Pulmonary hypertension (PH) refers to increased
pressure (>20 mmHg) in the pulmonary arteries. Based on its
etiology, pathophysiology and treatment, PH can be divided into
five clinical groups: Pulmonary arterial hypertension (PAH, group
I), PH in secondary to left-sided cardiac disease (group II), PH
resulting from chronic lung illness (group III), chronic
thromboembolic PH (group IV), and unclear and/or multifactorial PH
(group) (1). Different
etiologies are attributed to different forms of PH. Although
variations are observed among the different forms, pulmonary
vascular remodeling is the predominant pathological change
characterizing the disease (2).
Improved vascular remodeling is important for PH treatment. Various
targeted agents used to treat PH have significantly improved the
exercise tolerance and quality of life of patients with PH;
however, the mortality rate remains high (3). Only limited treatment options are
available for PH, with no therapies that can effectively reverse
the late structural vascular changes associated with PH. This
chronic, progressive and multi-causal condition affects ~1% of the
global population, with most >65 years of age, and >50% of
the affected patients exhibiting comorbid heart failure (HF)
(4). Therefore, understanding
the pathogenesis of PH is necessary to develop novel therapeutic
modalities.
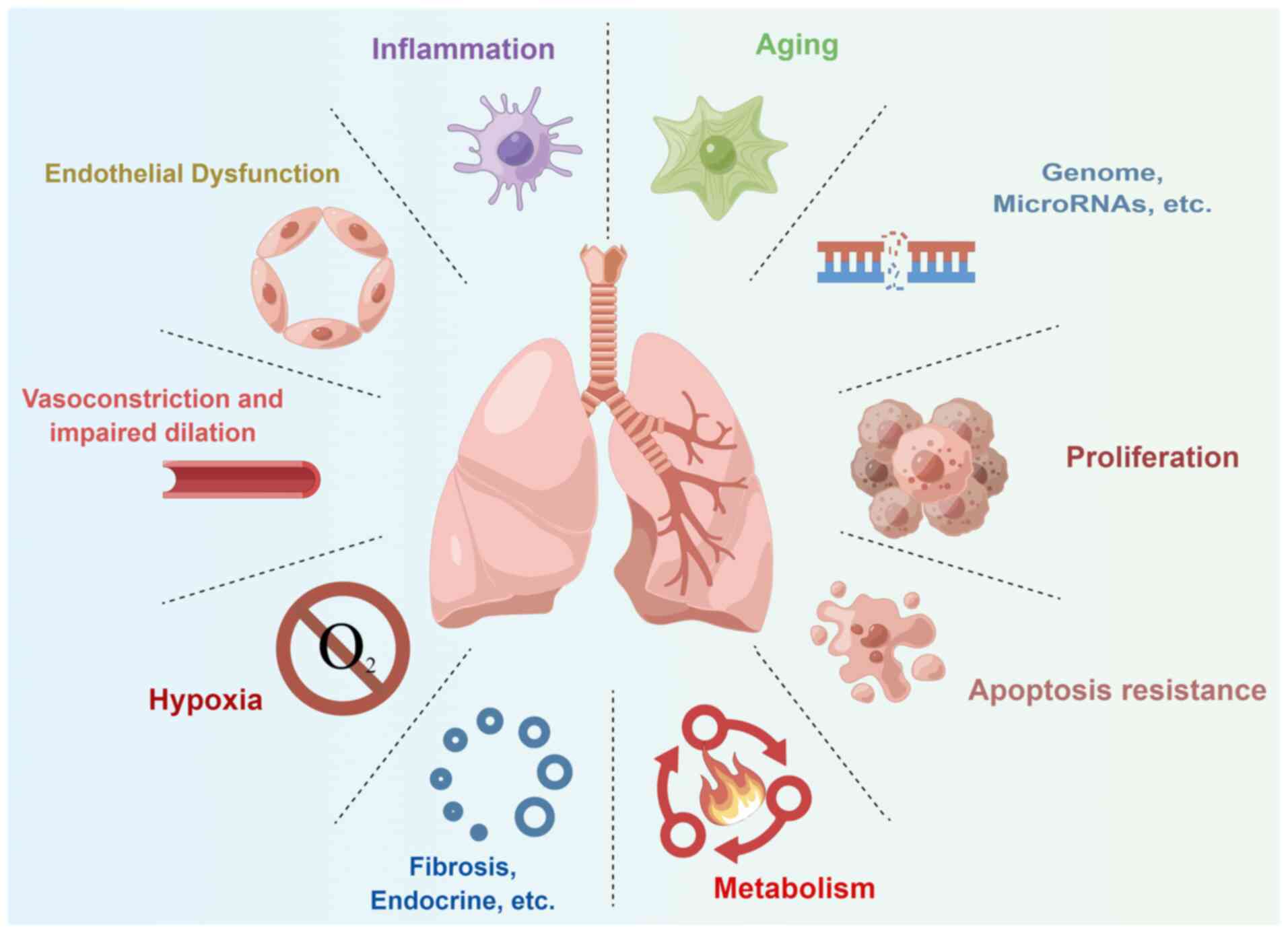
Over the years, numerous mechanisms have been
elucidated, from vasodilation, vasoconstriction and endothelial
dysfunction to more sophisticated regulatory mechanisms of cellular
signaling pathways, such as hypoxia, metabolism, proliferation,
apoptosis, aging and inflammation (Fig. 1). In 2020, five popular topics
associated with PH (hypoxia, cellular metabolism, inflammation,
abnormal proliferation and personalized medicine) were discussed,
highlighting the challenges and treatment opportunities at the 62nd
Thomas L. Petty Aspen Lung Conference (5). Metabolic abnormalities, including
abnormalities in the metabolism of glucose, fatty acids, glutamine,
and arginine, are universal features of PH widely observed in the
lungs and hearts of patients with PH and in the pulmonary vascular
cells isolated from patients with PH and PH rodent models (6-10). Glucose metabolism, the hub of
cellular energy metabolism, is involved in numerous metabolic
pathways and plays an important role in PH development. In the
present review, the roles of cellular metabolism in PH development
were discussed, focusing on the impact of impaired glucose
metabolism on the pulmonary vasculature. Furthermore, prospective
therapeutic techniques were outlined based on the discussed
translational findings.
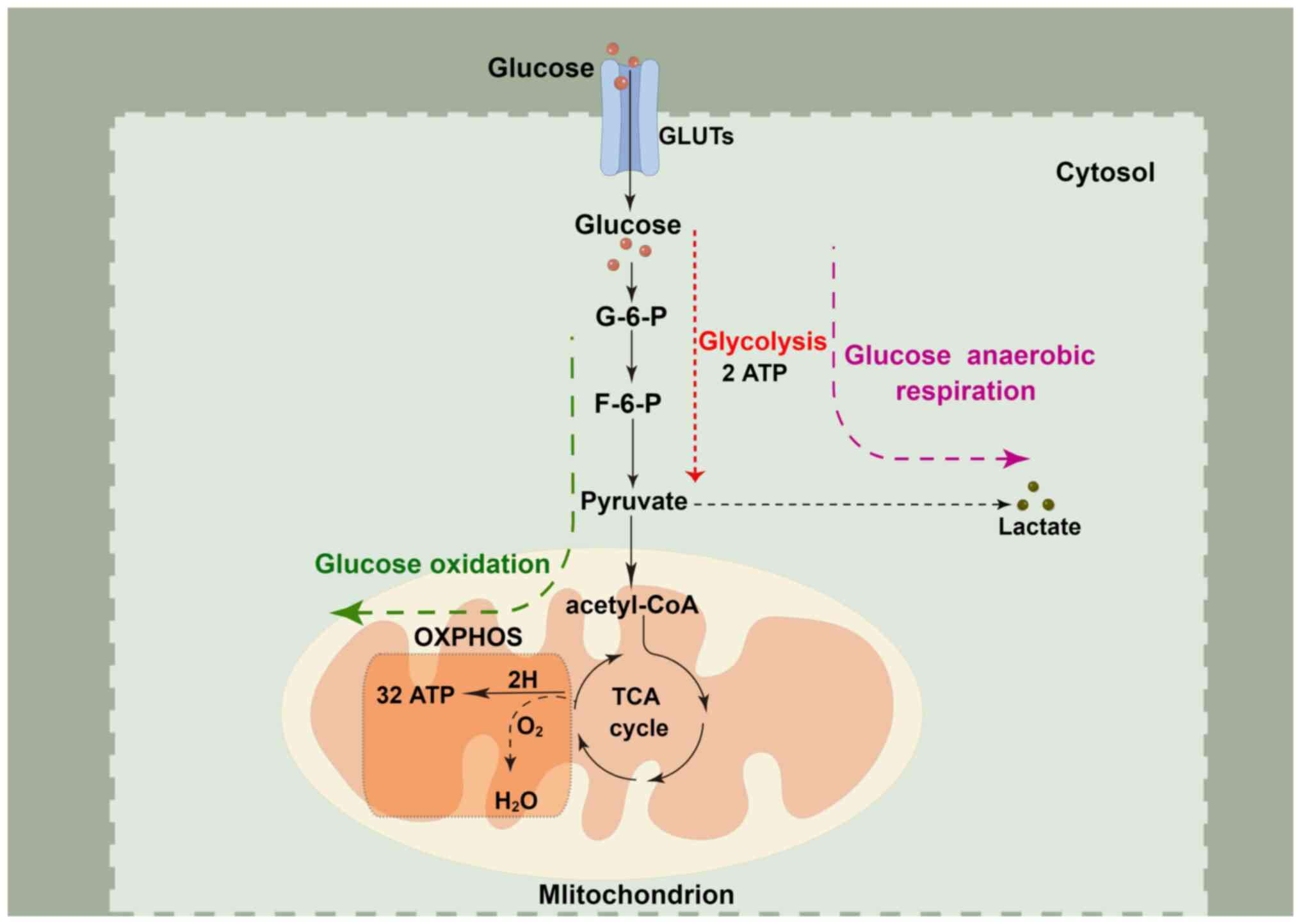
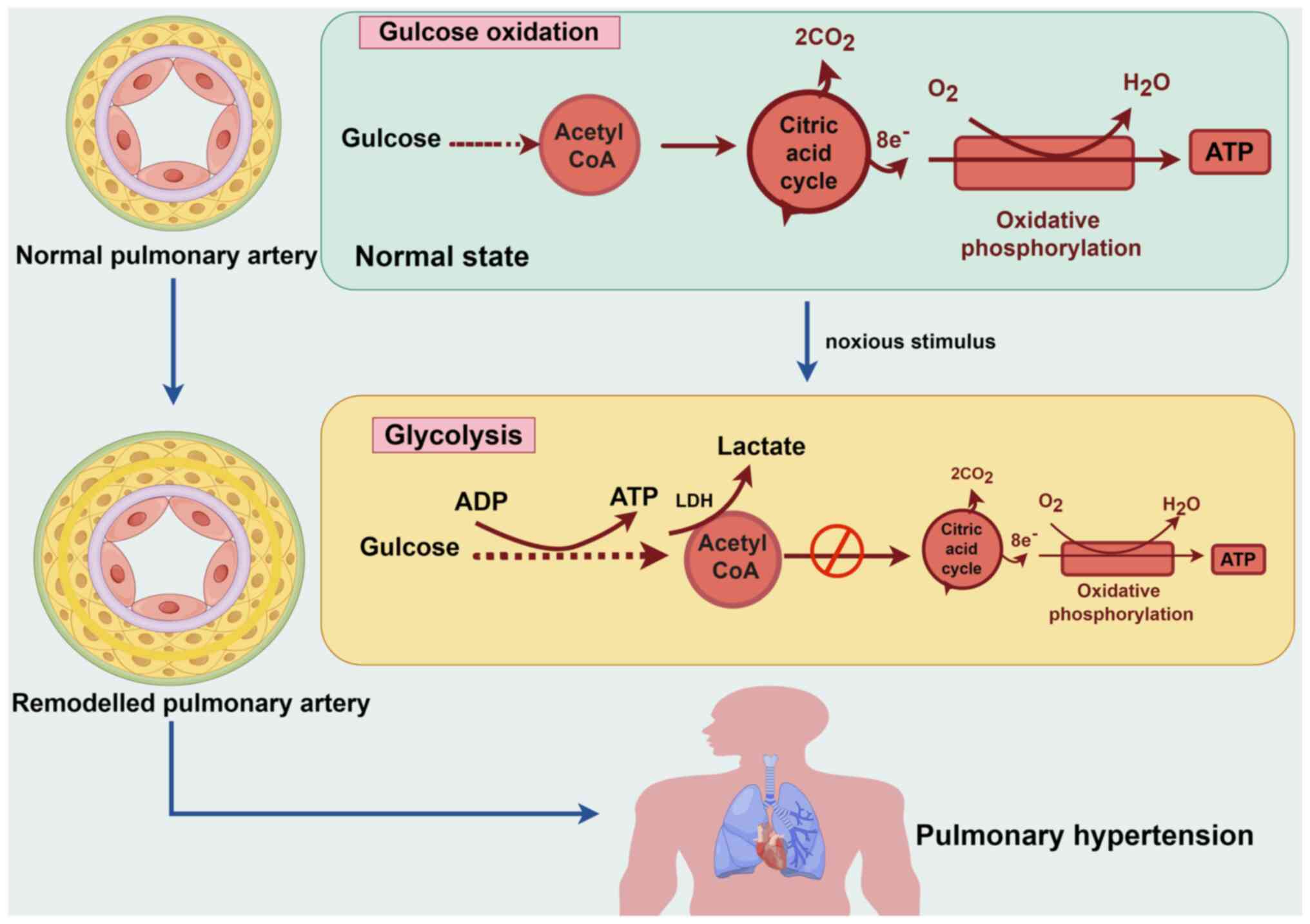
Glucose is the primary energy source for most
eukaryotic cells. Initially, glucose undergoes glycolysis in the
cytoplasm, where it is metabolized into pyruvate, yielding modest
quantities of the high-energy molecules, ATP and NADH. In the
presence of sufficient oxygen, pyruvate derived from glycolysis is
further converted to acetyl-CoA in the mitochondria, which
undergoes the tricarboxylic acid cycle and redox reactions along
the electron transport chain, known as oxidative phosphorylation
(OXPHOS). This process leads to the binding of the terminal
electron acceptor, molecular oxygen, to produce more ATP. However,
in the absence of sufficient oxygen, pyruvate does not undergo
OXPHOS and instead generates lactate via anaerobic respiration to
produce two ATP molecules (Fig.
2) (11). The net ATP
generated from OXPHOS of one glucose molecule is 32, which is
almost 16-times more than that generated from glycolysis and meets
the energy requirements for cell metabolism and proliferation.
Although vasodilators are important for PH
management, current research indicates that proliferative vascular
remodeling, rather than vasoconstriction, is the underlying cause
of PH (18). Pulmonary vascular
remodeling is essential for PH development and causes
histopathological changes similar to those in cancer, with the
aberrant proliferation and apoptotic resistance of PAECs and PASMCs
as the central components. The Warburg effect is a contributing
factor to PH, as its characteristic features, such as high glucose
uptake, lactate secretion, and anaerobic energy production, are
associated with PH (19,20). In patients with PH, positron
emission tomography (PET) shows significantly increased
fluorodeoxyglucose [(18F)FDG] uptake by the lungs and
right ventricle (21,22). Marsboom et al (23) and Zhao et al (24) reported that elevated
[18F]FDG uptake by the lungs is due to increased uptake
of [18F]FDG by PAECs and PASMCs. Blood outgrowth
endothelial cells of patients with heritable PAH (HPAH) and
idiopathic PAH (IPAH) carrying the bone morphogenetic protein
receptor 2 (BMPR2) mutation exhibit significantly higher
lactate secretion than that by the control cells. Lactate levels
are also high in the supernatants of HPAH and IPAH cells (25). Furthermore, ATP production is
significantly higher in PAH PASMCs than in non-PAH PASMCs under
hypoxia, suggesting increased anaerobic energy production in PAH
(26). Numerous studies on
patients with PH and rodent models have reported impaired glucose
OXPHOS and/or a shift to glycolysis in PAECs (23,27,28) and PASMCs (8,29,30), which are highly dependent on
glycolysis to promote proliferation. This metabolic shift helps to
maintain cellular energy homeostasis and reduce the dependency of
cells on oxygen, making it easier for the cells to survive in a
hypoxic environment (31).
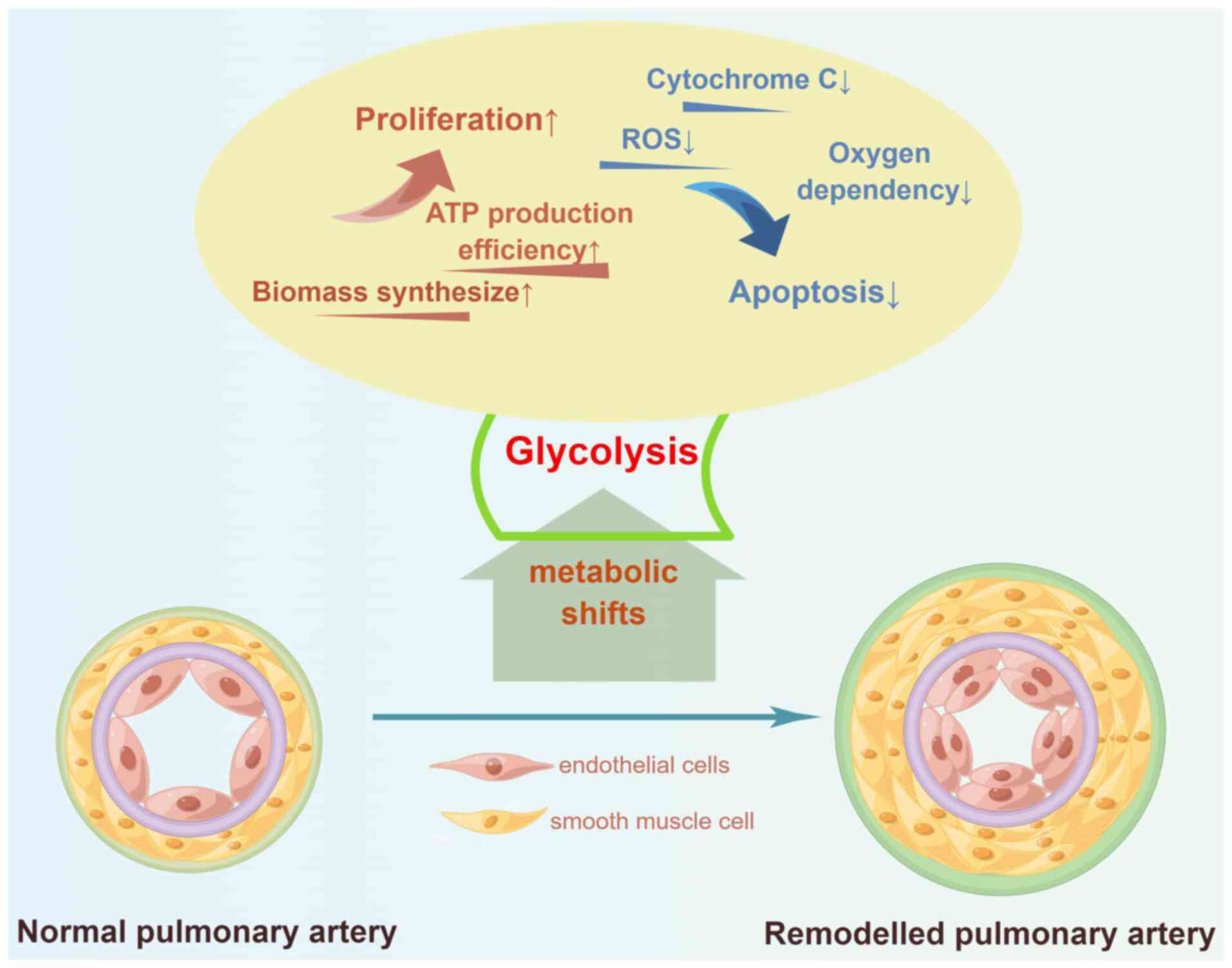
Increased glycolysis is always accompanied by the inhibition of
OXPHOS, which reduces the exposure to the mitochondrial
pro-apoptotic substances, reactive oxygen species (ROS) and
cytochrome C, thereby inhibiting apoptosis (6). Therefore, metabolic shift to
glycolysis promotes proliferation and apoptosis resistance in PAECs
and PASMCs, representing an attractive therapeutic approach for PH
(Fig. 3).
The precise molecular and biochemical changes that
drive the metabolic shift in PH remain unknown. Several essential
regulatory pathways have been identified. Specifically, stability
of hypoxia-inducible factor (HIF)-1α under hypoxia or normoxia is
the primary mechanism involved in the metabolic shift in PH
(32).
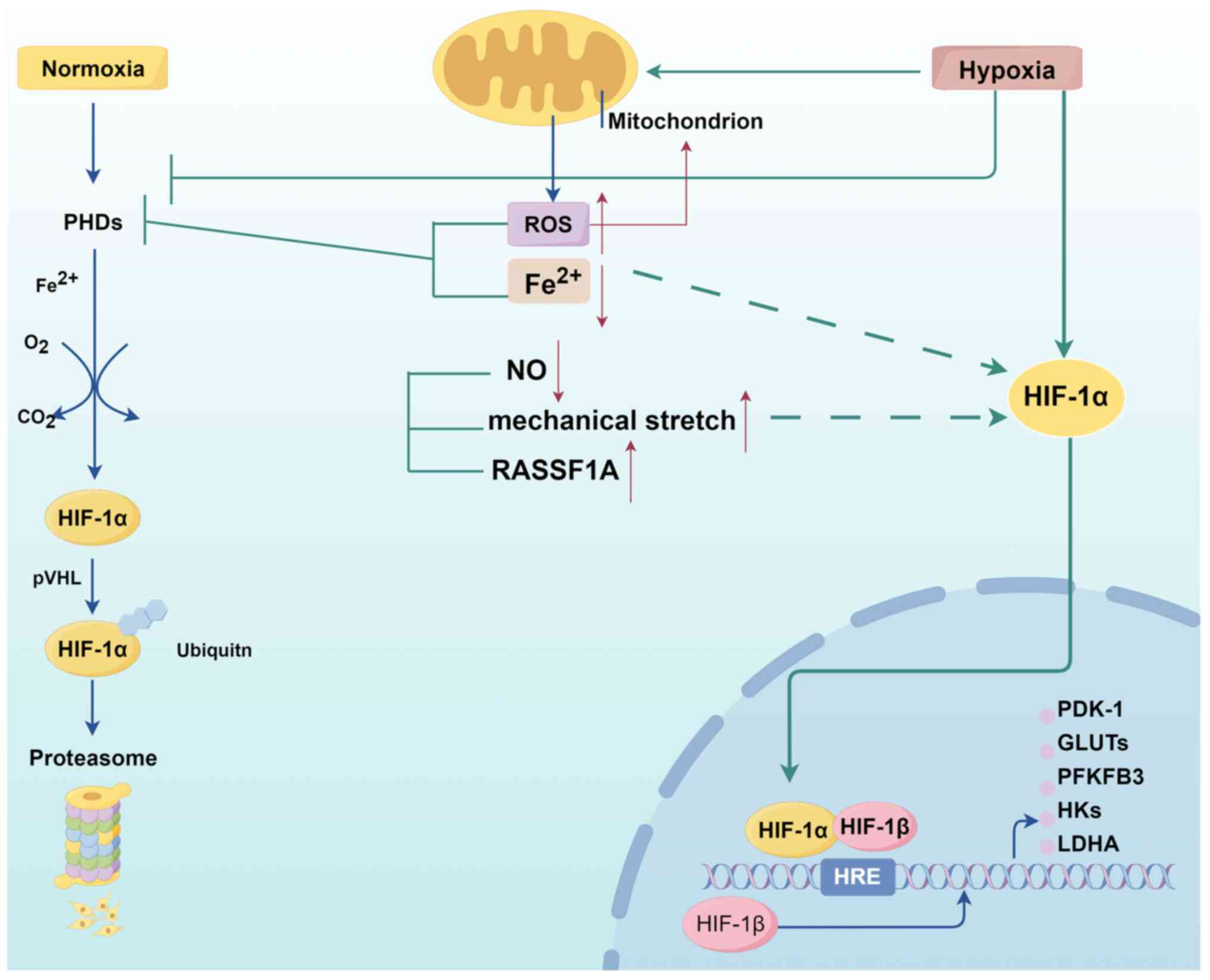
HIF-1α is one of the two subunits that assemble
HIF-1. Under normoxic conditions, prolyl hydroxylase (PHD) uses
O2 as a substrate to hydroxylate the proline residue in
HIF-1α, triggering its binding to von Hippel-Lindau and subsequent
degradation by the proteasome. Under hypoxia, PHD activity is
reduced, leading to HIF-1α stabilization and nuclear translocation.
In the nucleus, HIF-1α binds to constitutively expressed HIF-1β to
form the HIF-1 complex, which recruits cofactor proteins to
HIF-binding sites in the hypoxia response element and activates the
transcription of multiple target genes involved in cell
proliferation, angiogenesis, survival and metabolic processes
(33-35).
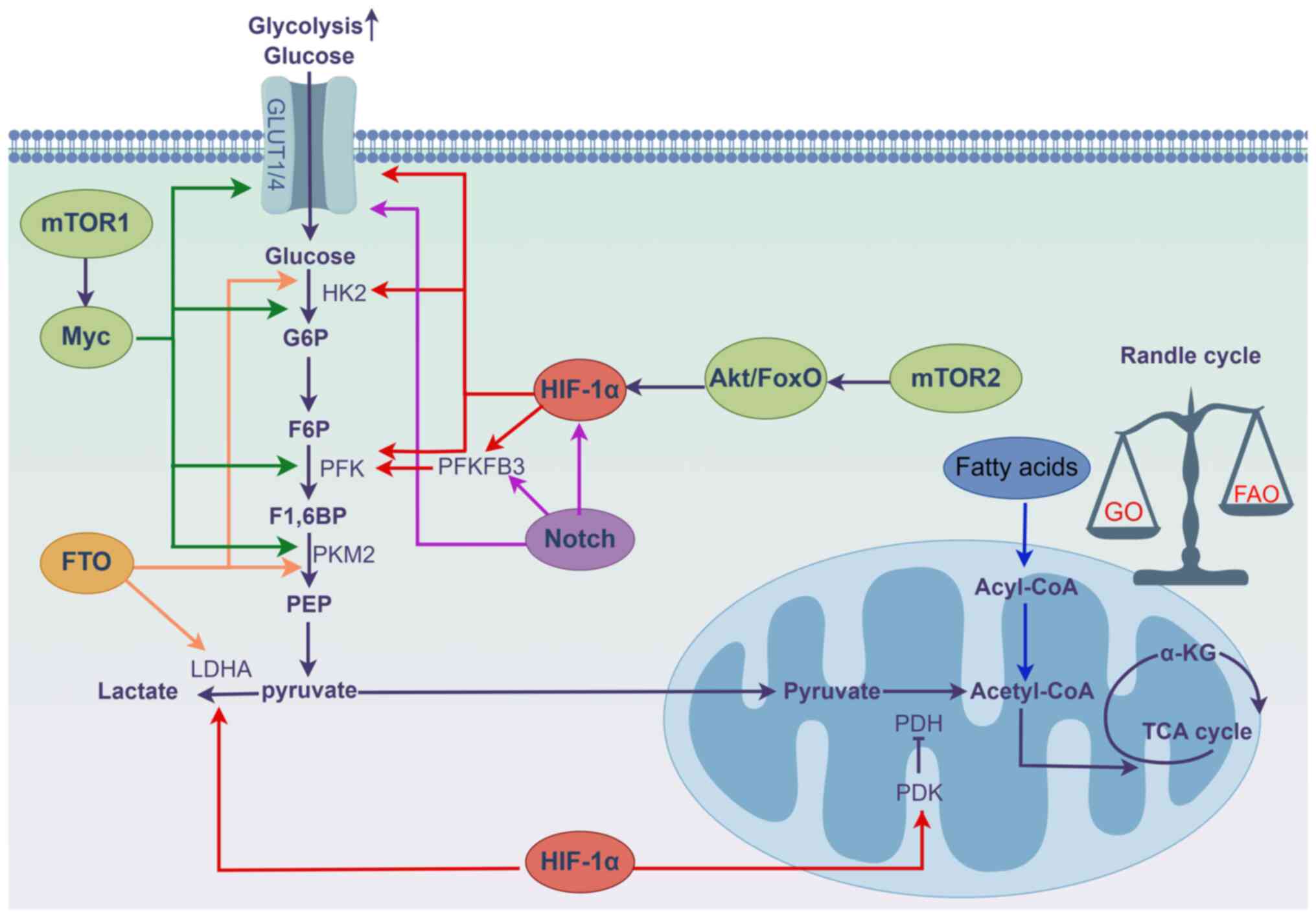
Activation of HIF-1α is closely related to the
metabolic shift in PH. HIF-1α activation plays a crucial role in
driving glycolysis by modulating various glycolysis-related
enzymes, including glucose transporters (GLUTs) (23,36,37), pyruvate dehydrogenase (PDH)
kinase 1 (PDK1) (38),
hexokinase 1/2 (HK1/2) (39) and
lactate dehydrogenase A (LDHA) (40,41). Specifically, upregulation of PDK1
by HIF-1α is a critical mediator linking intracellular hypoxia to
the metabolic shift. PDK1 inactivates PDH, a key enzyme for glucose
oxidation (GO) in the mitochondria, yielding pyruvate that cannot
be catabolized to acetyl-CoA and is accumulated in the cytoplasm to
undergo anaerobic respiration and produce lactate. Glycolysis
produces ATP with high efficiency but low output; therefore, HIF-1α
mediates the entry of large amounts of glucose into the cytosol for
glycolysis by upregulating the levels of GLUTs, HK2 and LDHA to
maintain energy homeostasis (42).
In addition to hypoxia, numerous factors contribute
to the upregulation of HIF-1α under normoxic conditions. These
include mitochondrial abnormalities (6), ROS (43), metal ions (44), nitric oxide (NO) production
(27) and mechanical stretch
(45), all of which act as
significant inducers of HIF-1α in pulmonary vascular cells
(Fig. 4). Mitochondria are the
cellular oxygen sensors that affect HIF-1α activity and regulate
cellular glucose metabolism by regulating the intracellular redox
status through the superoxide dismutase 2
(SOD2)-H2O2-HIF-1α-PDK-PDH pathway (46,47). In PH, epigenetic silencing of
SOD2 in mitochondria leads to decreased ROS
(H2O2) production, resulting in
pseudo-hypoxia in cells, which causes abnormal activation of HIF-1α
(48). Iron is a cofactor for
numerous enzymes, including PHD. Iron deficiency increases
morbidity and mortality in IPAH (49). In experimental PH, feeding rats
an iron-deficient diet induces the accumulation of HIF-1α and
promotes the upregulation of GLUT1, HK1 and PDK1 levels in their
pulmonary arteries, but iron replacement therapy (carboxymethyl
ferrous, 75 mg/kg) reduces the pulmonary artery pressure (PAP) and
alleviates pulmonary artery remodeling (44). Endothelin 1, a potent
vasoconstrictor, induces stabilization of the glycolytic switch
HIF-1α under normoxia by promoting ROS production and
calcium-dependent dephosphorylation of the receptor for activated C
kinase 1, leading to inhibition of PHD2 activity (50,51). NO generated by endothelial cells
is essential for vascular homeostasis, as evidenced by the lack of
NO in conditions associated with cardiopulmonary vascular diseases
(52). By preventing the
stabilization of HIF-1α by increasing PHD-mediated degradation, NO
also modulates the cellular response to hypoxia (53). Fijalkowska et al (27) reported that reduced NO production
leads to the loss of HIF-1α inhibition in PAECs under normoxic
conditions, thus contributing to the glycolytic shift. Previously,
another mechanism contributing to the stabilization of HIF-1α has
been reported in PASMCs. Ras association domain family 1A enhances
HIF-1α stability and nuclear entry, leading to the trans-activation
of target genes PDK1, HK2, and LDHA (54). In addition to the aforementioned
chemical triggers, mechanical stretch was identified by Wedgwood
et al (45) as an
independent regulator of HIF-1α. Mechanical stretch of PASMCs leads
to mitochondrial complex III-mediated ROS formation, which inhibits
PHD2 and activates HIF-1α (45).
These studies highlight the importance of the HIF-1
system in facilitating the metabolic shift, known as the Warburg
effect, in pulmonary vascular cells. Although hypoxia is an
important factor, it is not an absolute necessity for this effect.
Therefore, interventions aimed at inhibiting the activity of HIF-1α
or its downstream targets show potential as PH therapy.
Notch belongs to a family of single-channel
transmembrane receptor proteins. Four different Notch receptors
(Notch1-4) have been identified in mammals and can bind to five
different ligands: Three Delt-like (DLL1, DLL3 and DLL4) and two
Jagged (Jag1 and Jag2) ligands (55). After ligand engagement, the Notch
receptor is cut by γ-secretase, releasing the Notch intracellular
domain (NICD), which is transported to the nucleus as it modulates
nuclear localization signals. In the nucleus, NICD binds to the
DNA-binding protein, CSL (also known as the recombination signal
sequence-binding protein Jκ), which controls numerous cellular
processes, including cell growth, development, proliferation and
apoptosis, via the transcription of Notch targets (HEY1, HES1 and
Myc) and their downstream targets; this is an important pathway
that determines the cell fate (56,57). Notch1 and Notch3 levels are
upregulated in PAECs and PASMCs and mediate their proliferation and
apoptosis resistance, which are involved in the progression of PH
(58,59). A functional link is observed
between Notch signaling and cellular metabolism; hyperactivated
Notch undergoes a glycolytic switch in several cells (60-62). Under 5% O2,
Notch1-HES1 enhances the glycolytic pathway by inactivating p53 and
activating NF-κB signaling. Notch1-HES1 also enhances glycolysis by
directly binding to the promoters of key enzymes in glycolysis,
such as GLUT1, GLUT3 and fructose-2,6-biphosphatase 3 (PFKFB3), to
promote their transcription and expression (5,63). Moriyama et al (64) reported that treatment with DAPT
(a Notch inhibitor) increases the activities of PDH and COX IV (key
enzymes of OXPHOS) and inhibits the activity of LDH in hADMPCs,
reinforcing the important role of Notch in cellular metabolic
shift. In addition to acting independently, Notch promotes cellular
metabolic shift through synergistic effects with HIF-1α. Activation
of Notch1 significantly increases HIF-1α transcriptional activity,
and knockdown of HIF-1α partially attenuates glycolysis induced by
Notch1, suggesting that the cellular glycolytic pathway is
regulated by Notch signaling and HIF-1α in coordination (65).
mTOR, an atypical serine/threonine protein kinase
that is a member of the phosphatidylinositol kinase-related kinase
protein family, consists of two functional multi-protein complexes:
mTORC1 and mTORC2. mTORC1 is sensitive to rapamycin and consists of
mTOR, raptor (an mTOR-associated regulatory protein), and mLST8
(also known as GßL), whereas mTORC2 is insensitive to rapamycin and
consists of mTOR, Rictor (an mTOR chaperone that is insensitive to
rapamycin) and mLST8 (66). Both
cooperate to integrate various extracellular signals, such as
nutrients, energy and growth factors, and participate in biological
processes, such as gene transcription, protein translation and
ribosome synthesis, which play important roles in cell growth,
apoptosis, autophagy and metabolism (67,68).
FTO genes have obesity-associated alleles located on
chromosome 16q12.2 and are associated with dietary intake, appetite
regulation and energy metabolism (80,81). In 2007, FTO protein was
identified as an RNA demethylase (82). Methylation of the nitrogen atom
at the 6th adenine on the RNA chain is the most common mRNA
modification that regulates gene expression during the translation
of proteins. Since Jia et al (83) identified N6-methyladenosine (m6A)
in nuclear RNA as the primary substrate of FTO protein and
clarified that FTO proteins are RNA m6A demethylases, FTO-RNA
epigenetic modifications have been widely investigated. FTO protein
plays important regulatory roles in tumors (84), hypertrophic cardiomyopathy
(85), HF (86) and PH (87,88) through demethylation modifications
of RNA m6A. FTO increases the activity of transcription factors
c-Jun, JunB and C/EBPb via m6A demethylation modification, promotes
the expression of the glycolysis-related enzymes
phosphofructokinase, phosphoglycerate mutase 1 and HK1, and
facilitates glycolysis-dependent cell proliferation in cancer
(89). Furthermore, MDA-MB-231
cells transfected with miFTO inhibitors show significantly reduced
HK1 and PKM expression, which result in significantly decreased ATP
and lactate levels (90). These
findings suggest that FTO regulates cellular glycolysis through the
demethylation of RNA m6A. In line with the cancer theory of PH,
several studies have explored the role of FTO in PH. In
monocrotaline (MCT)-induced PH (MCT-PH) rats, the expression of FTO
in the lung tissue was significantly reduced, accompanied by an
increase in m6A methylation levels. The researchers then extracted
lung tissue RNA for methylated RNA immunoprecipitation sequencing,
and a total of 3,298 differentially methylated m6A sites were
screened based on false discovery rate ≤0.0001 and fold change ≥2.
Kyoto Encyclopedia of Genes and Genomes pathway analysis showed
that differentially methylated m6A sites were enriched in the
glycolytic/glycogenic pathway, in which the mRNA m6A levels of key
glycolytic enzymes HK3, glucose-6-phosphate isomerase, LDHA and PKM
were significantly upregulated (88). These results suggested that FTO
may affect the translation or transcription of key glycolytic
enzymes by regulating mRNA m6A levels and may be involved in the
development of PH.
Fatty acid oxidation (FAO) is a main source of ATP
production; however, it produces less ATP than that by GO with the
same oxygen consumption. Therefore, under normal conditions, most
cells rely on GO as their main energy source, except for rapidly
proliferating cells. There is a reciprocal regulation of fatty acid
and glucose metabolism, characterized by a competitive relationship
in which the dominance of one can lead to the inhibition of the
other, as evidenced by the increase in the proportion of acetyl CoA
produced from FAO that inhibits PDH activity, leading to the
suppression of GO, which is known as the glucose-fatty acid or
Randle cycle (91-93). FAO levels are elevated in
patients with PH and rodent models of PH (9,94). A non-targeted metabolomic
analysis performed on 21 patients with IPAH and 31 age-, body mass
index-, and sex-matched normal controls showed that FAO metabolites
are significantly upregulated in patients with IPAH compared with
those in the normal controls, suggesting that FAO is more abundant
in IPAH (95). Malonyl coenzyme
A decarboxylase is an important FAO regulatory enzyme that degrades
malonyl-CoA to acetyl-CoA and reduces the inhibitory effect of
malonyl-CoA on carnitine acyltransferase-1, the rate-limiting
enzyme of FAO. Malonyl coenzyme A decarboxylase knockout mice are
protected from hypoxic PH, and targeting carnitine
acyltransferase-1A with oxyfenicine attenuates Su/Hx-induced PH in
rats (96). All these effects of
improving PH by interfering with FAO are achieved through the
regulation of the Randle cycle, where FAO inhibition promotes GO,
drives cells from glycolysis to GO, and inhibits rapid cell
proliferation. In summary, FAO inhibition prevents glycolysis by
shifting the metabolism from FAO to GO, thereby facilitating PH
treatment (Fig. 5).
Glycolytic pathway exerts significant impact on PH,
serving as a target for PH treatment. Drugs targeting the key genes
and enzymes in the glycolytic pathway significantly improve PH in
rodents. Currently, some pharmacological agents targeting
glycolysis are undergoing clinical trials.
Influence of HIF-1α on glycolysis has been
extensively studied, with numerous studies focusing on
pharmacological agents targeting PHD or HIF-1α. Inhibitors that
directly or indirectly affect HIF-1α can be divided into five main
groups: Inhibitors that reduce HIF mRNA expression, such as
EZN-2968; inhibitors that prevent HIF-1α protein expression, such
as 2-methoxyestradiol (2-ME2); inhibitors that enhance the
degradation of HIF-1α protein, such as apigenin and bisphenol A;
inhibitors that inhibit the dimerization of HIF-1α and HIF-1β, such
as acriflavine and doxorubicin; and inhibitors of DNA binding of
HIF-1, such as echinomycin (97-99). Currently, clinical trials of
HIF-1α inhibitors are primarily focused on the treatment of cancer,
with no clinical trials related to PH currently registered on the
ClinicalTrials.gov registry. However, as a
signaling messenger, HIF-1α performs numerous important
physiological functions, and its inhibition may lead to adaptive
metabolic disorders in multiple organs and tissues (33). In a phase II trial, patients with
metastatic renal cell carcinoma were divided into two treatment
arms based on whether they stopped or continued sunitinib
treatment. Patients in treatment arm A received 1,500 mg panzem
(2-ME2) thrice daily, whereas those in treatment arm B received a
comparable dose of panzem plus sunitinib at the highest tolerated
dose for the patient. However, this study was halted after 17
patients experienced treatment toxicity in both arms, resulting in
fatigue (60%), diarrhea (53%), elevated aspartate aminotransferase
(41%), decreased appetite (35%), joint and muscle pain (35%), and
lack of objective response to treatment (100).
In a previous study, inhibition of the Notch
signaling pathway via gene ablation caused myocardial hypertrophy
and HF in adult mice (101),
which severely limited the development of PH pharmacological drugs
targeting HIF-1α or Notch. Therefore, investigation of downstream
glycolytic targets is important for novel drug development for PH.
Numerous inhibitors prevent or reverse PH in various cellular and
animal models of hypoxia, MCT and Su/Hx (Table I).
Several enzymes are involved in glycolysis.
Glycolysis is the main metabolic pathway for energy production in
the pulmonary vascular cells of patients with PH, and glycolytic
enzyme expression is increased in PH (25,113,114). Therefore, targeting
glycolysis-related molecules, such as GLUT1, PFKFB3 and PDK1, is a
promising strategy for PH treatment.
Glucose uptake is the first and most critical step
of glucose metabolism. GLUTs (also known as sodium-glucose
co-transporters) are a large group of membrane proteins that
facilitate glucose transport across the plasma membrane of
mammalian cells. To date, 14 members of the GLUT family have been
identified, of which GLUT1-4 are the most widely studied and
extremely important to maintain the normal physiological functions
in humans; their abnormal expression and function can lead to
various diseases (115-117). GLUT1-4 mRNA and protein levels
are significantly increased in the lungs, along with glycolysis, in
rodent models of PH (118-121). Empagliflozin and dapagliflozin
(GLUT2 inhibitors) significantly attenuate pulmonary vascular
remodeling, right ventricular (RV) systolic pressure (RVSP) and RV
hypertrophy index in rodents with PH (121,122). The Universitaire Ziekenhuizen
KU Leuven in Belgium is currently conducting a clinical trial
(NCT05731466) to investigate the effects of GLUT2 inhibitors on
RV-arterial coupling in patients with HF with preserved ejection
fraction and PH. A prospective randomized multi-center open-label
study previously investigated whether GLUT2 inhibitors improve the
left ventricular (LV) pump function and reduce the increase in LV
filling pressure (LVFP) and RVSP during exercise in patients with
type 2 diabetes mellitus. The study revealed that the addition of
dapagliflozin at a daily dose of 5 mg to conventional treatment
significantly improved both RVSP and LVFP during exercise in
patients with type 2 diabetes mellitus over a 6-month period
(123). This study highlighted
the potential use of GLUT2 inhibitors for PH treatment.
HKs play a critical role in glucose metabolism
regulation by catalyzing the first irreversible step of glycolysis.
In mammals, four HK isozymes (HK1-4) have been identified. HK1-3
are associated with the metabolic shift in PH, and HK2 is the most
extensively studied, whose inhibition exhibits potential for PH
therapy (124,125). Moreover, 3-bromopyruvate
(3-BrPA), a pyruvate analog targeting HK2, significantly improves
PH induced by hypoxia, MCT and Su/Hx in rodents (126-128). In addition to inhibiting the
glycolytic pathway, 3-BrPA also causes the opening of the
mitochondrial permeability transition pore and release of the
pro-apoptotic molecule, CytoC, into the cytoplasm by inhibiting the
binding of HK2 to the mitochondria, causing apoptosis via caspase 3
activation, which alleviates pulmonary vascular remodeling and RV
hypertrophy in animals with PH (129,130). However, studies on 3-BrPA for
PH treatment are currently in early stages. Therapeutic studies on
the HK2 selective inhibitors, ketoconazole and posaconazole, have
entered phase I clinical trials (NCT03763396). Considering the
promising results observed in experimental PH studies in rodents,
3-BrPA can be a promising drug for PH treatment after further
pharmacological, toxicological and clinical studies.
Notably, 2-DG is a glucose analog that enters the
cytoplasm through a deceptive mechanism; it mimics glucose to trick
the cells into internalizing it (131). On the one hand, 2-DG competes
with glucose for HK2, thereby inhibiting glycolysis. On the other
hand, unlike normal glucose, 2-DG is unable to generate energy via
subsequent catabolism, resulting in cell starvation and inhibition
of cell proliferation and other metabolic processes (132). It also suppresses PASMC
proliferation, which plays an important role in improving PH
(114,133). After completing the phase I
study, 2-DG entered the phase II study for prostate cancer
(NCT00633087); however, this study was stopped early due to slow
progress and insufficient data to measure outcomes. Therefore,
further research is essential to facilitate the application of 2-DG
for PH treatment.
In addition to being a potential therapeutic agent,
2-DG is a prospective diagnostic strategy to monitor the pulmonary
vasculature. [18F]FDG is a fluorinated derivative of
2-DG, in which the hydroxy group at the 2nd position of glucose is
replaced by the radioisotope, 18F. In patients with PH,
lung [18F]FDG uptake on PET imaging is positively
correlated with the condition severity (134,135).
Fructokinase 6-phosphate kinase 1 (PFK1), the second
rate-limiting enzyme of glycolysis catalyzing the formation of
fructose-1,6-bisphosphate from fructose 6-phosphate (F6P), is
regulated by PFKFB3 (136).
PFKFB3 does not play a direct role in the catalytic mechanism of
glycolysis. However, it is responsible for the synthesis of
fructose 2,6-bisphosphate, which is a potent allosteric activator
of PFK1. By catalyzing the conversion of F6P, PFKFB3 significantly
enhances the catalytic activity of PFK1. Therefore, PFKFB3 is
critical for glycolysis regulation (137). Similar to HK2 levels, PFKFB3
levels are significantly increased in the lung tissues of patients
with PAH and rodent models. Knockdown of PFKFB3 or use of
its small molecule isoenzyme inhibitor, 3-(3-pyrid
inyl)-1-(4-pyridinyl)-2-propen-1-one (3-PO), significantly reduces
the proliferation of PASMCs and PAECs, attenuates vascular
remodeling, and ameliorates PAH by inhibiting glycolysis (28,113,138). However, efficacy of 3-PO as a
therapeutic intervention for PH is currently being investigated in
experimental studies with cellular and animal models.
In total, four pyruvate kinase isoenzymes are
present in mammals: Pyruvate kinase M1, M2, L, and R (139). Among these, PKM2 is an
important regulator of anaerobic metabolism and a potential
therapeutic target for PH (140,141). PKM2 exists in two forms:
Low-activity dimer and high-activity tetramer. Dimeric form of PKM2
plays a critical role in the final rate-limiting step of the
glycolytic pathway by facilitating the production of pyruvate and
ATP (142,143). In addition to its primary
enzymatic function, dimeric form of PKM2 translocates to the
nucleus and functions as a transcriptional co-activator, enhancing
the activity of various transcription factors, such as HIF-1α and
signal transducer and activator of transcription 3, subsequently
increasing the expression levels of GLUT1, HK2 and LDHA (144-146). Targeted inhibition of PKM2
ameliorates MCT- and supra-coronary aortic banding-induced PH in
rodents (119,147). However, current research on
PKM2 inhibitors for PH treatment remains in its early stages.
PDH catalyzes the production of acetyl-CoA from
pyruvate, which is the central link between cytoplasmic glycolysis
and the mitochondrial tricarboxylic acid cycle. As a downstream
target of HIF-1α, PDK phosphorylates PDH, leading to its
inactivation, which results in pyruvate accumulation in the
cytoplasm, facilitating glycolysis. PDK levels are significantly
higher in PH lung tissues than in the healthy lung tissues
(42,148). PDK inhibitor dichloroacetate
(DCA) ameliorates PH caused by hypoxia (149), MCT (150) and serotonin transporter
overexpression (151) by
restoring OXPHOS in the glycolytic tissues of rodent models.
Importantly, unlike HK2 and PFKFB3 inhibitors, DCA has yielded
promising results in PH clinical trials. In 2010, Imperial College
London and University of Alberta in Canada conducted a phase I
two-center clinical trial of the efficacy of DCA for PH
(NCT01083524). A total of 30 patients with advanced IPAH were
enrolled and continuously administered oral DCA (3.0-12.5 mg/kg
twice daily) for 16 weeks. At the follow-up, DCA significantly
reduced the pulmonary vascular resistance and PAP and improved the
exercise tolerance in patients (42). The first-in-human study of a
mitochondria-targeting drug in PH revealed PDK as a druggable
target causing hemodynamic improvement in genetically susceptible
patients, thus facilitating the establishment of precision medicine
approaches for PH. However, extensive clinical trials are necessary
to assess the efficacy and safety of such drugs.
A reciprocal relationship is observed between FAO
and GO that is the core of the Randle cycle. The Randle cycle is
also observed in PH. Preventing FAO from producing acetyl-CoA
limits glycolysis by increasing PDH activity and enhancing GO via
the Randle cycle. TMZ, an inhibitor of mitochondrial enzyme
long-chain 3-ketoacyl CoA thiolase, increases GO and inhibits FAO
by activating the Randle cycle and is widely used to treat angina
pectoris (152), myocardial
infarction (153,154) and HF (155). Parra et al (156) reported that treatment of
HPASMCs with TMZ inhibits the increase in the mRNA levels of
glycolysis markers (HK2, PFKFB3 and GLUT1) and
suppresses hypoxia-induced HPASMC proliferation. Moreover,
TMZ-induced FAO inhibition triggers the accumulation of long-chain
fatty acids in the cytoplasm (lipotoxicity), resulting in
endoplasmic reticulum stress, ultimately leading to cell death and
alleviation of pulmonary vascular remodeling and RV hypertrophy
(157-159). A phase II clinical trial
(NCT02102672) sponsored by the Pontificia Universidad Catolica de
Chile evaluated the efficacy of TMZ in improving the RV function,
remodeling and functional class in patients with PAH. Over the
course of 3 months, participants in the study received TMZ at a
dose of 35 mg twice daily in addition to conventional PAH-specific
therapy. The Pontificia Universidad Catolica de Chile sponsored a
phase II clinical trial (NCT02102672) to evaluate the efficacy of
TMZ in improving the RV function, remodeling and functional class
in patients with PAH. Patients in the TMZ group exhibited a
significant decrease in the RV diastolic area, significant increase
in the 6-min walking distance, and modest but significant
improvement in RV remodeling (160). The aforementioned study serves
as a valuable reference for the application of TMZ to treat PH.
Ranolazine is an anti-anginal and anti-ischemic drug
that inhibits sodium-dependent calcium overload in the myocardium
without affecting the heart rate or blood pressure (161). Additionally, ranolazine
partially inhibits FAO (162).
However, unlike TMZ, ranolazine does not influence FAO-associated
enzymes; instead, it stimulates GO by activating PDH (163,164). Ranolazine also increases ATP
production, reduces the expression of glycolytic mediators, such as
GLUT1, HKI and LDHA, and decreases lactate production in a rat
pulmonary artery banding model (91). Cardiac function significantly
improves after ranolazine treatment. A randomized, double-blind,
placebo-controlled, multi-center, phase IV clinical trial assessing
the effects of ranolazine on RV dysfunction in patients with PAH
using cardiovascular magnetic resonance was sponsored by the
University of Pennsylvania (NCT01839110 and NCT02829034). The
participants orally received ranolazine (500 mg twice daily) with
stable PAH-specific treatment, which was subsequently increased to
1,000 mg twice daily after 2 weeks and continued for a total of 26
weeks. Only 9 patients completed the follow-up cardiovascular
magnetic resonance imaging, and six completed the placebo arm.
Notably, ranolazine only improves the RV function in precapillary
PAH and has no significant effect on other forms of PH (165). Therefore, further large-scale
studies are necessary to confirm the efficacy of ranolazine for PH
treatment. Previous studies and clinical trials focusing on glucose
metabolism are summarized in Tables
II and III,
respectively.
To date, most studies on PH have focused on
vasoconstriction, dilation and endothelial dysfunction. Recent
studies have revealed the significance of metabolic dysregulation
and reprogramming in driving excessive proliferation and apoptosis
resistance in pulmonary vascular cells, thereby leading to
pulmonary vascular remodeling. Metabolic shift is observed in both
patients with PH and animal models. Targeting glycolysis-related
pathways has been effective in mitigating PH, suggesting the role
of metabolic reprogramming in PH pathogenesis (Fig. 6). Furthermore, some clinical
studies have shown that modulation of cellular glucose metabolism
reduces the pulmonary vascular resistance and PAP and improves
exercise tolerance and RV function in patients with PH. In addition
to its mechanistic significance, glycolytic alterations in PH can
be used to develop a diagnostic approach based on the pulmonary
vasculature. PET using [18F]FDG as a radiotracer can
provide valuable information on the pulmonary vascular and RV
metabolic status, thereby aiding in the diagnosis and management of
PH. However, fundamental questions remain regarding the interplay
between mitochondrial dysfunction and metabolic switching and their
combined roles in PH pathogenesis. Therefore, further investigation
of the roles of metabolic abnormalities and mitochondrial
dysfunction in PH pathogenesis are necessary for the development of
novel therapeutic approaches for this condition.
Not applicable.
MC and WL conceived and designed the entire review
and wrote the paper. MC, HL, YLi and YLuo assisted with literature
collection and figure drawings. YH, XS and WL reviewed and edited
the manuscript. All authors read and approved the final version of
the manuscript. Data authentication is not applicable.
Not applicable.
Not applicable.
The authors declare that they have no competing
interests.
Not applicable.
The present study was supported by the National Natural Science
Foundation of China (grant no. 81970056), the Discipline
Construction Project of Guangdong Medical University (grant no.
4SG21233G), the Key platform of Department of Education of
Guangdong (grant no. 2021LSYS007), and Zhanjiang Science and
Technology Development Special Funding Competitive Allocation
Project (grant nos. 2022E05011, 2022A01196 and 2021A05158).
|
1
|
Humbert M, Kovacs G, Hoeper MM,
Badagliacca R, Berger RMF, Brida M, Carlsen J, Coats AJS,
Escribano-Subias P, Ferrari P, et al: 2022 ESC/ERS guidelines for
the diagnosis and treatment of pulmonary hypertension. Eur Respir
J. 61:22008792023. View Article : Google Scholar
|
|
2
|
Jia Z, Wang S, Yan H, Cao Y, Zhang X, Wang
L, Zhang Z, Lin S, Wang X and Mao J: Pulmonary vascular remodeling
in pulmonary hypertension. J Pers Med. 13:3662023. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kang M, Hart CM, Kempker JA, Veeraraghavan
S and Trammell AW: Pulmonary hypertension mortality trends in
United States 1999-2019. Ann Epidemiol. 75:47–52. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hoeper MM, Humbert M, Souza R, Idrees M,
Kawut SM, Sliwa-Hahnle K, Jing ZC and Gibbs JSR: A global view of
pulmonary hypertension. Lancet Respir Med. 4:306–322. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
George MP, Gladwin MT and Graham BB:
Exploring new therapeutic pathways in pulmonary hypertension.
metabolism, proliferation, and personalized medicine. Am J Respir
Cell Mol Biol. 63:279–292. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Culley MK and Chan SY: Mitochondrial
metabolism in pulmonary hypertension: Beyond mountains there are
mountains. J Clin Invest. 128:3704–3715. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Smolders VFED, Rodríguez C, Morén C,
Blanco I, Osorio J, Piccari L, Bonjoch C, Quax PHA, Peinado VI,
Castellà M, et al: Decreased glycolysis as metabolic fingerprint of
endothelial cells in chronic thromboembolic pulmonary hypertension.
Am J Respir Cell Mol Biol. 63:710–713. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Goncharov DA, Kudryashova TV, Ziai H,
Ihida-Stansbury K, DeLisser H, Krymskaya VP, Tuder RM, Kawut SM and
Goncharova EA: Mammalian target of rapamycin complex 2 (mTORC2)
coordinates pulmonary artery smooth muscle cell metabolism,
proliferation, and survival in pulmonary arterial hypertension.
Circulation. 129:864–874. 2014. View Article : Google Scholar
|
|
9
|
Singh N, Manhas A, Kaur G, Jagavelu K and
Hanif K: Inhibition of fatty acid synthase is protective in
pulmonary hypertension. Br J Pharmacol. 173:2030–2045. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Bertero T, Perk D and Chan SY: The
molecular rationale for therapeutic targeting of glutamine
metabolism in pulmonary hypertension. Expert Opin Ther Targets.
23:511–524. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Hantzidiamantis PJ and Lappin SL:
StatPearls; Treasure Island (FL): 2023
|
|
12
|
Warburg O, Wind F and Negelein E: The
metabolism of tumors in the body. J Gen Physiol. 8:519–530. 1927.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Vaupel P and Multhoff G: Revisiting the
Warburg effect: Historical dogma versus current understanding. J
Physiol. 599:1745–1757. 2021. View Article : Google Scholar
|
|
14
|
Archer SL: Pyruvate kinase and warburg
metabolism in pulmonary arterial hypertension: Uncoupled Glycolysis
and the cancer-like phenotype of pulmonary arterial hypertension.
Circulation. 136:2486–2490. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Archer SL: Acquired mitochondrial
abnormalities, including epigenetic inhibition of superoxide
dismutase 2, in pulmonary hypertension and cancer: Therapeutic
implications. Adv Exp Med Biol. 903:29–53. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Vaupel P, Schmidberger H and Mayer A: The
Warburg effect: Essential part of metabolic reprogramming and
central contributor to cancer progression. Int J Radiat Biol.
95:912–919. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Condon D, Agarwal S, Chakraborty A and de
Jesus Perez VA: The cancer hypothesis of pulmonary arterial
hypertension: The next ten years. Am J Physiol Lung Cell Mol
Physiol. 318:L1138–L1139. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Christou H and Khalil RA: Mechanisms of
pulmonary vascular dysfunction in pulmonary hypertension and
implications for novel therapies. Am J Physiol Heart Circ Physiol.
322:H702–H724. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ryanto GRT, Suraya R and Nagano T:
Mitochondrial dysfunction in pulmonary hypertension. Antioxidants
(Basel). 12:3722023. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Arai MA, Sakuraba K, Makita Y, Hara Y and
Ishibashi M: Evaluation of naturally occurring HIF-1 inhibitors for
pulmonary arterial hypertension. Chembiochem. 22:2799–2804. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ahmadi A, Ohira H and Mielniczuk LM: FDG
PET imaging for identifying pulmonary hypertension and right heart
failure. Curr Cardiol Rep. 17:5552015. View Article : Google Scholar
|
|
22
|
Abikhzer Y, Probst S and Rush C: Pulmonary
hypertension findings detected by F-18 FDG PET scan. Clin Nucl Med.
33:405–406. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Marsboom G, Wietholt C, Haney CR, Toth PT,
Ryan JJ, Morrow E, Thenappan T, Bache-Wiig P, Piao L, Paul J, et
al: Lung 18F-fluorodeoxyglucose positron emission tomography for
diagnosis and monitoring of pulmonary arterial hypertension. Am J
Respir Crit Care Med. 185:670–679. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zhao L, Ashek A, Wang L, Fang W, Dabral S,
Dubois O, Cupitt J, Pullamsetti SS, Cotroneo E, Jones H, et al:
Heterogeneity in lung (18)FDG uptake in pulmonary arterial
hypertension: Potential of dynamic (18)FDG positron emission
tomography with kinetic analysis as a bridging biomarker for
pulmonary vascular remodeling targeted treatments. Circulation.
128:1214–1224. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Caruso P, Dunmore BJ, Schlosser K, Schoors
S, Dos Santos C, Perez-Iratxeta C, Lavoie JR, Zhang H, Long L,
Flockton AR, et al: Identification of MicroRNA-124 as a major
regulator of enhanced endothelial cell glycolysis in pulmonary
arterial hypertension via PTBP1 (polypyrimidine tract binding
protein) and pyruvate kinase M2. Circulation. 136:2451–2467. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Akagi S, Nakamura K, Kondo M, Hirohata S,
Udono H, Nishida M, Saito Y, Yoshida M, Miyoshi T and Ito H:
Evidence for hypoxia-induced shift in ATP production from
glycolysis to mitochondrial respiration in pulmonary artery smooth
muscle cells in pulmonary arterial hypertension. J Clin Med.
12:50282023. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Fijalkowska I, Xu W, Comhair SAA, Janocha
AJ, Mavrakis LA, Krishnamachary B, Zhen L, Mao T, Richter A,
Erzurum SC and Tuder RM: Hypoxia inducible-factor1alpha regulates
the metabolic shift of pulmonary hypertensive endothelial cells. Am
J Pathol. 176:1130–1138. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Cao Y, Zhang X, Wang L, Yang Q, Ma Q, Xu
J, Wang J, Kovacs L, Ayon RJ, Liu Z, et al: PFKFB3-mediated
endothelial glycolysis promotes pulmonary hypertension. Proc Natl
Acad Sci USA. 116:13394–13403. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Boehme J, Sun X, Tormos KV, Gong W,
Kellner M, Datar SA, Kameny RJ, Yuan JXJ, Raff GW, Fineman JR, et
al: Pulmonary artery smooth muscle cell hyperproliferation and
metabolic shift triggered by pulmonary overcirculation. Am J
Physiol Heart Circ Physiol. 311:H944–H957. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Wujak M, Veith C, Wu CY, Wilke T, Kanbagli
ZI, Novoyatleva T, Guenther A, Seeger W, Grimminger F, Sommer N, et
al: Adenylate kinase 4-A Key regulator of proliferation and
metabolic shift in human pulmonary arterial smooth muscle cells via
Akt and HIF-1α signaling pathways. Int J Mol Sci. 22:103712021.
View Article : Google Scholar
|
|
31
|
Xu W and Erzurum SC: Endothelial cell
energy metabolism, proliferation, and apoptosis in pulmonary
hypertension. Compr Physiol. 1:357–372. 2011.PubMed/NCBI
|
|
32
|
Pullamsetti SS, Mamazhakypov A, Weissmann
N, Seeger W and Savai R: Hypoxia-inducible factor signaling in
pulmonary hypertension. J Clin Invest. 130:5638–5651. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Semenza GL: Hypoxia-inducible factors in
physiology and medicine. Cell. 148:399–408. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Paredes F, Williams HC and San Martin A:
Metabolic adaptation in hypoxia and cancer. Cancer Lett.
502:133–142. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Yu B, Wang X, Song Y, Xie G, Jiao S, Shi
L, Cao X, Han X and Qu A: The role of hypoxia-inducible factors in
cardiovascular diseases. Pharmacol Ther. 238:1081862022. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Mobasheri A, Richardson S, Mobasheri R,
Shakibaei M and Hoyland JA: Hypoxia inducible factor-1 and
facilitative glucose transporters GLUT1 and GLUT3: Putative
molecular components of the oxygen and glucose sensing apparatus in
articular chondrocytes. Histol Histopathol. 20:1327–1338.
2005.PubMed/NCBI
|
|
37
|
Mamun AA, Hayashi H, Yamamura A, Nayeem MJ
and Sato M: Hypoxia induces the translocation of glucose
transporter 1 to the plasma membrane in vascular endothelial cells.
J Physiol Sci. 70:442020. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Kim JW, Tchernyshyov I, Semenza GL and
Dang CV: HIF-1-mediated expression of pyruvate dehydrogenase
kinase: A metabolic switch required for cellular adaptation to
hypoxia. Cell Metab. 3:177–185. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Menendez MT, Teygong C, Wade K, Florimond
C and Blader IJ: siRNA screening identifies the host hexokinase 2
(HK2) gene as an important hypoxia-inducible transcription factor 1
(HIF-1) target gene in toxoplasma gondii-infected cells. mBio.
6:e004622015. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Cui XG, Han ZT, He SH, Wu XD, Chen TR,
Shao CH, Chen DL, Su N, Chen YM, Wang T, et al: HIF1/2α mediates
hypoxia-induced LDHA expression in human pancreatic cancer cells.
Oncotarget. 8:24840–24852. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Samec M, Liskova A, Koklesova L, Mersakova
S, Strnadel J, Kajo K, Pec M, Zhai K, Smejkal K, Mirzaei S, et al:
Flavonoids targeting HIF-1: Implications on cancer metabolism.
Cancers (Basel). 13:1302021. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Michelakis ED, Gurtu V, Webster L, Barnes
G, Watson G, Howard L, Cupitt J, Paterson I, Thompson RB, Chow K,
et al: Inhibition of pyruvate dehydrogenase kinase improves
pulmonary arterial hypertension in genetically susceptible
patients. Sci Transl Med. 9:eaao45832017. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Chen J, Zhang M, Liu Y, Zhao S, Wang Y,
Wang M, Niu W, Jin F and Li Z: Histone lactylation driven by
mROS-mediated glycolytic shift promotes hypoxic pulmonary
hypertension. J Mol Cell Biol. 14:mjac0732023. View Article : Google Scholar :
|
|
44
|
Cotroneo E, Ashek A, Wang L, Wharton J,
Dubois O, Bozorgi S, Busbridge M, Alavian KN, Wilkins MR and Zhao
L: Iron homeostasis and pulmonary hypertension: Iron deficiency
leads to pulmonary vascular remodeling in the rat. Circ Res.
116:1680–1690. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Wedgwood S, Lakshminrusimha S, Schumacker
PT and Steinhorn RH: Hypoxia inducible factor signaling and
experimental persistent pulmonary hypertension of the newborn.
Front Pharmacol. 6:472015. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Bonnet S, Michelakis ED, Porter CJ,
Andrade-Navarro MA, Thébaud B, Bonnet S, Haromy A, Harry G, Moudgil
R, McMurtry MS, et al: An abnormal mitochondrial-hypoxia inducible
factor-1alpha-Kv channel pathway disrupts oxygen sensing and
triggers pulmonary arterial hypertension in fawn hooded rats:
Similarities to human pulmonary arterial hypertension. Circulation.
113:2630–2641. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Archer SL, Gomberg-Maitland M, Maitland
ML, Rich S, Garcia JGN and Weir EK: Mitochondrial metabolism, redox
signaling, and fusion: A mitochondria-ROS-HIF-1alpha-Kv1.5
O2-sensing pathway at the intersection of pulmonary hypertension
and cancer. Am J Physiol Heart Circ Physiol. 294:H570–H578. 2008.
View Article : Google Scholar
|
|
48
|
Boucherat O, Vitry G, Trinh I, Paulin R,
Provencher S and Bonnet S: The cancer theory of pulmonary arterial
hypertension. Pulm Circ. 7:285–299. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Rhodes CJ, Howard LS, Busbridge M, Ashby
D, Kondili E, Gibbs JS, Wharton J and Wilkins MR: Iron deficiency
and raised hepcidin in idiopathic pulmonary arterial hypertension:
Clinical prevalence, outcomes, and mechanistic insights. J Am Coll
Cardiol. 58:300–309. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Lanspa SJ, Liu MW and Jenkins HJ Jr: Giant
bulla in pneumatosis cystoides intestinalis. J Clin Gastroenterol.
10:437–440. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Li M, Liu Y, Jin F, Sun X, Li Z, Liu Y,
Fang P, Shi H and Jiang X: Endothelin-1 induces hypoxia inducible
factor 1α expression in pulmonary artery smooth muscle cells. FEBS
Lett. 586:3888–3893. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Alqarni AA, Aldhahir AM, Alghamdi SA,
Alqahtani JS, Siraj RA, Alwafi H, AlGarni AA, Majrshi MS, Alshehri
SM and Pang L: Role of prostanoids, nitric oxide and endothelin
pathways in pulmonary hypertension due to COPD. Front Med
(Lausanne). 10:12756842023. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Jeffrey Man HS, Tsui AKY and Marsden PA:
Nitric oxide and hypoxia signaling. Vitam Horm. 96:161–192. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Dabral S, Muecke C, Valasarajan C,
Schmoranzer M, Wietelmann A, Semenza GL, Meister M, Muley T,
Seeger-Nukpezah T, Samakovlis C, et al: A RASSF1A-HIF1α loop drives
Warburg effect in cancer and pulmonary hypertension. Nat Commun.
10:21302019. View Article : Google Scholar
|
|
55
|
Siebel C and Lendahl U: Notch signaling in
development, tissue homeostasis, and disease. Physiol Rev.
97:1235–1294. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Gozlan O and Sprinzak D: Notch signaling
in development and homeostasis. Development. 150:dev2011382023.
View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Fernández-Chacón M, García-González I,
Mühleder S and Benedito R: Role of Notch in endothelial biology.
Angiogenesis. 24:237–250. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Dabral S, Tian X, Kojonazarov B, Savai R,
Ghofrani HA, Weissmann N, Florio M, Sun J, Jonigk D, Maegel L, et
al: Notch1 signalling regulates endothelial proliferation and
apoptosis in pulmonary arterial hypertension. Eur Respir J.
48:1137–1149. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Wang Y, Dai S, Cheng X, Prado E, Yan L, Hu
J, He Q, Lv Y, Lv Y and Du L: Notch3 signaling activation in smooth
muscle cells promotes extrauterine growth restriction-induced
pulmonary hypertension. Nutr Metab Cardiovasc Dis. 29:639–651.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Tien PC, Chen X, Elzey BD, Pollock RE and
Kuang S: Notch signaling regulates a metabolic switch through
inhibiting PGC-1α and mitochondrial biogenesis in dedifferentiated
liposarcoma. Oncogene. 42:2521–2535. 2023. View Article : Google Scholar :
|
|
61
|
Landor SKJ, Mutvei AP, Mamaeva V, Jin S,
Busk M, Borra R, Grönroos TJ, Kronqvist P, Lendahl U and Sahlgren
CM: Hypoand hyperactivated Notch signaling induce a glycolytic
switch through distinct mechanisms. Proc Natl Acad Sci USA.
108:18814–18819. 2011. View Article : Google Scholar
|
|
62
|
Sellers K, Allen TD, Bousamra M II, Tan J,
Méndez-Lucas A, Lin W, Bah N, Chernyavskaya Y, MacRae JI, Higashi
RM, et al: Metabolic reprogramming and Notch activity distinguish
between non-small cell lung cancer subtypes. Br J Cancer.
121:51–64. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Miyagawa K, Shi M, Chen PI, Hennigs JK,
Zhao Z, Wang M, Li CG, Saito T, Taylor S, Sa S, et al: Smooth
muscle contact drives endothelial regeneration by
BMPR2-Notch1-mediated metabolic and epigenetic changes. Circ Res.
124:211–224. 2019. View Article : Google Scholar :
|
|
64
|
Moriyama H, Moriyama M, Isshi H, Ishihara
S, Okura H, Ichinose A, Ozawa T, Matsuyama A and Hayakawa T: Role
of notch signaling in the maintenance of human mesenchymal stem
cells under hypoxic conditions. Stem Cells Dev. 23:2211–2224. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Moriyama H, Moriyama M, Ozawa T, Tsuruta
D, Iguchi T, Tamada S, Nakatani T, Nakagawa K and Hayakawa T: Notch
signaling enhances stemness by regulating metabolic pathways
through modifying p53, NF-κB, and HIF-1α. Stem Cells Dev.
27:935–947. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Liu GY and Sabatini DM: mTOR at the nexus
of nutrition, growth, ageing and disease. Nat Rev Mol Cell Biol.
21:183–203. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Saxton RA and Sabatini DM: mTOR signaling
in growth, metabolism, and disease. Cell. 169:361–371. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Sangüesa G, Roglans N, Baena M, Velázquez
AM, Laguna JC and Alegret M: mTOR is a key protein involved in the
metabolic effects of simple sugars. Int J Mol Sci. 20:11172019.
View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Wang AP, Li XH, Yang YM, Li WQ, Zhang W,
Hu CP, Zhang Z and Li YJ: A critical role of the mTOR/eIF2α pathway
in hypoxia-induced pulmonary hypertension. PLoS One.
10:e01308062015. View Article : Google Scholar
|
|
70
|
Krymskaya VP, Snow J, Cesarone G, Khavin
I, Goncharov DA, Lim PN, Veasey SC, Ihida-Stansbury K, Jones PL and
Goncharova EA: mTOR is required for pulmonary arterial vascular
smooth muscle cell proliferation under chronic hypoxia. FASEB J.
25:1922–1933. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Szwed A, Kim E and Jacinto E: Regulation
and metabolic functions of mTORC1 and mTORC2. Physiol Rev.
101:1371–1426. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Zhu Y, Shu D, Gong X, Lu M, Feng Q, Zeng
XB, Zhang H, Gao J, Guo YW, Liu L, et al: Platelet-derived TGF
(transforming growth factor)-β1 enhances the aerobic glycolysis of
pulmonary arterial smooth muscle cells by PKM2 (pyruvate kinase
muscle isoform 2) upregulation. Hypertension. 79:932–945. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Hudson CC, Liu M, Chiang GG, Otterness DM,
Loomis DC, Kaper F, Giaccia AJ and Abraham RT: Regulation of
hypoxia-inducible factor 1alpha expression and function by the
mammalian target of rapamycin. Mol Cell Biol. 22:7004–7014. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Lu H, Forbes RA and Verma A:
Hypoxia-inducible factor 1 activation by aerobic glycolysis
implicates the Warburg effect in carcinogenesis. J Biol Chem.
277:23111–23115. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Feng Y and Wu L: mTOR up-regulation of
PFKFB3 is essential for acute myeloid leukemia cell survival.
Biochem Biophys Res Commun. 483:897–903. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Wang C, Jiang J, Ji J, Cai Q, Chen X, Yu
Y, Zhu Z and Zhang J: PKM2 promotes cell migration and inhibits
autophagy by mediating PI3K/AKT activation and contributes to the
malignant development of gastric cancer. Sci Rep. 7:28862017.
View Article : Google Scholar : PubMed/NCBI
|
|
77
|
He L, Gomes AP, Wang X, Yoon SO, Lee G,
Nagiec MJ, Cho S, Chavez A, Islam T, Yu Y, et al: mTORC1 promotes
metabolic reprogramming by the suppression of GSK3-dependent Foxk1
phosphorylation. Mol Cell. 70:949–960.e4. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Dodd KM, Yang J, Shen MH, Sampson JR and
Tee AR: mTORC1 drives HIF-1α and VEGF-A signalling via multiple
mechanisms involving 4E-BP1, S6K1 and STAT3. Oncogene.
34:2239–2250. 2015. View Article : Google Scholar
|
|
79
|
Chi H: Sin1-mTORC2 signaling drives
glycolysis of developing thymocytes. J Mol Cell Biol. 11:91–92.
2019. View Article : Google Scholar
|
|
80
|
Lan N, Lu Y, Zhang Y, Pu S, Xi H, Nie X,
Liu J and Yuan W: FTO-a common genetic basis for obesity and
cancer. Front Genet. 11:5591382020. View Article : Google Scholar
|
|
81
|
Frayling TM, Timpson NJ, Weedon MN,
Zeggini E, Freathy RM, Lindgren CM, Perry JRB, Elliott KS, Lango H,
Rayner NW, et al: A common variant in the FTO gene is associated
with body mass index and predisposes to childhood and adult
obesity. Science. 316:889–894. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Jia G, Yang CG, Yang S, Jian X, Yi C, Zhou
Z and He C: Oxidative demethylation of 3-methylthymine and
3-methyluracil in single-stranded DNA and RNA by mouse and human
FTO. FEBS Lett. 582:3313–3319. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Jia G, Fu Y, Zhao X, Dai Q, Zheng G, Yang
Y, Yi C, Lindahl T, Pan T, Yang YG and He C: N6-methyladenosine in
nuclear RNA is a major substrate of the obesity-associated FTO. Nat
Chem Biol. 7:885–887. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Azzam SK, Alsafar H and Sajini AA: FTO m6A
demethylase in obesity and cancer: Implications and underlying
molecular mechanisms. Int J Mol Sci. 23:38002022. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Li W, Xing C, Bao L, Han S, Luo T, Wang Z
and Fan H: Comprehensive analysis of RNA m6A methylation in
pressure overload-induced cardiac hypertrophy. BMC Genomics.
23:5762022. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Zhang B, Jiang H, Wu J, Cai Y, Dong Z,
Zhao Y, Hu Q, Hu K, Sun A and Ge J: m6A demethylase FTO attenuates
cardiac dysfunction by regulating glucose uptake and glycolysis in
mice with pressure overload-induced heart failure. Signal Transduct
Target Ther. 6:3772021. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Hu L, Wang J, Huang H, Yu Y, Ding J, Yu Y,
Li K, Wei D, Ye Q, Wang F, et al: YTHDF1 regulates pulmonary
hypertension through translational control of MAGED1. Am J Respir
Crit Care Med. 203:1158–1172. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Zeng Y, Huang T, Zuo W, Wang D, Xie Y,
Wang X, Xiao Z, Chen Z, Liu Q, Liu N and Xiao Y: Integrated
analysis of m6A mRNA methylation in rats with
monocrotaline-induced pulmonary arterial hypertension. Aging
(Albany NY). 13:18238–18256. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Liu Y, Wang R, Zhang L, Li J, Lou K and
Shi B: The lipid metabolism gene FTO influences breast cancer cell
energy metabolism via the PI3K/AKT signaling pathway. Oncol Lett.
13:4685–4690. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Liu Y, Liang G, Xu H, Dong W, Dong Z, Qiu
Z, Zhang Z, Li F, Huang Y, Li Y, et al: Tumors exploit FTO-mediated
regulation of glycolytic metabolism to evade immune surveillance.
Cell Metab. 33:1221–1233.e11. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Fang YH, Piao L, Hong Z, Toth PT, Marsboom
G, Bache-Wiig P, Rehman J and Archer SL: Therapeutic inhibition of
fatty acid oxidation in right ventricular hypertrophy: Exploiting
Randle's cycle. J Mol Med (Berl). 90:31–43. 2012. View Article : Google Scholar
|
|
92
|
Randle PJ, Priestman DA, Mistry SC and
Halsall A: Glucose fatty acid interactions and the regulation of
glucose disposal. J Cell Biochem. 55(Suppl 1): S1–S11. 1994.
View Article : Google Scholar
|
|
93
|
Archer SL, Fang YH, Ryan JJ and Piao L:
Metabolism and bioenergetics in the right ventricle and pulmonary
vasculature in pulmonary hypertension. Pulm Circ. 3:144–152. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Lee MH, Sanders L, Kumar R,
Hernandez-Saavedra D, Yun X, Ford JA, Perez MJ, Mickael C, Gandjeva
A, Koyanagi DE, et al: Contribution of fatty acid oxidation to the
pathogenesis of pulmonary hypertension. Am J Physiol Lung Cell Mol
Physiol. 323:L355–L371. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Mey JT, Hari A, Axelrod CL, Fealy CE,
Erickson ML, Kirwan JP, Dweik RA and Heresi GA: Lipids and ketones
dominate metabolism at the expense of glucose control in pulmonary
arterial hypertension: A hyperglycaemic clamp and metabolomics
study. Eur Respir J. 55:19017002020. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Sutendra G, Bonnet S, Rochefort G, Haromy
A, Folmes KD, Lopaschuk GD, Dyck JRB and Michelakis ED: Fatty acid
oxidation and malonyl-CoA decarboxylase in the vascular remodeling
of pulmonary hypertension. Sci Transl Med. 2:44ra582010. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Greenberger LM, Horak ID, Filpula D, Sapra
P, Westergaard M, Frydenlund HF, Albaek C, Schrøder H and Ørum H: A
RNA antagonist of hypoxia-inducible factor-1alpha, EZN-2968,
inhibits tumor cell growth. Mol Cancer Ther. 7:3598–3608. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Zhao H, Jiang H, Li Z, Zhuang Y, Liu Y,
Zhou S, Xiao Y, Xie C, Zhou F and Zhou Y: 2-Methoxyestradiol
enhances radiosensitivity in radioresistant melanoma MDA-MB-435R
cells by regulating glycolysis via HIF-1α/PDK1 axis. Int J Oncol.
50:1531–1540. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Fallah J and Rini BI: HIF inhibitors:
Status of current clinical development. Curr Oncol Rep. 21:62019.
View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Bruce JY, Eickhoff J, Pili R, Logan T,
Carducci M, Arnott J, Treston A, Wilding G and Liu G: A phase II
study of 2-methoxyestradiol nanocrystal colloidal dispersion alone
and in combination with sunitinib malate in patients with
metastatic renal cell carcinoma progressing on sunitinib malate.
Invest New Drugs. 30:794–802. 2012. View Article : Google Scholar
|
|
101
|
Jabs M, Rose AJ, Lehmann LH, Taylor J,
Moll I, Sijmonsma TP, Herberich SE, Sauer SW, Poschet G, Federico
G, et al: Inhibition of endothelial notch signaling impairs fatty
acid transport and leads to metabolic and vascular remodeling of
the adult heart. Circulation. 137:2592–2608. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Chen T, Zhou Q, Tang H, Bozkanat M, Yuan
JXJ, Raj JU and Zhou G: miR-17/20 controls prolyl hydroxylase 2
(PHD2)/hypoxia-inducible factor 1 (HIF1) to regulate pulmonary
artery smooth muscle cell proliferation. J Am Heart Assoc.
5:e0045102016. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Wang S, Zeng H, Xie XJ, Tao YK, He X,
Roman RJ, Aschner JL and Chen JX: Loss of prolyl hydroxylase domain
protein 2 in vascular endothelium increases pericyte coverage and
promotes pulmonary arterial remodeling. Oncotarget. 7:58848–58861.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Han XJ, Zhang WF, Wang Q, Li M, Zhang CB,
Yang ZJ, Tan RJ, Gan LJ, Zhang LL, Lan XM, et al: HIF-1α promotes
the proliferation and migration of pulmonary arterial smooth muscle
cells via activation of Cx43. J Cell Mol Med. 25:10663–10673. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Dessouroux A, Akwa Y and Baulieu EE: DHEA
decreases HIF-1alpha accumulation under hypoxia in human pulmonary
artery cells: Potential role in the treatment of pulmonary arterial
hypertension. J Steroid Biochem Mol Biol. 109:81–89. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Ball MK, Waypa GB, Mungai PT, Nielsen JM,
Czech L, Dudley VJ, Beussink L, Dettman RW, Berkelhamer SK,
Steinhorn RH, et al: Regulation of hypoxia-induced pulmonary
hypertension by vascular smooth muscle hypoxia-inducible factor-1α.
Am J Respir Crit Care Med. 189:314–324. 2014. View Article : Google Scholar :
|
|
107
|
Docherty CK, Nilsen M and MacLean MR:
Influence of 2-methoxyestradiol and sex on hypoxia-induced
pulmonary hypertension and hypoxia-inducible factor-1-α. J Am Heart
Assoc. 8:e0116282019. View Article : Google Scholar
|
|
108
|
He Y, Fang X, Shi J, Li X, Xie M and Liu
X: Apigenin attenuates pulmonary hypertension by inducing
mitochondria-dependent apoptosis of PASMCs via inhibiting the
hypoxia inducible factor 1α-KV1.5 channel pathway. Chem Biol
Interact. 317:1089422020. View Article : Google Scholar
|
|
109
|
Jiang Y, Zhou Y, Peng G, Liu N, Tian H,
Pan D, Liu L, Yang X, Li C, Li W, et al: Topotecan prevents
hypoxia-induced pulmonary arterial hypertension and inhibits
hypoxia-inducible factor-1α and TRPC channels. Int J Biochem Cell
Biol. 104:161–170. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Koulmann N, Novel-Chaté V, Peinnequin A,
Chapot R, Serrurier B, Simler N, Richard H, Ventura-Clapier R and
Bigard X: Cyclosporin A inhibits hypoxia-induced pulmonary
hypertension and right ventricle hypertrophy. Am J Respir Crit Care
Med. 174:699–705. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Kurosawa R, Satoh K, Kikuchi N, Kikuchi H,
Saigusa D, Al-Mamun ME, Siddique MAH, Omura J, Satoh T, Sunamura S,
et al: Identification of celastramycin as a novel therapeutic agent
for pulmonary arterial hypertension. Circ Res. 125:309–327. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Abud EM, Maylor J, Undem C, Punjabi A,
Zaiman AL, Myers AC, Sylvester JT, Semenza GL and Shimoda LA:
Digoxin inhibits development of hypoxic pulmonary hypertension in
mice. Proc Natl Acad Sci USA. 109:1239–1244. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Kovacs L, Cao Y, Han W, Meadows L,
Kovacs-Kasa A, Kondrikov D, Verin AD, Barman SA, Dong Z, Huo Y and
Su Y: PFKFB3 in smooth muscle promotes vascular remodeling in
pulmonary arterial hypertension. Am J Respir Crit Care Med.
200:617–627. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Jiang L, Goncharov DA, Shen Y, Lin D,
Chang B, Pena A, DeLisser H, Goncharova EA and Kudryashova TV:
Akt-dependent glycolysis-driven lipogenesis supports proliferation
and survival of human pulmonary arterial smooth muscle cells in
pulmonary hypertension. Front Med (Lausanne). 9:8868682022.
View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Yan N: A glimpse of membrane transport
through structures-advances in the structural biology of the GLUT
glucose transporters. J Mol Biol. 429:2710–2725. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Thorens B and Mueckler M: Glucose
transporters in the 21st century. Am J Physiol Endocrinol Metab.
298:E141–E145. 2010. View Article : Google Scholar :
|
|
117
|
Ismail A and Tanasova M: Importance of
GLUT transporters in disease diagnosis and treatment. Int J Mol
Sci. 23:86982022. View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Broderick TL and King TM: Upregulation of
GLUT-4 in right ventricle of rats with monocrotaline-induced
pulmonary hypertension. Med Sci Monit. 14:BR261–BR264.
2008.PubMed/NCBI
|
|
119
|
Li W, Chen W, Peng H, Xiao Z, Liu J, Zeng
Y, Huang T, Song Q, Wang X and Xiao Y: Shikonin improves pulmonary
vascular remodeling in monocrotaline-induced pulmonary arterial
hypertension via regulation of PKM2. Mol Med Rep. 27:602023.
View Article : Google Scholar
|
|
120
|
Liu A, Li B, Yang M, Shi Y and Su J:
Targeted treprostinil delivery inhibits pulmonary arterial
remodeling. Eur J Pharmacol. 923:1747002022. View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Chowdhury B, Luu AZ, Luu VZ, Kabir MG, Pan
Y, Teoh H, Quan A, Sabongui S, Al-Omran M, Bhatt DL, et al: The
SGLT2 inhibitor empagliflozin reduces mortality and prevents
progression in experimental pulmonary hypertension. Biochem Biophys
Res Commun. 524:50–56. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Tang Y, Tan S, Li M, Tang Y, Xu X, Zhang
Q, Fu Q, Tang M, He J, Zhang Y, et al: Dapagliflozin, sildenafil
and their combination in monocrotaline-induced pulmonary arterial
hypertension. BMC Pulm Med. 22:1422022. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Kayano H, Koba S, Hirano T, Matsui T,
Fukuoka H, Tsuijita H, Tsukamoto S, Hayashi T, Toshida T, Watanabe
N, et al: Dapagliflozin influences ventricular hemodynamics and
exercise-induced pulmonary hypertension in type 2 diabetes
patients-a randomized controlled trial. Circ J. 84:1807–1817. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Zapater JL, Lednovich KR, Khan MW, Pusec
CM and Layden BT: Hexokinase domain-containing protein-1 in
metabolic diseases and beyond. Trends Endocrinol Metab. 33:72–84.
2022. View Article : Google Scholar
|
|
125
|
Wilson JE: Isozymes of mammalian
hexokinase: Structure, subcellular localization and metabolic
function. J Exp Biol. 206:2049–2057. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Chen F, Wang H, Lai J, Cai S and Yuan L:
3-Bromopyruvate reverses hypoxia-induced pulmonary arterial
hypertension through inhibiting glycolysis: In vitro and in vivo
studies. Int J Cardiol. 266:236–241. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Zhang YL, Zhang R, Shen YF, Huang KY, He
YY, Zhao JH and Jing ZC: 3-Bromopyruvate attenuates experimental
pulmonary hypertension via inhibition of glycolysis. Am J
Hypertens. 32:426–432. 2019. View Article : Google Scholar
|
|
128
|
Liu J, Wang W, Wang L, Qi XM, Sha YH and
Yang T: 3-Bromopyruvate alleviates the development of
monocrotaline-induced rat pulmonary arterial hypertension by
decreasing aerobic glycolysis, inducing apoptosis, and suppressing
inflammation. Chin Med J (Engl). 133:49–60. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
129
|
Gao S, Chen X, Jin H, Ren S, Liu Z, Fang X
and Zhang G: Overexpression of ErbB2 renders breast cancer cells
susceptible to 3-BrPA through the increased dissociation of
hexokinase II from mitochondrial outer membrane. Oncol Lett.
11:1567–1573. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Mathupala SP, Ko YH and Pedersen PL:
Hexokinase II: Cancer's double-edged sword acting as both
facilitator and gatekeeper of malignancy when bound to
mitochondria. Oncogene. 25:4777–4786. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Pajak B, Siwiak E, Sołtyka M, Priebe A,
Zieliński R, Fokt I, Ziemniak M, Jaśkiewicz A, Borowski R,
Domoradzki T and Priebe W: 2-Deoxy-d-glucose and its analogs: From
diagnostic to therapeutic agents. Int J Mol Sci. 21:2342019.
View Article : Google Scholar
|
|
132
|
Laussel C and Léon S: Cellular toxicity of
the metabolic inhibitor 2-deoxyglucose and associated resistance
mechanisms. Biochem Pharmacol. 182:1142132020. View Article : Google Scholar : PubMed/NCBI
|
|
133
|
Lu Y, Chen R, Ma JY, Wang LP, Qiu LL, Wang
CP, Yan JC and Liu PJ: Platelet derived growth factor-BB regulates
phenotype transformation of pulmonary artery smooth muscle cells
via SIRT3 affecting glycolytic pathway. Zhonghua Xin Xue Guan Bing
Za Zhi. 47:993–999. 2019.In Chinese. PubMed/NCBI
|
|
134
|
Maier A, Liao SL, Lescure T, Robson PM,
Hirata N, Sartori S, Narula N, Vergani V, Soultanidis G, Morgenthau
A, et al: Pulmonary artery 18F-fluorodeoxyglucose uptake
by PET/CMR as a marker of pulmonary hypertension in sarcoidosis.
JACC Cardiovasc Imaging. 15:108–120. 2022. View Article : Google Scholar
|
|
135
|
Frille A, Steinhoff KG, Hesse S, Grachtrup
S, Wald A, Wirtz H, Sabri O and Seyfarth HJ: Thoracic
[18F]fluorodeoxyglucose uptake measured by positron emission
tomography/computed tomography in pulmonary hypertension. Medicine
(Baltimore). 95:e39762016. View Article : Google Scholar : PubMed/NCBI
|
|
136
|
Van Schaftingen E, Jett MF, Hue L and Hers
HG: Control of liver 6-phosphofructokinase by fructose
2,6-bisphosphate and other effectors. Proc Natl Acad Sci USA.
78:3483–3486. 1981. View Article : Google Scholar : PubMed/NCBI
|
|
137
|
Rider MH, Bertrand L, Vertommen D, Michels
PA, Rousseau GG and Hue L:
6-Phosphofructo-2-kinase/fructose-2,6-bisphosphatase: Head-to-head
with a bifunctional enzyme that controls glycolysis. Biochem J.
381:561–579. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
138
|
Wang L, Zhang X, Cao Y, Ma Q, Mao X, Xu J,
Yang Q, Zhou Y, Lucas R, Fulton DJ, et al: Mice with a specific
deficiency of Pfkfb3 in myeloid cells are protected from
hypoxia-induced pulmonary hypertension. Br J Pharmacol.
178:1055–1072. 2021. View Article : Google Scholar
|
|
139
|
Zahra K, Dey T, Ashish, Mishra SP and
Pandey U: Pyruvate kinase M2 and cancer: The role of PKM2 in
promoting tumorigenesis. Front Oncol. 10:1592020. View Article : Google Scholar : PubMed/NCBI
|
|
140
|
Dasgupta A, Wu D, Tian L, Xiong PY,
Dunham-Snary KJ, Chen KH, Alizadeh E, Motamed M, Potus F, Hindmarch
CCT and Archer SL: Mitochondria in the pulmonary vasculature in
health and disease: Oxygen-sensing, metabolism, and dynamics. Compr
Physiol. 10:713–765. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
141
|
Shimauchi T, Boucherat O, Yokokawa T,
Grobs Y, Wu W, Orcholski M, Martineau S, Omura J, Tremblay E,
Shimauchi K, et al: PARP1-PKM2 axis mediates right ventricular
failure associated with pulmonary arterial hypertension. JACC Basic
Transl Sci. 7:384–403. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
142
|
Arora S, Joshi G, Chaturvedi A, Heuser M,
Patil S and Kumar R: A perspective on medicinal chemistry
approaches for targeting pyruvate kinase M2. J Med Chem.
65:1171–1205. 2022. View Article : Google Scholar
|
|
143
|
Chhipa AS and Patel S: Targeting pyruvate
kinase muscle isoform 2 (PKM2) in cancer: What do we know so far?
Life Sci. 280:1196942021. View Article : Google Scholar : PubMed/NCBI
|
|
144
|
Hua Q, Mi B, Xu F, Wen J, Zhao L, Liu J
and Huang G: Hypoxia-induced lncRNA-AC020978 promotes proliferation
and glycolytic metabolism of non-small cell lung cancer by
regulating PKM2/HIF-1α axis. Theranostics. 10:4762–4778. 2020.
View Article : Google Scholar :
|
|
145
|
Chen D, Wei L, Liu ZR, Yang JJ, Gu X, Wei
ZZ, Liu LP and Yu SP: Pyruvate kinase M2 increases angiogenesis,
neurogenesis, and functional recovery mediated by upregulation of
STAT3 and focal adhesion kinase activities after ischemic stroke in
adult mice. Neurotherapeutics. 15:770–784. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
146
|
Pei L, Le Y, Chen H, Feng J, Liu Z, Zhu J,
Wang C, Chen L, Dou X and Lu D: Cynaroside prevents macrophage
polarization into pro-inflammatory phenotype and alleviates cecal
ligation and puncture-induced liver injury by targeting PKM2/HIF-1α
axis. Fitoterapia. 152:1049222021. View Article : Google Scholar
|
|
147
|
Xiong PY, Motamed M, Chen KH, Dasgupta A,
Potus F, Tian L, Martin A, Mewburn J, Jones O, Thébaud A and Archer
SL: Inhibiting pyruvate kinase muscle isoform 2 regresses group 2
pulmonary hypertension induced by supra-coronary aortic banding.
Acta Physiol (Oxf). 234:e137642022. View Article : Google Scholar : PubMed/NCBI
|
|
148
|
Piao L, Sidhu VK, Fang YH, Ryan JJ, Parikh
KS, Hong Z, Toth PT, Morrow E, Kutty S, Lopaschuk GD and Archer SL:
FOXO1-mediated upregulation of pyruvate dehydrogenase kinase-4
(PDK4) decreases glucose oxidation and impairs right ventricular
function in pulmonary hypertension: Therapeutic benefits of
dichloroacetate. J Mol Med (Berl). 91:333–346. 2013. View Article : Google Scholar
|
|
149
|
Michelakis ED, McMurtry MS, Wu XC, Dyck
JRB, Moudgil R, Hopkins TA, Lopaschuk GD, Puttagunta L, Waite R and
Archer SL: Dichloroacetate, a metabolic modulator, prevents and
reverses chronic hypoxic pulmonary hypertension in rats: Role of
increased expression and activity of voltage-gated potassium
channels. Circulation. 105:244–250. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
150
|
McMurtry MS, Bonnet S, Wu X, Dyck JRB,
Haromy A, Hashimoto K and Michelakis ED: Dichloroacetate prevents
and reverses pulmonary hypertension by inducing pulmonary artery
smooth muscle cell apoptosis. Circ Res. 95:830–840. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
151
|
Guignabert C, Tu L, Izikki M, Dewachter L,
Zadigue P, Humbert M, Adnot S, Fadel E and Eddahibi S:
Dichloroacetate treatment partially regresses established pulmonary
hypertension in mice with SM22alpha-targeted overexpression of the
serotonin transporter. FASEB J. 23:4135–4147. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
152
|
Ciapponi A, Pizarro R and Harrison J:
WITHDRAWN: Trimetazidine for stable angina. Cochrane Database Syst
Rev. 3:CD0036142017.PubMed/NCBI
|
|
153
|
Wu Z, Yu L and Li X and Li X: Protective
mechanism of trimetazidine in myocardial cells in myocardial
infarction rats through ERK signaling pathway. Biomed Res Int.
2021:99245492021. View Article : Google Scholar : PubMed/NCBI
|
|
154
|
Ferrari R, Ford I, Fox K, Challeton JP,
Correges A, Tendera M, Widimský P and Danchin N; ATPCI
investigators: Efficacy and safety of trimetazidine after
percutaneous coronary intervention (ATPCI): A randomised,
double-blind, placebo-controlled trial. Lancet. 396:830–838. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
155
|
Shu H, Peng Y, Hang W, Zhou N and Wang DW:
Trimetazidine in heart failure. Front Pharmacol. 11:5691322021.
View Article : Google Scholar : PubMed/NCBI
|
|
156
|
Parra V, Bravo-Sagua R, Norambuena-Soto I,
Hernández-Fuentes CP, Gómez-Contreras AG, Verdejo HE, Mellado R,
Chiong M, Lavandero S and Castro PF: Inhibition of mitochondrial
fission prevents hypoxia-induced metabolic shift and cellular
proliferation of pulmonary arterial smooth muscle cells. Biochim
Biophys Acta Mol Basis Dis. 1863:2891–2903. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
157
|
Dobbins RL, Szczepaniak LS, Bentley B,
Esser V, Myhill J and McGarry JD: Prolonged inhibition of muscle
carnitine palmitoyltransferase-1 promotes intramyocellular lipid
accumulation and insulin resistance in rats. Diabetes. 50:123–130.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
158
|
Leamy AK, Egnatchik RA and Young JD:
Molecular mechanisms and the role of saturated fatty acids in the
progression of non-alcoholic fatty liver disease. Prog Lipid Res.
52:165–174. 2013. View Article : Google Scholar
|
|
159
|
Ma Y, Temkin SM, Hawkridge AM, Guo C, Wang
W, Wang XY and Fang X: Fatty acid oxidation: An emerging facet of
metabolic transformation in cancer. Cancer Lett. 435:92–100. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
160
|
Verdejo HE, Rojas A, López-Crisosto C,
Baraona F, Gabrielli L, Maracaja-Coutinho V, Chiong M, Lavandero S
and Castro PF: Effects of trimetazidine on right ventricular
function and ventricular remodeling in patients with pulmonary
artery hypertension: A randomised controlled trial. J Clin Med.
12:15712023. View Article : Google Scholar : PubMed/NCBI
|
|
161
|
Cavallino C, Facchini M, Veia A, Bacchni
S, Rosso R, Rognoni A, Rametta F, Lupi A and Bongo AS: New
anti-anginal drugs: Ranolazine. Cardiovasc Hematol Agents Med Chem.
13:14–20. 2015. View Article : Google Scholar
|
|
162
|
McKelvey KJ, Wilson EB, Short S, Melcher
AA, Biggs M, Diakos CI and Howell VM: Glycolysis and fatty acid
oxidation inhibition improves survival in glioblastoma. Front
Oncol. 11:6332102021. View Article : Google Scholar : PubMed/NCBI
|
|
163
|
McCormack JG, Barr RL, Wolff AA and
Lopaschuk GD: Ranolazine stimulates glucose oxidation in normoxic,
ischemic, and reperfused ischemic rat hearts. Circulation.
93:135–142. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
164
|
Clarke B, Wyatt KM and McCormack JG:
Ranolazine increases active pyruvate dehydrogenase in perfused
normoxic rat hearts: Evidence for an indirect mechanism. J Mol Cell
Cardiol. 28:341–350. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
165
|
Han QJ, Forfia P, Vaidya A, Ramani G,
deKemp RA, Mach RH, Mankoff DA, Bravo PE, DiCarli M, Chan SY, et
al: Effects of ranolazine on right ventricular function, fluid
dynamics, and metabolism in patients with precapillary pulmonary
hypertension: Insights from a longitudinal, randomized,
double-blinded, placebo controlled, multicenter study. Front
Cardiovasc Med. 10:11187962023. View Article : Google Scholar : PubMed/NCBI
|
|
166
|
Wu J, Liu T, Shi S, Fan Z, Hiram R, Xiong
F, Cui B, Su X, Chang R, Zhang W, et al: Dapagliflozin reduces the
vulnerability of rats with pulmonary arterial hypertension-induced
right heart failure to ventricular arrhythmia by restoring calcium
handling. Cardiovasc Diabetol. 21:1972022. View Article : Google Scholar : PubMed/NCBI
|
|
167
|
Luo L, Xiao L, Lian G, Wang H and Xie L:
miR-125a-5p inhibits glycolysis by targeting hexokinase-II to
improve pulmonary arterial hypertension. Aging (Albany NY).
12:9014–9030. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
168
|
Kassa B, Kumar R, Mickael C, Sanders L,
Vohwinkel C, Lee MH, Gu S, Poth JM, Stenmark KR, Zhao YY, et al:
Endothelial cell PHD2-HIF1α-PFKFB3 contributes to right ventricle
vascular adaptation in pulmonary hypertension. Am J Physiol Lung
Cell Mol Physiol. 321:L675–L685. 2021. View Article : Google Scholar
|
|
169
|
Qi L, Lv T, Cheng Y, Yu M, Han H, Kong H,
Xie W, Wang H, Zhang Y and Huang Z: Fasudil dichloroacetate (FDCA),
an orally available agent with potent therapeutic efficiency on
monocrotaline-induced pulmonary arterial hypertension rats. Bioorg
Med Chem Lett. 29:1812–1818. 2019. View Article : Google Scholar : PubMed/NCBI
|