Introduction
Neonatal hypoxic-ischemic brain damage is caused by
perinatal asphyxia. The incidence of hypoxic-ischemic brain damage
in developed countries is estimated to be 1-8% for new births every
year (1,2). Currently, mild therapeutic
hypothermia is used to cure perinatal asphyxia and to reduce the
damage caused by hypoxic brain damage (3). Children with severe perinatal
asphyxia may experience notable complications, such as severe
hypoxic ischemic organ damage, which can have long-lasting effects
on their future health and wellbeing (4).
Neuronal cell death is the main pathophysiological
alteration that occurs after hypoxic brain damage and contributes
to long-term neurological disorders. Previous studies have shown
that hypoxic brain damage induces a serious form of cell death that
occurs concurrently or sequentially (5,6).
Cerebral hypoxia has been reported to rapidly elevate the levels of
reactive oxygen species (ROS), leading to the direct modification
of cellular macromolecules, such as cell membranes, lipids and DNA.
Consequently, oxidative stress and inflammatory responses are
induced (7). Ferroptosis, a form
of cell death driven by peroxidation, is associated with hypoxic
brain damage. Lin et al (8) observed elevated iron levels in the
brain tissues of neonatal patients with hypoxic brain damage.
Additionally, the modulation of iron metabolism through the use of
desferrioxamine and erythropoietin may potentially improve the
prognosis of hypoxic brain damage (9).
Numerous critical brain processes rely on copper, an
essential trace element, as evidenced by various studies (10-12). Studies on patients with ischemic
stroke have shown an increase in plasma copper concentrations
(13,14), indicating copper dyshomeostasis
following hypoxic-ischemic brain damage. Previous studies have
demonstrated the protective effects of supplemental copper against
ischemic brain injury (15,16); however, the precise mechanism by
which copper supplementation affects hypoxia-induced brain damage
remains unclear. Copper serves as the catalytic center for
antioxidant enzymes. Previous studies have shown that copper
secretion can protect cells from ferroptosis (17,18), suggesting that supplementation
with copper could have beneficial effects on ferroptosis-induced
cell death. The aim of the present study was to evaluate the
effects of copper supplementation on hypoxia-induced neuronal
damage and ferroptosis. The current study may offer valuable
insights into a therapeutic approach for hypoxia-induced cellular
damage by utilizing copper supplementation.
Materials and methods
Materials
Copper dichloride (CuCl2) was purchased
from MilliporeSigma. Anti-copper transporting α polypeptide (ATP7A)
(cat. no. PA5-103110), anti-copper transporting β polypeptide
(ATP7B) (cat. no. PA1-16583), anti-copper transporter 1 (CTR1)
(cat. no. PA1-16586) and BODIPY™ 581/591 C11 (cat. no. D3861) were
purchased from Invitrogen (Thermo Fisher Scientific, Inc.).
Anti-copper chaperone for superoxide dismutase (CCS) (cat. no.
ab167170), anti-superoxide dismutase (SOD)1 (cat. no. ab51254),
anti-β actin (cat. no. ab6276), Goat Anti-Rabbit IgG H&L (HRP)
(cat. no. ab205718) and Goat Anti-Mouse IgG H&L (HRP) (cat. no.
ab205719) were purchased from Abcam. Anti-XIAP (cat. no. 2042S) and
anti-glutathione peroxidase 4 (GPX4) (cat. no. 52455S) were
purchased from Cell Signaling Technology, Inc. The Cell Counting
Kit (CCK)-8 assay kit was purchased from APeXBIO Technology LLC.
Ferrostatin-1 (Fer-1; cat. no. HY-100579) was purchased from
MedChemExpress and FerroOrange (cat. no. F374) was purchased from
Dojindo Laboratories, Inc. ROS (cat. no. S0033S), malondialdehyde
(MDA; cat. no. S0131S), hydrogen peroxide
(H2O2; cat. no. S0038) and Cu/Zn-SOD and
Mn-SOD Assay Kit with WST-8 (cat. no. S0103) kits were purchased
from Beyotime Institute of Biotechnology.
Cell culture and exposure to hypoxia
HT22 mouse hippocampal neuronal cells were procured
from the American Type Culture Collection. Following resuscitation,
the cells were cultured in high-glucose DMEM (Gibco; Thermo Fisher
Scientific, Inc.) supplemented with 10% fetal bovine serum (Gibco;
Thermo Fisher Scientific, Inc.), 1% streptomycin and 1% penicillin
at 37°C with 5% CO2.
A microaerophilic incubation system (DWS-H85; Don
Whitley Scientific Limited) filled with 1% O2 and 5%
CO2 was used to generate an in vitro model of
hypoxic exposure for 48 h, as described previously (19). The culture medium was pre-treated
in the microaerophilic incubation system for 9 h before use.
For Fer-1 and copper treatment, HT22 cells were
cultured with media containing 1 μM Fer-1 and 5 μM
CuCl2 for 48 h at 37°C.
CCK-8 analysis
The CCK-8 assay kit was used to determine the
effects of a hypoxic challenge on cell viability. This kit uses a
water-soluble tetrazolium salt to quantify the number of live cells
by producing an orange formazan dye upon bioreduction in the
presence of an electron carrier. HT22 cells (8×103
cells/well) were incubated for 24 h in a 96-well plate. Following
exposure to hypoxic conditions (12, 24, 36, 48 and 72 h), 10
μl CCK-8 reagent was added and the cells were then incubated
for 2 h at 37°C. Finally, the optical density (OD) was measured at
450 nm. Cell viability was calculated as follows: (OD value of
experimental groups-OD value of blank groups)/(OD value of control
groups-OD value of blank groups).
Transmission electron microscopy
(TEM)
After 48 h of exposure to hypoxia, HT22 cells were
collected, prefixed in 2.5% glutaraldehyde phosphate (0.1 M, pH
7.4) overnight at 4°C, and postfixed in 2% buffered osmium
tetroxide at 4°C for 15 min. The fixed cells were dehydrated with
70, 80, 90 and 100% ethanol (each for 15 min). Subsequently, the
cells were embedded in Epon812 (Merck KGaA) at room temperature for
30 min, and ultrathin sections (60 nm) were cut and stained with
uranyl acetate and lead citrate at room temperature for 30 min.
Images were captured using TEM (FEI; Thermo Fisher Scientific,
Inc.).
Western blot analysis
A lysis buffer (RIPA Lysis Buffer; cat. no. P0013C;
Beyotime Institute of Biotechnology) containing protease inhibitors
was used to extract proteins from HT22 cells and western blotting
was performed to detect protein expression. After protein
extraction, the BCA detection kit (cat. no. 23225; Thermo Fisher
Scientific, Inc.) was used to quantify the protein concentration.
Protein samples (30 μg) were then subjected to sodium
dodecyl sulfate-polyacrylamide gel electrophoresis on 10% gels and
were transferred onto polyvinylidene difluoride membranes, which
were incubated in 5% bovine serum albumin (cat. no. ST023; Beyotime
Institute of Biotechnology) at room temperature for 1 h.
Subsequently, the membranes were incubated with primary antibodies
at 4°C overnight (anti-ATP7A, 1:1,000; anti-ATP7B, 1:1,000;
anti-CTR1,1:500; anti-CCS, 1:1,000; anti-SOD1, 1:1,000; anti-XIAP,
1:1,000; anti-GPX4, 1:1,000; anti-β-actin, 1:1,000). The membranes
were then washed three times with Tris-buffered saline-0.1% Tween
20, and were incubated with the corresponding horseradish
peroxidase-conjugated antibodies (Anti-Rabbit IgG antibody, 1:500;
Anti-Mouse IgG antibody, 1:500) for 2 h at room temperature. A
chemiluminescence system (Bio-Rad Laboratories, Inc.) was used to
visualize the protein bands. After being normalized to β-actin,
ImageJ (version 1.51; National Institutes of Health) was adopted to
evaluate the protein expression; protein expression levels were
normalized to those in control cells.
ROS levels
Intracellular ROS levels were measured using a ROS
assay kit. HT22 cells (2×107) were collected after 48 h
of exposure to hypoxia, were resuspended in 2 ml DMEM and were
incubated with 10 μM DCFH-DA for 20 min at 37°C. Finally,
the cells were measured at 488 nm excitation and 525 nm emission
using a fluorescence spectrophotometer (BioTek; Agilent
Technologies, Inc.). The relative ROS content was normalized to the
number of cells.
H2O2 levels
HT22 cells were collected after being exposed to
hypoxia (1% O2) for 48 h. H2O2
levels were detected using an assay kit. Briefly, a standard curve
was constructed using standard H2O2 solutions
and the corresponding OD values. HT22 cells were prepared using
RIPA lysis buffer (cat. no. P0013C; Beyotime Institute of
Biotechnology) and the corresponding OD values were detected at 520
nm. Accurate H2O2 levels were calculated
using the standard curve.
MDA, SOD and SOD1 assays
A MDA assay kit was used to quantify the generation
of MDA, whereas a Cu/Zn-SOD and Mn-SOD assay kit (cat. no. S0103;
Beyotime Institute of Biotechnology) was used to measure the levels
of SOD and SOD1 enzyme activity. Briefly, HT22 cells were
trypsinized according to the manufacturer's instructions. After
sonication (20 kHz) was carried out in five 1-min cycles on ice
(30-sec sonication and 30-sec rest), the lysed cells were
centrifuged at 16,114 × g for 15 min at 4°C to remove the debris.
The total protein, and MDA, SOD and SOD1 levels in the supernatant
were measured and normalized to mg protein, according to the
manufacturer's protocols.
Copper concentration
The intracellular copper concentration in HT22 cells
was measured using graphite furnace atomic absorption spectroscopy
(GF-ASS). Briefly, HT22 cells were exposed to hypoxia for 24 and 48
h, after which, the medium was refreshed with complete medium
containing 5 μM CuCl2 for 5 h at 37°C in an
incubator. Subsequently, the cells were cultured and centrifuged at
6,500 × g for 10 min to obtain cell clumps, which were then dried
and weighed, followed by incubation with 100 μl nitric acid
at 100°C for 1 h for nitrolysis, and then the cell digestion
solution (40 μl) was mixed with 360 μl diluent (1%
nitric acid + 0.1% Triton + 98.9% ddH2O), and
subsequently, 20 μl of the resulting mixture was introduced
into the GF-ASS (PinAAcle900T; PerkinElmer, Inc.) for the detection
of copper. Finally, the copper concentration in HT22 cells was
normalized to the sample weight.
Intracellular ferrous ions and lipid
reactive oxygen species (LOS)
The fluorescence levels of total intracellular
ferrous ions and LOS in HT22 cells were detected using FerroOrange
and BODIPY 581/591 C11 respectively. After treatment with 5
μM CuCl2 at 37°C for 48 h, cells in confocal
dishes were resuspended in 2 ml fresh Hank's balanced salt solution
(cat. no. PB180323; Procell Life Science & Technology Co.,
Ltd.). The cells were then incubated with 1 μM C11-BODIPY or
FerroOrange for 20 min at 37°C. After two washes, intracellular
FerroOrange and C11-BODIPY (581/591) fluorescence imaging was
performed using a fluorescence microscope. The maximum absorbance
of excitation and emission wavelengths of FerroOrange is 543/580
nm, and that of C11-BODIPY (581/591) is 488/510 nm. The images were
analyzed using ImageJ.
Small interfering RNA (siRNA)
transfection
For siRNA-induced knockdown, CCS siRNA and a normal
control (NC) siRNA were designed and synthesized by Shanghai
GenePharma Co., Ltd. The sequences were as follows: CCS siRNA,
forward (F) 5′-GGUAUGGGCAGUAGCCAAUTT-3′, reverse (R)
5′-AUUGGCUACUGCCCAUACCTT-3′; NC siRNA, 5′-FUUCUCCGAACGUGUCACGUTT-3′
and R 5′-ACGUGACACGUUCGGAGAATT-3′. Cells (1×107) were
transiently transfected with 400 nM siRNA using 1%
Lipofectamine® 2000 (cat. no. 11668030; Invitrogen;
Thermo Fisher Scientific, Inc.) in Opti-MEM (cat. no. 31985070;
Invitrogen; Thermo Fisher Scientific, Inc.) according to the
manufacturer's instructions. After 6 h of incubation at 37°C, the
medium was replaced and other experiments were conducted 8 h later.
Transfection efficiency was measured using western blot
analysis.
RNA extraction and reverse
transcription-quantitative (q)PCR
According to the manufacturer's instructions, total
RNA was isolated from cells using TRIzol® reagent (cat
no. 15596018; Invitrogen; Thermo Fisher Scientific, Inc.) and was
then reverse transcribed into cDNA using PrimeScript Master Mix
(cat. no. RR036A; Takara Biotechnology Co., Ltd.) according to the
manufacturer's protocol (37°C for 15 min and 85°C for 5 sec). qPCR
was performed using TB Green Premix Ex Taq II (cat. no. RR820A;
Takara Biotechnology Co., Ltd.) on an Applied Biosystems
QuantStudio 5 instrument (Applied Biosystems; Thermo Fisher
Scientific, Inc.), and the qPCR thermocycling conditions were as
follows: 95°C for 30 sec, followed by 40 cycles at 95°C for 30 sec
and 60°C for 34 sec. Quantification of qPCR data was performed
using the 2−ΔΔCq method (20). All data were normalized to the
expression levels of β-actin. The specific primers used were as
follows: GPX4, F 5′-GATGGAGCCCATTCCTGAACC-3′, R
5′-CCCTGTACTTATCCAGGCAGA-3′; β-actin, F 5′-GGCTGTATTCCCCTCCATCG-3′
and R 5′-CCAGTTGGTAACAATGCCATGT-3′.
Cell apoptosis assay
Apoptosis was detected by flow cytometry using an
Annexin V-FITC kit (cat. no. C1062S; Beyotime Institute of
Biotechnology). According to the manufacturer's instructions, after
48 h of exposure to hypoxia, HT22 cells were harvested via
centrifugation (1,000 × g, 5 min) at room temperature and
resuspended in Annexin V-FITC binding solution, Subsequently, the
samples were mixed with Annexin V-FITC staining solution, followed
by PI staining solution. The mixture was then incubated at room
temperature for 20 min and analyzed using a CytoFLEX flow cytometer
(Beckman Coulter, Inc.). CytExpert 2.4 software (Beckman Coulter,
Inc.) was used for data analysis.
Statistical analysis
All experiments were conducted in triplicate and all
data were analyzed using GraphPad Prism 9 (Dotmatics). Data are
presented as the mean ± SEM. One-way ANOVA was performed to compare
more than two groups followed by Tukey's post hoc test, whereas
unpaired Student's t-test was used to compare the differences
between two groups. P<0.05 was consider to indicate a
statistically significant difference.
Results
Hypoxia induces ferroptosis in
neurons
The CCK-8 assay was performed on HT22 cells after
exposure to hypoxia for 12, 24, 36, 48 and 72 h to assess the
impact of hypoxia on neurons. The results indicated that the
viability of HT22 cells was notably slower following hypoxic
exposure compared with that of the control group, suggesting
potential damage to the cells caused by hypoxia (Fig. 1A). Additionally, TEM analysis
revealed distinctive morphological changes in HT22 cells exposed to
hypoxia for 48 h, including smaller mitochondria and increased
membrane density (Fig. 1B).
FerroOrange staining showed that cells subjected to hypoxia
exhibited notable accumulation of ferrous ions. This effect was
attenuated by administering Fer-1, as evidenced by a reduction in
ferrous ion staining (Fig. 1C and
D). Increased levels of ferrous ions induce oxidative stress,
particularly LOS, thereby indicating that ferroptosis may serve a
potential role in the pathogenesis of hypoxia. As expected,
increased LOS levels were detected in HT22 cells exposed to hypoxia
using the fluorescent probe C11-BODIPY, whereas a substantial
decrease in LOS accumulation was observed when the cells were
exposed to hypoxia with Fer-1 treatment (Fig. 1E and F). The expression levels of
GPX4 were detected as a marker of ferroptosis to provide additional
evidence of hypoxia-induced ferroptosis in HT22 cells. The results
revealed a significant decrease in the protein expression levels of
GPX4 in HT22 cells subjected to hypoxia compared with those in the
control group (Fig. 1G and H).
In addition, the alteration in the mRNA expression levels of GPX4
was consistent with that in the protein levels (Fig. 1I). Additionally, the present
study measured apoptotic cells using an Annexin V-FITC/PI apoptosis
detection kit; the results indicated a slight but insignificant
increase in apoptosis following 48 h of exposure to hypoxia
(Fig. S1). These findings
suggested that hypoxia may induce ferroptosis and potentially
intensify oxidative stress.
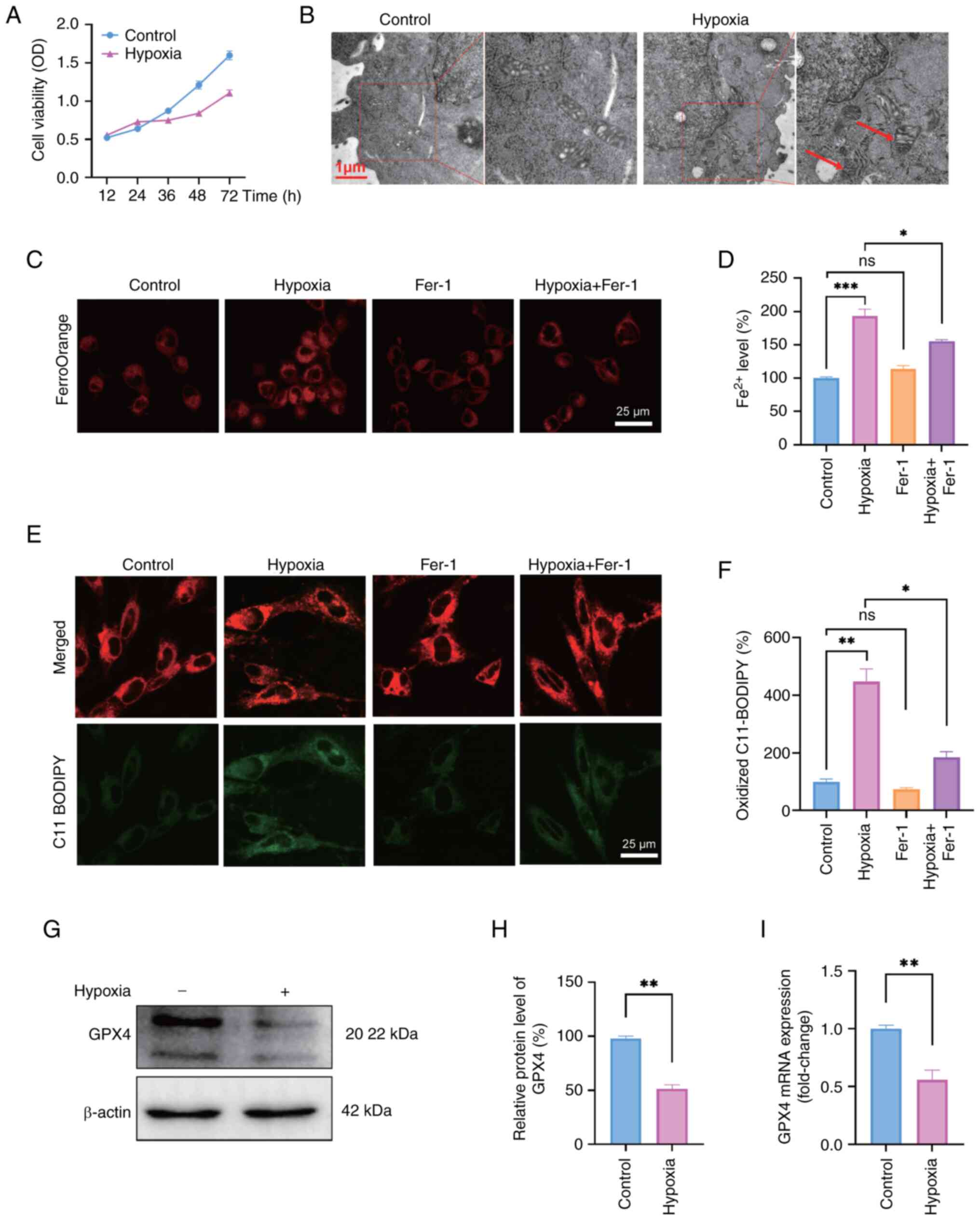 | Figure 1Hypoxia induces ferroptosis in
neurons. (A) HT22 cell viability at different time points was
detected by Cell Counting Kit-8 assay. (B) Images of the
ultrastructure of mitochondria in HT22 cells exposed to normoxia
and hypoxia (1% O2) for 48 h were captured under a
transmission electron microscope. Red arrows indicate shrunken
mitochondria. Scale bars, 1 μm. (C) Ferrous ion levels
detected by FerroOrange staining and (D) corresponding
semi-quantification by ImageJ. Scale bars, 25 μm. (E) Lipid
reactive oxygen species staining with the fluorescent probe
C11-BODIPY and (F) corresponding semi-quantification. Scale bars,
25 μm. (G) Western blot analysis and (H) semi-quantification
of the protein expression levels of the ferroptosis biomarker GPX4
following exposure of cells to hypoxia for 48 h. (I) mRNA
expression levels of GPX4 after exposure to hypoxia for 48 h.
One-way ANOVA was used for multiple-group comparisons, and unpaired
Student's t-test for two-group comparisons. Data are presented as
the mean ± SEM (n=3). *P<0.05,
**P<0.01, ***P<0.001. Fer-1,
ferrostatin-1; GPX4, glutathione peroxidase 4; ns, not significant;
OD, optical density. |
Neurons are subjected to oxidative stress
under hypoxia
The intracellular ROS levels were measured after
exposure to hypoxia for 48 h to investigate the effects of hypoxia
on oxidative stress in HT22 cells. These results indicated a
significant increase in intracellular ROS levels (Fig. 2A). H2O2
levels were also significantly increased in HT22 cells following
exposure to hypoxia for 48 h (Fig.
2B). Furthermore, MDA, a final product of polyunsaturated fatty
acid peroxidation and a marker of oxidative stress, was measured in
HT22 cells and was significantly increased in response to hypoxia
compared with that in the control group (Fig. 2C). SOD serves an essential role
in the first line of antioxidant defense. The present study
examined the activities of SOD and SOD1 enzymes in HT22 cells, and
revealed that their activities were markedly reduced in the hypoxia
group compared with those in the control group (Fig. 2D and E). These results suggested
that hypoxia may weaken the overall antioxidant capacity of
neurons.
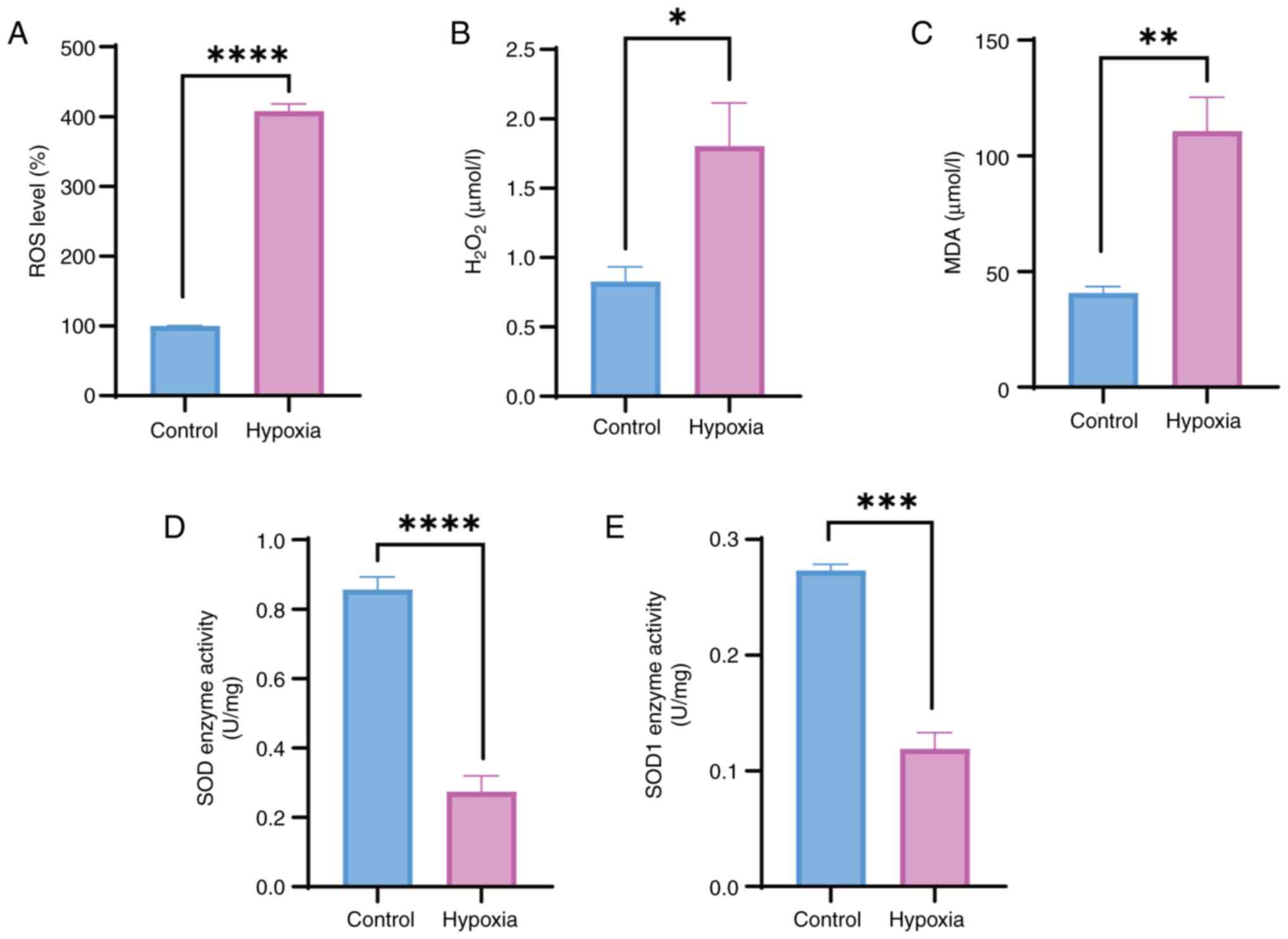 | Figure 2Hypoxia leads to oxidative stress in
neurons. Detection of (A) ROS levels, (B)
H2O2, (C) MDA, and (D) SOD and (E) SOD1
enzyme activity in HT22 cells exposed to normoxia or hypoxia (1%
O2) for 48 h. Unpaired Student's t-test was used for
analysis. Data are presented as the mean ± SEM (n=3).
*P<0.05, **P<0.01,
***P<0.001, ****P<0.0001.
H2O2, hydrogen peroxide; MDA,
malondialdehyde; ns, not significant; ROS, reactive oxygen species;
SOD, superoxide dismutase. |
HT22 cells exposed to hypoxic conditions
exhibit abnormal copper metabolism
The copper concentration in HT22 cells exposed to
hypoxia for 24 and 48 h was quantified to investigate the impact of
hypoxia on the copper metabolism of HT22 cells. The results
indicated a significant decrease in the copper concentration after
both 24 and 48 h of exposure to hypoxia (Fig. 3A). The expression levels of four
copper transport proteins, namely, CTR1, ATP7A, ATP7B and CCS, were
assessed to further elucidate the mechanisms underlying this
alteration in intracellular copper concentration. Western blot
analysis indicated that the expression levels of ATP7B, CTR1 and
CCS were elevated after 24 h of hypoxic exposure compared with
those in the control group, whereas the expression of ATP7A showed
no significant difference (Fig.
3B-F). However, as the duration of hypoxic exposure increased,
the expression levels of ATP7B decreased, and the expression of
CTR1 remained relatively stable after 48 h of hypoxia compared to
that in the control group (Fig.
3B-F). These results suggested distinct copper levels and
copper transporter alterations following exposure to hypoxia,
potentially implicating alterations in hypoxia-induced cell
damage.
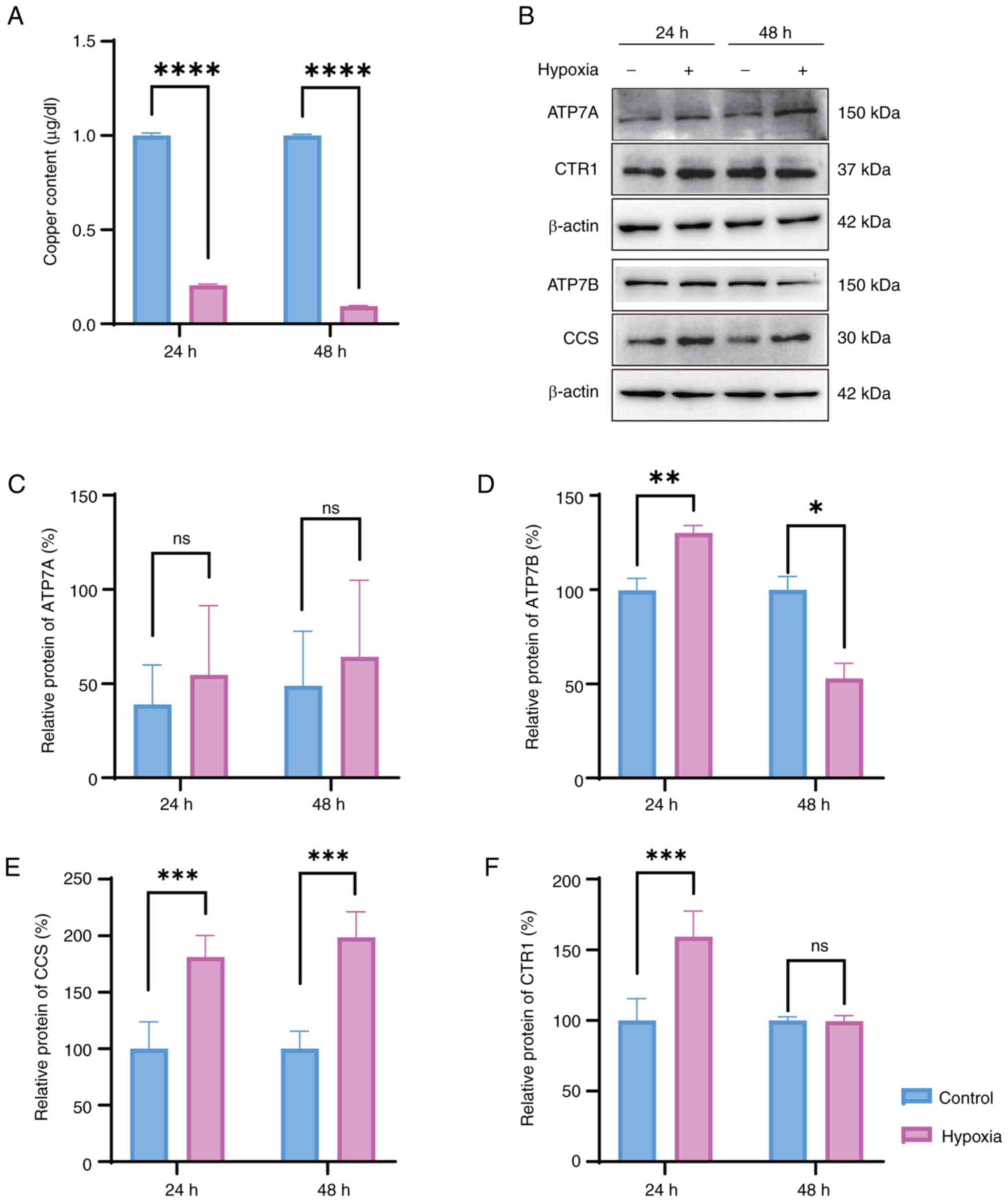 | Figure 3Copper metabolism is involved in the
survival of HT22 cells exposed to hypoxia. (A) Copper content of
HT22 cells exposed to hypoxia for 24 and 48 h, as determined by
graphite furnace atomic absorption spectroscopy. (B) Western blot
analysis and semi-quantification of the protein expression levels
of copper transport proteins, including (C) ATP7A, (D) ATP7B, (E)
CCS and (F) CTR1 was determined following exposure to hypoxia (1%
O2) for 24 and 48 h. Unpaired Student's t-test was used
for analysis. Data are presented as the mean ± SEM (n=3).
*P<0.05, **P<0.01,
***P<0.001, ****P<0.0001. ATP7A, copper
transporting α polypeptide; ATP7B, copper transporting β
polypeptide; CCS, copper chaperone for superoxide dismutase; CTR1,
copper transporter 1; ns, not significant. |
Copper could improve the viability of
HT22 cells exposed to hypoxia by enhancing their anti-oxidative
ability
Based on the aforementioned experimental results,
the present study assessed the effect of different concentrations
of copper supplementation on the viability of HT22 cells after
hypoxic exposure. Copper treatment after hypoxia resulted in a
dose-dependent increase in cell viability when a dose ≤5 μM
was administered (Fig. 4A); this
suggested that an appropriate concentration of copper may promote
the survival of cells exposed to hypoxia. Subsequently, the copper
concentration in HT22 cells subjected to hypoxia and treated with 5
μM CuCl2 was analyzed. The findings indicated a
significant decrease in copper concentration in the hypoxia group
compared with that in the control group, which is consistent with
prior research (Fig. 3A).
Conversely, HT22 cells exposed to hypoxia and treated with medium
supplemented with 5 μM CuCl2 exhibited elevated
copper levels in comparison to the hypoxia group (Fig. 4B). The present study also
examined the activities of SOD and SOD1 enzymes to investigate the
potential of copper supplementation in mitigating oxidative stress
induced by hypoxia. The results revealed a significant increase in
SOD and SOD1 enzyme activity in HT22 cells in the hypoxia + copper
treatment group compared with that in the hypoxia group (Fig. 4C and D). Moreover,
H2O2 and MDA were examined,
H2O2 and MDA levels were significantly
decreased in the hypoxia + copper group compared with those in the
hypoxia group (Fig. 4E and F).
Overall, the present study indicated that an appropriate dose of
copper may protect HT22 cells from hypoxia-induced oxidative
stress.
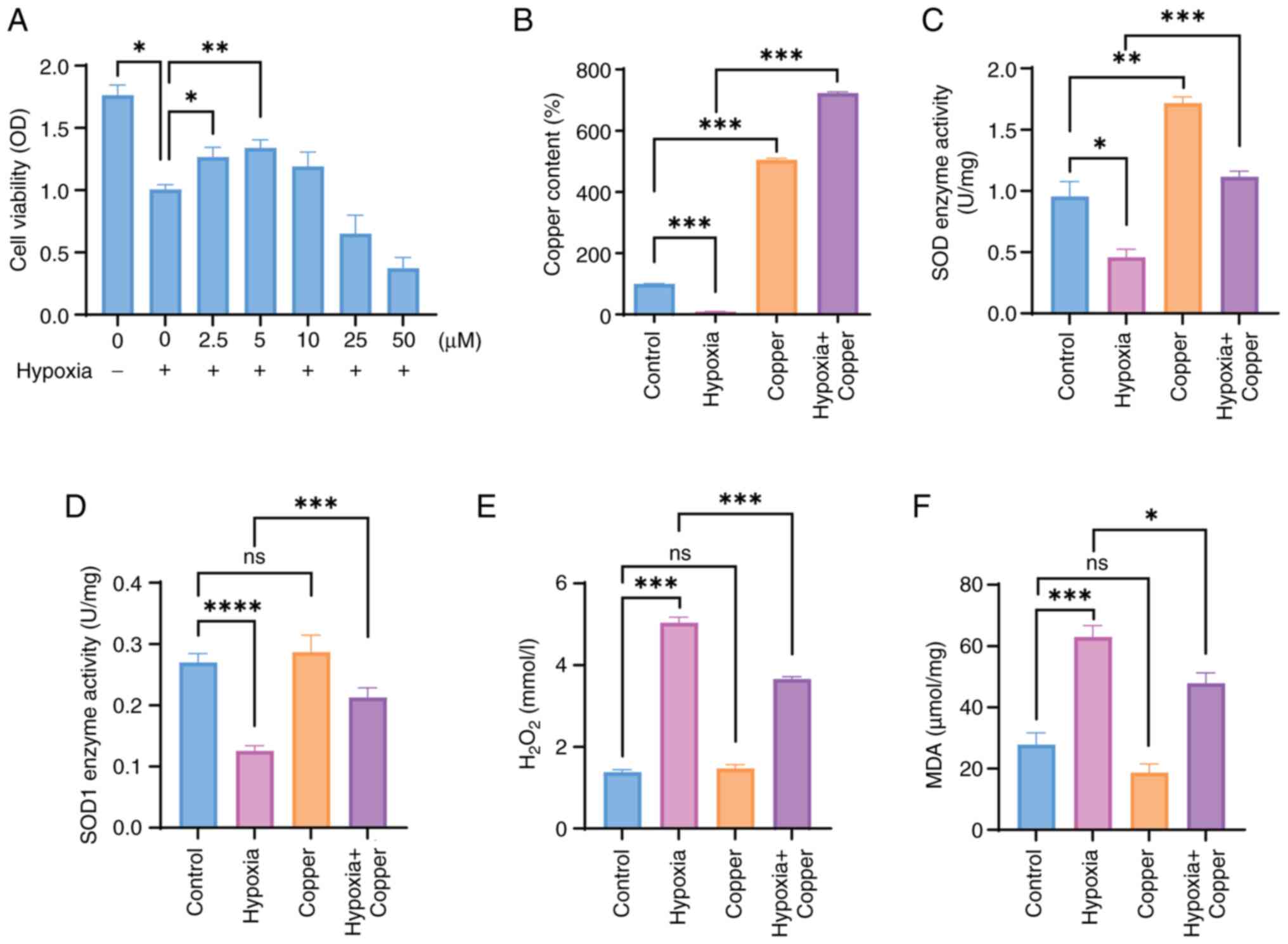 | Figure 4Copper can improve the viability of
HT22 cells exposed to hypoxia through enhancing their
anti-oxidative ability. (A) Viability of HT22 cells exposed to
hypoxia (1% O2) and different copper concentrations for
48 h, as detected by Cell Counting Kit-8 assay. (B) Copper content
of HT22 cells after 48 h was detected by graphite furnace atomic
absorption spectroscopy. (C) SOD and (D) SOD1 enzyme activity
analysis. Detection of (E) H2O2 and (F) MDA
in HT22 cells exposed to normoxia or hypoxia (1% O2) for
48 h, with or without CuCl2 (5 μM) exposure.
One-way ANOVA was used for multiple-group comparisons. Data are
presented as the mean ± SEM (n=3). *P<0.05,
**P<0.01, ***P<0.001,
****P<0.0001. H2O2, hydrogen
peroxide; OD, optical density; MDA, malondialdehyde; ns, not
significant; SOD, superoxide dismutase. |
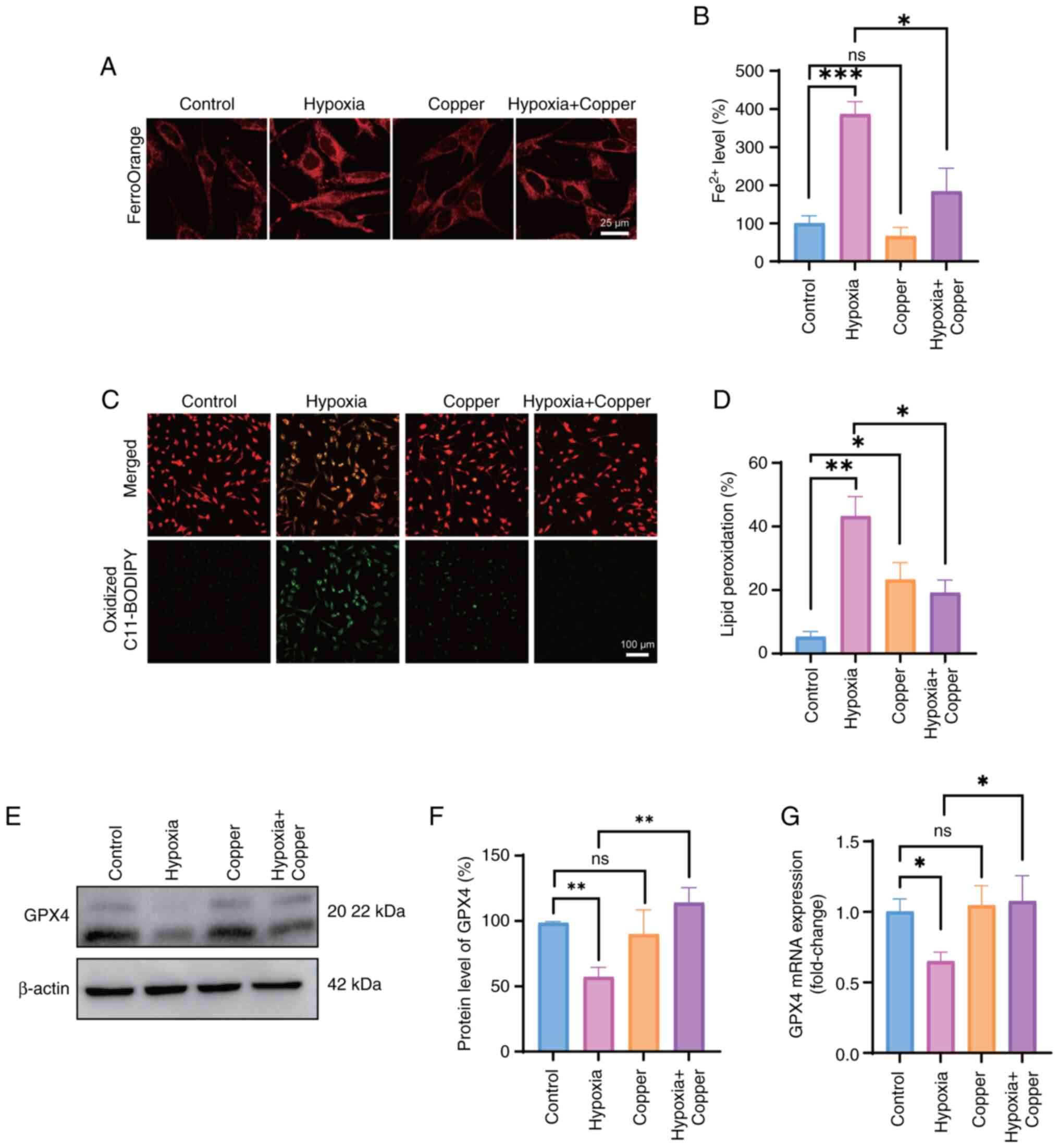
Copper supplementation reduces
hypoxia-induced ferroptosis
Given the decrease in copper concentration under
hypoxic conditions, the present study investigated whether copper
supplementation could alleviate hypoxia-induced ferroptosis and
oxidative stress. First, the levels of intracellular ferrous ions
were evaluated using a FerroOrange fluorescence probe. Despite the
increase in intracellular ferrous ions in response to hypoxia,
copper supplementation reduced the accumulation of intracellular
ferrous ions under hypoxia, suggesting that copper may protect
cells against ferroptosis (Fig. 5A
and B). Next, the levels of LOS in HT22 cells exposed to
hypoxia and treated with 5 μM copper were assessed, and it
was revealed that the levels of LOS in the hypoxia + copper group
were significantly decreased compared with those in the hypoxia
group (Fig. 5C and D).
Decreased GPX4 expression is notably associated with
ferroptosis (21). Following the
confirmation of hypoxia-induced ferroptosis in HT22 cells based on
the aforementioned findings, an investigation was conducted to
determine the effects of copper supplementation on ferroptosis. The
effects of supplementation of the medium with 5 μM
CuCl2 in the hypoxia + copper group on GPX4 expression
were examined. The expression levels of GPX4 in the hypoxia +
copper group were significantly increased compared with those in
the hypoxia group, indicating that copper supplementation may
alleviate neuronal ferroptosis (Fig.
5E-G).
Copper alleviates hypoxia-induced
neuronal injury through the CCS/SOD1/GPX4 axis
The present study analyzed the expression of various
copper-related proteins involved in cellular oxidative stress to
examine the neuroprotective effects of copper in mitigating
hypoxia-induced neuronal damage. Using a hypoxic model of HT22
cells supplemented with copper, western blot analysis revealed
upregulation of XIAP expression, downregulation of CCS expression,
and no significant change in the expression levels of SOD1 compared
with those in the hypoxia group (Fig. 6A-D). Subsequently, siRNA
targeting CCS was effectively transfected into cells (Fig. S2), followed by western blot
analysis to assess the expression levels of CCS, SOD1 and GPX4
after exposure to hypoxia. Compared with in the hypoxia + Copper
group, the protein expression levels of GPX4 were decreased in the
hypoxia + Copper + siRNA group, whereas those of SOD1 remained
significantly unchanged following siRNA knockdown of CCS (Fig. 6E-H). In addition, SOD1 activity
and MDA content were detected after the knockdown of CCS (Fig. 6I-J). Consistent with the western
blot analysis results, the rescue effects of copper supplementation
after hypoxia were attenuated, as reflected by diminished increases
in SOD1 enzyme activity and an increase in MDA content. In
conclusion, the neuroprotective effects of copper may be mediated
through the CCS/SOD1/GPX4 axis.
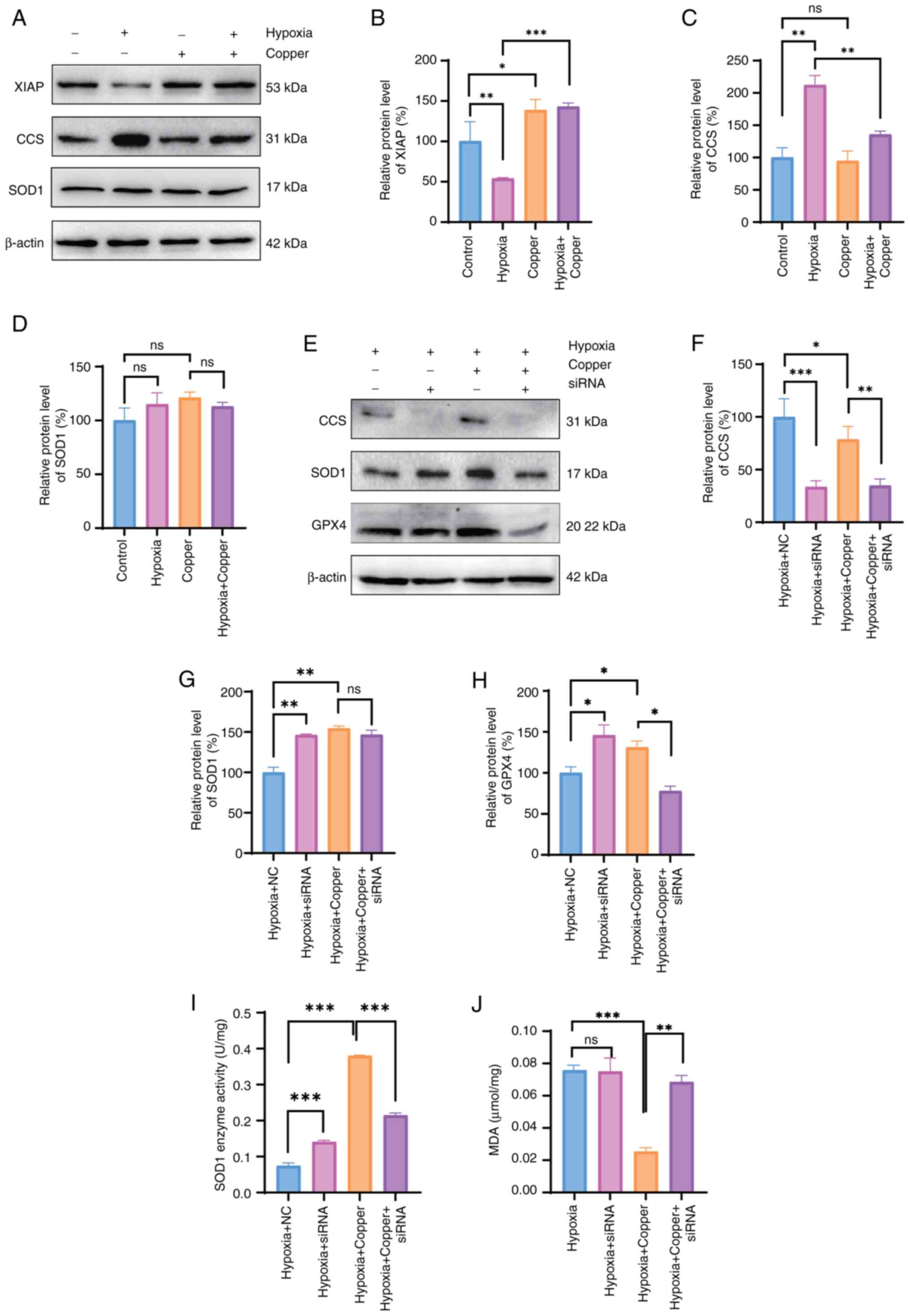 | Figure 6Copper alleviates hypoxia-induced
neuronal injury through the CCS/SOD1/GPX4 axis. (A) Western blot
analysis, and semi-quantification of the protein expression levels
of copper transport proteins, including (B) XIAP, (C) CCS and (D)
SOD1 following exposure to hypoxia (1% O2) and copper
supplementation for 48 h. (E) Western blot analysis, and
semi-quantification of the protein expression levels of copper
transport proteins, including (F) CCS, (G) SOD1 and (H) GPX4 was
determined following exposure to hypoxia (1% O2) for 48
h, with or without copper supplementation and CCS siRNA
transfection. (I) SOD1 enzyme activity and (J) MDA content of HT22
cells following exposure to hypoxia (1% O2) for 48 h,
with or without copper supplementation and CCS siRNA transfection.
One-way ANOVA was used for multiple-group comparisons. Data are
presented as the mean ± SEM (n=3). *P<0.05,
**P<0.01, ***P<0.001. CCS, copper
chaperone for superoxide dismutase; GPX4, glutathione peroxidase 4;
MDA, malondialdehyde; NC, normal control; ns, not significant;
siRNA, small interfering RNA; SOD1, superoxide dismutase 1. |
Discussion
The neuronal damage induced by hypoxia observed in
the present study aligns with the findings reported in prior
research that hypoxia could lead to neuronal oxidative stress and
ferroptosis (22). The
maintenance of central nervous system function requires appropriate
levels of copper, as demonstrated by reports of impaired cognitive
and motor functions in mammals with a copper deficiency (23). Studies have indicated a decrease
in copper concentration in neurological disorders marked by
hypoxia, suggesting the potential involvement of copper metabolism
in the pathogenesis of hypoxia-induced neurological impairments
(24,25). In the present study, a notable
reduction in the copper concentration within neurons was observed
following exposure to hypoxia. This decline in copper levels may
contribute to the development of neurological impairment under
hypoxic conditions, suggesting that copper could serve as a
biomarker for brain damage induced by hypoxia.
Copper homeostasis mainly depends on the regulation
of copper-related proteins, among which CTR1 is structurally and
functionally conserved in humans, and is responsible for the
majority of copper uptake into cells, whereas ATP7A and ATP7B act
as major transporters for exporting copper from neurons (26,27). The expression levels of CTR1 were
increased after exposure to hypoxia for 24 h but did not change
after 48 h, which may be related to copper depletion in the cells.
ATP7B expression was increased at 24 h but was significantly
decreased at 48 h, which may be related to reduced cellular copper
efflux after exposure to hypoxia for 48 h. The CCS chaperone
facilitates the delivery of copper to SOD1 to detoxify ROS and
maintain Cu homeostasis (28).
It has been demonstrated that organisms deficient in SOD1 exhibit
elevated oxidative stress levels (29). The present study observed a
significant upregulation in CCS expression and a decrease in SOD1
enzyme activity following exposure to hypoxia. These changes were
accompanied by increased levels of oxidative stress markers,
including ROS, H2O2 and MDA.
Oxidative stress resulting from hypoxia-induced
injury is characterized by an excess of ROS, which can affect all
neuronal cells (30). The
present investigation detected elevated intracellular levels of
ROS, H2O2 and MDA following exposure to
hypoxia. SOD serves a crucial role in mitigating oxidative damage
to tissues (31). The present
findings indicated a significant decrease in SOD and SOD1 activity
in HT22 cells subjected to hypoxic conditions, thus indicating that
oxidative stress may have a critical role in neuronal injury
following exposure to hypoxia.
Ferroptosis is a unique type of cell death resulting
from iron accumulation and lipid peroxidation that can be blocked
by Fer-1 (32). In the present
study, ferroptosis was observed in neurons following hypoxia. It
has previously been demonstrated that GPX4 is a key regulator of
ferroptosis (33), and that the
expression of GPX4 in neurons is significantly decreased after
hypoxia. Additionally, the present study indicated a reduction in
mitochondrial size, decreased mitochondrial ridges, increased
bilayer membrane density, elevated levels of ferrous ions and lipid
peroxidation in neuronal cells in response to hypoxia. The
alterations in ferrous ions and lipid peroxidation were effectively
reversed by Fer-1, which is consistent with the findings of a
previous study (34).
In a prior study, treatment with copper was shown to
reduce neuronal ferroptosis and oxidative stress (35). Notably, in the present study, the
levels of H2O2 and MDA in the hypoxia +
copper group were significantly reduced compared with those in the
hypoxia group, indicating that copper can alleviate oxidative
stress after hypoxic exposure. XIAP enhances E3 ubiquitination of
the CCS by enhancing the delivery of copper to SOD1 to redistribute
cellular copper and regulate copper homeostasis (36,37). SOD1 is a major antioxidant in
cells, and copper is required for its maturation and enzymatic
activity (38). The results of
the current study indicated upregulation of XIAP protein expression
and downregulation of CCS protein expression in response to copper
supplementation in a hypoxic model, suggesting an increased
requirement for copper in neurons under hypoxic conditions. Prior
research has demonstrated the crucial role of the CCS in the
antioxidant function of SOD1, as evidenced by a significant
reduction in SOD1 activity in CCS-knockout mouse models (39,40). The present study also revealed
that SOD1 activity was significantly reduced following CCS
knockdown by siRNA without affecting its expression levels.
Furthermore, GPX4 expression and MDA levels were comparable to
those in the hypoxia group after copper treatment, suggesting that
CCS/SOD1 may be involved in GPX4-mediated ferroptosis during
neuronal hypoxic damage.
These findings indicated that copper could serve as
a therapeutic agent for the prevention and treatment of hypoxic
brain damage (41). Collective
evidence has suggested that neuronal ferroptosis under hypoxic
conditions may be attributable to disrupted copper metabolism and
depletion. Further research, including in vivo studies and
the elucidation of the precise mechanisms involved, is warranted to
better understand the role of copper in ferroptosis following
exposure to hypoxia.
In conclusion, exposure to hypoxia may disrupt
copper metabolism, reduce copper levels in neurons, worsen
oxidative stress and promote ferroptosis. Copper administration can
help alleviate oxidative stress and prevent neuronal ferroptosis,
potentially serving as a therapeutic strategy for hypoxic brain
damage.
Supplementary Data
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
GZ, TW, JW and YZ conceived the study. ZZ and JW
analyzed the data. ZC, RG, KJ, JW ST and LS developed the model and
performed molecular biology experiments. RG was responsible for the
supplementary work of the experiment. JW and RG confirm the
authenticity of all the raw data. All authors read and approved the
final version of the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgements
The authors would like to thank Dr Weihua Yu (Fourth
Military Medical University, Xi'an, China) for his suggestions and
technical support for the laboratory experiments and data
analysis.
Funding
This study was supported by the National Natural Science
Foundation of China International Cooperation Program (grant no.
81920108030), the National Natural Science Foundation of China
General Program (grant nos. 82271913, 82204089 and 82302116), the
Fourth Military Medical University ZhuFeng project (grant no.
2020rcfczg), and the Air Force Medical University Pilotage
Operation New Flight Program Project (grant no. 2023rcjfzyk).
References
|
1
|
Douglas-Escobar M and Weiss MD:
Hypoxic-ischemic encephalopathy: A review for the clinician. JAMA
Pediatr. 169:397–403. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ahearne CE, Boylan GB and Murray DM: Short
and long term prognosis in perinatal asphyxia: An update. World J
Clin Pediatr. 5:67–74. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Whitelaw A and Thoresen M: Therapeutic
hypothermia for hypoxic-ischemic brain injury is more effective in
newborn infants than in older patients: Review and hypotheses. Ther
Hypothermia Temp Manag. 13:170–174. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Golubnitschaja O, Yeghiazaryan K, Cebioglu
M, Morelli M and Herrera-Marschitz M: Birth asphyxia as the major
complication in newborns: Moving towards improved individual
outcomes by prediction, targeted prevention and tailored medical
care. EPMA J. 2:197–210. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Arumugam TV, Baik SH, Balaganapathy P,
Sobey CG, Mattson MP and Jo DG: Notch signaling and neuronal death
in stroke. Prog Neurobiol. 165-167:103–116. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ji X, Zhou Y, Gao Q, He H, Wu Z, Feng B,
Mei Y, Cheng Y, Zhou W, Chen Y and Xiong M: Functional
reconstruction of the basal ganglia neural circuit by human
striatal neurons in hypoxic-ischaemic injured brain. Brain.
146:612–628. 2023. View Article : Google Scholar
|
|
7
|
Akyuva Y and Nazıroğlu M: Resveratrol
attenuates hypoxia-induced neuronal cell death, inflammation and
mitochondrial oxidative stress by modulation of TRPM2 channel. Sci
Rep. 10:64492020. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lin W, Zhang T, Zheng J, Zhou Y, Lin Z and
Fu X: Ferroptosis is involved in hypoxic-ischemic brain damage in
neonatal rats. Neuroscience. 487:131–142. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Oorschot DE, Sizemore RJ and Amer AR:
Treatment of neonatal hypoxic-ischemic encephalopathy with
erythropoietin alone, and erythropoietin combined with hypothermia:
History, current status, and future research. Int J Mol Sci.
21:14872020. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Davies KM, Hare DJ, Cottam V, Chen N,
Hilgers L, Halliday G, Mercer JFB and Double KL: Localization of
copper and copper transporters in the human brain. Metallomics.
5:43–51. 2013. View Article : Google Scholar
|
|
11
|
Scheiber IF, Mercer JFB and Dringen R:
Metabolism and functions of copper in brain. Prog Neurobiol.
116:33–57. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Alemany S, Vilor-Tejedor N, Bustamante M,
Álvarez-Pedrerol M, Rivas I, Forns J, Querol X, Pujol J and Sunyer
J: Interaction between airborne copper exposure and ATP7B
polymorphisms on inattentiveness in scholar children. Int J Hyg
Environ Health. 220:51–56. 2017. View Article : Google Scholar
|
|
13
|
Zhang J, Cao J, Zhang H, Jiang C, Lin T,
Zhou Z, Song Y, Li Y, Liu C, Liu L, et al: Plasma copper and the
risk of first stroke in hypertensive patients: A nested
case-control study. Am J Clin Nutr. 110:212–220. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zhang M, Li W, Wang Y, Wang T, Ma M and
Tian C: Association between the change of serum copper and ischemic
stroke: A systematic review and meta-analysis. J Mol Neurosci.
70:475–480. 2020. View Article : Google Scholar
|
|
15
|
Gromadzka G, Tarnacka B, Flaga A and
Adamczyk A: Copper dyshomeostasis in neurodegenerative
diseases-therapeutic implications. Int J Mol Sci. 21:212392592020.
View Article : Google Scholar
|
|
16
|
Yang S, Li X, Yan J, Jiang F, Fan X, Jin
J, Zhang W, Zhong D and Li G: Disulfiram downregulates ferredoxin 1
to maintain copper homeostasis and inhibit inflammation in cerebral
ischemia/reperfusion injury. Sci Rep. 14:151752024. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Sun X, Zhang X, Yan H, Wu H, Cao S, Zhao
W, Dong T and Zhou A: Protective effect of curcumin on
hepatolenticular degeneration through copper excretion and
inhibition of ferroptosis. Phytomedicine. 113:1545392022.
View Article : Google Scholar
|
|
18
|
Li F, Wu X, Liu H, Liu M, Yue Z, Wu Z, Liu
L and Li F: Copper depletion strongly enhances ferroptosis via
mitochondrial perturbation and reduction in antioxidative
mechanisms. Antioxidants (Basel). 11:20842022. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Guan R, Yang C, Zhang J, Wang J, Chen R
and Su P: Dehydroepiandrosterone alleviates hypoxia-induced
learning and memory dysfunction by maintaining synaptic
homeostasis. CNS Neurosci Ther. 28:1339–1350. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
21
|
Yang WS, SriRamaratnam R, Welsch ME,
Shimada K, Skouta R, Viswanathan VS, Cheah JH, Clemons PA, Shamji
AF, Clish CB, et al: Regulation of ferroptotic cancer cell death by
GPX4. Cell. 156:317–331. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhu K, Zhu X, Liu S, Yu J, Wu S and Hei M:
Glycyrrhizin attenuates hypoxic-ischemic brain damage by inhibiting
ferroptosis and neuroinflammation in neonatal rats via the
HMGB1/GPX4 pathway. Oxid Med Cell Longev. 2022:84385282022.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Bakkar N, Starr A, Rabichow BE, Lorenzini
I, McEachin ZT, Kraft R, Chaung M, Macklin-Isquierdo S, Wingfield
T, Carhart B, et al: The M1311V variant of ATP7A is associated with
impaired trafficking and copper homeostasis in models of motor
neuron disease. Neurobiol Dis. 149:1052282021. View Article : Google Scholar
|
|
24
|
Huuskonen MT, Tuo QZ, Loppi S, Dhungana H,
Korhonen P, McInnes LE, Donnelly PS, Grubman A, Wojciechowski S,
Lejavova K, et al: The Copper bis(thiosemicarbazone) complex
CuII(atsm) is protective against cerebral ischemia
through modulation of the inflammatory milieu. Neurotherapeutics.
14:519–532. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Nikseresht S, Hilton JBW, Kysenius K,
Liddell JR and Crouch PJ: Copper-ATSM as a treatment for ALS:
Support from mutant SOD1 models and beyond. Life (Basel).
10:2712020.PubMed/NCBI
|
|
26
|
Chen L, Min J and Wang F: Copper
homeostasis and cuproptosis in health and disease. Signal Transduct
Target Ther. 7:3782022. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Batzios S, Tal G, DiStasio AT, Peng Y,
Charalambous C, Nicolaides P, Kamsteeg EJ, Korman SH, Mandel H,
Steinbach PJ, et al: Newly identified disorder of copper metabolism
caused by variants in CTR1, a high-affinity copper transporter. Hum
Mol Genet. 31:4121–4130. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Dong X, Zhang Z, Zhao J, Lei J, Chen Y, Li
X, Chen H, Tian J, Zhang D and Liu C and Liu C: The rational design
of specific SOD1 inhibitors via copper coordination and their
application in ROS signaling research. Chem Sci. 7:6251–6262. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Fischer LR, Igoudjil A, Magrané J, Li Y,
Hansen JM, Manfredi G and Glass JD: SOD1 targeted to the
mitochondrial intermembrane space prevents motor neuropathy in the
Sod1 knockout mouse. Brain. 134:196–209. 2011. View Article : Google Scholar
|
|
30
|
Meyer C, Rao NS, Vasanthi SS, Pereira B,
Gage M, Putra M, Holtkamp C, Huss J and Thippeswamy T: Peripheral
and central effects of NADPH oxidase inhibitor, mitoapocynin, in a
rat model of diisopropylfluorophosphate (DFP) toxicity. Front Cell
Neurosci. 17:11958432023. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Borgstahl GEO and Oberley-Deegan RE:
Superoxide dismutases (SODs) and SOD mimetics. Antioxidants
(Basel). 7:1562018. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Tang D, Chen X, Kang R and Kroemer G:
Ferroptosis: Molecular mechanisms and health implications. Cell
Res. 31:107–125. 2021. View Article : Google Scholar :
|
|
33
|
Jiang X, Stockwell BR and Conrad M:
Ferroptosis: Mechanisms, biology and role in disease. Nat Rev Mol
Cell Biol. 22:266–282. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Doll S, Freitas FP, Shah R, Aldrovandi M,
da Silva MC, Ingold I, Goya Grocin A, Xavier da Silva TN, Panzilius
E, Scheel CH, et al: FSP1 is a glutathione-independent ferroptosis
suppressor. Nature. 575:693–698. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Mezzaroba L, Alfieri DF, Colado Simão AN
and Vissoci Reiche EM: The role of zinc, copper, manganese and iron
in neurodegenerative diseases. Neurotoxicology. 74:230–241. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Wang L, Ge Y and Kang YJ: Featured
article: Effect of copper on nuclear translocation of copper
chaperone for superoxide dismutase-1. Exp Biol Med (Maywood).
241:1483–1488. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Brady GF, Galbán S, Liu X, Basrur V,
Gitlin JD, Elenitoba-Johnson KSJ, Wilson TE and Duckett CS:
Regulation of the copper chaperone CCS by XIAP-mediated
ubiquitination. Mol Cell Biol. 30:1923–1936. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Schmidt PJ, Kunst C and Culotta VC: Copper
activation of superoxide dismutase 1 (SOD1) in vivo. Role for
protein-protein interactions with the copper chaperone for SOD1. J
Biol Chem. 275:33771–33776. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Wong PC, Waggoner D, Subramaniam JR,
Tessarollo L, Bartnikas TB, Culotta VC, Price DL, Rothstein J and
Gitlin JD: Copper chaperone for superoxide dismutase is essential
to activate mammalian Cu/Zn superoxide dismutase. Proc Natl Acad
Sci USA. 97:2886–2891. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Beckman JS, Esétvez AG, Barbeito L and
Crow JP: CCS knockout mice establish an alternative source of
copper for SOD in ALS. Free Radic Biol Med. 33:1433–1435. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Giampietro R, Spinelli F, Contino M and
Colabufo NA: The pivotal role of copper in neurodegeneration: A new
strategy for the therapy of neurodegenerative disorders. Mol Pharm.
15:808–820. 2018. View Article : Google Scholar : PubMed/NCBI
|