Introduction
Mitochondria are known as the powerhouses of the
cell and influence key signaling pathways related to cellular
homeostasis, proliferation and apoptosis (1-3).
The study of mitochondrial dynamics and biogenesis has attracted
significant attention in recent years due to its vital role in
elucidating diverse biological phenomena, including the process of
apoptosis in cancer cells (4-7).
Mitochondrial homeostasis is regulated by two opposing processes:
Fusion and fission (8).
Mitochondria can fuse to form larger networks or undergo fission
into smaller mitochondria (1).
These distinct alterations in mitochondrial morphology can trigger
different metabolic and regulatory processes, thereby enhancing the
chemoresistance of cancer cells (5,6,9).
Evidence suggests a close association between
dysregulated mitochondrial homeostasis and tumorigenesis, offering
a novel perspective for comprehending intricate oncogenic
processes. Mitochondrial fission has been observed in neoplastic
cells across various solid tumors. Research has demonstrated that
dysregulated mitochondrial homeostasis, characterized by increased
fission or weakened fusion, is commonly found in numerous types of
cancer, resulting in mitochondrial fragmentation (10-13). The process of mitochondrial
fusion, which involves the merging of mitochondrial membranes, is
facilitated by mitofusin1 (MFN1), mitofusin2 (MFN2) and optic
atrophy 1 (OPA1). By contrast, mitochondrial fission is regulated
by DRP1, which forms a ring-like structure on the outer
mitochondrial membrane to facilitate the constriction and division
of mitochondria. Notably, in most types of cancer, the expression
of the key mitochondrial fission gene DRP1 is upregulated, while
the expression of the mitochondrial fusion gene MFN2 is
downregulated (14-17). These finding suggest a potential
role for mitochondrial homeostasis in tumor progression.
Dysregulated mitochondrial homeostasis may play a
pivotal role in cancer chemoresistance. The mechanisms underlying
chemoresistance in tumors are complex, involving multiple cellular
processes and molecular pathways. One hypothesis suggests that
mitochondrial homeostasis contributes to the acquisition of
anti-apoptotic capabilities. The release of cytochrome c
from the mitochondrial outer membrane, triggered by changes in
membrane permeability, initiates a cascade leading to programmed
cell death (18). Changes in
mitochondrial homeostasis directly affect the permeability of the
mitochondrial outer membrane, potentially inhibiting the shifts in
membrane potential induced by chemotherapeutic agents and thereby
granting cells anti-apoptotic properties (4).
In addition to mitochondrial homeostasis, cancer
heterogeneity plays a crucial role in chemoresistance. In
osteosarcoma, a highly heterogeneous malignant tumor that
predominantly affects adolescents (19), its heterogeneity driven by the
plasticity of osteosarcoma cells, contributes to the high
chemoresistance observed in this disease. Osteosarcoma cells can be
categorized into two subpopulations: osteosarcoma stem cells (OSCs)
and non-osteosarcoma stem cells (non-OSCs). 'OSCs' refer to
osteosarcoma cells that exhibit stem cell properties, such as
self-renewal, differentiation capacity, chemoresistance and high
tumorigenic potential (20). By
contrast, 'non-OSCs' refers to osteosarcoma cells that do not
possess these stem cell properties (20). The high chemoresistance exhibited
by OSCs presents a significant challenge for eradication,
complicating the clinical management behind osteosarcoma (21,22). However, the underlying mechanism
of the difference in chemoresistance between OSCs and non-OSCs are
still unknown remain to be elucidated.
Given the potential role of mitochondrial
homeostasis in cancer chemoresistance, the present study aimed to
investigate the disparities in mitochondrial dynamic changes
between OSCs and non-OSCs and study the involvement of these
alterations in mechanisms underlying chemoresistance. Exploring
mitochondrial dynamics in osteosarcoma could elucidate
chemoresistance mechanisms and enhance therapeutic strategies,
potentially improving patient outcomes.
Materials and methods
Cell culture
The human osteosarcoma cell line MG-63 was obtained
from the Cell Bank of the Chinese Academy of Sciences (cat. no.
TCHu124) and maintained as monolayer cultures in Dulbecco's
modified Eagle's medium/F12 (DF12; cat. no. D8900; MilliporeSigma)
supplemented with 5% fetal bovine serum (FBS; MilliporeSigma),
penicillin (100 U/ml) and streptomycin (100 U/ml) in an incubator
at 37°C with 5% CO2. The human embryonic kidney cells
293T were obtained from the Cell Bank of the Chinese Academy of
Sciences (cat. no. SCSP-502) and maintained as monolayer cultures
in Dulbecco's modified Eagle's medium-high glucose (cat. no. D5648;
MilliporeSigma) supplemented with 5% fetal bovine serum (FBS;
MilliporeSigma), penicillin (100 U/ml) and streptomycin (100 U/ml)
in an incubator at 37°C with 5% CO2. For OSCs, the MG-63
was cultured in serum-free DF12 supplemented with 5 factors (5F),
including 10 μg/ml human insulin, 5 μg/ml human
transferrin, 10 μM 2-aminoethanol, 10 nM sodium selenite, 10
μM mercaptoethanol, 5 mg/ml bovine serum albumin and 5 ng/ml
transforming growth factor-β, as previously described (23,24).
Vectors and cell transfection
The knockout of DRP1 by CRISPR/Cas9 in the
MG-63 cells was performed using the 2nd Lenti-Crispr-vector system
(cat. no. 49535; Addgene, Inc.). Lentivirus (5 μg) was
amplified from 293T packaging cells with pSPAX2 and pMD2G (cat.
nos. 12260 and 12259; Addgene, Inc.) helper plasmids (quantity of
plasmids ratio was pSPAX2:pMD2G:Lentivirus=3.75:1.25:5). The
virus-containing supernatants were collected at 48 h following
transfection. The supernatants, with 5% PEG8000 were centrifuged at
4,000 × g for 2 h at 4°C to concentrate the lentiviral particles,
diluted in 200 μl PBS and then stored at -80°C. The MG-63
cells were then transfected with the lentivirus (40 μl
lentivirus in 2 ml DF12; with a multiplicity of infection of 10 for
lentiviral vectors) for 8 h at 37°C then selected with 1
μg/ml puromycin (cat. no. A1113803; Gibco; Thermo Fisher
Scientific, Inc.). The time interval between transduction and
subsequent experimentation was 48 h to allow sufficient expression
of the transgene. The single guide (sg)RNA-DRP1 primers were as
follows: Sequence 1, AUAUUCUGUUUUCAGAGCAG and sequence 2,
GAGCUCAGUGCUAGAAAGCC.
RNA isolation and reverse
transcription-quantitative (RT-q) PCR
Total RNA was extracted from the cells
(1×106 cells per well in a 6-well plate) using an
EZ-press RNA Purification kit (cat. no. B0004DP; EZBioscience) and
equal amounts of RNA were reverse-transcribed into cDNA using the
First Strand cDNA Synthesis kit, ReverTraAce (cat. no. FSQ-201;
Toyobo Life Science). RNA extraction and cDNA synthesis were
performed according to the manufacturer's protocols. RT-qPCR was
performed using a LightCycler 480 SYBR-Green I Master (Roche
Diagnostics) according to the manufacturer's instructions. The
thermocycling conditions were applied at 95°C for 5 min, followed
by 40 cycles of 95°C for 10 sec (denaturation), 60°C for 20 sec
(annealing) and then at 72°C for 20 sec (extension). mRNA
expression was normalized to GAPDH using the 2−ΔΔCq
method (25). All the
experiments were carried out at least three times independently.
The primers used for RT-qPCR are listed in Table SI.
Western blot analysis
The cells were lysed by RIPA lysis buffer (cat. no.
P0013K; Beyotime Institute of Biotechnology) containing a protease
inhibitor cocktail (cat. no. 04693132001; Roche Diagnostics) on ice
for 30 min. The protein concentration was determined using the BCA
kit assay (cat. no. 23225; Thermo Fisher Scientific, Inc.). Total
cellular proteins were extracted and analyzed by immunoblotting as
described previously (23). In
brief, a total of 30 μg of protein per lane were separated
on 10% SDS-polyacrylamide gel and then transferred to PVDF
membranes. Non-fat dried milk (5%; cat. no. 1172GR500; BioFroxx)
dissolved in TBST (Tris-buffered saline with 0.5% Tween-20, cat.
no. 1115GR500; BioFroxx) was used to block at room temperature for
1 h. The corresponding primary antibodies were incubated overnight
at 4°C, followed by the addition of HRP-labeled secondary
antibodies at room temperature for 1 h. Enhanced Chemiluminescence
(ECL; cat. no. 34580; Thermo Fisher Scientific, Inc.) was employed
for visualizing results Subsequently, images were acquired using a
CCD system. Densitometry analysis was performed using ImageJ
(Version v1.8.0; National Institutes of Health) software. The
antibodies used for western blotting are listed in Table SII.
Chemoresistance assay
A CCK-8 assay was employed to evaluate the
chemoresistance of non-OSCs and OSCs to doxorubicin (DOX) or
cisplatin (CIS; cat. nos. HY-15142 and HY-17394; MedChemExpress).
Cells were plated in 96-well plates at a density of 5,000
cells/well. Different concentrations (0-25 μM) of CIS or
(0-100 μM) DOX were added to the medium for 24 h. After
treatments, 10% CCK-8 solution was added into mediums incubated for
2 h and optical density (OD) values were evaluated at 450 nm using
a microplate reader. CIS and DOX were dissolved in dimethyl
sulfoxide (DMSO) and the equivalent amount of DMSO was added to the
control group for consistency in the present study.
Flow cytometry assay
The intracellular reactive oxygen species (ROS) were
detected by the Reactive Oxygen Species Assay kit (cat. no. S0033S;
Beyotime Institute of Biotechnology) which includes
2′,7′-Dichlorodihydrofluorescein diacetate (DCFH-DA) detection.
According to the kit protocol, cells were treated with 10 μM
DCFH-DA diluted in DF12 for 20 min at 37°C. Fluorescence was
detected at 488 nm excitation and 525 nm emission using a Beckman
MoFlo Astrios EQs flow cytometer (Beckman Coulter, Inc.). The flow
cytometry data were analyzed using FlowJo v10 software (FlowJo
LLC).
Analysis of NADPH and ATP
The intracellular NADPH levels were determined using
an NADP+/NADPH Assay kit with water-soluble tetrazolium
salt 8 (cat. no. S0179; Beyotime Institute of Biotechnology). The
absorbance values were measured at 450 nm by recording luminescence
using a BioTek Synergy LX multimode reader (BioTek; Agilent
Technologies, Inc.). The intracellular ATP levels were determined
using an ATP assay kit (cat. no. S0026, Beyotime Institute of
Biotechnology). Luminescence was recorded using the same multimode
reader with an integration time of 10 sec per well. All the
analyses were conducted according to the instructions of the
manufacturer.
Fluorescent staining for
mitochondria
Cells were seeded in glass-bottom dishes (Standard
Imaging, Inc.) and treated with CIS or DOX. OSCs were initially
cultured in normal petri dishes and then transferred to
glass-bottom dishes for image capture. Cells were stained with PK
Mito Red (PKMR) dye (cat. no. PKMR-2; GenVivo, Inc.) at 37°C for 15
min. Images were acquired using a Multimodality Structured
Illumination Microscopy (Multi-SIM) imaging system
(NanoInsights-Tech) equipped with a 100, 1.49NA oil objective and a
Kinetix camera (Photometrics). The PKMR was excited at a wavelength
of 561 nm and the resulting images were acquired in 3D-SIM mode
with a laser power of 50 mW and an exposure time of either 1 msec
(for OSCs) or 2 msec (for non-OSCs). Subsequently, the acquired
images were reconstructed using SIM Imaging Analyser software
(Version 2.23.9, NanoInsights-Tech). During image acquisition,
cells were maintained in a humidified chamber at 37°C under 5%
CO2.
3D rendering
The acquired images were reconstructed using Imaris
v9.6.0 (Oxford Instruments plc). The original images were observed
in 3D view mode and the surface module was used for 3D rendering.
The rendering parameter surface detail was 0.0612. The background
subtraction mode was selected and the diameter of the largest
sphere was 1 μm (for OSCs)/4 μm (for non-OSCs). The
threshold and filter surfaces were adjusted according to the actual
conditions of each image. After rendering, mitochondrial parameters
including length, volume and sphericity of the mitochondrial
network were obtained in a single cell for drawing the density
distribution map.
Statistical analysis
The data were presented as the mean ± standard
deviation. A one-way analysis of variance (ANOVA) followed by
Tukey's multiple comparisons test was conducted to analyze the
among multiple groups. An unpaired two-tailed Student's t-test was
used to compare the data between two groups. P<0.05 was
considered to indicate a statistically significant difference.
Results
OSCs exhibit distinct mitochondrial
morphology compared with non-OSCs
To investigate the mitochondrial morphology of OSCs,
OSCs were cultured in a serum-free culture medium as previously
described (23,24). When cultured in DF12 supplemented
with 5% FBS, cells exhibited characteristics of non-OSCs and
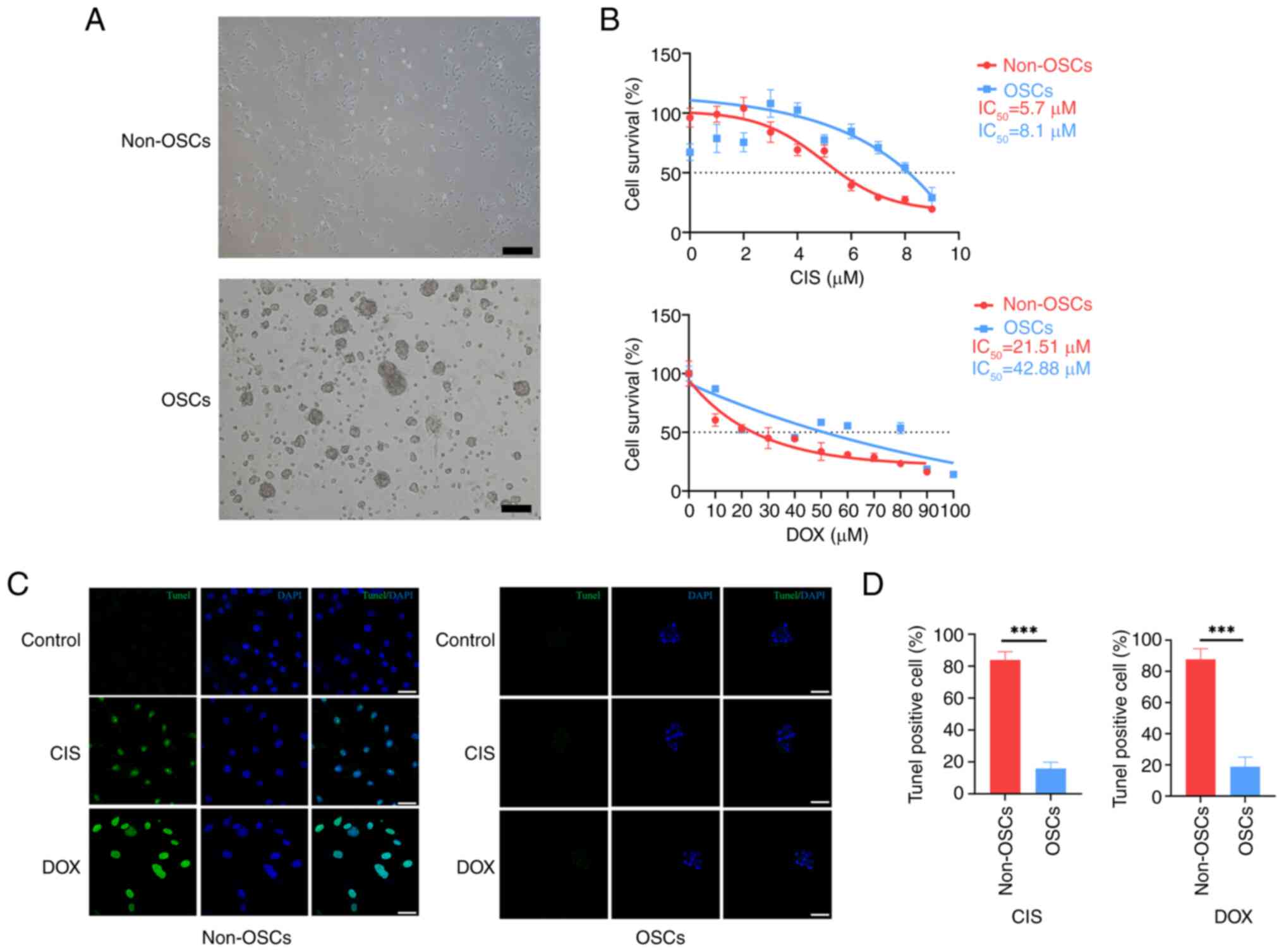
demonstrated a monolayer phenotype (Fig. 1A). By contrast, under serum-free
conditions, these cells exhibited characteristics of OSCs and
displayed a sphere phenotype (Fig.
1A). To assess the chemoresistance of OSCs compared with
non-OSCs, the cells were treated with primary clinical
chemotherapeutics CIS and DOX. The results indicated that OSCs
exhibited a higher level of chemoresistance to CIS or DOX treatment
compared with non-OSCs (Fig.
1B). TUNEL staining was employed to assess the apoptotic status
of the cells. Non-OSCs exhibited significant apoptosis upon
treatment with CIS or DOX compared with the untreated group
(Fig. 1C). However, only a few
apoptotic cells were detected in the OSCs compared with the
non-OSCs group (Fig. 1C).
Compared with non-OSCs, OSCs showed significantly increased
resistance to apoptosis (Fig.
1D). Overall, these results indicated that the OSCs showed
higher chemoresistance compared with non-OSCs.
For an improved characterization and quantitative
analysis of mitochondrial morphology and dynamic alteration,
particularly fusion and fission status, high-quality mitochondrial
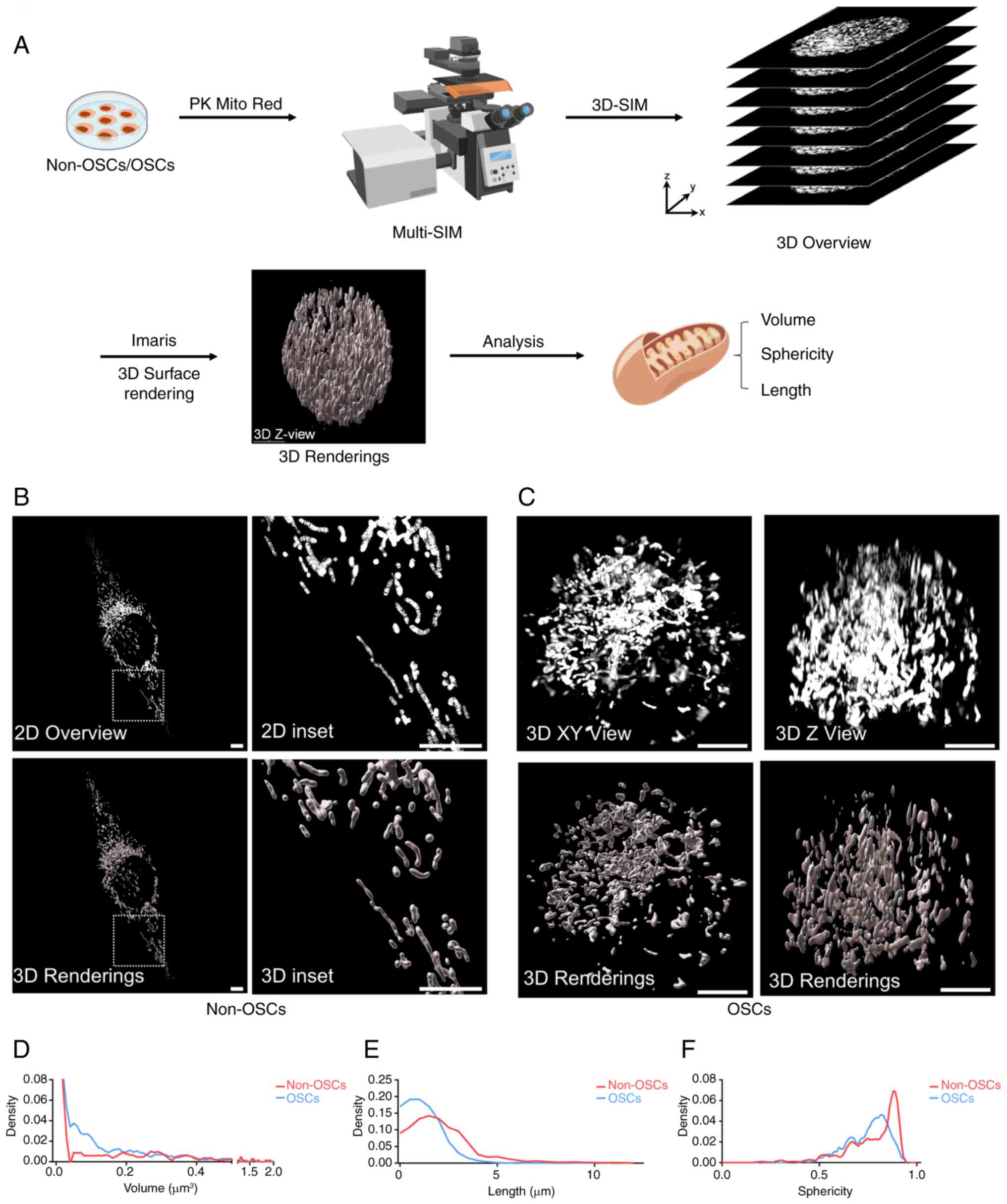
images are essential. A schematic illustrated the process of
capturing and rendering 3D images of cells (Fig. 2A). The complete structure and
network of mitochondria within non-OSCs were visualized (Fig. 2B). This revealed a diverse range
of morphologies in non-OSCs, including punctate shapes, linear
forms and interconnected networks. Additionally, OSCs exhibited
mitochondrial morphologies distinct from those observed in non-OSCs
(Fig. 2C). For an improved
characterization and quantitative analysis of mitochondrial
morphology, videos were created to show the full range of
mitochondrial morphologies in both individual non-OSCs and OSCs
(Videos S1-S4). To further
elucidate the differences in mitochondrial morphologies between
non-OSCs and OSCs, the present study analyzed and quantified 10-20
cells for each group, measuring various morphological parameters of
whole mitochondria, including length, volume and sphericity.
Mitochondria in OSCs exhibited a predominant fusion pattern and
reduced sphericity. These findings indicated that mitochondria
within OSCs were characterized by increased size and enhanced
fusion compared with non-OSCs.
OSCs exhibit stable mitochondrial
homeostasis compared with non-OSCs upon chemotherapy
The diverse morphologies of mitochondria could
influence mitochondrial functions, including cell survival,
apoptosis, metabolism, ROS management and mitophagy (26,27). CIS or DOX triggers apoptosis
through mitochondrial pathways involving the release of cytochrome
c, generation of ROS and permeabilization mediated by
Bax/Bak (28). Following
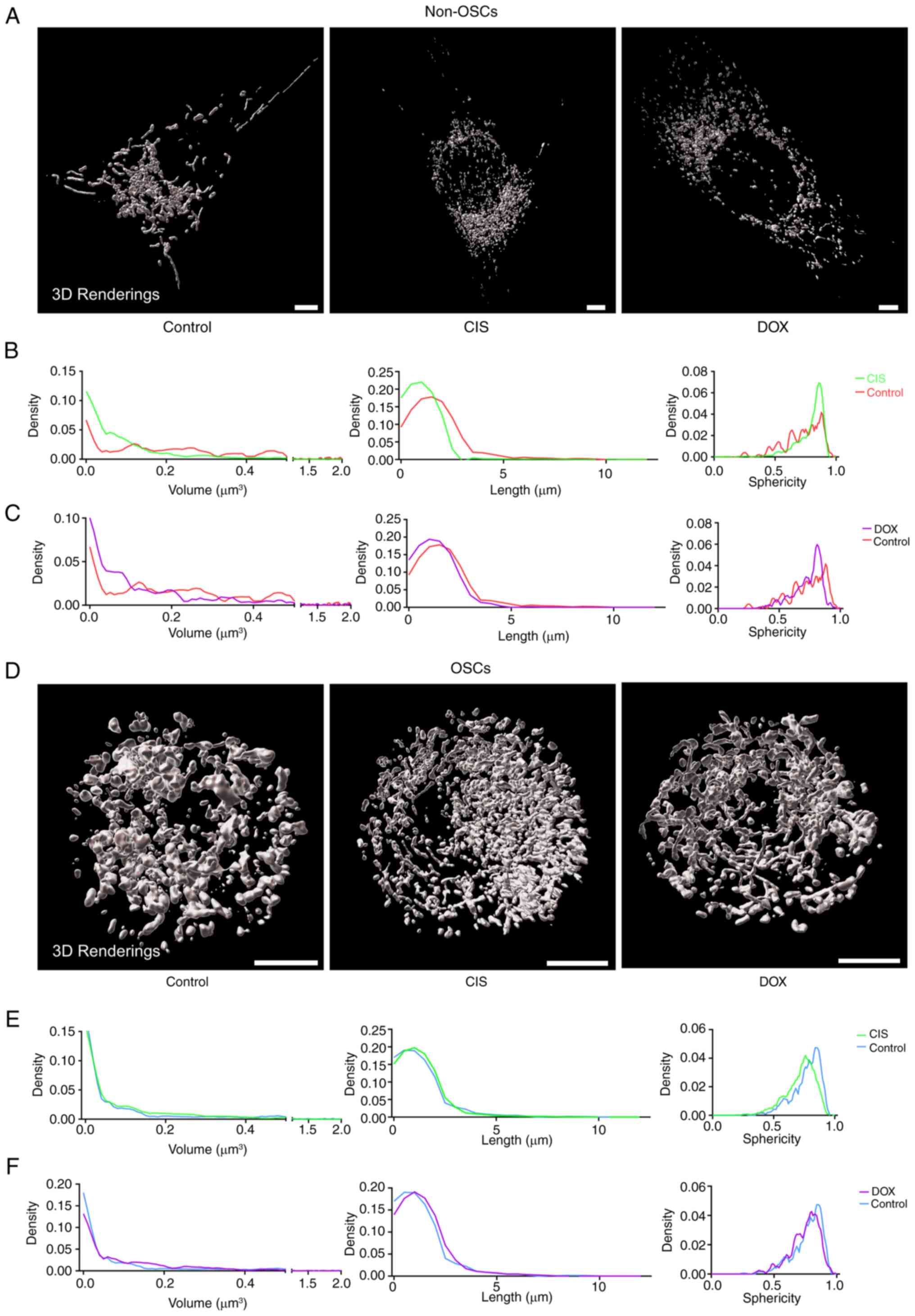
treatment with CIS or DOX, non-OSCs showed higher levels of
mitochondrial fragmentation and fission compared with the untreated
group (Fig. 3A). Additionally,
non-OSCs treated with CIS or DOX showed smaller mitochondrial
volume and shorter length, with more homogeneous sphericity
distribution compared with the untreated group (Fig. 3B and C). The mitochondrial
networks of OSCs did not exhibit significant alterations and their
overall morphology remained stable following CIS or DOX treatment
(Fig. 3D). Moreover, CIS or DOX
treatment did not affect the mitochondrial volume, length and
sphericity distribution. These results suggested that CIS or DOX
can induce mitochondrial fragmentation and fission in non-OSCs,
while having no impact on OSCs. Mitochondria in OSCs exhibited more
stable homeostasis compared with non-OSCs when exposed to
chemotherapeutics.
OSCs show mitochondrial functional
homeostasis, compared with non-OSCs, under chemotherapy
Disruption of mitochondrial homeostasis impairs
mitochondrial function, affects cellular metabolism and can
potentially lead to apoptosis (29). Mitochondria play a crucial role
in regulating the metabolism and bioenergetics of cancer cells and
their dysfunction can disrupt the production of essential
metabolites such as ATP and NADPH (14). To assess mitochondrial function,
specific assays were used to measure parameters related to ATP
production, NADPH levels and ROS levels. Non-OSCs exhibited
significantly reduced intracellular ATP levels following CIS or DOX
treatment compared with the untreated group (Fig. 4A). The intracellular ATP levels
in OSCs were unaffected by CIS or DOX treatment, compared with the
untreated group (Fig. 4A).
NADPH/NADP directly affects the redox balance within cells
(30). Non-OSCs after the
treatment with CIS or DOX showed a significant downregulation of
the intracellular NADPH/(NADP+ + NADPH) ratio compared
with the untreated group (Fig.
4B). However, there was no significant decrease in the
NADPH/(NADP+ + NADPH) ratio observed in OSCs following
CIS or DOX treatment (Fig. 4B).
Intracellular ROS was detected by flow cytometry using DCFH-DA
staining. The results demonstrated a significant increase in
intracellular ROS accumulation in non-OSCs following CIS and DOX
treatment, whereas no substantial ROS accumulation was observed in
OSCs following exposure to either CIS or DOX (Fig. 4D).
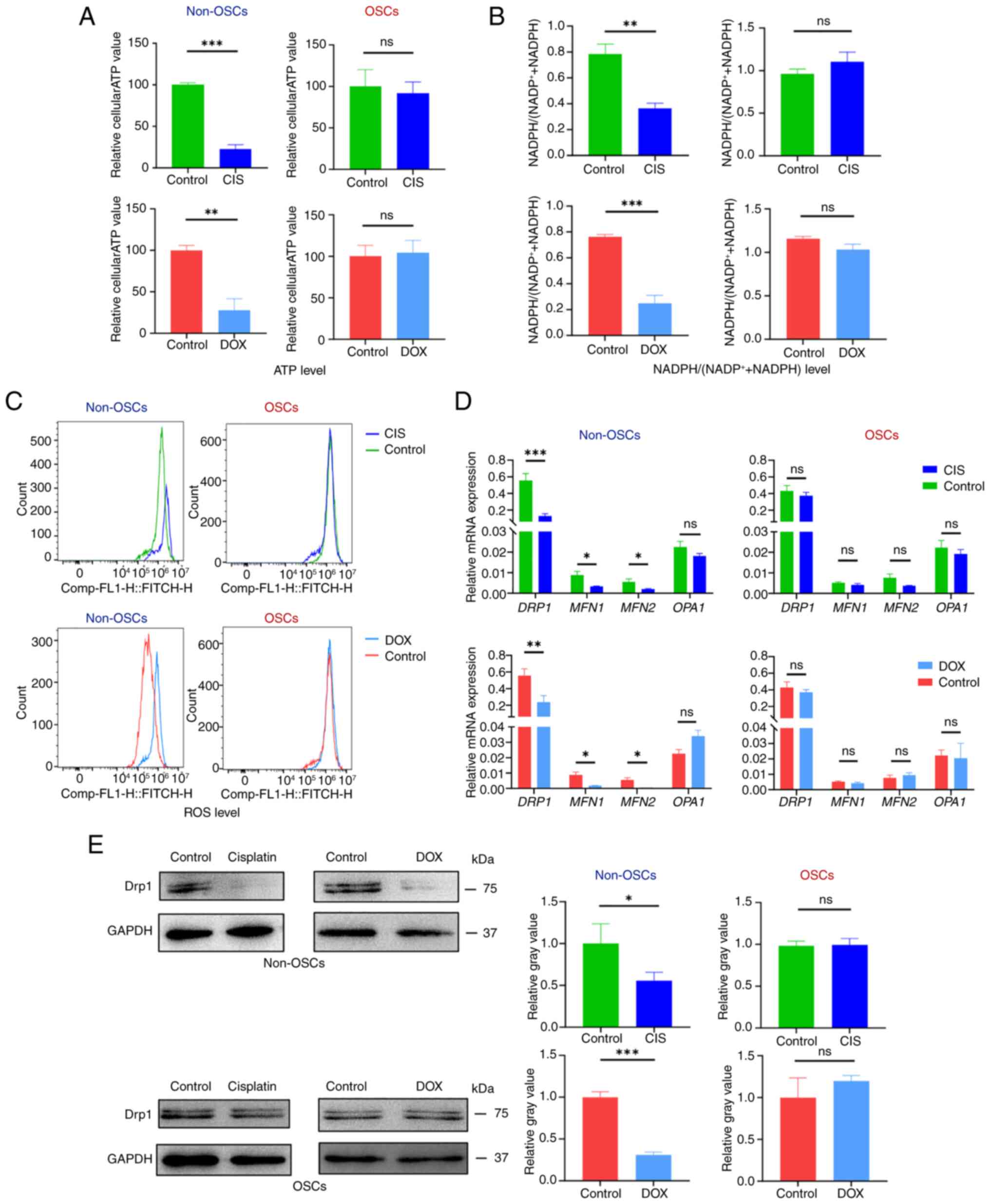 | Figure 4OSCs resist drug-induced
mitochondrial functional damage. (A) Relative intracellular ATP
generation per cell in non-OSCs and OSCs treated with CIS or DOX.
(B) Relative intracellular NADPH/(NADP++NADPH) assay in
non-OSCs and OSCs treated with CIS or DOX (C) ROS labeling with
DCFH-DA in non-OSCs and OSCs treated with CIS or DOX. (D) Reverse
transcription-quantitative PCR detecting the expression of
mitochondrial fusion and fission-related genes (DRP1, MFN1, MFN2,
OPA1) treated with CIS or DOX. (E) Western blot analysis showing
DRP1 protein expression and relative gray value. Data are presented
as the mean ± SD of three independent experiments (n=3);
*P<0.05, **P<0.01,
***P<0.001, ns, not significance. OSCs, osteosarcoma
stem cells; CIS, cisplatin; DOX, doxorubicin; ROS, reactive oxygen
species; DCFH-DA, 2′,7′-Dichlorodihydrofluorescein diacetate; DRP1,
dynamin-related protein 1. |
The regulation of mitochondrial dynamics involves
specific proteins, including MFN1, MFN2, OPA1 and DRP1. The results
of the present study showed that non-OSCs treated with CIS or DOX
exhibited significant downregulation of MFN1, MFN2 and DRP1
expression while showing no significant change in OPA1 expression.
Conversely, in OSCs treated with CIS or DOX, the expression levels
of MFN1, MFN2, DRP1 and OPA1 remained unchanged (Fig. 4D). Western blot analysis was used
to detect the protein expression level of DRP1, which showed that
non-OSCs had significantly downregulated DRP1 expression following
CIS and DOX treatment, while OSCs showed no significant change in
DRP1 expression (Fig. 4E). The
results suggested that CIS or DOX can induce mitochondrial
dysfunction in non-OSCs but not in OSCs. Meanwhile, the expression
of MFN1, MFN2 and fission-related DRP1 was downregulated following
CIS or DOX treatment; however, this suppression of these genes in
OSCs was not observed.
In summary, under CIS or DOX treatment, OSCs
maintained mitochondrial stability without significant alterations
in ATP or NADPH levels, ROS accumulation, or gene expression.
Conversely, non-OSCs exhibited impaired mitochondrial function,
characterized by reduced levels of ATP and NADPH, elevated ROS
accumulation and suppressed gene expression. These results
indicated that OSCs can maintain mitochondrial functional
homeostasis during chemotherapy, whereas non-OSCs lack this
ability.
DRP1 regulates mitochondrial morphology
in OSCs
Changes in mitochondrial morphology are a continuous
process, where mitochondria constantly undergo fusion and fission.
Disruption of these processes can lead to dysregulated of
mitochondrial homeostasis. The present study used two different
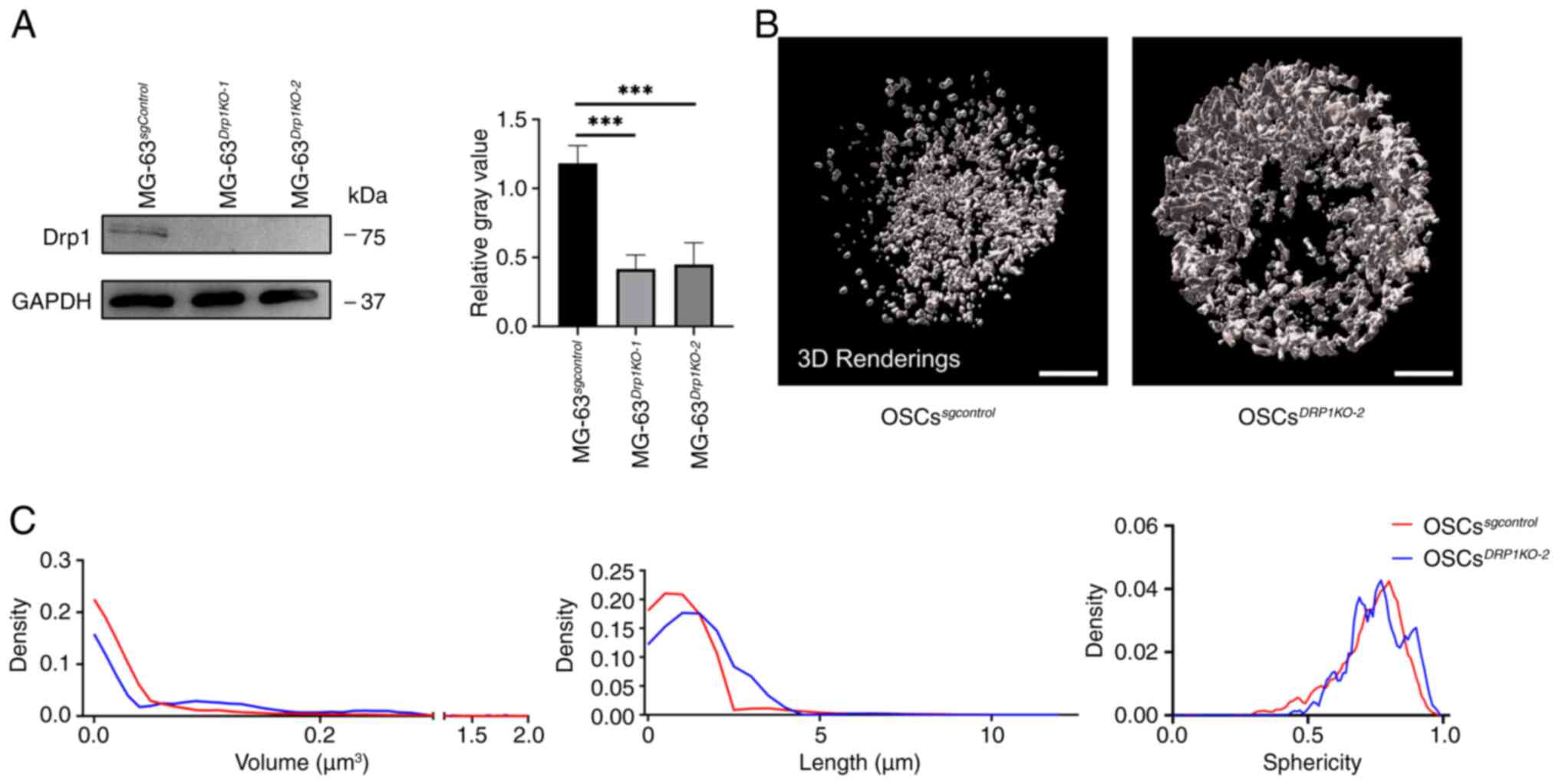
sgRNA sequences to knock out DRP1 (OSCsDRP1-KO,
and OSCsDRP1-KO2) and western blotting was
employed to determine the knockout efficiency (Fig. 5A). The two sgRNA sequences
demonstrated high knockdown efficiency and
OSCsDRP1-KO2 was selected for further
experiments. After sequencing MG-63DRP1-KO2, a
4-base pair deletion in exon 2 of the DRP1 gene was
identified, which caused a frameshift mutation that led to the
production of a premature stop codon in exon 5, truncating the
protein. This mutation affected the GTPase domain, which is crucial
for GTP binding and hydrolysis, both of which are essential for the
protein's role in mitochondrial and peroxisomal fission (Fig. S1). The mitochondrial morphology
in OSCsDRP1-KO2 showed increased fusion (Fig. 5B). Further quantitative analysis
revealed an increase in mitochondrial volume and length in
OSCsDRP1-KO2 compared with the
OSCssgcontrol. Additionally, the cellular
sphericity distribution did not show any significant change
(Fig. 5C). These findings
indicate that DRP1 is essential for regulating mitochondrial
fission and fusion processes and knockdown of DRP1 leads to
significant alterations in the morphology of the mitochondrial
network.
DRP1 affects OSC chemoresistance
To investigate the relationship between changes in
mitochondrial morphology and chemoresistance,
OSCsDRP1-KO2 and OSCssgcontrol
were treated with CIS or DOX at various concentrations. The results
indicated that half-maximal inhibitory concentration
(IC50) was significantly decreased in
OSCsDRP1-KO2 following the treatments with CIS or
DOX (Fig. 6A). TUNEL staining
for apoptosis revealed a significant increase in fluorescence in
the OSCsDRP1-KO2 compared with the
OSCssgcontrol treated with CIS or DOX (Fig. 6B). Furthermore, parameters
representing mitochondrial function were assessed.
OSCsDRP1-KO2 exhibited a significant decrease in
average intracellular ATP production (Fig. 6C) following CIS or DOX treatment
compared with the untreated group. Intracellular
NADPH/(NADP++NADPH) in OSCsDrp-1KO2
showed a significant downregulation following CIS or DOX treatment
compared with the untreated group (Fig. 6D). Additionally, knocking out
DRP1 in OSCs led to significant intracellular ROS accumulation and
reduced chemoresistance following CIS and DOX treatment compared
with OSCssgcontrol (Fig. 6E). These results underscore the
critical role of DRP1 in maintaining chemoresistance and
mitochondrial function in OSCs.
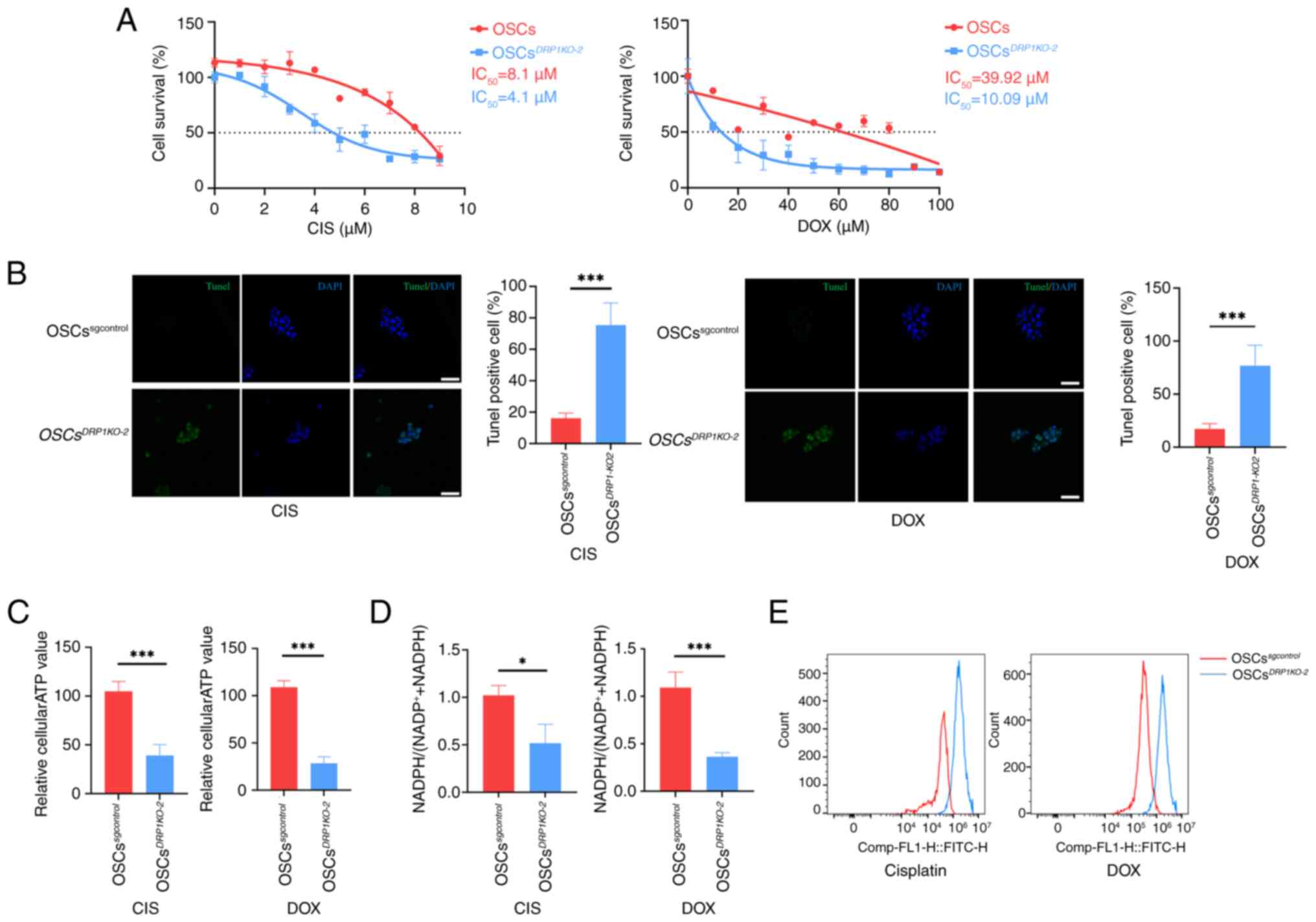 | Figure 6DRP1 affects OSCs chemoresistance.
(A) Survival curves and IC50 statistics for OSCs and
OSCsDRP1KO-2 cells treated with CIS and DOX. (B)
TUNEL staining of apoptosis in OSCs and
OSCsDRP1KO-2 treated with CIS and DOX and
statistics of TUNEL positive cells; green: TUNEL, blue: DAPI, scale
bar, 50 μm). (C) Relative intracellular ATP generation per
cell in OSCs and OSCsDRP1KO-2 treated with CIS or
DOX. (D) Relative intracellular NADPH/(NADP++NADPH)
assay in OSCs and OSCsDRP1KO-2 treated with CIS
or DOX. (E) ROS labeling with DCFH-DA in OSCs and
OSCsDRP1KO-2 treated with CIS or DOX. Data are
presented as the mean ± SD of three independent experiments (n=3);
*P<0.05, ***P<0.001. DRP1,
dynamin-related protein 1; OSCs, osteosarcoma stem cells;
IC50, half-maximal inhibitory concentration; CIS,
cisplatin; DOX, doxorubicin; ROS, reactive oxygen species. |
Discussion
Chemoresistance in cancer cells is a major cause of
poor prognosis in patients. Our previous studies have shown that
non-OSCs can be reprogrammed into OSCs, which subsequently acquire
higher chemoresistance (23,24,31). However, the mechanisms underlying
this increased chemoresistance in OSCs remain unclear. The present
study discovered a potential mechanism of chemoresistance in
osteosarcoma cells. OSCs counteract apoptosis and mitochondrial
dysfunction induced by chemotherapy by maintaining mitochondrial
homeostasis through the fission gene DRP1, suggesting a potential
target for eliminating OSCs.
Cancer stem cells (CSCs) demonstrate significantly
enhanced chemoresistance compared with non-CSCs, attributed to
multiple complex mechanisms (32). Elevated expression of ATP-binding
cassette (ABC) transporters (33) and augmented DNA damage repair
capabilities were notable (34).
Additionally, enhanced chemoresistance is attributed to alterations
in the expression levels of anti-apoptotic proteins, such as Bcl-2
and BAX (35), along with the
reprogramming of metabolic pathways (36). As well as these mechanisms, the
present study investigated the mechanisms of chemoresistance in
OSCs from the perspective of mitochondrial stability. OSCs
exhibited more stable mitochondrial homeostasis compared with
non-OSCs, both in terms of morphology and function. Cancer cells
with enlarged and fused mitochondria typically demonstrate
heightened chemoresistance through the maintenance of elevated ATP
levels and the mitigation of oxidative stress (37). The present study demonstrated
that OSCs were able to maintain stable ATP and NADPH production
under CIS or DOX-induced stress, whereas non-OSCs could not. Vlashi
et al (38) similarly
demonstrated metabolic differences between CSCs and non-CSCs in
glioma; CSCs were able to maintain higher ATP levels under stress
and exhibited enhanced mitochondrial repair capabilities. The
accumulation of ROS could induce apoptosis by causing a decrease in
mitochondrial membrane potential, leading to the release of
cytochrome c from the mitochondria into the cytosol, which
then triggered a cascade of apoptotic events. Another study found
that breast cancer CSCs could resist radiotherapy by clearing ROS
through the synthesis of glutathione genes, thereby inhibiting
apoptosis (39). The present
study found that OSCs were able to resist chemotherapeutics by
preventing the accumulation of ROS by maintaining mitochondrial
homeostasis, which further elucidated the mechanism behind the
difficulty of targeting CSCs for elimination in clinical
settings.
Regarding mitochondrial morphology, there are few
widely recognized conclusions about the differences between CSCs
and non-CSCs and the results from various studies remained
controversial. Civenni et al (40) indicated that BRD4 promotes
mitochondrial fission and sustains the survival of CSCs by
regulating the expression of the mitochondrial fission factor.
Inhibiting BRD4 impedes mitochondrial fission, leading to
mitochondrial dysfunction and the senescence and exhaustion of
CSCs. The same conclusions were validated in brain tumor-initiating
cells (BTICs), where BTICs exhibited a more fragmented
mitochondrial morphology compared with non-BTICs, indicating
increased mitochondrial fission within BTICs. DRP1, a key mediator
protein of mitochondrial fission, was activated in BTICs but
inhibited in non-BTICs (41).
However, mitochondrial dynamics in breast cancer involve different
mechanisms. Wu et al (42) indicate that
epithelial-mesenchymal transition (EMT) promotes mitochondrial
fusion by upregulating the expression of MFN1, enhancing
antioxidant capacity and thus sustaining the self-renewal and
expansion of CSCs. The study demonstrated that mitochondrial
dynamics in breast CSCs tend towards fusion. Research in esophageal
squamous cell carcinoma demonstrated that CSCs induced
mitochondrial fission through the activation of the key autophagy
protein Parkin, which is activated by EMT and leads to mitophagy
(43). The aforementioned
results indicate that mitochondrial morphology is complex in CSCs
across different tissues and can be regulated by a variety of
mechanisms. The findings of the present study revealed significant
disparities in mitochondrial morphology between non-OSCs and OSCs.
Notably, the mitochondria in OSCs exhibited a higher degree of
fusion, resulting in a more interconnected mitochondrial network
compared with non-OSCs. Notably, the mitochondrial network with
more interconnection showed more stable homeostasis under the
stress of chemotherapy CIS and DOX. This increased mitochondrial
fusion helped reduce chemosensitivity in OSCs, thereby promoting
chemoresistance. Moreover, the regulation of this mitochondrial
network was facilitated by the expression of the mitochondrial
fission protein DRP1. The present study revealed the critical role
of DRP1 in OSCs, particularly in maintaining their chemoresistance
through the regulation of mitochondrial dynamics. The high
expression of DRP1 is crucial for sustaining the proliferation and
survival of OSCs, making it a potential therapeutic target. These
findings provide a significant basis for the future development of
cancer treatment strategies targeting DRP1.
CIS and DOX can both induce DNA damage. CIS forms
DNA cross-links, inhibiting DNA replication and transcription,
while DOX intercalates into DNA, disrupting the function of
topoisomerase II and leading to DNA strand breaks (44). In the present study, OSCs
exhibited no differences in resistance to these two chemicals,
suggesting that mitochondrial dynamics may involve a similar
mechanism and resistance effect in countering chemotherapeutics
that induce apoptosis through DNA damage. The results supplemented
the finding on mitochondrial dynamics between OSCs and non-OSCs,
offering a new direction for further research into the
chemoresistance of OSCs. In fact, the role of MFN1 in the
chemoresistance of OSCs was also detected. However, MFN1 was
expressed at a very low level in osteosarcoma (data not shown).
Live-cell 3D imaging presented challenges due to
various limitations, including probe photobleaching, rapid
mitochondrial mobility and, most critically, phototoxicity,
especially for suspension cells. Phototoxic effects accumulate from
repeated scanning during Z-stacking, leading to continuous swelling
artifacts. Moreover, the results displayed the 3D mitochondrial
structure of OSCs, suspension cells, without any occurrence of
phototoxicity or continuous swelling artifacts. Another classical
approach to studying mitochondrial dynamics involves the use of
electron microscopy, which, although offering high resolution, can
only observe fixed cells and cannot replicate the mitochondrial
state in living cells. By contrast, the method of the present study
allowed for the observation of mitochondria in living cells and
enabled continuous imaging to monitor dynamic changes in the
mitochondrial network. The present study provided a new
experimental approach for exploring changes in mitochondrial
dynamics within live cells.
In conclusion, the present study showed a novel
mechanism of chemoresistance in OSCs from a mitochondrial dynamics
perspective. This provided new insights into chemoresistance in
CSCs and suggested potential therapeutic targets for the
elimination of OSCs in clinical settings.
Supplementary Data
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
BT performed the experiments, analyzed the data,
conceived the study, wrote the original draft, reviewed and edited
the manuscript. YW performed the experiments, analyzed the data,
wrote the original draft, reviewed and edited the manuscript. XD
performed the experiments. YZ wrote, reviewed and edited the
manuscript, supervised and administered the project and was
responsible for funding acquisition.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no conflicts of
interest.
Acknowledgements
The authors appreciate the support of Ms. Jialing Xu
(Core Facilities of Life Sciences, School of Life Sciences, Sun
Yat-sen University, Guangdong, China) for equipment support and
technical assistance.
Funding
The present study was supported by a grant from the Programs of
Guangdong Science and Technology (grant no. 2019B1515210015), China
Postdoctoral Science Foundation (grant no. 2023M744083) and
National Natural Science Foundation of China (grant no.
31871413).
References
|
1
|
Friedman JR and Nunnari J: Mitochondrial
form and function. Nature. 505:335–343. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Labbé K, Murley A and Nunnari J:
Determinants and functions of mitochondrial behavior. Annu Rev Cell
Dev Biol. 30:357–391. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Xing J, Qi L, Liu X, Shi G, Sun X and Yang
Y: Roles of mitochondrial fusion and fission in breast cancer
progression: A systematic review. World J Surg Oncol. 20:3312022.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Giacomello M, Pyakurel A, Glytsou C and
Scorrano L: The cell biology of mitochondrial membrane dynamics.
Nat Rev Mol Cell Biol. 21:204–224. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Wai T and Langer T: Mitochondrial dynamics
and metabolic regulation. Trends Endocrinol Metab. 27:105–117.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Eisner V, Picard M and Hajnóczky G:
Mitochondrial dynamics in adaptive and maladaptive cellular stress
responses. Nat Cell Biol. 20:755–765. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Vyas S, Zaganjor E and Haigis MC:
Mitochondria and Cancer. Cell. 166:555–566. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Boulton DP and Caino MC: Mitochondrial
fission and fusion in tumor progression to metastasis. Front Cell
Dev Biol. 10:8499622022. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Quintana-Cabrera R and Scorrano L:
Determinants and outcomes of mitochondrial dynamics. Mol Cell.
83:857–876. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Chan DC: Mitochondrial dynamics and its
involvement in disease. Annu Rev Pathol. 15:235–259. 2020.
View Article : Google Scholar
|
|
11
|
Rodrigues T and Ferraz LS: Therapeutic
potential of targeting mitochondrial dynamics in cancer. Biochem
Pharmacol. 182:1142822020. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zacharioudakis E and Gavathiotis E:
Mitochondrial dynamics proteins as emerging drug targets. Trends
Pharmacol Sci. 44:112–127. 2023. View Article : Google Scholar :
|
|
13
|
Kumar S, Ashraf R and C KA: Mitochondrial
dynamics regulators: Implications for therapeutic intervention in
cancer. Cell Biol Toxicol. 38:377–406. 2022. View Article : Google Scholar
|
|
14
|
Zeng X, Zhang YD, Ma RY, Chen YJ, Xiang
XM, Hou DY, Li XH, Huang H, Li T and Duan CY: Activated Drp1
regulates p62-mediated autophagic flux and aggravates inflammation
in cerebral ischemia-reperfusion via the ROS-RIP1/RIP3-exosome
axis. Mil Med Res. 9:252022.PubMed/NCBI
|
|
15
|
Chuang KC, Chang CR, Chang SH, Huang SW,
Chuang SM, Li ZY, Wang ST, Kao JK, Chen YJ and Shieh JJ:
Imiquimod-induced ROS production disrupts the balance of
mitochondrial dynamics and increases mitophagy in skin cancer
cells. J Dermatol Sci. 98:152–162. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Jiang Y, Krantz S, Qin X, Li S, Gunasekara
H, Kim YM, Zimnicka A, Bae M, Ma K, Toth PT, et al: Caveolin-1
controls mitochondrial damage and ROS production by regulating
fission-fusion dynamics and mitophagy. Redox Biol. 52:1023042022.
View Article : Google Scholar
|
|
17
|
Wu Z, Xiao C, Long J, Huang W, You F and
Li X: Mitochondrial dynamics and colorectal cancer biology:
Mechanisms and potential targets. Cell Commun Signal. 22:912024.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Harrington JS, Ryter SW, Plataki M, Price
DR and Choi AMK: Mitochondria in health, disease and aging. Physiol
Rev. 103:2349–2422. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhang Y, Mai Q, Zhang X, Xie C and Zhang
Y: Microenvironment signals and mechanisms in the regulation of
osteosarcoma. Osteosarcoma-Biology, Behavior and Mechanisms. Honoki
K and Weiss KR: InTech; 2017, View
Article : Google Scholar
|
|
20
|
Tian B, Du X, Zheng S and Zhang Y: The
role of tumor microenvironment in regulating the plasticity of
osteosarcoma cells. Int J Mol Sci. 23:161552022. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Arima Y, Nobusue H and Saya H: Targeting
of cancer stem cells by differentiation therapy. Cancer Sci.
111:2689–2695. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Batlle E and Clevers H: Cancer stem cells
revisited. Nat Med. 23:1124–1134. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhang Y, Pan Y, Xie C and Zhang Y: miR-34a
exerts as a key regulator in the dedifferentiation of osteosarcoma
via PAI-1-Sox2 axis. Cell Death Dis. 9:7772018. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Pan Y and Zhang Y, Tang W and Zhang Y:
Interstitial serum albumin empowers osteosarcoma cells with FAIM2
transcription to obtain viability via dedifferentiation. In Vitro
Cell Dev Biol Anim. 56:129–144. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
26
|
Youle RJ and Karbowski M: Mitochondrial
fission in apoptosis. Nat Rev Mol Cell Biol. 6:657–663. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Sheridan C and Martin SJ: Mitochondrial
fission/fusion dynamics and apoptosis. Mitochondrion. 10:640–648.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Fulda S: Regulation of apoptosis pathways
in cancer stem cells. Cancer Lett. 338:168–173. 2013. View Article : Google Scholar
|
|
29
|
Vasileiou PVS, Evangelou K, Vlasis K,
Fildisis G, Panayiotidis MI, Chronopoulos E, Passias PG,
Kouloukoussa M, Gorgoulis VG and Havaki S: Mitochondrial
homeostasis and cellular senescence. Cells. 8:6862019. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Lin W, Lu X, Yang H, Huang L, Huang W,
Tang Y, Liu S, Wang H and Zhang Y: Metabolic heterogeneity protects
metastatic mucosal melanomas cells from ferroptosis. Int J Mol Med.
50:1242022. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Zhang H, Wu H, Zheng J, Yu P, Xu L, Jiang
P, Gao J, Wang H and Zhang Y: Transforming growth factor β1 signal
is crucial for dedifferentiation of cancer cells to cancer stem
cells in osteosarcoma. Stem Cells. 31:433–446. 2013. View Article : Google Scholar
|
|
32
|
Chu X, Tian W, Ning J, Xiao G, Zhou Y,
Wang Z, Zhai Z, Tanzhu G, Yang J and Zhou R: Cancer stem cells:
Advances in knowledge and implications for cancer therapy. Signal
Transduct Target Ther. 9:1702024. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Li Y, Wang Z, Ajani JA and Song S: Drug
resistance and Cancer stem cells. Cell Commun Signal. 19:192021.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Garcia-Mayea Y, Mir C, Masson F, Paciucci
R and LLeonart ME: Insights into new mechanisms and models of
cancer stem cell multidrug resistance. Semin Cancer Biol.
60:166–180. 2020. View Article : Google Scholar
|
|
35
|
Zheng Q, Zhang M, Zhou F, Zhang L and Meng
X: The breast cancer stem cells traits and drug resistance. Front
Pharmacol. 11:5999652021. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
De Angelis ML, Francescangeli F, La Torre
F and Zeuner A: Stem cell plasticity and dormancy in the
development of cancer therapy resistance. Front Oncol. 9:6262019.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Genovese I, Carinci M, Modesti L, Aguiari
G, Pinton P and Giorgi C: Mitochondria: Insights into crucial
features to overcome cancer chemoresistance. Int J Mol Sci.
22:47702021. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Vlashi E, Lagadec C, Vergnes L, Matsutani
T, Masui K, Poulou M, Popescu R, Della Donna L, Evers P, Dekmezian
C, et al: Metabolic state of glioma stem cells and nontumorigenic
cells. Proc Natl Acad Sci USA. 108:16062–16067. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Diehn M, Cho RW, Lobo NA, Kalisky T, Dorie
MJ, Kulp AN, Qian D, Lam JS, Ailles LE, Wong M, et al: Association
of reactive oxygen species levels and radioresistance in cancer
stem cells. Nature. 458:780–783. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Civenni G, Bosotti R, Timpanaro A, Vàzquez
R, Merulla J, Pandit S, Rossi S, Albino D, Allegrini S, Mitra A, et
al: Epigenetic control of mitochondrial fission enables
self-renewal of stem-like tumor cells in human prostate cancer.
Cell Metab. 30:303–318.e6. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Xie Q, Wu Q, Horbinski CM, Flavahan WA,
Yang K, Zhou W, Dombrowski SM, Huang Z, Fang X, Shi Y, et al:
Mitochondrial control by DRP1 in brain tumor initiating cells. Nat
Neurosci. 18:501–510. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Wu MJ, Chen YS, Kim MR, Chang CC, Gampala
S, Zhang Y, Wang Y, Chang CY, Yang JY and Chang CJ:
Epithelial-Mesenchymal transition directs stem cell polarity via
regulation of mitofusin. Cell Metab. 29:993–1002 e6. 2019.
View Article : Google Scholar
|
|
43
|
Whelan KA, Chandramouleeswaran PM, Tanaka
K, Natsuizaka M, Guha M, Srinivasan S, Darling DS, Kita Y, Natsugoe
S, Winkler JD, et al: Autophagy supports generation of cells with
high CD44 expression via modulation of oxidative stress and
Parkin-mediated mitochondrial clearance. Oncogene. 36:4843–4858.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Duyndam MC, van Berkel MP, Dorsman JC,
Rockx DA, Pinedo HM and Boven E: Cisplatin and doxorubicin repress
Vascular Endothelial Growth Factor expression and differentially
down-regulate Hypoxia-inducible Factor I activity in human ovarian
cancer cells. Biochem Pharmacol. 74:191–201. 2007. View Article : Google Scholar : PubMed/NCBI
|