxidative stress and renal injury in
diabetes. J Am Soc Nephrol. 27:2021–2034. 2016. View Article : Google Scholar :
|
|
67
|
Pavenstädt H, Kriz W and Kretzler M: Cell
biology of the glomerular podocyte. Physiol Rev. 83:253–307. 2003.
View Article : Google Scholar
|
|
68
|
Nagata M: Podocyte injury and its
consequences. Kidney Int. 89:1221–1230. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Reidy K, Kang HM, Hostetter T and Susztak
K: Molecular mechanisms of diabetic kidney disease. J Clin Invest.
124:2333–2340. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Shankland SJ: The podocyte's response to
injury: role in proteinuria and glomerulosclerosis. Kidney Int.
69:2131–2147. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Ying Q and Wu G: Molecular mechanisms
involved in podocyte EMT and concomitant diabetic kidney diseases:
An update. Ren Fail. 39:474–483. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
May CJ, Saleem M and Welsh GI: Podocyte
dedifferentiation: a specialized process for a specialized cell.
Front Endocrinol (Lausanne). 5:1482014. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Guo Y, Xiong Z and Guo X: Histone
demethylase KDM6B regulates human podocyte differentiation in
vitro. Biochem J. 476:1741–1751. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Wan J, Hou X, Zhou Z, Geng J, Tian J, Bai
X and Nie J: WT1 ameliorates podocyte injury via repression of
EZH2/β-catenin pathway in diabetic nephropathy. Free Radic Biol
Med. 108:280–299. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Nishad R, Meshram P, Singh AK, Reddy GB
and Pasupulati AK: Activation of Notch1 signaling in podocytes by
glucose-derived AGEs contributes to proteinuria. BMJ Open Diabetes
Res Care. 8:e0012032020. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Liebisch M and Wolf G: AGE-Induced
Suppression of EZH2 mediates injury of podocytes by reducing
H3K27me3. Am J Nephrol. 51:676–692. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Lin CL, Hsu YC, Huang YT, Shih YH, Wang
CJ, Chiang WC and Chang PJ: A KDM6A-KLF10 reinforcing feedback
mechanism aggravates diabetic podocyte dysfunction. EMBO Mol Med.
11:e98282019. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Muñoz IM and Rouse J: Control of histone
methylation and genome stability by PTIP. EMBO Rep. 10:239–245.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Patel SR, Kim D, Levitan I and Dressler
GR: The BRCT-domain containing protein PTIP links PAX2 to a histone
H3, lysine 4 methyltransferase complex. Dev Cell. 13:580–592. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Cao A, Li J, Asadi M, Basgen JM, Zhu B, Yi
Z, Jiang S, Doke T, El Shamy O, Patel N, et al: DACH1 protects
podocytes from experimental diabetic injury and modulates
PTIP-H3K4Me3 activity. J Clin Invest. 131:e1412792021. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Zhang T, Zhang Y, Xu H, Lan J, Feng Z,
Huang R, Geng J, Chi H and Bai X: LINC00355 Mediates CTNNBIP1
promoter methylation and promotes endoplasmic reticulum
stress-induced podocyte injury in diabetic nephropathy. Antioxid
Redox Signal. 39:225–240. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Qi R and Yang C: Renal tubular epithelial
cells: The neglected mediator of tubulointerstitial fibrosis after
injury. Cell Death Dis. 9:11262018. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Legouis D, Ricksten SE, Faivre A,
Verissimo T, Gariani K, Verney C, Galichon P, Berchtold L, Feraille
E, Fernandez M, et al: Altered proximal tubular cell glucose
metabolism during acute kidney injury is associated with mortality.
Nat Metab. 2:732–743. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Edwards A, Palm F and Layton AT: A model
of mitochondrial O(2) consumption and ATP generation in rat
proximal tubule cells. Am J Physiol Renal Physiol. 318:F248–F259.
2020. View Article : Google Scholar
|
|
85
|
Wang Y, Jin M, Cheng CK and Li Q: Tubular
injury in diabetic kidney disease: Molecular mechanisms and
potential therapeutic perspectives. Front Endocrinol (Lausanne).
14:12389272023. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Liu Y: Cellular and molecular mechanisms
of renal fibrosis. Nat Rev Nephrol. 7:684–696. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Sun L, Liu L, Jiang J, Liu K, Zhu J, Wu L,
Lu X, Huang Z, Yuan Y, Crowley SD, et al: Transcription factor
Twist1 drives fibroblast activation to promote kidney fibrosis via
signaling proteins Prrx1/TNC. Kidney Int. Aug 22–2024.Epub ahead of
print. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Bai M, Xu S, Jiang M, Guo Y, Hu D, He J,
Wang T, Zhang Y, Guo Y, Zhang Y, et al: Meis1 targets protein
tyrosine phosphatase receptor J in fibroblast to retard chronic
kidney disease progression. Adv Sci (Weinh). Aug 20–2024.Epub ahead
of print. View Article : Google Scholar
|
|
89
|
Kim DH, Sung M, Park MS, Sun EG, Yoon S,
Yoo KH, Radhakrishnan K, Jung SY, Bae WK, Cho SH and Chung IJ:
Galectin 3-binding protein (LGALS3BP) depletion attenuates hepatic
fibrosis by reducing transforming growth factor-β1 (TGF-β1)
availability and inhibits hepatocarcinogenesis. Cancer Commun
(Lond). Jul 28–2024.Epub ahead of print. View Article : Google Scholar
|
|
90
|
Fesneau O, Thevin V, Pinet V, Goldsmith C,
Vieille B, M'Homa Soudja S, Lattanzio R, Hahne M, Dardalhon V,
Hernandez-Vargas H, et al: An intestinal T(H)17 cell-derived subset
can initiate cancer. Nat Immunol. 25:1637–1649. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Gifford CC, Tang J, Costello A, Khakoo NS,
Nguyen TQ, Goldschmeding R, Higgins PJ and Samarakoon R: Negative
regulators of TGF-β1 signaling in renal fibrosis; pathological
mechanisms and novel therapeutic opportunities. Clin Sci (Lond).
135:275–303. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
You JB, Cao Y, You QY, Liu ZY, Wang XC,
Ling H, Sha JM and Tao H: The landscape of histone modification in
organ fibrosis. Eur J Pharmacol. 977:1767482024. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Zou J, Yu C, Zhang C, Guan Y, Zhang Y,
Tolbert E, Zhang W, Zhao T, Bayliss G, Li X, et al: Inhibition of
MLL1-menin interaction attenuates renal fibrosis in obstructive
nephropathy. FASEB J. 37:e227122023. View Article : Google Scholar
|
|
94
|
Hu HH, Chen DQ, Wang YN, Feng YL, Cao G,
Vaziri ND and Zhao YY: New insights into TGF-β/Smad signaling in
tissue fibrosis. Chem Biol Interact. 292:76–83. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Wang J, Pan J, Li H, Long J, Fang F, Chen
J, Zhu X, Xiang X and Zhang D: lncRNA ZEB1-AS1 was suppressed by
p53 for renal fibrosis in diabetic nephropathy. Mol Ther Nucleic
Acids. 12:741–750. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Irifuku T, Doi S, Sasaki K, Doi T,
Nakashima A, Ueno T, Yamada K, Arihiro K, Kohno N and Masaki T:
Inhibition of H3K9 histone methyltransferase G9a attenuates renal
fibrosis and retains klotho expression. Kidney Int. 89:147–157.
2016. View Article : Google Scholar
|
|
97
|
Ike T, Doi S, Nakashima A, Sasaki K,
Ishiuchi N, Asano T and Masaki T: The hypoxia-inducible factor-α
prolyl hydroxylase inhibitor FG4592 ameliorates renal fibrosis by
inducing the H3K9 demethylase JMJD1A. Am J Physiol Renal Physiol.
323:F539–F552. 2022. View Article : Google Scholar
|
|
98
|
Wang S, Zuo A, Jiang W, Xie J, Lin H, Sun
W, Zhao M, Xia J, Shao J, Zhao X, et al: JMJD1A/NR4A1 signaling
regulates the procession of renal tubular epithelial interstitial
fibrosis induced by AGEs in HK-2. Front Med (Lausanne).
8:8076942022. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Han X and Sun Z: Epigenetic Regulation of
KL (Klotho) via H3K27me3 (Histone 3 Lysine [K] 27 Trimethylation)
in renal tubule cells. Hypertension. 75:1233–1241. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Niculae A, Gherghina ME, Peride I, Tiglis
M, Nechita AM and Checherita IA: Pathway from acute kidney injury
to chronic kidney disease: Molecules involved in renal fibrosis.
Int J Mol Sci. 24:140192023. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Cohen C, Mhaidly R, Croizer H, Kieffer Y,
Leclere R, Vincent-Salomon A, Robley C, Anglicheau D, Rabant M,
Sannier A, et al: WNT-dependent interaction between inflammatory
fibroblasts and FOLR2+ macrophages promotes fibrosis in chronic
kidney disease. Nat Commun. 15:7432024. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Huang R, Fu P and Ma L: Kidney fibrosis:
From mechanisms to therapeutic medicines. Signal Transduct Target
Ther. 8:1292023. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Liu Y: New insights into
epithelial-mesenchymal transition in kidney fibrosis. J Am Soc
Nephrol. 21:212–222. 2010. View Article : Google Scholar
|
|
104
|
Hewitson TD, Holt SG, Tan SJ, Wigg B,
Samuel CS and Smith ER: Epigenetic modifications to H3K9 in renal
tubulointerstitial cells after unilateral ureteric obstruction and
TGF-β1 Stimulation. Front Pharmacol. 8:3072017. View Article : Google Scholar
|
|
105
|
Zhou X, Zang X, Ponnusamy M, Masucci MV,
Tolbert E, Gong R, Zhao TC, Liu N, Bayliss G, Dworkin LD and Zhuang
S: Enhancer of Zeste Homolog 2 inhibition attenuates renal fibrosis
by maintaining Smad7 and phosphatase and tensin homolog expression.
J Am Soc Nephrol. 27:2092–2108. 2016. View Article : Google Scholar :
|
|
106
|
An C, Jiao B, Du H, Tran M, Song B, Wang
P, Zhou D and Wang Y: Jumonji domain-containing protein-3 (JMJD3)
promotes myeloid fibroblast activation and macrophage polarization
in kidney fibrosis. Br J Pharmacol. 180:2250–2265. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Steffes MW, Osterby R, Chavers B and Mauer
SM: Mesangial expansion as a central mechanism for loss of kidney
function in diabetic patients. Diabetes. 38:1077–1081. 1989.
View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Thomas HY and Ford Versypt AN:
Pathophysiology of mesangial expansion in diabetic nephropathy:
Mesangial structure, glomerular biomechanics, and biochemical
signaling and regulation. J Biol Eng. 16:192022. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Kriz W: Maintenance and breakdown of
glomerular tuft architecture. J Am Soc Nephrol. 29:1075–1077. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Avraham S, Korin B, Chung JJ, Oxburgh L
and Shaw AS: The Mesangial cell-the glomerular stromal cell. Nat
Rev Nephrol. 17:855–864. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Alicic RZ, Rooney MT and Tuttle KR:
Diabetic kidney disease: Challenges, progress, and possibilities.
Clin J Am Soc Nephrol. 12:2032–2045. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Baccora MH, Cortes P, Hassett C, Taube DW
and Yee J: Effects of long-term elevated glucose on collagen
formation by mesangial cells. Kidney Int. 72:1216–1225. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Wu P, Ren Y, Ma Y, Wang Y, Jiang H,
Chaudhari S, Davis ME, Zuckerman JE and Ma R: Negative regulation
of Smad1 pathway and collagen IV expression by store-operated
Ca(2+) entry in glomerular mesangial cells. Am J Physiol Renal
Physiol. 312:F1090–F1100. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Kuo FC, Chao CT and Lin SH: The dynamics
and plasticity of epigenetics in diabetic kidney disease:
therapeutic applications Vis-à-Vis. Int J Mol Sci. 23:8432022.
View Article : Google Scholar
|
|
115
|
Boi R, Ebefors K and Nyström J: The role
of the mesangium in glomerular function. Acta Physiol (Oxf).
239:e140452023. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Zhong W, Hong C, Dong Y, Li Y, Xiao C and
Liu X: ASH2L aggravates fibrosis and inflammation through HIPK2 in
high glucose-induced glomerular mesangial cells. Genes (Basel).
13:22442022. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Zhong W, Hong C, Zhang Y, Li Y, Xiao C and
Liu X: ASH2L-mediated H3K4me3 drives diabetic nephropathy through
HIPK2 and Notch1 pathway. Transl Res. 264:85–96. 2024. View Article : Google Scholar
|
|
118
|
Chen YX, Zhu SY, Huang C, Xu CY, Fang XD
and Tu WP: LncRNA Dlx6os1 accelerates diabetic nephropathy
progression by epigenetically repressing SOX6 via Recruiting EZH2.
Kidney Blood Press Res. 47:177–184. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Hung PH, Hsu YC, Chen TH, Ho C and Lin CL:
The histone demethylase inhibitor GSK-J4 Is a therapeutic target
for the kidney fibrosis of diabetic kidney disease via DKK1
Modulation. Int J Mol Sci. 23:94072022. View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Chen H, Huang Y, Zhu X, Liu C, Yuan Y, Su
H, Zhang C, Liu C, Xiong M, Qu Y, et al: Histone demethylase UTX is
a therapeutic target for diabetic kidney disease. J Physiol.
597:1643–1660. 2019. View Article : Google Scholar :
|
|
121
|
Jia Y, Reddy MA, Das S, Oh HJ, Abdollahi
M, Yuan H, Zhang E, Lanting L, Wang M and Natarajan R:
Dysregulation of histone H3 lysine 27 trimethylation in
transforming growth factor-β1-induced gene expression in mesangial
cells and diabetic kidney. J Biol Chem. 294:12695–12707. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Satchell SC and Braet F: Glomerular
endothelial cell fenestrations: An integral component of the
glomerular filtration barrier. Am J Physiol Renal Physiol.
296:F947–F956. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Savage CO: The biology of the glomerulus:
endothelial cells. Kidney Int. 45:314–319. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Lassén E and Daehn IS: Molecular
mechanisms in early diabetic kidney disease: Glomerular endothelial
cell dysfunction. Int J Mol Sci. 21:94562020. View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Thakar S, Katakia YT, Ramakrishnan SK,
Pandya Thakkar N and Majumder S: Intermittent high glucose elevates
nuclear localization of EZH2 to Cause H3K27me3-dependent repression
of KLF2 leading to endothelial inflammation. Cells. 10:25482021.
View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Takizawa F, Mizutani S, Ogawa Y and Sawada
N: Glucose-independent persistence of PAI-1 gene expression and
H3K4 tri-methylation in type 1 diabetic mouse endothelium:
implication in metabolic memory. Biochem Biophys Res Commun.
433:66–72. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Huang T, Li X, Wang F, Lu L, Hou W, Zhu M
and Miao C: The CREB/KMT5A complex regulates PTP1B to modulate high
glucose-induced endothelial inflammatory factor levels in diabetic
nephropathy. Cell Death Dis. 12:3332021. View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Alvandi Z and Bischoff J:
Endothelial-Mesenchymal transition in cardiovascular disease.
Arterioscler Thromb Vasc Biol. 41:2357–2369. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
129
|
Glaser SF, Heumüller AW, Tombor L, Hofmann
P, Muhly-Reinholz M, Fischer A, Günther S, Kokot KE, Hassel D,
Kumar S, et al: The histone demethylase JMJD2B regulates
endothelial-to-mesenchymal transition. Proc Natl Acad Sci USA.
117:4180–4187. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Pandya Thakkar N, Pereira BMV, Katakia YT,
Ramakrishnan SK, Thakar S, Sakhuja A, Rajeev G, Soorya S, Thieme K
and Majumder S: Elevated H3K4me3 Through MLL2-WDR82 upon
hyperglycemia causes jagged ligand dependent notch activation to
interplay with differentiation state of endothelial cells. Front
Cell Dev Biol. 10:8391092022. View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Vastenhouw NL and Schier AF: Bivalent
histone modifications in early embryogenesis. Curr Opin Cell Biol.
24:374–386. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
132
|
Conway E, Healy E and Bracken AP: PRC2
mediated H3K27 methylations in cellular identity and cancer. Curr
Opin Cell Biol. 37:42–48. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
133
|
Miller SA, Damle M, Kim J and Kingston RE:
Full methylation of H3K27 by PRC2 is dispensable for initial
embryoid body formation but required to maintain differentiated
cell identity. Development. 148:dev1963292021. View Article : Google Scholar : PubMed/NCBI
|
|
134
|
Lavarone E, Barbieri CM and Pasini D:
Dissecting the role of H3K27 acetylation and methylation in PRC2
mediated control of cellular identity. Nat Commun. 10:16792019.
View Article : Google Scholar : PubMed/NCBI
|
|
135
|
Soofi A, Kutschat AP, Azam M, Laszczyk AM
and Dressler GR: Regeneration after acute kidney injury requires
PTIP-mediated epigenetic modifications. JCI insight. 5:e1302042020.
View Article : Google Scholar : PubMed/NCBI
|
|
136
|
El-Dahr SS and Saifudeen Z: Epigenetic
regulation of renal development. Semin Cell Dev Biol. 91:111–118.
2019. View Article : Google Scholar
|
|
137
|
Jin J, Liu XM, Shao W and Meng XM: Nucleic
acid and protein methylation modification in renal diseases. Acta
Pharmacol Sin. 45:661–673. 2024. View Article : Google Scholar
|
|
138
|
Yu C and Zhuang S: Histone
methyltransferases as therapeutic targets for kidney diseases.
Front Pharmacol. 10:13932019. View Article : Google Scholar : PubMed/NCBI
|
|
139
|
Ho TCS, Chan AHY and Ganesan A: Thirty
Years of HDAC Inhibitors: 2020 insight and hindsight. J Med Chem.
63:12460–12484. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
140
|
Bhat KP, Ümit Kaniskan H, Jin J and Gozani
O: Epigenetics and beyond: Targeting writers of protein lysine
methylation to treat disease. Nat Rev Drug Discov. 20:265–286.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
141
|
Kourtidou C and Tziomalos K: The role of
histone modifications in the pathogenesis of diabetic kidney
disease. Int J Mol Sci. 24:60072023. View Article : Google Scholar : PubMed/NCBI
|
|
142
|
Jones PA, Issa JP and Baylin S: Targeting
the cancer epigenome for therapy. Nat Rev Genet. 17:630–641. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
143
|
Zhang Q, Chen X, Cao J, Yang W, Wan G,
Feng Q, Zhou S, Yang H, Wang N, Liu Z, et al: Discovery of a Novel
Covalent EZH2 inhibitor based on tazemetostat scaffold for the
treatment of ovarian cancer. J Med Chem. 66:1725–1741. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
144
|
Vejmelkova K, Pokorna P, Noskova K,
Faustmannova A, Drabova K, Pavelka Z, Bajciova V, Broz M, Tinka P,
Jezova M, et al: Tazemetostat in the therapy of pediatric
INI1-negative malignant rhabdoid tumors. Sci Rep. 13:216232023.
View Article : Google Scholar : PubMed/NCBI
|
|
145
|
Ni J, Hou X, Wang X, Shi Y, Xu L, Zheng X,
Liu N, Qiu A and Zhuang S: 3-deazaneplanocin A protects against
cisplatin-induced renal tubular cell apoptosis and acute kidney
injury by restoration of E-cadherin expression. Cell Death Dis.
10:3552019. View Article : Google Scholar : PubMed/NCBI
|
|
146
|
Tellez CS, Picchi MA, Juri D, Do K, Desai
DH, Amin SG, Hutt JA, Filipczak PT and Belinsky SA: Chromatin
remodeling by the histone methyltransferase EZH2 drives lung
pre-malignancy and is a target for cancer prevention. Clin
Epigenetics. 13:442021. View Article : Google Scholar : PubMed/NCBI
|
|
147
|
De La Rosa J, Urdiciain A, Zazpe I, Zelaya
MV, Meléndez B, Rey JA, Idoate MA and Castresana JS: The
synergistic effect of DZ-NEP, panobinostat and temozolomide reduces
clonogenicity and induces apoptosis in glioblastoma cells. Int J
Oncol. 56:283–300. 2020.
|
|
148
|
Li Y, Ren Y, Wang Y, Tan Y, Wang Q, Cai J,
Zhou J, Yang C, Zhao K, Yi K, et al: A Compound AC1Q3QWB
Selectively Disrupts HOTAIR-Mediated Recruitment of PRC2 and
Enhances Cancer Therapy of DZNep. Theranostics. 9:4608–4623. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
149
|
Miranda TB, Cortez CC, Yoo CB, Liang G,
Abe M, Kelly TK, Marquez VE and Jones PA: DZNep is a global histone
methylation inhibitor that reactivates developmental genes not
silenced by DNA methylation. Mol Cancer Ther. 8:1579–1588. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
150
|
Gounder M, Schöffski P, Jones RL, Agulnik
M, Cote GM, Villalobos VM, Attia S, Chugh R, Chen TW, Jahan T, et
al: Tazemetostat in advanced epithelioid sarcoma with loss of
INI1/SMARCB1: An international, open-label, phase 2 basket study.
Lancet Oncol. 21:1423–1432. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
151
|
Dalpatraj N, Naik A and Thakur N: GSK-J4:
An H3K27 histone demethylase inhibitor, as a potential anti-cancer
agent. Int J Cancer. 153:1130–1138. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
152
|
Chang Y, Zhang X, Horton JR, Upadhyay AK,
Spannhoff A, Liu J, Snyder JP, Bedford MT and Cheng X: Structural
basis for G9a-like protein lysine methyltransferase inhibition by
BIX-01294. Nat Struct Mol Biol. 16:312–317. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
153
|
Chae YC, Kim JY, Park JW, Kim KB, Oh H,
Lee KH and Seo SB: FOXO1 degradation via G9a-mediated methylation
promotes cell proliferation in colon cancer. Nucleic Acids Res.
47:1692–1705. 2019. View Article : Google Scholar :
|
|
154
|
Kim Y, Kim YS, Kim DE, Lee JS, Song JH,
Kim HG, Cho DH, Jeong SY, Jin DH, Jang SJ, et al: BIX-01294 induces
autophagy-associated cell death via EHMT2/G9a dysfunction and
intracellular reactive oxygen species production. Autophagy.
9:2126–2139. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
155
|
Yokoyama A and Cleary ML: Menin critically
links MLL proteins with LEDGF on cancer-associated target genes.
Cancer Cell. 14:36–46. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
156
|
Caslini C, Yang Z, El-Osta M, Milne TA,
Slany RK and Hess JL: Interaction of MLL amino terminal sequences
with menin is required for transformation. Cancer Res.
67:7275–7283. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
157
|
Newman DJ and Cragg GM: Natural products
as sources of new drugs over the nearly four decades from 01/1981
to 09/2019. J Nat Prod. 83:770–803. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
158
|
Sasaki K, Doi S, Nakashima A, Irifuku T,
Yamada K, Kokoroishi K, Ueno T, Doi T, Hida E, Arihiro K, et al:
Inhibition of SET domain-containing lysine methyltransferase 7/9
ameliorates renal fibrosis. J Am Soc Nephrol. 27:203–215. 2016.
View Article : Google Scholar
|
|
159
|
Yu X, Zhao Q and Zhang H, Fan C, Zhang X,
Xie Q, Xu C, Liu Y, Wu X, Han Q and Zhang H: Gambogenic acid
inhibits LPS-simulated inflammatory response by suppressing NF-κB
and MAPK in macrophages. Acta Biochim Biophys Sin (Shanghai).
48:454–461. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
160
|
Chen X, Zhang X, Cai H, Yang W, Lei H, Xu
H, Wang W, Zhu Q, Kang J, Yin T, et al: Targeting USP9x/SOX2 axis
contributes to the anti-osteosarcoma effect of neogambogic acid.
Cancer Lett. 469:277–286. 2020. View Article : Google Scholar
|
|
161
|
Xu L, Meng X, Xu N, Fu W, Tan H, Zhang L,
Zhou Q, Qian J, Tu S, Li X, et al: Gambogenic acid inhibits
fibroblast growth factor receptor signaling pathway in
erlotinib-resistant non-small-cell lung cancer and suppresses
patient-derived xenograft growth. Cell Death Dis. 9:2622018.
View Article : Google Scholar : PubMed/NCBI
|