Introduction
Lung cancer ranks among the most frequently
diagnosed cancers worldwide, affecting over 2 million patients each
year (1). A total of ~90% of
these patients are diagnosed with non-small cell lung cancer
(NSCLC). Lung adenocarcinoma (LUAD) is the predominant subtype of
NSCLC, accounting for ~40% of all lung cancer cases. Notably, the
epidermal growth factor receptor (EGFR) mutation is the most
prevalent oncogenic mutation in LUAD, particularly among
individuals of East Asian descent, women and non-smokers (2). At present, the primary approach for
treating NSCLC revolves around identifying targetable driver
mutations and immune checkpoints. However, patients who do not
possess these characteristics are left with limited options, such
as chemotherapy and radiotherapy, but with severe side effects and
unpredictable outcomes. Therefore, the pursuit of more-effective
treatments for patients lacking these features remains a paramount
concern (3).
The repurposing of well-characterized and
well-tolerated drugs for cancer therapy has emerged as an appealing
alternative to the lengthy and expensive process of new drug
development. In recent years, there has been a growing trend of
repurposing non-anticancer medications for treating cancers, such
as the antidiabetic drugs, metformin and thiazolidinediones, for
treating various cancers (4) and
the antibiotic, nitroxoline, for treating pancreatic cancer
(5). Repurposing offers the
potential to discover novel treatments for diseases more
cost-effectively and with shorter development timelines. This is
especially valuable when preclinical safety data are already
available, allowing for the swift evaluation of new therapeutic
applications in clinical trials (6).
Tumors maintain an inflammatory microenvironment,
and anti-inflammatory medications have demonstrated potential in
cancer therapy. Loratadine and its primary metabolite,
desloratadine, are potent antagonists of the human histamine
receptor H1 (HRH1) and were originally intended to treat allergies
and allergic rhinitis (7).
Previously, multiple studies reported improved survival associated
with the use of loratadine or desloratadine in patients with
melanomas (8) and breast cancer
(9). In addition to those cancer
types, loratadine and desloratadine also showed consistent
associations with improved prognoses, especially in other
immunogenic tumors, including gastric, colorectal, pancreatic,
lung, prostate, kidney and bladder cancers, as well as the
non-solid tumor, Hodgkin lymphoma (10). Apart from clinical assessments of
loratadine's impact on the prognosis of patients with cancer,
several in vitro and in vivo studies demonstrated the
anticancer potential of loratadine and other H1-antihistamines on
various cancer cells. For instance, Chen et al (11) indicated that the combination of
thioridazine and loratadine displayed favorable
anti-gastrointestinal cancer effects by inducing cell apoptosis
(11). Desloratadine was shown
to inhibit the growth and invasion of bladder cancer cells by
suppressing the epithelial-to-mesenchymal transition and
interleukin-6 expression (12).
Additionally, another H1-antihistamine, terfenadine, was reported
to induce apoptosis and autophagy in melanoma cells (13). In contrast to those tumor types,
the anticancer potential and the underlying mechanisms of
loratadine against NSCLC remain unknown. Transforming growth factor
β-activated kinase 1 (TAK1) acts as an upstream kinase for both
NF-κB and c-Jun N-terminal kinase (JNK)-activator protein-1 (AP-1)
signaling pathways (14) and has
been implicated in the anti-inflammatory effects of loratadine
(15). These signaling pathways
have also been associated with poor prognosis, advanced clinical
stages and metastasis in NSCLC (16), indicating that loratadine may
hold therapeutic potential for NSCLC treatment.
The current study explored loratadine's potential
anticancer properties in various LUAD cell lines. These cell lines
carry either the wild-type (WT) or mutant EGFR. The study delved
into the mechanisms underlying these effects in both in
vitro experiments and a subcutaneous xenograft model. Our
findings indicated that loratadine effectively reduced cell
viability and proliferation in various LUAD cell lines, both in
vitro and in vivo, suggesting its potential as an
anticancer agent for lung cancer therapy. Additionally, it was
observed that loratadine promoted autophagy-mediated apoptotic cell
death by suppressing the activation of signal transducer and
activator of transcription 3 (STAT3), JNK and p38 through
phosphatase 2A (PP2A)-dependent or -independent mechanisms.
Materials and methods
Data collection from bioinformatics
analyses
The PRECOG online database (https://precog.stanford.edu/, accessed on Oct. 12,
2023) showed the survival meta-Z score of HRH1 expression among 37
types of cancer. The Depmap portal online database (https://depmap.org/portal/; accessed on Apr. 29, 2022)
presented the correlation between the HRH1 expression in lung
cancer cell lines and the cytotoxicity of loratadine (17). The UALCAN online database
(http://ualcan.path.uab.edu, accessed on
Nov. 10, 2023) (18) was
utilized to compute protein expression levels of PP2A-C, along with
clinicopathologic parameters such as the clinical stage and tumor
grade in LUAD, within the Clinical Proteomic Tumor Analysis
Consortium (CPTAC) dataset. The Kaplan-Meier (KM) plotter database
(https://kmplot.com/analysis/, accessed
on Nov. 8, 2023) was employed to assess the impact of PP2A levels
on the survival of LUAD subjects, utilizing data obtained from Gene
Expression Omnibus (https://www.ncbi.nlm.nih.gov/geo/), including
GSE102287, GSE14814, GSE19188, GSE29013, GSE30219, GSE31210,
GSE31908, GSE37745, GSE43580, GSE50081, GSE77803, GSE8894, GSE68465
and GSE3141; and The Cancer Genome Atlas (https://www.cancer.gov/ccg/research/genome-sequencing/tcga),
TCGA-LUAD, datasets. Patients were categorized into two groups
based on high and low expression levels of PP2A-C, as determined by
the best cutoff values for gene expression.
Chemicals and materials
Loratadine (cat. no. 15625), desloratadine (cat. no.
16931), fexofenadine (cat. no. 18191), and the PP2A inhbitor,
okadaic acid (OA; cat. no. 10011490) were purchased from Cayman
Chemical Company. Histamine (cat. no. H7125), chloroquine (CQ; cat.
no. C6628) and acridine orange (AO; cat. no. A9231) were obtained
from MilliporeSigma. Antibodies of cleaved-PARP (cat. no. 5625),
PARP (cat. no. 9532), cleaved-caspase-3 (cat. no. 9664), caspase-3
(cat. no. 9665), cleaved-caspase-8 (cat. no. 8582),
cleaved-caspase-9 (cat. no. 7237), caspase-9 (cat. no. cs9508), p62
(cat. no. 5114), LC3 (cat. no. 4108), PP2A-C (cat. no. 2259),
phosphorylated (p)-STAT3 (Tyr705; cat. no. 9145), STAT3 (cat. no.
9139), p-JNK (cat. no. 9251), p-extraceellular signal-regulated
kinase (ERK; cat. no. 4370), ERK (cat. no. 4695), p-p38 (cat. no.
4511) and p38 (cat. no. 9212) were obtained from Cell Signaling
Technology, Inc. HRH1 (cat. no. sc-374621), beclin (cat. no.
sc-48341), JNK (cat. no. sc-7345) and GAPDH (cat. no. sc-32233)
antibodies were obtained from Santa Cruz Biotechnology, Inc. An
antibody specific for the phosphorylated form of PP2A-Cα (Tyr307)
was purchased from Thermo Fisher Scientific, Inc. Lentiviral soups
of small hairpin (sh)HRH1 were obtained from the RNA Technology
Platfrom and Gene Manipulation Core at Academic Sinica. Targeting
sequences of shRNAs were as follows: HRH1 shRNA-1:
GCTCTGGTTCTATGCCAAGAT and HRH1 shRNA-2: CCTCTGCTGGATCCCTTATTT.
Cell lines and cell culture
The study utilized five LUAD cell lines. A549, H23,
H1975 and HCC827 were purchased from the American Type Culture
Collection (ATCC). PC9 cells were obtained from the National Cancer
Center Hospital (Tokyo, Japan). Cancer cell lines were cultured in
RPMI-1640 medium with 10% fetal bovine serum (Gibco; Thermo Fisher
Scientific, Inc.) and 1% penicillin-streptomycin-glutamine (Thermo
Fisher Scientific, Inc.). Incubation for all cell types occurred at
37°C in a humidified incubator with 5% CO2. All cell
lines were verified by short tandem repeat profiling analysis
(Mission Biotech, Co., Ltd.). Cells were regularly checked for
mycoplasma infection using EZ-PCR mycoplasma detection kit
(Sartorius AG). All cell lines were performed with mycoplasma-free
cells.
Cell survival assay
In total, 3,000 HRH1-knockdown or control LUAD cells
were seeded into single wells of a 96-well plate after treating
cells with varying concentrations of loratadine or desloratadine
for either 24 or 48 h. Cell viability was assessed using 100
μl medium contain 10% Cell Counting Kit-8 (CCK-8; cat. no.
96992; MilliporeSigma) in a well. The assay was performed at 37°C,
and absorbance (OD 450 nm) was measured every 1 h for up to 4 h.
All measurements were performed using a CLARIOstar plate reader
(BMG Labtech GmbH).
Plate colony-forming assay
LUAD cells were initially seeded in six-well plates
at a density of 103 cells/well and allowed to incubate
for 24 h. Following this, cells were treated with loratadine or
desloratadine for an additional 24 h. Subsequently, the culture
medium was refreshed every 2 days. After 7~14 days of incubation
until a colony contained >50 cells, cells were fixed with 4%
paraformaldehyde at room temperature for 20 min, and then stained
with 0.1% crystal violet at room temperature for 30 min. Images of
violet-stained colonies were captured and quantified by dissolving
the violet stain in 10% acetic acid. To remove the stain, plates
were placed on a shaker for 15 min, and the destaining solution was
measured using a iMark™ microplate absorbance reader (Bio-Rad
Laboratories, Inc.) at 595 nm.
Western blot assay
Proteins were extracted using a PRO-PREP protein
extraction solution (iNtRON Biotechnology, Inc.), and protein
concentrations were determined as previously described (19). In total, 20 μg of protein
lysate was loaded into individual wells of 10% SDS-PAGE, and the
entire gel was subsequently transferred onto a PVDF membrane
(Bio-Rad Laboratories, Inc.). The blocking reagent (WesternF1;
LionBIO, Inc.) was applied at room temperature for 1 min. The
primary antibody was diluted at a 1:1,000 ratio in 1% BSA TBST
buffer (0.1% Tween-20) and incubated overnight at 4°C. The
anti-rabbit/mouse secondary antibody (cat. no. 12-348/12-349;
MilliporeSigma) was diluted at a 1:2,000 ratio in TBST buffer and
incubated for 1 h at room temperature. Separated proteins were then
probed with respective antibodies and visualized using an enhanced
chemiluminescence (ECL) system (Pierce Biotechnology; Thermo Fisher
Scientific, Inc.). The intensity of band was measured by ImageJ
version 1.54f (National Institutes of Health).
Analysis of the sub-G1
phase
In total, 3.0×105 LUAD cells were
initially seeded in 6-cm dishes. Following overnight incubation, 10
and 30 μM of loratadine were introduced. Cells were
collected 24 h after treatment, fixed using 70% ice-cold ethanol
for 5 min on ice, and then stained with a propidium iodide (PI)
staining solution at room temperature for 1 h in the dark. Stained
cells were left at room temperature in the dark for 0.5 h.
Subsequently, flow cytometry (CytoFLEX; Beckman Coulter, Inc.) was
employed to measure the cells, and their distribution into
different intensities indicated the DNA content in distinct phases
of the cell cycle. The sub-G1 phase was identified as
the population with a DNA intensity lower than the threshold
corresponding to the lowest DNA intensity in the
G0/G1 peak. Each experiment was replicated
three times. The analysis was proceeded by CytExpert 2.0 software
(Beckman Coulter, Inc).
Terminal deoxynucleotide transferase dUTP
nick end labeling (TUNEL) stain
In total, 3×105 LUAD cells were initially
placed in 6-cm dishes. Following overnight incubation, 10 and 30
μM of loratadine were introduced. Cells were collected at 24
h after treatment, fixed with 4% paraformaldehyde at room
temperature for 20 min, and then treated with 70% ice-cold ethanol
for 5 min on ice. Subsequently, they were digested with proteinase
K (20 μg/ml). Terminal ends of DNA fragments in dead cells
were labeled with BrdU using the terminal deoxynucleotidyl
transferase (TdT) enzyme. To detect signals, a fluorescein-labeled
anti-BrdU antibody (Enzo Life Sciences, Inc.) was utilized, and
measurements were taken using flow cytometry (CytoFLEX) and
analyzed by CytExpert 2.0 software. Tumor tissue slides intended
for TUNEL staining underwent dewaxing with Neo-Clear®
(MilliporeSigma) and subsequent rehydration with a series of
ethanol solutions. The staining procedure then followed the same
steps as those used for cell staining after ethanol fixation.
Detection of acidic vesicular organelles
(AVOs) via AO staining
Autophagy is the process of enclosing cytoplasmic
proteins within a lytic compartment, marked by the formation and
buildup of AVOs. To visualize these acidic cellular compartments,
AO was employed, which emits bright-red fluorescence within acidic
vesicles while displaying green fluorescence in the cytoplasm and
nuclei. LUAD cells (3×105 per well) were plated in 6-cm
dishes and exposed to 10 or 30 μM loratadine for 24 h.
Following this, AO was introduced at a final concentration of 1
mg/ml for a 15-min incubation period at room temperature.
Subsequently, images were captured using a fluorescence microscope
(Zeiss AG).
Lentiviral production and infection
The second generation lentivirus system was obtained
from RNA Technology Platform and Gene Manipulation Core (Academia
Sinica). Three lentiviral packaging vectors were introduced into
293T packaging cells (ATCC) through a calcium phosphate
transfection method. To elaborate, 106 293T cells were
transfected with 10 μg of an HRH1 shRNA-expressing plasmid
(pLKO-shHRH1-puro), in conjunction with 10 μg of pCMVDR8.91
(the packaging vector) and 1 μg of pMD.G (the envelope
vector). Following a 16-h incubation at 37°C, transfection medium
was replaced with fresh culture medium. After 48 h, the medium
containing the lentivirus was collected from transfection, and cell
debris was separated by centrifugation at 380 × g for 5 min at room
temperature. The virus titer was measure by calculating the virus
particle to generate infected colony by selecting with 5
μg/ml puromycin. A multiplicity of infection of 5 was
applied for further experiments. Subsequently, LUAD cells were
exposed to the fresh lentivirus-containing medium, which was
supplemented with 8 μg/ml polybrene, for a 24-h period. The
knockdown efficiency was assessed using a reverse-transcription
quantitative polymerase chain reaction (RT-qPCR).
RT-qPCR
Total RNA was isolated from LUAD cells with
HRH1-knockdown using the TRIzol® reagent (Thermo Fisher
Scientific, Inc.). Subsequently, it was reverse-transcribed into
complementary (c)DNA using the iScript™ cDNA Synthesis kit (Bio-Rad
Laboratories, Inc.) according to the mnufacturer's protocol. The
generated cDNA was then utilized in an RT-qPCR, by applying the
SYBR qPCR supermix reagent (Bio-Rad Laboratories, Inc.). The
thermocycling condition was an initial denaturation at 95°C for 10
min and a 40 cycles of two steps PCR at 95°C for 15 sec and 60°C
for 30 sec, then a dissociation stage for melting curve analysis.
Results were recorded with a 7300 Real-Time PCR system (Applied
Biosystems; Thermo Fisher Scientific, Inc.) and the
2−ΔΔCq method was used for quantification (20). Primers used in the RT-qPCR are
listed as follows: HRH1 forward, 5′-GCC GAGAGGACAAGTGTGA-3′ and
reverse, 5′-GGAGACTCCTTCCCTGGTTT-3′; and GAPDH forward,
5′-CTGGAGAAACCTGCCAAGTATGAT-3′; and reverse,
5′-TTCTTACTCCTTGGAGGCCATGTA-3′.
In vivo xenograft model
In this xenograft tumor model, a total of 14
8-week-old non-obese diabetic (NOD)-SCID male mice were employed.
The body weight at the beginning was 20-25 g. Mice were housed in
an individually ventilated cage (IVC) system under controlled
conditions: Temperature maintained at 20°C, total air exchange,
relative humidity at 55±5%, a 12/12-h light/dark cycle, and with
ad libitum access to sterilized water and food. A total of 5
million PC9 cells were subcutaneously injected into the back of
each mouse in a 100-μl phosphate-buffered saline suspension.
Following cell transplantation, loratadine [10 mg/kg body weight
(BW)] was administered orally by gavage on a daily basis. There
were seven mice in each group. Treatment was administered on
weekdays, with weekends serving as rest days, until the conclusion
of the experiment. The tumor size, measured in cubic millimeters
with the formula 1/2 ab2 (where 'a' represents length
and 'b' represents width), was recorded weekly. Additionally, BWs
of the mice were recorded on a weekly basis. Each group consisted
of seven mice. At the end of the experiment, the mice were humanely
euthanized by CO2 asphyxiation within their home cages.
A controlled CO2 flow rate of 3 l/min was maintained for
at least 5 min to ensure complete and irreversible loss of
consciousness. This resulted in an air displacement rate of ~40% of
the chamber volume per min. Primary tumor specimens were resected,
images were captured, fixed, and sectioned for hematoxylin and
eosin (H&E) and immunohistochemical (IHC) staining. All animal
experiments were conducted in accordance (approval no.
wan-lac-110-026) with guidelines of the Institutional Animal Care
and Use Committee (IACUC) of Wan Fang Hospital (Taipei,
Taiwan).
IHC of tumor specimens
All tumor tissue samples were fixed in a 10%
buffered formaldehyde solution at room temperature overnight,
embedded in paraffin blocks, and cut into 5-μm sections.
Paraffin-embedded LUAD tissue sections were deparaffinized using
Neo-Clear® (MilliporeSigma) and rehydrated through a
gradient of ethanol concentrations. Subsequently, slides underwent
high-pressure incubation in antigen retrieval buffer (pH 6.0; DAKO;
Agilent Technologies, Inc.) for 30 min, followed by blocking with
super block buffer (ScyTek Laboratories, Inc.) for 1 h, then
overnight incubation at 4°C with the anti-Ki-67 (cat. no. 9027S;
Cell Signaling Technology, Inc.), anti-CD31 (cat. no. 77699S; Cell
Signaling Technology, Inc.), or anti-LC3 antibody. Primary
antibodies were diluted with with antibody dilution buffer (cat.
no. ADB250; Ventana; Roche Tissue Diagnostics) at 1:100 ratio.
Next, a HRP rabbit/mouse reagent was applied and incubated with the
slides at room temperature for 1 h, with the Dako REAL EnVision
system (cat. no. K5007; DAKO; Agilent Technologies, Inc.) being
used to visualize the binding of the antibody to the tissue.
Hematoxylin was used as a counterstain. Finally, the cover slides
were sealed, and slides were scanned using the light motic digital
pathology system, MoticEasyScan Pro 6.
Statistical analysis
The in vitro and in vivo study results
are presented as the mean ± standard deviation (SD). Pearson's
correlation coefficients were used to assess the relationship
between gene expression and drug sensitivity. Survival analysis was
conducted using the Kaplan-Meier method combined with the log-rank
test. The Z-value of PPP2CA indicates the SD from the median across
lung cancer samples, while log2 spectral count ratio values from
CPTAC were normalized within individual sample profiles and
subsequently across all samples. Comparisons involving more than
three groups and varying drug concentrations were analyzed using
ANOVA with Bonferroni post hoc tests. Comparisons between two
groups were performed using an unpaired, two-tailed Student's
t-test, with statistical significance threshold set at a
P<0.05.
Results
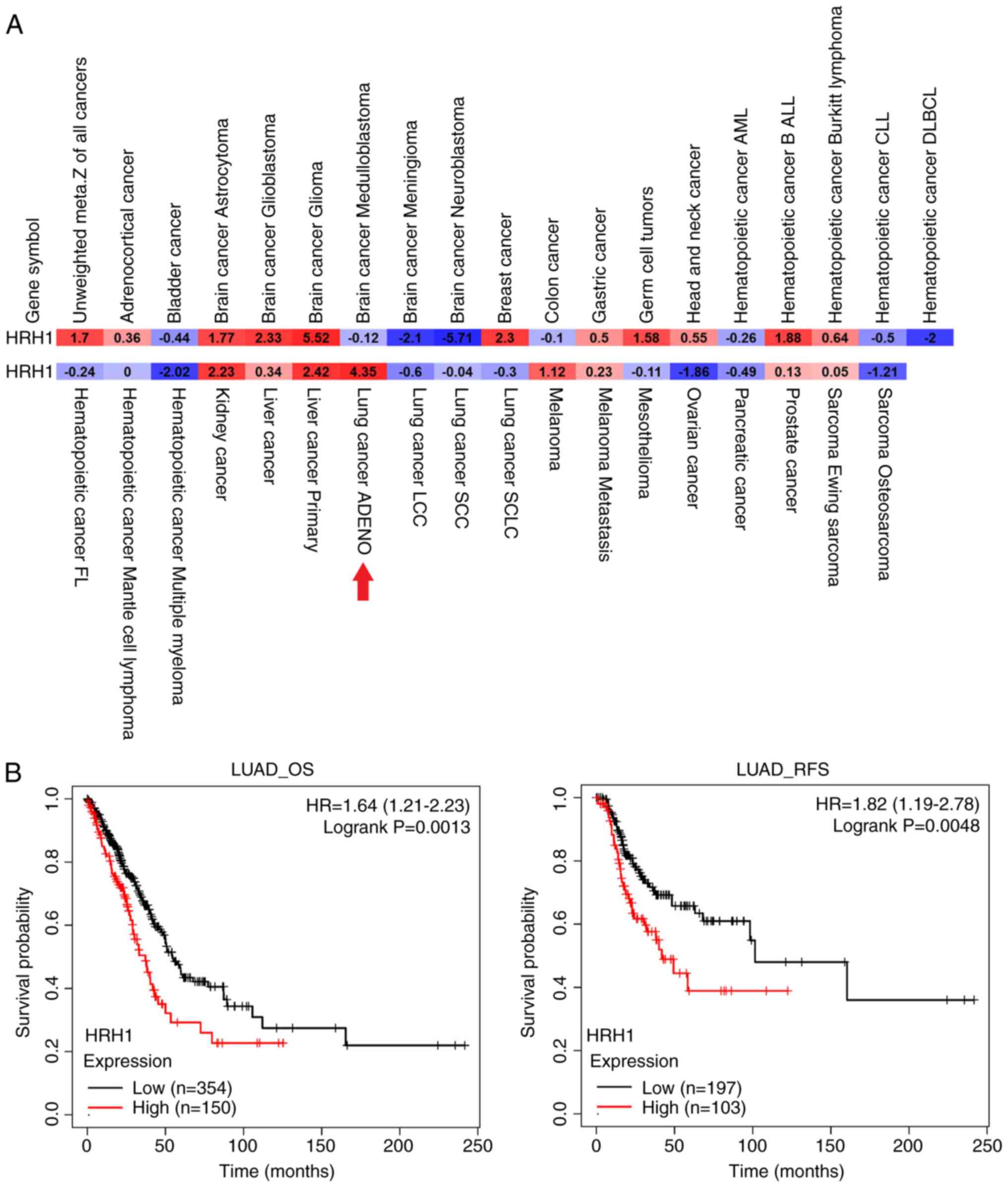
HRH1 expression is correlated with poor
prognoses in patients with LUAD
Because the use of HRH1 antagonists was reported to
reduce the risk of liver cancer development (21) and improve survival rates in
patients with melanoma and breast cancer (8,9),
an analysis of HRH1 expression levels and their prognostic
significance was initiated in 39 different cancer types using the
pan-cancer prognostic database, PRECOG. Results indicated that HRH1
exhibited prognostic potential for unfavorable outcomes in various
human cancers, including LUAD (Fig.
1A). Furthermore, an analysis of associations of HRH1
expression with overall survival (OS) and relapse-free survival
(RFS) was conducted in patients with LUAD using the KM plotter
(Fig. 1B). It was found that
higher levels of HRH1 were associated with poorer OS and RFS in
patients with LUAD. Collectively, these clinical data suggest that
HRH1 expression may promote the progression of LUAD, indicating
that HRH1 antagonists hold promise as a potential treatment for
LUAD.
HRH1 antagonists induce proliferation
inhibition and apoptotic cell death in human LUAD cells harboring
different EGFR statuses
To further explore the extensive therapeutic
potential of HRH1 antagonist loratadine in the context of LUAD, an
investigation was conducted into the impacts of various
concentrations of loratadine and its metabolite, desloratadine, on
the viability of LUAD cells with distinct EGFR statuses. These
statuses included WT EGFR in A549 and H23 cells, EGFR exon 19
deletion in PC9 and HCC827 cells, and L858R/T790M mutations in
H1975 cells. The present results revealed that both loratadine and
desloratadine exhibited cytotoxic effects on all tested LUAD cell
lines in the concentration range of 30~60 μM, as assessed by
a CCK-8 viability assay. Notably, loratadine exhibited a
more-potent inhibitory effect on the viability of LUAD cells
compared with desloratadine (Fig. 2A
and B). As a result, subsequent experiments were conducted
using a specific concentration range of loratadine. Unlike the
toxic effects observed on LUAD cells, loratadine treatment at the
same concentrations resulted in no significant toxicity or only
minor effects on BEAS-2B normal lung epithelial cells (data not
shown). The effect of loratadine on the long-term growth of LUAD
cells was further assessed through a colony-formation assay. This
assay revealed a concentration-dependent reduction in the number of
cancer cell colonies following loratadine treatment (Fig. 2C and D). These findings suggested
that loratadine holds promise as a therapeutic agent for managing
LUAD, in cases of both WT and mutant EGFR. To investigate the mode
of the antiproliferative effects induced by loratadine, H23 and PC9
cells were treated with 10 and 30 μM loratadine for 24 and
48 h. A flow cytometry-based cell cycle analysis revealed increased
accumulation of cells in the sub-G1 phase after 24 h of
treatment with 30 μM loratadine (Fig. 2E). Furthermore, DNA fragmentation
was evaluated using TUNEL staining and increased percentages of
TUNEL-positive cells were observed in H23 and PC9 cells treated
with 30 μM loratadine compared with control cells (Fig. 2F). Additionally, expression
levels of apoptotic markers, including cleaved caspase-8/-9/-3 and
PARP, were concentration-dependently induced after 24 h of
treatment with different loratadine concentrations (10~30
μM) (Fig. 2G). Results
collectively represented typical characteristics of apoptotic cell
death and underscored loratadine's ability to induce apoptosis in
LUAD cells.
The antiproliferative ability of
loratadine against LUAD cells is independent of HRH1
In addition to loratadine, the effects of another
commonly used HRH1 antagonist, fexofenadine, were tested on LUAD
proliferation. Surprisingly, it was observed that fexofenadine,
despite having a different chemical structure than loratadine, did
not exhibit a significant cytotoxic effect on various LUAD cell
lines, including A549, H23, H1975 and PC9 cells (Fig. 3A). Moreover, treatment of various
LUAD cell lines with the HRH1 ligand histamine at different
concentrations showed no significant impact on cell proliferation
rates, except for 10 μM histamine in H1975 cells. (Fig. 3B). Furthermore, it was observed
that there was no significant correlation (r=-0.439,
P=0.453) between the HRH1 protein expression level and 50%
inhibitory concentration (IC50) values of loratadine in
the LUAD cells tested (Fig. 3C).
To further validate the aforementioned results, a correlation
analysis was performed using DepMap to investigate the relationship
between HRH1 RNA expression in lung cancer cell lines and the
cytotoxicity of loratadine (Fig.
3D). The analysis revealed no significant correlation between
HRH1 expression and loratadine sensitivity (Pearson correlation
r=-0.111, P=0.291). These results suggested that
loratadine-induced cytotoxicity was independent of HRH1. To further
investigate whether the antiproliferative effect of loratadine was
mediated via HRH1, stable shHRH1 clones were established in various
LUAD cell lines with different EGFR statuses, including H23 and PC9
cells (Fig. S1A and B).
Regardless of whether cells expressed the control vector or HRH1
shRNAs, a similar trend of proliferation inhibition was observed
upon loratadine treatment. However, certain concentrations showed
significant differences compared with the control, such as 3 and 10
μM in H23-shHRH1-1 cells, 10 and 30 μM in
PC9-shHRH1-1 cells, and 30 μM in PC9-shHRH1-2 cells
(Fig. 3E and F). In contrast to
HRH1-knockdown, overexpressing HRH1 in A549 cells (Fig. S1C) did not significantly affect
the cytotoxic effect of loratadine (Fig. 3G). Taken together, these results
suggested that HRH1 does not appear to be the major factor involved
in loratadine-induced cell death.
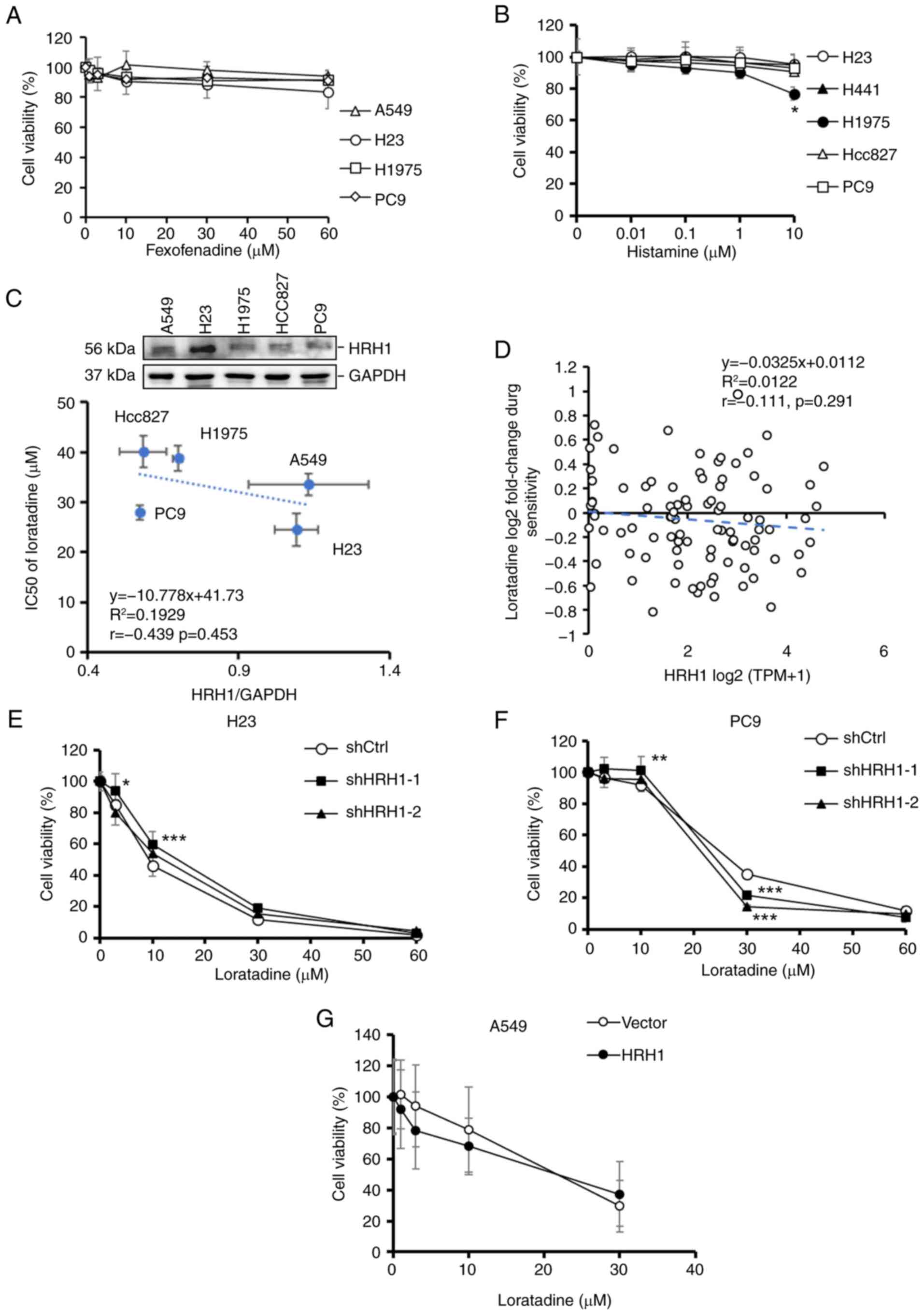 | Figure 3Antiproliferative ability of
loratadine on LUAD cells is independent of HRH1. (A and B) LUAD
cells with different epidermal growth factor receptor statuses were
exposed to varying concentrations of (A) fexofenadine and (B)
histamine for 24 h, and their cell viability was assessed with a
Cell Counting Kit-8 assay. Values are expressed as a percentage of
vehicle-treated cells, with vehicle-treated cells set as 100%. Data
are presented as the mean ± SD. (C) Upper panel: Endogenous HRH1
levels in various LUAD cell lines were determined via a western
blot analysis. Lower panel: Correlations between HRH1 protein
expression levels and the IC50 of loratadine in LUAD
cell lines. (D) Correlations between HRH1 mRNA expression and the
IC50 of loratadine using data from lung cancer cell
lines in the DepMap database. (E-G) Cell viability of (E) H23, (F)
PC9 and (G) A549 cells, which were respectively subjected to HRH1
knockdown and overexpression, followed by loratadine treatment at
various concentrations for an additional 24 h. Values are expressed
as a percentage of vehicle-treated cells, with vehicle-treated
cells set as 100%. Data are presented as the mean ± SD. Data from
panels A, B, and E-G were analyzed using two-way ANOVA.
*P<0.05, **P<0.01 and
***P<0.001 vs. the control group. LUAD, lung
adenocarcinoma; HRH1, histamine receptor H1; IC50, 50%
inhibitory concentration; sh-, short hairpin. |
Loratadine induces autophagy-mediated
apoptotic cell death in LUAD cells
Autophagy, a form of type-II programmed cell death,
is currently a highly studied field in cancer biology. Terfenadine,
an HRH1 antagonist, was reported to induce both apoptosis and
autophagy in melanoma cells (13). Since terfenadine and loratadine
both belong to the class of cationic amphiphilic antihistamines
(22), it was investigated
whether autophagy plays a role in the cell death induced by
loratadine in LUAD cells. Initially, the potential for autophagy
induction by loratadine was assessed through detecting AVOs using
AO staining. It was found that loratadine-treated H23 and PC9 cells
displayed AVOs, which were identified as red fluorescent spots. The
number of AVOs was higher in loratadine-treated cells than in
untreated cells (Fig. 4A).
Furthermore, the conversion of LC3-I to LC3-II and levels of beclin
1 and p62, an autophagosome component or target, was examined using
western blotting. Indeed, loratadine treatment of H23 and PC9 cells
caused the LC3-I to -II conversion and p62 downregulation in a
concentration-dependent manner (Fig.
4B). Additionally, loratadine treatment led to an increase in
beclin 1 expression (data not shown). These results collectively
indicated that loratadine treatment induces autophagy in LUAD
cells. Next, to explore the interplay between autophagy and
apoptosis induced by loratadine, the autophagy inhibitor, CQ, was
employed. Pretreatment of H23 and PC9 cells with CQ significantly
reversed the cleavage of caspase-3 and PARP induced by loratadine,
compared with loratadine treatment alone (Fig. 4C). Moreover, changes in H23 and
PC9 cell death was examined following treatment with loratadine,
both with and without CQ. CCK-8 assays revealed that CQ
significantly mitigated loratadine-induced cell death (Fig. 4D), suggesting that autophagy
induction enhanced the apoptotic effect initiated by loratadine in
LUAD cells.
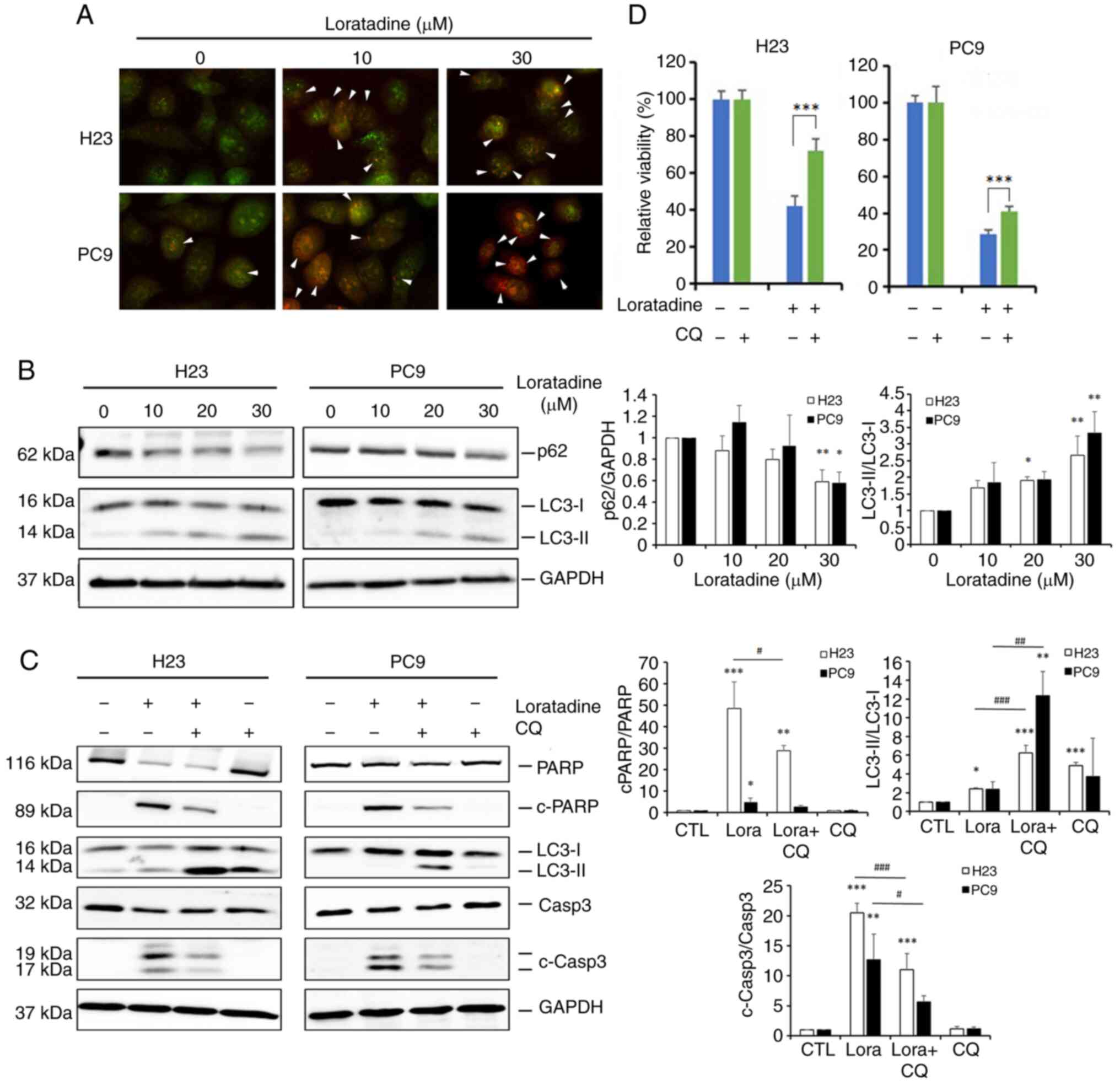 | Figure 4Loratadine induces autophagy-mediated
apoptotic cell death in lung adenocarcinoma cells. (A) H23 and PC9
cells were treated with loratadine (10 and 30 μM) for 24 h.
Lysosomal membrane stability was measured by acridine orange
staining under a fluorescence microscope. Autophagy was indicated
by cells exhibiting bright-red fluorescence (white arrowheads). The
image was captured at a magnification of ×40. (B) H23 and PC9 cells
were treated with loratadine at the indicated concentrations for 24
h. LC3 conversion (LC3-I to LC3-II) and expression of p62 was
detected by western blot analysis. GAPDH was used as a loading
control. (C and D) H23 and PC9 cells were pretreated with CQ (20
μM) for 1 h, followed by loratadine (30 μM) treatment
for 24 h. LC3 conversion, c-PARP and c-caspase-3 levels, and cell
viability in both cell lines were detected by western blot analysis
and Cell Counting Kit-8 assay, respectively. Values, expressed as
the percentage of cell inhibition, considered vehicle- or
CQ-treated cells as 100%. Data are presented as the mean ± SD. Data
from panels B and C were analyzed using one-way ANOVA.
*P<0.05, **P<0.01 and
***P<0.001 vs. the control group.
#P<0.05, ##P<0.01 and
###P<0.001 vs. the loratadine-treated group. LC3,
light chain 3; CQ, chloroquine; PARP, poly (ADP ribose) polymerase;
c-, cleaved. |
Loratadine exhibits a significant
antitumor effect in the PC9 xenograft model by suppressing
angiogenesis and promoting autophagy and apoptosis
To assess the in vivo inhibitory properties
of loratadine, xenograft mouse models were created using PC9 cells.
Loratadine was administered through oral gavage five times a week,
as illustrated in Fig. 5A. On
day 21, the average tumor volume in mice treated with loratadine
was smaller compared with mice received the vehicle, as depicted in
Fig. 5B. Moreover, tumor weights
of xenografts removed from the loratadine-treated group were
significantly lower than those from the control group (Fig. 5C and D). The average weight of
tumors treated with loratadine was reduced by ~56% compared with
tumors treated with the vehicle (Fig. 5D). Furthermore, the treatment
dosage of loratadine (10 mg/kg BW) did not affect BWs of mice
(Fig. 5E). A histopathological
analysis of tumor tissues by H&E staining unveiled a notable
region of cell death in the loratadine-treated group, encompassing
~33.6±6.2% of the cross-sectional area (P=0.002), in contrast to
the vehicle-treated group, where it was only 16±11% (Fig. 5F and G). In support of the
anticancer mechanism in loratadine-treated LUAD cells,
proliferative, autophagic, angiogenic and apoptotic signals within
tumor tissues were assessed through IHC staining for Ki67, LC3 and
cluster of differentiation 31 (CD31), as well as immunofluorescent
staining for BrdU. It was revealed that the proliferation marker,
Ki67, and the angiogenic marker, CD31, exhibited reduced expression
in the loratadine-treated group compared with the vehicle-treated
group. Conversely, the autophagy marker, LC3, displayed a
significant increase in tumor tissues treated with loratadine
(Fig. 5H and I). Additionally,
TUNEL staining demonstrated an increase in apoptotic cells in the
loratadine-treated group compared with the vehicle-treated group
(Fig. 5J). In summary, the
anticancer efficacy of loratadine in the PC9 xenograft model is
attributed to its ability to inhibit proliferation and
angiogenesis, as well as to induce autophagy and apoptosis.
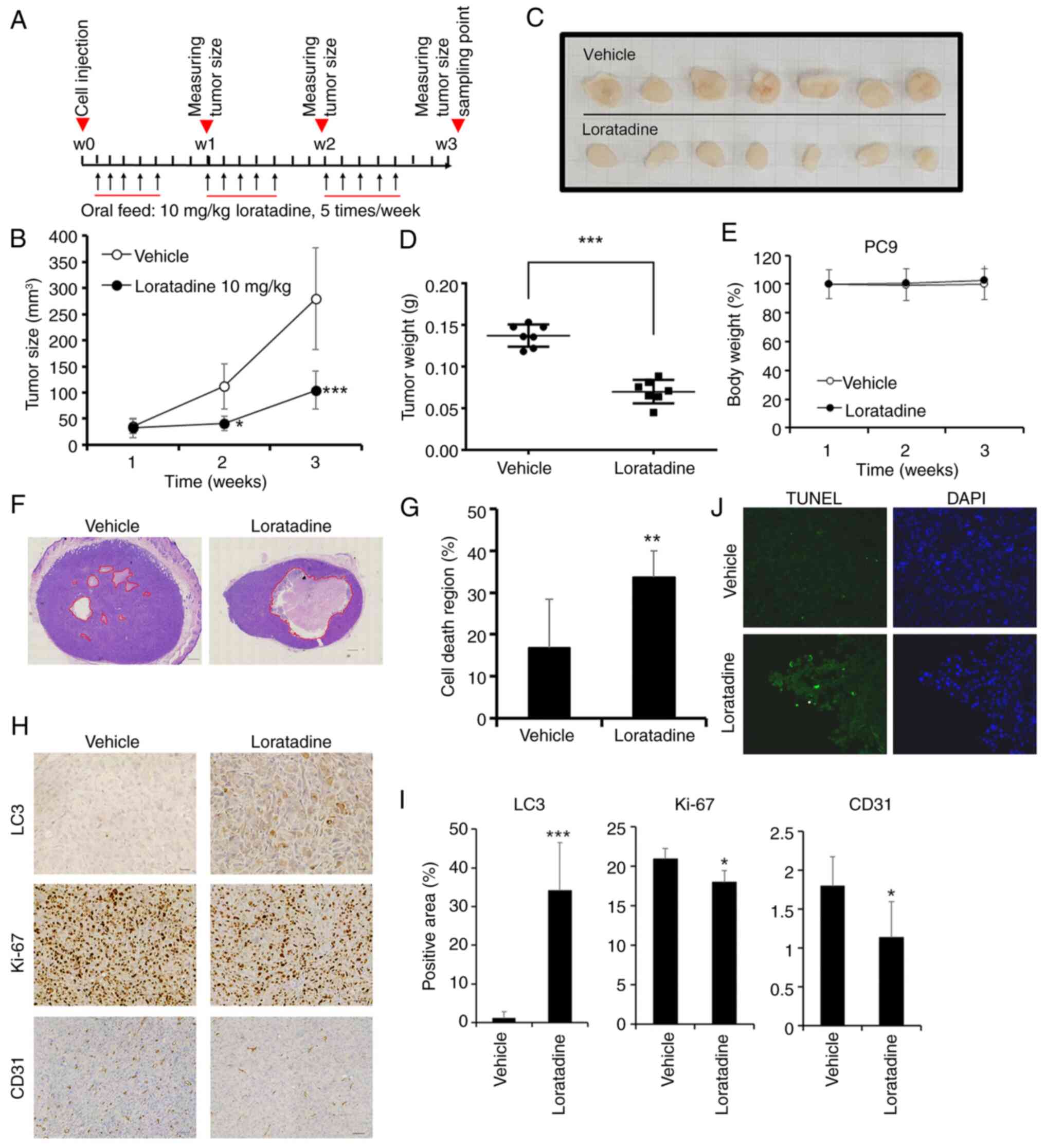 | Figure 5Anticancer growth effects of
loratadine in a PC9 xenograft model. (A) Timeline of the in
vivo study design for investigating anticancer activities of
loratadine. PC9 cells were subcutaneously injected into the back of
NOD-SCID mice which were subsequently orally fed loratadine (10
mg/kg body weight), five times per week for a duration of 21 days
(B) Average tumor volume of vehicle-treated (open circles, n=7)
versus loratadine-treated (filled circles, n=7) NOD-SCID mice. Data
were analyzed by two-way ANOVA. (C) Gross appearance of
subcutaneous tumors after treatment with vehicle or loratadine for
21 days. (D) Tumor weights were compared between loratadine-treated
(n=7) and vehicle-treated (n=7) tumor-bearing mice at the end of
the study. (E) Body weight of each mice was measured weekly. Values
were normalized by using the average value of first week as 100%.
(F) Cross-section of tumor tissues from animals treated with
loratadine or vehicle were stained with H&E. Scale bar, 600
μm. (G) Measurement of the area of the cell death region in
cross-sections of tumor tissues from both vehicle- and
loratadine-treated groups. (H) PC9 tumor tissue sections from
animals treated with vehicle or loratadine were IHC-stained for
LC3, Ki-67, or CD31, and counterstained with hematoxylin. Scale
bar, 60 μm. (I) Quantitative analysis of LC3, Ki-67, and
CD31 IHC staining intensities using ImageJ software. (J)
Fluorescent TUNEL-stained death part of tumor tissues with
loratadine treatment compared with the vehicle. DAPI stain
presented the cell distribution and the morphology of nucleus.
Values represent the mean ± SD. *P<0.05,
**P<0.01 and ***P<0.001 compared with
the vehicle-treated group. IHC, immunohistochemical; LC3, light
chain 3; CD31, cluster of differentiation 31. |
Loratadine triggers autophagy-mediated
apoptotic cell death via inducing the deactivation of the p38, JNK
and STAT3 pathways
Previous research indicated the association of the
mitogen-activated protein kinase (MAPK) and Akt signaling pathways
with autophagy and apoptosis in cancer (23-25). In addition, the anticancer
mechanisms of cationic amphiphilic antihistamines were linked to
STAT3 inhibition (26).
Consequently, it was investigated whether the MAPK, Akt and STAT3
pathways play roles in loratadine-induced autophagy and apoptosis
in LUAD cells. Loratadine treatment for 8 and 24 h resulted in the
inhibition of MAPK signals, specifically p38 and JNK, but not ERK,
in H23 (Fig. 6A) and PC9
(Fig. 6B) cells. Furthermore,
loratadine was found to inhibit STAT3 activation (Y705) in both
cell lines (Fig. 6A and B). By
contrast, loratadine treatment had no inhibitory effect on Akt
phosphorylation in either LUAD cell line (Fig. S2). To examine whether the p38,
JNK and STAT3 pathways were involved in loratadine-induced
autophagy and apoptosis in LUAD cells, H23 and PC9 cells were
pretreated with inhibitors: 10 μM SB203580 (a p38
inhibitor), 5 μM JNK-in-8 (a JNK inhibitor), or 30 μM
C188 (a STAT3 inhibitor) for 1 h. Subsequently, cells were treated
with 30 μM loratadine for another 24 h and analyzed using
western blotting. The cleavage of PARP and LC3 turnover induced by
loratadine were markedly amplified by the inhibitors SB203580,
JNK-in-8 and C188 (Fig. 6C and
D). Functionally, inhibition of the p38, JNK and STAT3 pathways
further enhanced loratadine's suppressive effects on the viability
of H23 and PC9 cells (Fig. 6E and
F). Additionally, treatment with these inhibitors alone
partially reduced the viability of both LUAD cell lines (Fig. S3). These findings collectively
highlight the critical roles of the p38, JNK and STAT3 pathways in
facilitating loratadine-induced, autophagy-mediated apoptosis in
LUAD cells.
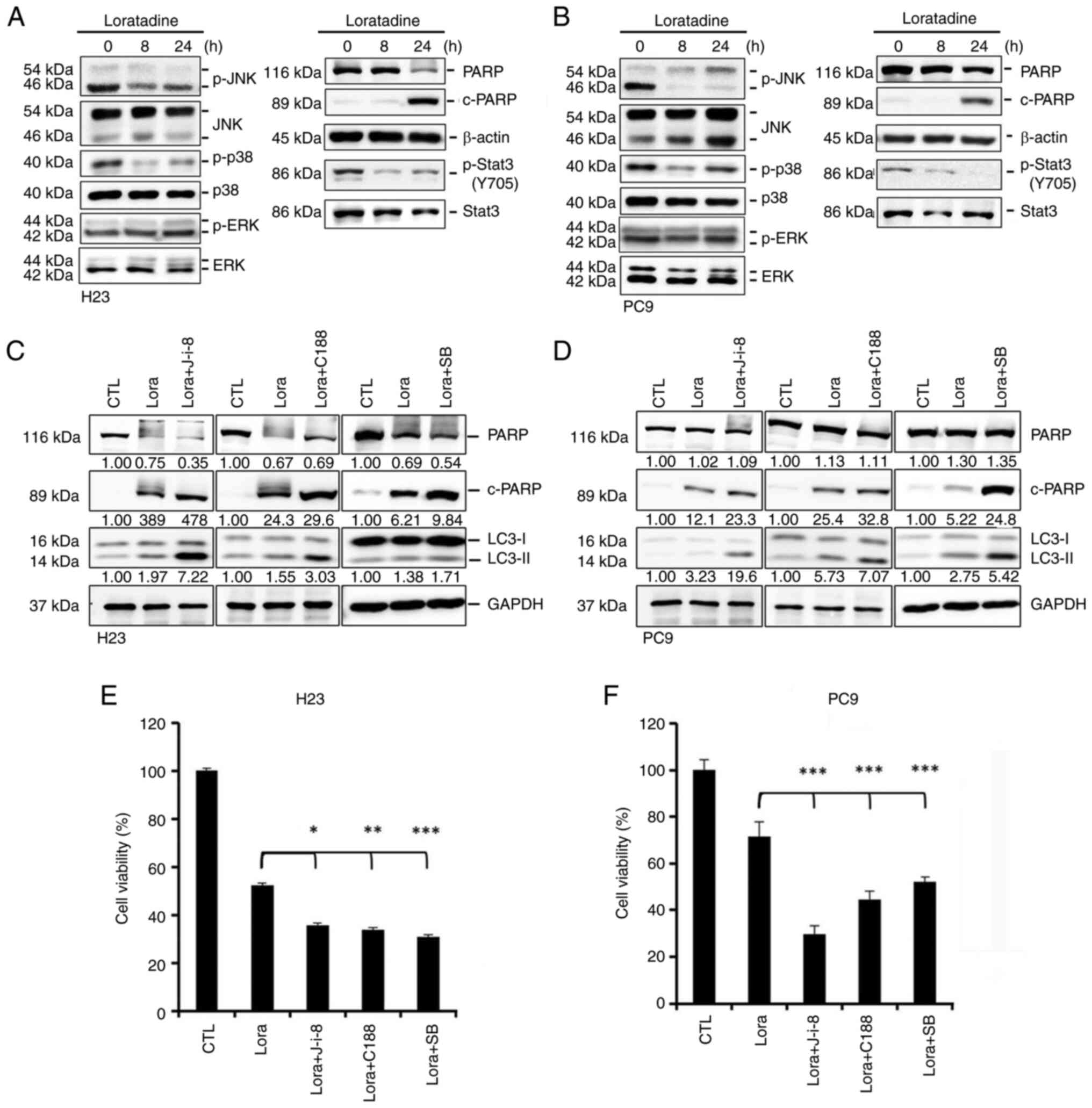 | Figure 6Loratadine induces autophagy-mediated
apoptotic cell death via deactivating p38, c-JNK and 3 STAT3
pathways in lung adenocarcinoma cells. (A and B) Phosphorylation
levels of JNK1/2, p38, ERK1/2 and STAT3 and the expression level of
c-PARP were assessed using western blot analysis after treating (A)
H23 or (B) PC9 cells with 30 μM loratadine for the indicated
time points. (C and D) H23 and PC9 cells were pretreated with and
without 5 μM JNK-in-8, 30 μM C188, or 10 μM
SB203580 for 1 h followed by 30 μM loratadine treatment for
an additional 24 h. Expression levels of c-PARP and LC3 conversion
were determined by western blot analysis. GAPDH was used as a
loading control. (E and F) H23 and PC9 cells were treated as
aforementioned and cell viability changes of both cells were
determined by a Cell Counting Kit-8 assay. Values are expressed as
the percentage of cell inhibition, with vehicle-treated cells
considered 100%. Data are presented as the mean ± SD. Data were
analyzed by one-way ANOVA. *P<0.05,
**P<0.01 and ***P<0.001 compared with
the loratadine-treated group. c-, cleaved; JNK, Jun N-terminal
kinase; STAT3, signal transducer and activator of transcription 3;
ERK1/2, extracellular signal-regulated kinase 1/2; PARP, poly (ADP
ribose) polymerase; LC3, light chain 3; p-, phosphorylated. |
The deactivation of p38 and JNK signals
mediated by loratadine is dependent on PP2A activation, whereas the
deactivation of STAT3 is independent of PP2A
It is worth noting that PP2A, a serine/threonine
phosphatase, was shown to deactivate MAPKs (27) and STAT3 (28). Hence, it was hypothesized that
loratadine might activate PP2A to deactivate MAPKs and STAT3,
ultimately leading to suppression of LUAD growth. Indeed, treatment
of H23 and PC9 cells with loratadine was observed to reduce the
phosphorylation of PP2A at Tyr307 (Fig. 7A and B), a modification known to
decrease its activity (29).
Moreover, it was observed that pretreatment with the PP2A
inhibitor, OA, was able to reverse the loratadine-induced
deactivation of p38 and JNK, and cleavage of PARP in both H23 and
PC9 cells (Fig. 7C and D). By
contrast, the inhibition of p-STAT3 caused by loratadine was not
reversed by OA pretreatment (7C and D). These findings suggested
that the apoptotic effect induced by loratadine in LUAD cells may
occur through activation of PP2A, which in turn negatively
regulates p38 and JNK activities. In clinical settings, a
significant reduction in PP2A protein levels was evident in LUAD
tissues compared with normal tissues, as indicated by proteomic
data sourced from the UALCAN database (18). Furthermore, a subgroup analysis
was conducted considering various clinical and pathological factors
related to LUAD and it was found that PP2A protein levels exhibited
a decrease in LUAD clinical stages 1 to 3 and tumor grades 2 to 3
compared with normal samples (Fig.
7E). In addition, associations between PP2A and the OS of
patients with LUAD were analyzed using the KM-plotter. As depicted
in the left panel of Fig. 7F,
patients with LUAD with high PP2A expression exhibited an extended
OS compared with those with low PP2A expression. Moreover, high
PP2A in patients with LUAD with negative surgical margins also had
longer OS times compared with those with low PP2A expression
(Fig. 7F, right panel).
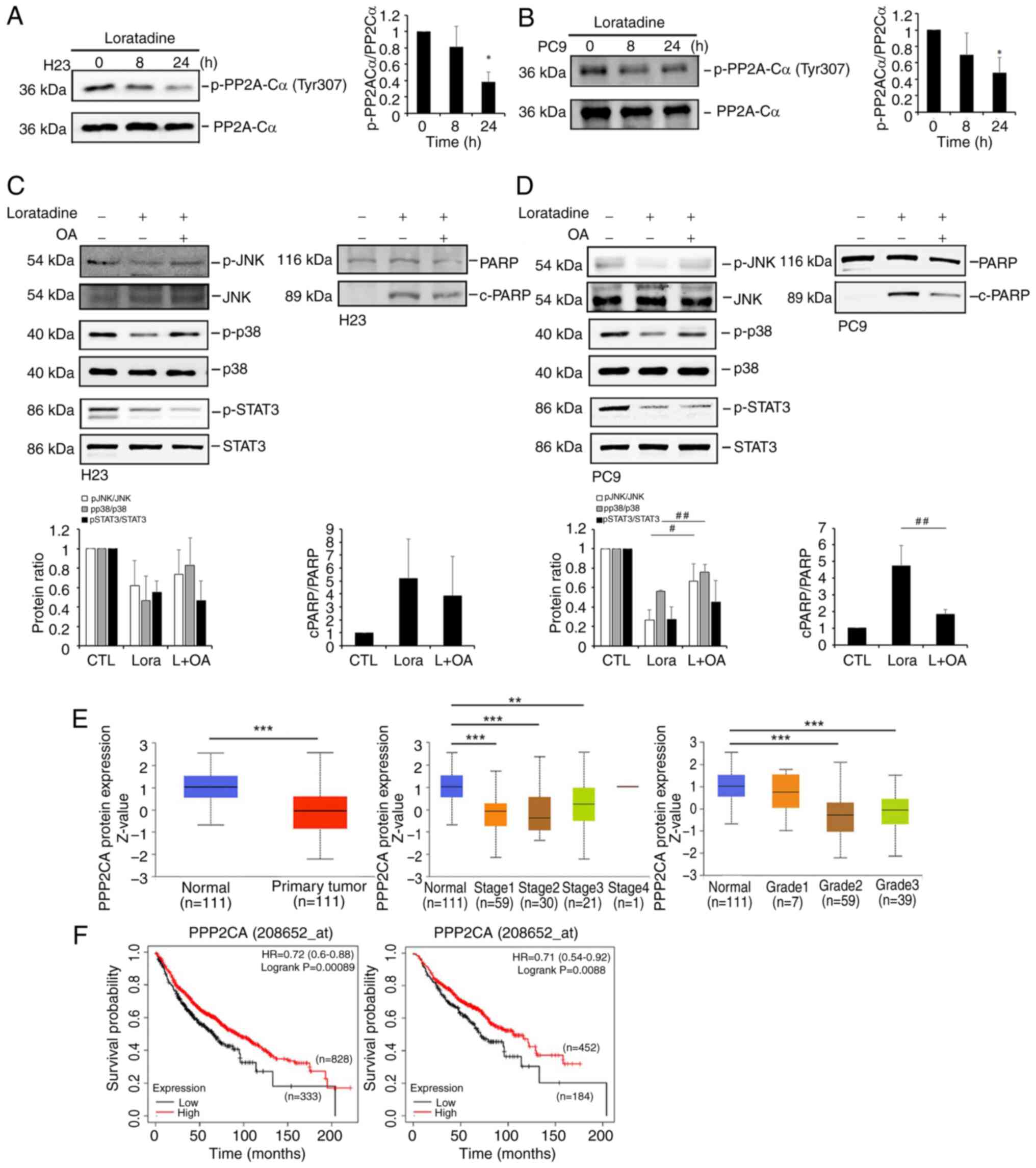 | Figure 7Loratadine-induced PP2A activation is
responsible for the deactivation of p38 and c-JNK, but not for the
STAT3 pathways in LUAD cells. (A and B) Phosphorylation level of
PP2A-Cα at Tyr307 was assessed using western blot analysis after
treating (A) H23 or (B) PC9 cells with 30 μM loratadine for
8 and 24 h. (C and D) Pretreatment of H23 and PC9 cells with 5 nM
OA for 1 h followed by 30 μM loratadine treatment for an
additional 24 h. Phosphorylation levels of p38, JNK, STAT3 and
c-PARP were determined by western blot analysis. (E) UALCAN portal
analysis of LUAD samples from the CPTAC dataset. Comparison of
PP2A-C (PPP2CA) protein expression levels between normal and tumor
tissues (left panel). Expression levels of PP2A-C protein levels in
tumor tissues obtained from patients with LUAD at different
clinical stages (middle panel) and tumor grades (right panel).
**P<0.01 and ***P<0.001 compared with
the normal group. (F) Association between PPP2CA expression with
overall survival in patients with LUAD (left panel) or a
sub-population with negative surgical margins (right panel) as
determined using a Kaplan-Meier plotter database. Gene expression
levels were dichotomized into high and low values using the best
cutoff value. P<0.05 was considered to indicate a statistically
significant difference. Data are presented as the mean ± SD. Data
from panels A-D were analyzed using one-way ANOVA.
*P<0.05 compared with the vehicle-treated group.
#P<0.05 and ##P<0.01 vs. the
loratadine-treated group. PP2A, protein phosphatase 2A; c-,
cleaved; JNK, Jun N-terminal kinase; STAT3, signal transducer and
activator of transcription 3; LUAD, lung adenocarcinoma; OA,
okadaic acid; PARP, poly (ADP ribose) polymerase; HR, hazard ratio;
p-, phosphorylated. |
Discussion
A nationwide pharmacoepidemiological cohort study
showed that the use of loratadine, a cationic amphiphilic H1
antihistamine, was correlated with significantly reduced all-cause
mortality among patients with non-localized NSCLC (22), suggesting that loratadine may
exhibit therapeutic potential for NSCLC. In the present study, it
was found that loratadine inhibited the proliferation of LUAD cells
harboring either WT or mutant EGFR in both in vitro and
in vivo experiments. Conversely, fexofenadine, a
non-cationic amphiphilic H1 antihistamine, did not exhibit a
significant anticancer growth effect in LUAD cells. This result may
echo previous clinical observations which indicated that loratadine
use was associated with significantly reduced mortality compared
with the use of the fexofenadine in patients with NSCLC (22). Previously, histamine was
demonstrated to play an important role in modulating the
proliferation of various cancer cells and was shown to act on HRH1
(30,31). However, the results of the
present study demonstrated that histamine treatment did not affect
the proliferation of various LUAD cells. Furthermore, both
knockdown and overexpression of HRH1 had slight impacts on the
cytotoxic effect of loratadine against LUAD cells. Collectively,
these findings suggest that loratadine-induced cytotoxicity in LUAD
cells is independent of HRH1. Similar to the current findings,
another cationic amphiphilic H1 antihistamine, terfenadine, was
shown to induce cell death independently of HRH1 in melanoma cells
(32). Moreover, there is a
recent study presenting the HRH1-independent anti-inflammatory
effects of HRH1 antagonists by targeting a member of MAPK kinase
kinase (MAP3K), TAK1, and suppressing consequent AP-1 signaling
pathway activation (15).
Mechanistically, the present study demonstrated that
autophagy and apoptosis were both induced in H23 and PC9 cells. The
interplay between apoptosis and autophagy in loratadine-induced
cytotoxicity was also emphasized. Autophagy was reported to act as
either a guardian or executor of apoptosis in NSCLC, depending on
the surrounding microenvironment, therapeutic interventions and
stage of the carcinoma (33).
The current research revealed that inhibition of autophagy by an
autophagy inhibitor reduced loratadine-induced apoptotic cell death
in LUAD cells, indicating that loratadine-induced autophagy serves
as a pro-death mechanism rather than a prosurvival one. The role of
autophagy in promoting or inhibiting apoptosis varies depending on
specific molecular factors, numerous of which remain poorly
understood. Mutations in the p53 tumor suppressor gene are among
the most common genetic alterations in LUAD, occurring in 45-70% of
cases (33). The nuclear
transcriptional activity of WT p53 activates multiple target genes
that promote both autophagy and apoptosis (33). Conversely, deletion, depletion,
mutations, or pharmacological inhibition of p53 induces autophagy
in human cells, protecting them from apoptosis under hypoxic or
nutrient-starvation conditions, thereby suggesting an
anti-apoptotic function for autophagy (34). Notably, a recent study revealed
that loratadine treatment enhances p53 expression in a Lewis lung
carcinoma xenograft model (35).
Additionally, high level p53 activation was shown in H23 and PC9
LUAD cells (36) which were used
in the present study. These findings suggest that patients with
LUAD with WT p53 may be better suited for loratadine treatment
compared with those with mutant p53. Various factors function in
both apoptosis and autophagy. For instance, the interaction between
caspases and autophagy-related (ATG) proteins in regulating
apoptosis was previously documented. ATG4D can be cleaved by
caspase-3 and recruited to mitochondria to stimulate apoptosis
(37). Caspase-9 was shown to
induce autophagy by increasing LC3 lipidation through interaction
with ATG7 (38). While
loratadine was demonstrated to activate caspase-9 and -3 in LUAD
cells, further investigation is needed in the future to elucidate
regulation of the crosstalk between caspases and ATGs by
loratadine. Additionally, the Farnesoid X receptor (FXR)-oxidative
stress-induced growth inhibitor 1 (OSGIN1) axis has been reported
to promote autophagy, influencing various inflammatory-related
disorders such as pancreatitis (39) and chronic obstructive pulmonary
disease (40). Interestingly,
both upregulation and downregulation of OSGIN1 were shown to
enhance autophagy responses triggered by tobacco smoking in the
human airway epithelium (41).
In NSCLC, elevated levels of FXR and OSGIN1 were found to
contribute to disease progression by activating STAT3 signaling
(42) and modulating microtubule
dynamics (43), respectively.
However, it remains to be determined whether loratadine-induced
autophagic responses play a role in the FXR-OSGIN1 axis-mediated
progression of NSCLC.
Extensive research has highlighted the pivotal role
of MAPKs in converting extracellular stimuli into a wide range of
cellular responses, including cell growth, proliferation, apoptosis
and autophagy. Among MAPKs, JNK and p38 MAPK were shown to mediate
certain antiapoptotic processes (44). For example, JNK activation was
reported to mitigate endoplasmic reticulum stress-induced cell
death by enhancing expression levels of various antiapoptotic
proteins, including cellular inhibitor of apoptosis protein 1
(cIAP1), cIAP2, X-linked inhibitor of apoptosis protein and
baculoviral AIP repeat-containing 6 (45). Transient JNK activation was
demonstrated to delay caspase-9 activation by directly interacting
with apoptotic protease-activating factor-1 and cytochrome c,
inhibiting the formation of the apoptosome complex (46). Additionally, JNK-mediated
phosphorylation of Bad at Thr201 promotes the dissociation of the
antiapoptotic protein Bcl-xL from Bad (47). On the other hand, p38 MAPK
signaling was implicated in promoting the survival or proliferation
of various cancer cell lines, in addition to being associated with
poor prognoses in cancer (48,49). The inhibitory phosphorylation of
caspase-9 at Thr125 by p38 MAPK was reported to restrain apoptosis
(50). The present study
revealed that loratadine treatment can suppress activation of p38
and JNK while inducing caspase-8/-9/-3-mediated apoptosis in LUAD
cells. Further investigation is warranted to explore the crosstalk
between MAPK deactivation and caspase activation in
loratadine-induced cell death of LUAD cells.
In addition to MAPKs, STAT3 was identified as
another target of loratadine in modulating cell death in LUAD
cells. Our findings align with a recent study which demonstrated
that cationic amphiphilic antihistamines induce lysosomal
H+ efflux and cytosolic acidification in cancer cells,
subsequently rendering cancer cells more susceptible to apoptosis
through STAT3 inactivation. Furthermore, it was observed that a
STAT3 inhibitor could sensitize HeLa cancer cells to apoptosis
induced by cationic amphiphilic antihistamines (26). To confirm whether the
loratadine-induced inhibition of STAT3 in LUAD cells is also linked
to lysosomal H+ efflux and cytosolic acidification,
further validation is required. Additionally, the present study
indicated that both a STAT3 inhibitor and JNK and p38 inhibitors
could enhance autophagy-mediated apoptosis in LUAD cells. This
suggests that not only STAT3 but also JNK and p38 signaling
pathways are critical components of loratadine's anticancer
mechanisms in LUAD cells.
One of the main serine-threonine phosphatases, PP2A,
plays a tumor-suppressive role, as it is often genetically altered
or functionally inactivated in numerous solid cancers and leukemia,
leading to the promotion of tumor progression through inhibition of
PP2A activity (51). Mutations
in the genes encoding the regulatory β-subunit of PP2A have also
been linked to LUAD proliferation (52). In the present study, it was
observed that PP2A was downregulated in LUAD tissues in advanced
stages and was correlated with favorable prognoses of patients with
LUAD, suggesting that PP2A may play a tumor-suppressive role in
modulating LUAD development. Previous research demonstrated that
penfluridol, a clinically relevant cationic amphiphilic drug (CAD),
can activate PP2A to deactivate the MAPK pathway, resulting in
caspase-mediated apoptosis of leukemia cells (53). In the present study, it was also
observed that the CAD antihistamine, loratadine, could activate
PP2A in LUAD cells, regardless of whether they harbored WT or
mutant EGFR. This activation led to the deactivation of downstream
signaling pathways such as JNK and p38, ultimately triggering
apoptotic cell death. The inhibition of p-STAT3 caused by
loratadine was found to be independent of PP2A; however, it was
still implicated in loratadine-mediated cell death. It was
previously reported that PP2A activation can enhance the
sensitivity of chemotherapy drugs in A549 cells with WT EGFR
(54) and increase the
sensitivity of EGFR tyrosine kinase inhibitors (TKIs) in
TKI-resistant LUAD cells with mutant EGFR (55). Vasculogenic mimicry (VM) refers
to a novel microvascular structure resembling a three-dimensional
channel formed by tumor cells without the involvement of
endothelial cells, providing essential nutrients and oxygen to
support tumor growth. Research has shown that the presence of VM
contributes to increased chemotherapy resistance, distant
metastases and poor prognosis in NSCLC. Zhang et al
(56) demonstrated that
activating PP2A plays a crucial role in inhibiting VM formation, as
well as reducing invasion and metastasis in NSCLC. In clinical
settings, the association between loratadine use and reduced
mortality appears to be more pronounced among patients with NSCLC
who have records of concurrent chemotherapy compared with those
without chemotherapy (22).
Therefore, loratadine may hold promise as a potential tool to
overcome chemoresistance or TKI-resistance in various LUAD cells by
targeting PP2A. In the current study, a preliminary evaluation of
the synergistic effects of loratadine was conducted in combination
with varying concentrations of the EGFR TKI gefitinib and the
chemotherapy drug cisplatin on HCC827 and A549 cells, respectively.
Our observations revealed that most combinations did not
demonstrate synergistic inhibitory effects on the viability of A549
or HCC827 cells (combination index >1). However, specific
combinations, such as 10 μM loratadine with 33 μM
cisplatin or 30 μM loratadine with 1 μM gefitinib,
exhibited synergistic inhibitory effects (combination index <1)
on A549 and HCC827 cells, respectively (data not shown). Further
studies are needed to optimize the concentrations of these drug
combinations for effective LUAD treatment.
The present study marks the first time, to the best
of our knowledge, that treatment of LUAD cells carrying WT or
mutant EGFR with loratadine, a CAD antihistamine, was shown to
induce autophagy-mediated apoptosis independent of HRH1. This
effect was achieved by triggering PP2A-mediated deactivation of the
JNK and p38 signaling pathways. Moreover, STAT3 inhibition was also
involved in loratadine-mediated cell death of LUAD. A schematic
representation of this mechanism is provided in Fig. 8. Based on these findings, it is
proposed to repurpose the safe and cost-effective loratadine for
treating LUAD. This repurposing approach may enhance the
anti-neoplastic response in combination with chemotherapy or
EGFR-targeted therapy, offering a novel therapeutic strategy for
LUAD.
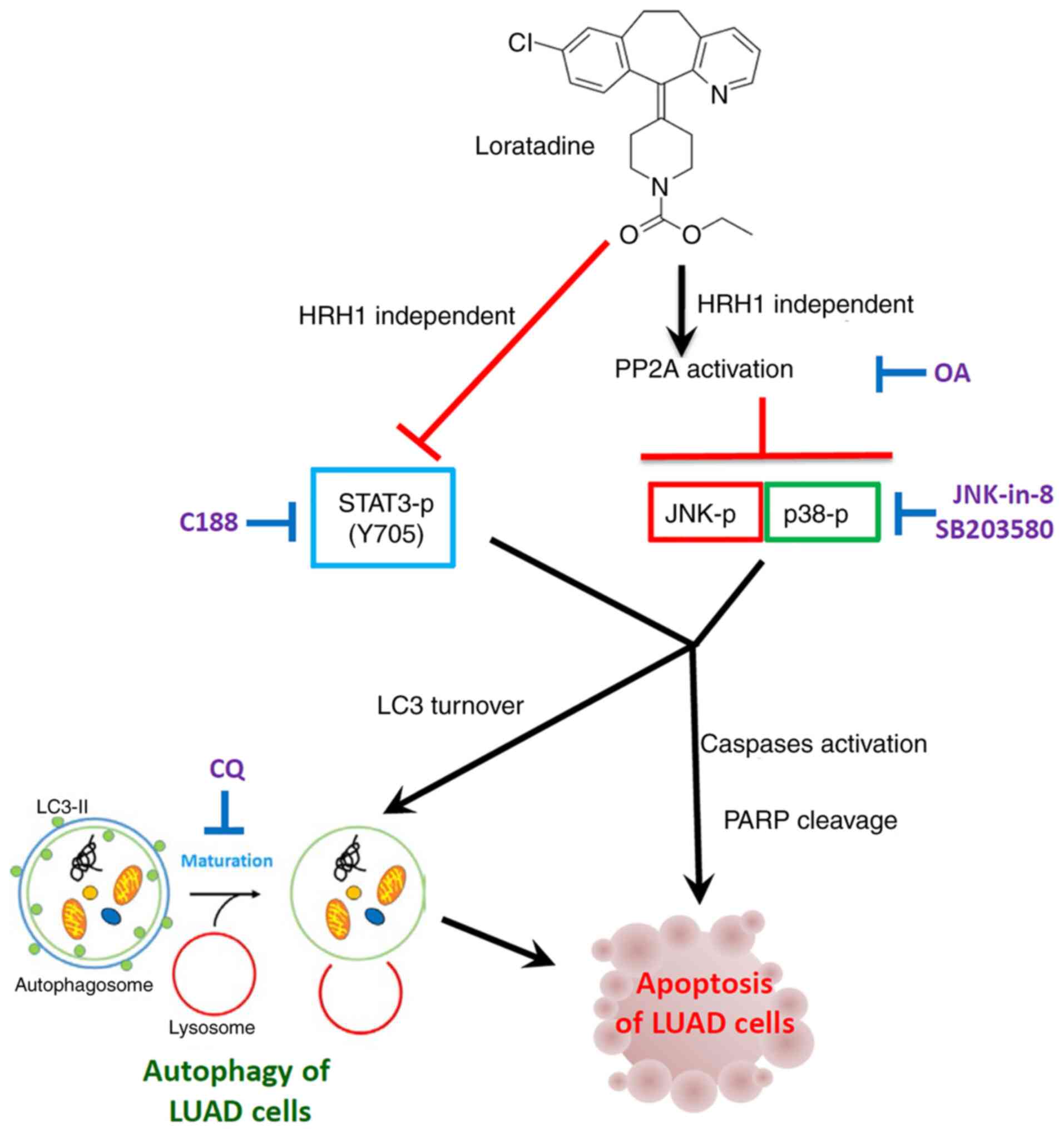 | Figure 8Working model showing the molecular
mechanism underlying the ability of loratadine to induce apoptotic
cell death of LUAD cells. The anticancer growth effect of
loratadine was attributed to its induction of
autophagy-mediated-apoptosis independent of HRH1. Mechanistically,
loratadine initiates activation of PP2A, leading to the
deactivation of p-JNK and p-p38 pathways, ultimately culminating in
autophagy-mediated apoptotic cell death. The inhibition of p-STAT3
caused by loratadine is independent of PP2A; nonetheless, it
remains implicated in loratadine-mediated cell death. LUAD, lung
adenocarcinoma; HRH1, histamine receptor H1; PP2A, protein
phosphatase 2A; P-, phosphorylated; JNK, Jun N-terminal kinase;
STAT3, signal transducer and activator of transcription 3; CQ,
chloroquine; LC3, light chain 3; OA, okadaic acid; PARP, poly (ADP
ribose) polymerase. |
Supplementary Data
Availability of data and materials
The data generated in the present study are included
in the figures and/or tables of this article.
Authors' contributions
WYH, TCL, MHC and JHC designed and conceived the
study. WYH, MHC, KLL and JHC supervised the study. CHT and TCL
performed the in vitro experiments and acquired the data.
MHC, KLL and FKH contributed to the bioinformatic and statistical
analyses. TCL, WJL and MHC performed the in vivo xenograft
studies and analyzed data from the animal model. WYH, TCL, MHC and
JHC wrote and revised the manuscript. All authors reviewed the
results, read and approved the final version of the manuscript. MHC
and TCL confirm the authenticity of all the raw data.
Ethics approval and consent to
participate
All animal experiments were carried out in
accordance with guidelines of a protocol approved (approval
no.wan-lac-110-026) by the Institutional Animal Care and Use
Committee (IACUC) at Wan Fang Hospital (Taipei, Taiwan).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgements
Not applicable.
Funding
The present study was supported by the TMU Research Center of
Cancer Translational Medicine from The Featured Areas Research
Center Program within the framework of the Higher Education Sprout
Project by the Ministry of Education in Taiwan. The present study
was also supported (grant no. 111-wf-eva-01) from Wan Fang Hospital
(Taipei Medical University, Taipei, Taiwan).
References
|
1
|
Sung H, Ferlay J, Siegel RL, Laversanne M,
Soerjomataram I, Jemal A and Bray F: Global Cancer Statistics 2020:
GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36
Cancers in 185 Countries. CA Cancer J Clin. 71:209–249. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Dearden S, Stevens J, Wu YL and Blowers D:
Mutation incidence and coincidence in non small-cell lung cancer:
Meta-analyses by ethnicity and histology (mutMap). Ann Oncol.
24:2371–2376. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Yang SR, Schultheis AM, Yu H, Mandelker D,
Ladanyi M and Buttner R: Precision medicine in non-small cell lung
cancer: Current applications and future directions. Semin Cancer
Biol. 84:184–198. 2022. View Article : Google Scholar
|
|
4
|
Shafiei-Irannejad V, Samadi N, Salehi R,
Yousefi B and Zarghami N: New insights into antidiabetic drugs:
Possible applications in cancer treatment. Chem Biol Drug Des.
90:1056–1066. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Veschi S, De Lellis L, Florio R, Lanuti P,
Massucci A, Tinari N, De Tursi M, di Sebastiano P, Marchisio M,
Natoli C and Cama A: Effects of repurposed drug candidates
nitroxoline and nelfinavir as single agents or in combination with
erlotinib in pancreatic cancer cells. J Exp Clin Cancer Res.
37:2362018. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Baker NC, Ekins S, Williams AJ and Tropsha
A: A bibliometric review of drug repurposing. Drug Discov Today.
23:661–672. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
May JR and Dolen WK: Management of
allergic rhinitis: A review for the community pharmacist. Clin
Ther. 39:2410–2419. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Fritz I, Wagner P, Bottai M, Eriksson H,
Ingvar C, Krakowski I, Nielsen K and Olsson H: Desloratadine and
loratadine use associated with improved melanoma survival. Allergy.
75:2096–2099. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Fritz I, Wagner P, Broberg P, Einefors R
and Olsson H: Desloratadine and loratadine stand out among common
H(1)-antihistamines for association with improved breast cancer
survival. Acta Oncol. 59:1103–1109. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Fritz I, Wagner P and Olsson H: Improved
survival in several cancers with use of H(1)-antihistamines
desloratadine and loratadine. Transl Oncol. 14:1010292021.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Chen T, Hu Y, Liu B, Huang X, Li Q, Gao N,
Jin Z, Jia T, Guo D and Jin G: Combining thioridazine and
loratadine for the treatment of gastrointestinal tumor. Oncol Lett.
14:4573–4580. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ma J, Qi J, Li S, Zhang C, Wang H, Shao L,
Yuan X and Sha Q: Desloratadine, a novel antigrowth reagent for
bladder cancer. Technol Cancer Res Treat. 19:15330338209265912020.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Nicolau-Galmés F, Asumendi A,
Alonso-Tejerina E, Pérez-Yarza G, Jangi SM, Gardeazabal J,
Arroyo-Berdugo Y, Careaga JM, Díaz-Ramón JL, Apraiz A and Boyano
MD: Terfenadine induces apoptosis and autophagy in melanoma cells
through ROS-dependent and -independent mechanisms. Apoptosis.
16:1253–1267. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Guo Q, Jin Y, Chen X, Ye X, Shen X, Lin M,
Zeng C, Zhou T and Zhang J: NF-κB in biology and targeted therapy:
New insights and translational implications. Signal Transduct
Target Ther. 9:532024. View Article : Google Scholar
|
|
15
|
Jang J, Hunto ST, Kim JW, Lee HP, Kim HG
and Cho JY: Anti-Inflammatory activities of an anti-histamine drug,
loratadine, by suppressing TAK1 in AP-1 pathway. Int J Mol Sci.
23:39862022. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zhu J, Li Q, He JT and Liu GY: Expression
of TAK1/TAB1 expression in non-small cell lung carcinoma and
adjacent normal tissues and their clinical significance. Int J Clin
Exp Pathol. 8:15801–15807. 2015.
|
|
17
|
Shimada K, Bachman JA, Muhlich JL and
Mitchison TJ: shinyDepMap, a tool to identify targetable cancer
genes and their functional connections from cancer dependency map
data. Elife. 10:e571162021. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Chandrashekar DS, Bashel B, Balasubramanya
SAH, Creighton CJ, Ponce-Rodriguez I, Chakravarthi B and Varambally
S: UALCAN: A portal for facilitating tumor subgroup gene expression
and survival analyses. Neoplasia. 19:649–658. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Chien MH, Lin YW, Wen YC, Yang YC, Hsiao
M, Chang JL, Huang HC and Lee WJ: Targeting the SPOCK1-snail/slug
axis-mediated epithelial-to-mesenchymal transition by apigenin
contributes to repression of prostate cancer metastasis. J Exp Clin
Cancer Res. 38:2462019. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
21
|
Shen YC, Hsu HC, Lin TM, Chang YS, Hu LF,
Chen LF, Lin SH, Kuo PI, Chen WS, Lin YC, et al: H1-Antihistamines
reduce the risk of hepatocellular carcinoma in patients with
hepatitis B virus, hepatitis C virus, or dual hepatitis B
virus-hepatitis C Virus infection. J Clin Oncol. 40:1206–1219.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ellegaard AM, Dehlendorff C, Vind AC,
Anand A, Cederkvist L, Petersen NHT, Nylandsted J, Stenvang J,
Mellemgaard A, Osterlind K, et al: Repurposing cationic amphiphilic
antihistamines for cancer treatment. EBioMedicine. 9:130–139. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Chen C, Gao H and Su X: Autophagy-related
signaling pathways are involved in cancer (Review). Exp Ther Med.
22:7102021. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Anjum J, Mitra S, Das R, Alam R, Mojumder
A, Emran TB, Islam F, Rauf A, Hossain MJ, Aljohani ASM, et al: A
renewed concept on the MAPK signaling pathway in cancers:
Polyphenols as a choice of therapeutics. Pharmacol Res.
184:1063982022. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Muilenburg D, Parsons C, Coates J,
Virudachalam S and Bold RJ: Role of autophagy in apoptotic
regulation by Akt in pancreatic cancer. Anticancer Res. 34:631–637.
2014.PubMed/NCBI
|
|
26
|
Liu B, Chen R, Zhang Y, Huang J, Luo Y,
Rosthoj S, Zhao C and Jäättelä M: Cationic amphiphilic
antihistamines inhibit STAT3 via Ca(2+)-dependent lysosomal H(+)
efflux. Cell Rep. 42:1121372023. View Article : Google Scholar
|
|
27
|
Bryant JP, Levy A, Heiss J and
Banasavadi-Siddegowda YK: Review of PP2A tumor biology and
antitumor effects of PP2A inhibitor LB100 in the nervous system.
Cancers (Basel). 13:30872021. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Liu CC, Lin SP, Hsu HS, Yang SH, Lin CH,
Yang MH, Hung MC and Hung SC: Suspension survival mediated by
PP2A-STAT3-Col XVII determines tumour initiation and metastasis in
cancer stem cells. Nat Commun. 7:117982016. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Chen J, Martin BL and Brautigan DL:
Regulation of protein serine-threonine phosphatase type-2A by
tyrosine phosphorylation. Science. 257:1261–1264. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Tilly BC, Tertoolen LG, Remorie R, Ladoux
A, Verlaan I, de Laat SW and Moolenaar WH: Histamine as a growth
factor and chemoattractant for human carcinoma and melanoma cells:
Action through Ca2(+)-mobilizing H1 receptors. J Cell Biol.
110:1211–1215. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Kennedy L, Hodges K, Meng F, Alpini G and
Francis H: Histamine and histamine receptor regulation of
gastrointestinal cancers. Transl Gastrointest Cancer. 1:215–227.
2012.PubMed/NCBI
|
|
32
|
Jangi SM, Ruiz-Larrea MB, Nicolau-Galmés
F, Andollo N, Arroyo-Berdugo Y, Ortega-Martínez I, Díaz-Pérez JL
and Boyano MD: Terfenadine-induced apoptosis in human melanoma
cells is mediated through Ca2+ homeostasis modulation and tyrosine
kinase activity, independently of H1 histamine receptors.
Carcinogenesis. 29:500–509. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Liu G, Pei F, Yang F, Li L, Amin AD, Liu
S, Buchan JR and Cho WC: Role of autophagy and apoptosis in
non-small-cell lung cancer. Int J Mol Sci. 18:3672017. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Nikoletopoulou V, Markaki M, Palikaras K
and Tavernarakis N: Crosstalk between apoptosis, necrosis and
autophagy. Biochim Biophys Acta. 1833:3448–3459. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Liu X, Zhong R, Huang J, Chen Z, Xu H, Lin
L, Cai Q, He M, Lao S, Deng H, et al: Loratidine is associated with
improved prognosis and exerts antineoplastic effects via apoptotic
and pyroptotic crosstalk in lung cancer. J Exp Clin Cancer Res.
43:52024. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Ummanni R, Mannsperger HA, Sonntag J,
Oswald M, Sharma AK, König R and Korf U: Evaluation of reverse
phase protein array (RPPA)-based pathway-activation profiling in 84
non-small cell lung cancer (NSCLC) cell lines as platform for
cancer proteomics and biomarker discovery. Biochim Biophys Acta.
1844:950–959. 2014. View Article : Google Scholar
|
|
37
|
Betin VM and Lane JD: Atg4D at the
interface between autophagy and apoptosis. Autophagy. 5:1057–1059.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Han J, Hou W, Goldstein LA, Stolz DB,
Watkins SC and Rabinowich H: A complex between Atg7 and Caspase-9:
A novel mechanism of cross-regulation between autophagy and
apoptosis. J Biol Chem. 289:6485–6497. 2014. View Article : Google Scholar :
|
|
39
|
Zheng Y, Sun W, Wang Z, Liu J, Shan C, He
C, Li B, Hu X, Zhu W, Liu L, et al: Activation of pancreatic Acinar
FXR Protects against pancreatitis via osgin1-mediated restoration
of efficient autophagy. Research (Wash D C).
2022:97840812022.PubMed/NCBI
|
|
40
|
Sukkar MB and Harris J: Potential impact
of oxidative stress induced growth inhibitor 1 (OSGIN1) on airway
epithelial cell autophagy in chronic obstructive pulmonary disease
(COPD). J Thorac Dis. 9:4825–4827. 2017. View Article : Google Scholar
|
|
41
|
Wang G, Zhou H, Strulovici-Barel Y,
Al-Hijji M, Ou X, Salit J, Walters MS, Staudt MR, Kaner RJ and
Crystal RG: Role of OSGIN1 in mediating smoking-induced autophagy
in the human airway epithelium. Autophagy. 13:1205–1220. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Jin X, Shang B, Wang J, Sun J, Li J, Liang
B, Wang X, Su L, You W and Jiang S: Farnesoid X receptor promotes
non-small cell lung cancer metastasis by activating Jak2/STAT3
signaling via transactivation of IL-6ST and IL-6 genes. Cell Death
Dis. 15:1482024. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Xie X, Laster KV, Li J, Nie W, Yi YW, Liu
K, Seong YS, Dong Z and Kim DJ: OSGIN1 is a novel TUBB3 regulator
that promotes tumor progression and gefitinib resistance in
non-small cell lung cancer. Cell Mol Life Sci. 80:2722023.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Yue J and López JM: Understanding MAPK
signaling pathways in apoptosis. Int J Mol Sci. 21:23462020.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Brown M, Strudwick N, Suwara M, Sutcliffe
LK, Mihai AD, Ali AA, Watson JN and Schröder M: An initial phase of
JNK activation inhibits cell death early in the endoplasmic
reticulum stress response. J Cell Sci. 129:2317–2328. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Tran TH, Andreka P, Rodrigues CO, Webster
KA and Bishopric NH: Jun kinase delays caspase-9 activation by
interaction with the apoptosome. J Biol Chem. 282:20340–20350.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Yu C, Minemoto Y, Zhang J, Liu J, Tang F,
Bui TN, Xiang J and Lin A: JNK suppresses apoptosis via
phosphorylation of the proapoptotic Bcl-2 family protein BAD. Mol
Cell. 13:329–340. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Chen L, Mayer JA, Krisko TI, Speers CW,
Wang T, Hilsenbeck SG and Brown PH: Inhibition of the p38 kinase
suppresses the proliferation of human ER-negative breast cancer
cells. Cancer Res. 69:8853–8861. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Ricote M, García-Tuñón I, Bethencourt F,
Fraile B, Onsurbe P, Paniagua R and Royuela M: The p38 transduction
pathway in prostatic neoplasia. J Pathol. 208:401–407. 2006.
View Article : Google Scholar
|
|
50
|
Seifert A and Clarke PR: p38alpha- and
DYRK1A-dependent phosphorylation of caspase-9 at an inhibitory site
in response to hyperosmotic stress. Cell Signal. 21:1626–1633.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Janssens V, Goris J and Van Hoof C: PP2A:
The expected tumor suppressor. Curr Opin Genet Dev. 15:34–41. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Yu H, Zaveri S, Sattar Z, Schaible M,
Gandara BP, Uddin A, McGarvey LR, Ohlmeyer M and Geraghty P:
Protein phosphatase 2A as a therapeutic target in pulmonary
diseases. Medicina (Kaunas). 59:15522023. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Wu SY, Wen YC, Ku CC, Yang YC, Chow JM,
Yang SF, Lee WJ and Chien MH: Penfluridol triggers cytoprotective
autophagy and cellular apoptosis through ROS induction and
activation of the PP2A-modulated MAPK pathway in acute myeloid
leukemia with different FLT3 statuses. J Biomed Sci. 26:632019.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Hung MH, Wang CY, Chen YL, Chu PY, Hsiao
YJ, Tai WT, Chao TT, Yu HC, Shiau CW and Chen KF: SET antagonist
enhances the chemosensitivity of non-small cell lung cancer cells
by reactivating protein phosphatase 2A. Oncotarget. 7:638–655.
2016. View Article : Google Scholar :
|
|
55
|
Tohmé R, Izadmehr S, Gandhe S, Tabaro G,
Vallabhaneni S, Thomas A, Vasireddi N, Dhawan NS, Ma'ayan A, Sharma
N, et al: Direct activation of PP2A for the treatment of tyrosine
kinase inhibitor-resistant lung adenocarcinoma. JCI Insight.
4:e1256932019. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Zhang Y, Wang X, Li A, Guan Y, Shen P, Ni
Y and Han X: PP2A regulates metastasis and vasculogenic mimicry
formation via PI3K/AKT/ZEB1 axis in non-small cell lung cancers. J
Pharmacol Sci. 150:56–66. 2022. View Article : Google Scholar : PubMed/NCBI
|