Introduction
Stroke is among the most prevalent causes of
mortality and disability worldwide (1), and can be classified into two
types, ischemic stroke and cerebral hemorrhage. At present, the
principal treatment of ischemic stroke lies in restoring the blood
supply to the ischemic area without delay. However, while restoring
blood perfusion to the ischemic area, brain tissue damage is
aggravated and leads to more serious neurological dysfunction,
known as cerebral ischemia/reperfusion (I/R) injury (CIRI)
(2). A pathophysiological trait
of ischemic stroke is the disruption of the blood-brain barrier
(BBB), characterized by increased permeability due to the breakdown
of tight junctions (TJs) between brain microvascular endothelial
cells (BMECs) and the increased vesicular transport in BMECs,
causing unregulated influx of blood-derived cells, macromolecules
and fluid. This ultimately leads to destructive cytotoxicity and
vascular-origin edema, as well as life-threatening hemorrhagic
transformation (3-5). In contrast to patients with mild
BBB injury, patients with severe BBB injury exhibit higher National
Institutes of Health Stroke Scale scores, worse functional outcomes
and a greater risk of mortality (6,7).
Therefore, alleviating the BBB injury can improve the neurological
prognosis of patients with stroke (8).
Ferroptosis was initially reported in 2012 as a new
form of regulated cell death that is distinct from apoptosis during
the processes of tumor and embryonic development (9). Research has indicated that
ferroptosis is associated with BBB injury caused by CIRI (10). In 2017, research demonstrated
that the inhibition of ferroptosis in a middle cerebral artery
occlusion (MCAO) model protected mice from CIRI (11). Furthermore, dietary iron
supplementation can enhance the rebuilding of the BBB and defend
neurons following ischemic stroke (12,13). In addition, co-injection of
ceruloplasmin and Fe2+ has been shown to significantly
reduce BBB permeability, DNA damage, neuronal cell death and the
degree of neurological deficits (14).
p23 is a protein with a molecular weight of 23 kDa,
which is widely expressed in the heart, brain, kidney, lung and
other tissues (15). Structural
studies have reported that the N-terminus of the p23 protein
contains a β-sheet domain consisting of two inverted β-sheets, and
its C-terminus is an unfolded tail (16,17). Furthermore, yeast p23 has been
reported to regulate the normal function of the Golgi apparatus,
ribosome synthesis and DNA repair (18). Deletion of the p23 gene can lead
to abnormal skin and lung tissue development, and perinatal death
in mouse embryos (19). Several
studies have shown that p23 can bind to a variety of proteins and
serve a regulatory role in various diseases by regulating the
biological functions of these target proteins. For example, the
helical motif at the C-terminus of p23 can bind to the
glucocorticoid receptor, thereby regulating its activity (20). In gastric cancer, p23 bound to
gedunin is more efficiently cleaved by activated caspase 7, thereby
promoting tumor cell apoptosis (21). However, to the best of our
knowledge, the biological significance of p23 in ferroptosis caused
by CIRI-induced BBB injury has not yet been studied.
The present study aimed to determine whether
ferroptosis occurs in BMECs during ischemic stroke, and to
investigate the regulation of p23 on BBB function and ferroptosis
in BMECs during ischemic stroke, so as to explore its specific
mechanism.
Materials and methods
Animals and ethics approval
Male Sprague-Dawley rats were acquired from the
Experimental Animal Center of Central South University (Changsha,
China). GPX4 FosCreERT2 mice (GPX4
FosCreERT2/− and GPX4 FosCreERT2/+, GPX4
FosCreERT2/− mice were used as controls for GPX4
FosCreERT2/+ mice to rule out the influence of the Cre
recombinase or other experimental factors) were produced using
GPX4-flox mice and FosCreERT2 mice (both obtained from
GemPharmatech Co., Ltd.). It was confirmed that the expression
levels of GPX4 were reduced in the brain tissues of GPX4
FosCreERT2/+ mice (Fig.
S1). The rats that were aged 8 weeks and weighed 280±20 g
underwent Ferrostatin-1 (Fer-1) or short hairpin (sh)RNA
treatments, whereas the rats that were aged 6 weeks and weighed
130-150 g were treated with for adeno-associated virus. The mice
were aged 8-12 weeks and weighed 22±25 g. All animals were kept in
a room with a controlled temperature of 25°C and humidity at 40-60%
under a 12-h light/dark cycle, with free access to food and
water.
A total of 78 rats and 30 GPX4 FosCreERT2
mice were used in the present study. For the Evans Blue
extravasation test in vivo, 3 rats were allocated to the
sham group, and 6 rats were allocated to the I/R 12-h group and the
I/R 24-h group. For the ZO-1 and p23 immunofluorescence assay in
vivo, 3 rats were allocated to the sham group and 6 rats were
allocated to the I/R 12-h group. To assess the effects of Fer-1 on
ZO-1 expression in vivo, 3 rats were allocated to the sham +
vehicle group and the sham + Fer-1 group, whereas 6 rats were
allocated to the I/R + vehicle group and I/R + Fer-1 group. For the
p23 shRNA test in vivo, the sham + shNC group and sham + p23
shRNA group were each allocated 3 rats, and the I/R + shNC group
and I/R + p23 shRNA group were each allocated 6 rats. For the p23
overexpression (OE) test in vivo, the sham + vector group
and sham + OE p23 group were each allocated 3 rats, and the I/R +
vector group and I/R + OE p23 group were each allocated 6 rats. For
the GPX4 FosCreERT2 immunofluorescence assay in
vivo, 3 GPX4 FosCreERT2/− and 3 GPX4
FosCreERT2/+ mice were allocated to the sham + OE-vector
group, and the I/R + OE-vector group and I/R + OE-p23 group were
each allocated 6 GPX4 FosCreERT2/− mice and 6 GPX4
FosCreERT2/+ mice. All experiments and animal procedures
were approved by the Experimental Animal Center of Central South
University (approval no. 2018sydw0222).
Cell culture and treatment
The mouse BMEC line bEnd.3 was procured from The
Cell Bank of Type Culture Collection of The Chinese Academy of
Sciences. The cells were cultured in DMEM (Gibco; Thermo Fisher
Scientific, Inc.) supplemented with 10% FBS (Gibco; Thermo Fisher
Scientific, Inc.) and 1% penicillin-streptomycin (Gen-View
Scientific, Inc.) at 37°C and 5% CO2. bEnd.3 cells were
used to establish an in vitro model of the BBB, and the
oxygen-glucose deprivation/reoxygenation (OGD/R) experiment was
used to simulate I/R injury in vitro. As for the
administration of Fer-1 (Sigma-Aldrich; Merck KGaA), bEnd.3 cells
were exposed to 10 µM Fer-1 at 37°C for 2 h before OGD/R.
For the cycloheximide (CHX) chase assay, CHX (10 µM; cat.
no. NSC-185; Selleck Chemicals) was applied in OGD/R-insulted b.End
3 cells for 3-12 h at 37°C to inhibit the proteasome pathway for
the degradation of TJ proteins. The GPX4 agonist PKUMDL-LC-101-D04
(200 µM; CAS no. 2143896-83-5; MedChemExpress) was used to
treat the cells for 1 h at 37°C after OGD/R and p23 siRNA
transfection, whereas the GPX4 inhibitor RAS-selective lethal 3
(RSL3; 200 nM; CAS no. 1219810-16-8; MedChemExpress) was used to
treat the cells for 16 h at 37°C after OGD/R and p23 oe.
Small interfering RNA (siRNA)
transfection
siRNA transfection was performed using the riboFECT™
CP Transfection Kit (Guangzhou RiboBio Co., Ltd.) according to the
manufacturer's instructions. Briefly, 1×106 cells/well
were plated in 6-well plates and cultured for 12-24 h until the
cell density reached 50-60%. The transfection complex was prepared
by diluting 5 µl 20 µM siRNA storage solution with
120 µl 1X riboFECT CP Buffer, adding 12 µl riboFECT
CP Reagent and incubating at room temperature for 15 min. The
transfection complex was then added to the medium and cultured in a
CO2 incubator at 37°C for 24 h. A negative control (NC)
siRNA that does not target any known human, mouse or rat genes, and
is not chemically modified, was used as a NC (cat. no.
siN0000001-1-10; Guangzhou RiboBio Co., Ltd.). The of p23 siRNA
sequence was as follows: 5′-GGTCCTGTTTGAACAAAGA-3′. The cell
samples were subjected to OGD. After 3 h of OGD exposure, the cells
were cultured in normal medium containing 10% FBS within a cell
incubator containing 5% CO2 at 37°C for 12 h.
Plasmid transfection
The p23 OE plasmid, truncated p23 plasmids and blank
vector plasmid (GV486; both from Shanghai GeneChem Co., Ltd.) were
transfected into cells using the Neofect™ DNA transfection reagent
(Neofect Biotech Co., Ltd.) according to the manufacturer's
protocol. The truncated p23 sequences are provided in Table I. A total of 1×106
cells/well were plated in 6-well plates and cultured for 12-24 h
until they reached a cell density of 60-80%. The DNA diluent was
prepared by adding 100 µl Opti-MEM serum-free (Gibco; Thermo
Fisher Scientific, Inc.) to 2 µg plasmid. Subsequently, 2
µl Neofect was added to the DNA diluent and incubated at
room temperature for 15-30 min. Finally, the complex was added to
the cells in 6-well plates and cultured in a CO2
incubator at 37°C for 48 h.
 | Table Ip23 truncated sequences. |
Table I
p23 truncated sequences.
| Truncation | Sequence,
5′-3′ |
|---|
| p23(1-160) |
ATGGACTACAAGGATGACGATGAC
AAGGATTACAAAGACGACGATGATAAGGACTATAAGGATGA |
|
TGACGACAAAATGCAGCCTGCTTCTGCAAAGTGGTACGATCGAAGGGACTATGTATTCATTGAATT |
|
TTGTGTTGAAGACAGTAAAGATGTTAATGTAAACTTTGAAAAATCCAAACTTACTTTCAGTTGTCT |
|
TGGAGGAAGCGATAATTTTAAGCATTTAAATGAAATTGATCTTTTTCATTGTATCGATCCAAATGATT |
|
CCAAGCATAAAAGAACAGACAGATCGATTTTATGTTGTTTGCGAAAAGGAGAATCCGGCCAGTCA |
|
TGGCCTAGGTTAACAAAGGAAAGGGCAAAGCTTAATTGGCTCAGTGTGGACTTCAATAATTGGAA |
|
AGACTGGGAGGATGACTCAGATGAAGACATGTCGAATTTTGACCGTTTCTCTGAGATGATGGATCA |
|
CATGGGTGGTGATGAGGATGTAGATTTACCAGAAGTAGATGGAGCAGATGATGATTCACAAGACA |
|
GTGATGATGAAAAGATGCCAGATCTGGAGTAA |
| p23(1-90) |
ATGGACTACAAGGATGACGATGACAAGGATTACAAAGACGACGATGATAAGGACTATAAGGATGA |
|
TGACGACAAAATGCAGCCTGCTTCTGCAAAGTGGTACGATCGAAGGGACTATGTATTCATTGAATT |
|
TTGTGTTGAAGACAGTAAAGATGTTAATGTAAACTTTGAAAAATCCAAACTTACTTTCAGTTGTCT |
|
TGGAGGAAGCGATAATTTTAAGCATTTAAATGAAATTGATCTTTTTCATTGTATCGATCCAAATGAT |
|
TCCAAGCATAAAAGAACAGACAGATCGATTTTATGTTGTTTGCGAAAAGGAGAATCCGGCCAGTC |
|
ATGGCCTAGGTTAACATAA |
| p23(91-160) |
ATGGACTACAAGGATGACGATGACAAGGATTACAAAGACGACGATGATAAGGACTATAAGGATGAT |
|
GACGACAAAAAGGAAAGGGCAAAGCTTAATTGGCTCAGTGTGGACTTCAATAATTGGAAAGACTG |
|
GGAGGATGACTCAGATGAAGACATGTCGAATTTTGACCGTTTCTCTGAGATGATGGATCACATGGG |
|
TGGTGATGAGGATGTAGATTTACCAGAAGTAGATGGAGCAGATGATGATTCACAAGACAGTGATGA |
|
TGAAAAGATGCCAGATCTGGAGTAA |
Cell viability assay
Cell viability was assessed using the Cell Counting
Kit-8 (CCK-8; cat. no. GK3607; Gen-View Scientific, Inc.). A total
of 5,000 cells were seeded in a 96-well plate and were cultured for
~24 h. Following treatment, CCK-8 was added to the medium in each
well at a 1:10 ratio and the cells were incubated at 37°C for 1-4
h. Subsequently, the colorimetric absorbance of each well was
measured at 450 nm using a microplate reader and a ratio was
calculated compared with the control group.
Reverse-transcription quantitative PCR
(RT-qPCR)
Following extraction with TRIzol® reagent
(Invitrogen; Thermo Fisher Scientific, Inc.), 1 µg RNA from
cells or animal brain samples was reverse transcribed using 5X Evo
M-MLV RT Reaction Mix (Accurate Biology) according to the
manufacturer's protocol. Subsequently, qPCR was carried out
according to the manufacturer's protocol using the SYBR Premix Ex
Taq II kit (Accurate Biology) with the following amplification
conditions: Pre-denaturation at 95°C for 30 sec, followed by 40
cycles at 95°C for 5 sec and 60°C for 34 sec. The primer sequences
used are presented in Table II.
The relative quantification of mRNA was analyzed using the
2−ΔΔCq method (22).
| Table IIPrimer sequences used in the present
study. |
Table II
Primer sequences used in the present
study.
| Primer | Forward sequence,
5′-3′ | Reverse sequence,
5′-3′ |
|---|
| GPX4 |
ATAAGAACGGCTGCGTGGTGAAG |
TAGAGATAGCACGGCAGGTCCTTC |
| ZO-1 |
CTGGTGAAGTCTCGGAAAAATG |
CATCTCTTGCTGCCAAACTATC |
| Occludin |
TGCTTCATCGCTTCCTTAGTAA |
GGGTTCACTCCCATTATGTACA |
| p23 |
AGGAAAGGGCAAAGCTTAATTG |
TCATCTCAGAGAAACGGTCAAA |
| COX-2 |
ATTCCAAACCAGCAGACTCATA |
CTTGAGTTTGAAGTGGTAACCG |
| β-actin |
CTACCTCATGAAGATCCTGACC |
CACAGCTTCTCTTTGATGTCAC |
Western blotting
Proteins from cells or animal brain samples were
extracted using radioimmunoprecipitation assay lysis buffer
(Guangzhou Dingguo Biotechnology Co., Ltd.) containing 1% PMSF, and
were quantified using the BCA method. Subsequently, 20-30 µg
cell proteins or 50-80 µg animal proteins were transferred
to a PVDF membrane following SDS-PAGE on 10-15% gels. Subsequently,
the membranes were blocked with TBS-1% Tween 20 (TBST) containing
5% skim milk powder at room temperature for 2 h. The membranes were
then incubated with primary antibodies against β-actin (cat. no.
A1978; dilution, 1:5,000; Sigma-Aldrich; Merck KGaA), p23 (cat. no.
160150; dilution 1:200; Cayman Chemical Company), zonula
occludens-1 (ZO-1; cat. no. 402200; dilution 1:2,000; Invitrogen;
Thermo Fisher Scientific, Inc.), occludin (cat. no. 404700;
dilution 1:1,000; Invitrogen; Thermo Fisher Scientific, Inc.), GPX4
(cat. no. ab125066; dilution 1:3,000; Abcam), cyclooxygenase
(COX)-2 (cat. no. 160106; dilution 1:200; Cayman Chemical Company),
heat shock protein 90 (HSP90; cat. no. 60318-1-lg; dilution
1:2,000; Proteintech Group, Inc.) and Flag (cat. no. 20543-1-AP;
dilution 1:2,000; Proteintech Group, Inc.) at 4°C overnight. The
membranes were washed with TBST three times at 10-min intervals and
were then incubated with the HRP-conjugated goat anti-rabbit IgG
(cat. no. BA1055; dilution 1:5,000; Boster Biological Technology)
or goat anti-mouse IgG (cat. no. BA1056; dilution 1:5,000; Boster
Biological Technology) secondary antibodies at room temperature for
1 h, followed by three further washes with TBST at 10-min
intervals. The color reaction was developed using the Clarity
Western ECL Substrate (cat. no. 170-5061; Bio-Rad Laboratories,
Inc.), and band density was measured using ImageJ software V 1.8.0
(National Institutes of Health) and was normalized to β-actin.
Immunofluorescence
After fixing with 4% paraformaldehyde for 10 min at
room temperature, 5,000 bEnd.3 cells grown in 96-well plates were
washed three times with PBS (5 min each) and were blocked with BSA
(Gen-View Scientific, Inc.) at 37°C for 1 h. The cells were then
incubated with a primary antibody against ZO-1 (cat. no. 61-7300;
dilution 1:50; Invitrogen; Thermo Fisher Scientific, Inc.), p23
(dilution 1:20; cat. no. 160150; Cayman Chemical Company), CD31
(cat. no. ab9498; dilution 1:1,000; Abcam) and GPX4 (dilution
1:100; cat. no. ab125066; Abcam) at 4°C overnight. The cells were
then incubated with a TRITC-conjugatedsecondary antibody (cat. no.
AS040; dilution 1:1,000; ABclonal Biotech Co., Ltd.) at 37°C in the
dark for 1 h. Nuclei were stained with 1X DAPI working solution at
room temperature for 5 min, washed three times with PBS-1% Tween 80
(PBST) and images were captured using a fluorescence microscope
(Olympus Corporation).
Experimental MCAO model
The rats or GPX4 FosCreERT2 mice were
anaesthetized through an intraperitoneal injection of 50 mg/kg
sodium pentobarbital (China National Pharmaceutical Group). An
incision was made in the neck to expose the external carotid,
internal carotid, right common carotid and common carotid arteries,
and the internal carotid artery was clamped. Following the ligation
of the external carotid artery, a small incision was made into the
external carotid artery. Monofilaments coated with poly-L-lysine
were inserted via the incision, through the internal carotid artery
and ultimately to the middle cerebral artery. Following occlusion
(2 h for rats and 1 h for mice), the nylon monofilament was
withdrawn for reperfusion for 24 h. The sham group underwent the
same procedure as the MCAO group without insertion of the
monofilament. The survival and health status of the animals was
observed every 3 h. The cortex of the infarcted hemisphere was
collected for subsequent experiments at 12 or 24 h
post-reperfusion. The animals were euthanized by rapid dislocation
of the head and neck when it was time to collect the specimens, or
when the animals reached the following humane endpoints: They were
too weak to eat or drink for up to 24 h; they were in a moribund
state, i.e., without anesthesia or sedation, they were showing
mental depression accompanied by hypothermia (<37°C). The loss
of respiratory movement (the rise and fall of the chest and
abdomen), and reflex actions of the eyes, nose and feet were used
to verify the death of the animals. Notably, after MCAO treatment,
12 rats and 8 mice were sacrificed before sample collection, and 6
rats and 4 mice died spontaneously.
Lateral ventricle injection
After anesthetizing with 50 mg/kg sodium
pentobarbital and preserving the skin on the top of the head, the
rats were positioned horizontally and prone on a stereotaxic frame
(RWD Life Science). Subsequently, 40 nmol/kg Fer-1, with saline
used as a vehicle control; or p23 shRNA [forward (5′-3′): CCG GGA
TCC TAG ACC ATG GAT TTA ACT CGA GTT AAA TCC ATG GTC TAG GAT CTT TTT
G, reverse (5′-3′): AAT TCA AAA AGA TCC TAG ACC ATG GAT TTA ACT CGA
GTT AAA TCC ATG GTC TAG GAT C; pRNAT-U6.1/Neo, Guangzhou RiboBio
Co., Ltd.], with shNC [forward (5′-3′): TAC GAT ACA AGG CTG TTA GAG
AG, reverse (5′-3′): TAG AAG GCA CAG TCG AGG used as the control]
was infused into the right lateral ventricle at a flow rate of 0.5
µl/min using a syringe pump (KD Scientific) in conjunction
with a Hamilton syringe. The stereotaxic coordinates for the right
lateral ventricle were -1.5 mm laterally, -1.2 mm anteroposteriorly
and -4.5 mm dorsoventrally, with the bregma point serving as the
origin. The p23 OE adeno-associated virus (AAV-9; Shanghai GeneChem
Co., Ltd.) was injected into the right lateral ventricle -1.2 mm
laterally, -0.8 mm anteroposteriorly and -3.5 mm dorsoventrally,
with empty AAV-9 used as the vector control. Following the
injection, the skull was sealed using bone wax. OE was induced 2-3
weeks after injection. Similarly, the lateral ventricle injection
of GPX4 FosCreERT2 mice was performed as that performed
in rats; the stereotaxic coordinates were as follows: -1.2 mm
laterally, -0.8 mm anteroposteriorly and -3.5 mm dorsoventrally.
Subsequently, the animals underwent MCAO modeling and the brain
tissue was collected for testing.
In vivo BBB permeability assay
In rats, the permeability of the BBB was evaluated
through Evans Blue extravasation. A 2% Evans Blue solution was
prepared using normal saline prior to the experiment. A total of 1
h before the conclusion of reperfusion, 2% Evans Blue (2 ml/kg) was
injected into the tail vein of the rats. After 1 h, the rats were
anesthetized with 50 mg/kg sodium pentobarbital and underwent
cardiac perfusion with PBS. The brains were then collected and 2-mm
thick coronal sections were made.
OGD/R
Cells were subjected to a 3-h culture in glucose-
and serum-free medium in an anaerobic incubator (37°C, 95%
N2, 5% CO2). After 3 h of OGD exposure, the
cells were cultured in normal medium containing 10% FBS within a
cell incubator containing 5% CO2 at 37°C for 12 and 24
h. The control group cells were cultured under normoxic condition,
while the 0 h OGD/R group underwent OGD without reperfusion. The
cell samples were then collected for subsequent experiments.
In vitro permeability assay
The bEnd.3 cells were cultured on the upper chamber
of gelatin-coated Transwell inserts (0.4 µm; Costar;
Corning, Inc.) and were exposed to hypoxic conditions (0%
O2) at 37°C for 3 h. Subsequently, 165 µg/ml
Evans Blue -0.1% BSA (Sigma Aldrich; Merck KGaA) was added to the
upper chamber 1 h prior to measurements. The intensities of the
diffused Evans Blue -0.1% BSA in the lower chamber were
subsequently measured at 650 nm using a microplate reader. The
results are presented as the ratio of the Evans Blue -0.1% BSA
concentration in the lower chamber to the total concentration of
Evans Blue -0.1% BSA added to the upper chamber at the onset of the
experiment.
Reactive oxygen species (ROS) assay
Using the fluorescent probe
diacetyldichlorofluorescein (DCFH-DA) ROS detection kit (cat. no.
S0033S; Beyotime Institute of Biotechnology), DCFH-DA was diluted
at a ratio of 1:1,000 with serum-free medium to a final
concentration of 10 µM. Subsequently, the cell culture
medium was removed from the cells, an appropriate volume of diluted
DCFH-DA was added and incubation was carried out in a cell
incubator at 37°C for 20 min. The cells were then washed with PBS
three times, and a 488-nm excitation wavelength and 525-nm emission
wavelength were used to detect the optical density using a
microplate reader.
Malondialdehyde (MDA) assay
The MDA assay was conducted using a Lipid
Peroxidation MDA Assay Kit (cat. no. S0131S; Beyotime Institute of
Biotechnology). Following homogenization, the brain or cell samples
were centrifuged at 13,000 × g and 4°C for 10 min to obtain the
supernatant for detection. Subsequently, the prepared
thiobarbituric acid working solution was added and heated at 100°C
for 15 min. Eventually, the mixtures were centrifuged at 13,000 × g
and 4°C for 10 min to obtain the supernatant, which was transferred
to a 96-well plate to measure absorbance at 532 nm using a
microplate reader. The content of MDA was normalized with respect
to the protein amount of the samples, which was determined using
the BCA method.
Glutathione (GSH) assay
The GSH assay was conducted using a GSH and reduced
glutathione (GSSG) assay kit (cat. no. S0053; Beyotime Institute of
Biotechnology) according to the manufacturer's protocol. After
OGD/R treatment, the cells were collected and resuspended with
protein removal agent M. After rupturing the cell membrane through
two freeze-thaw cycles, the cells were centrifuged at 10,000 × g at
4°C for 10 min and the supernatant was used for the determination
of total GSH. Part of the supernatant was taken for GSSG detection:
GSH removal auxiliary solution and GSH removal reagent working
solution were added, and the reaction was performed at 25°C for 60
min. The protein removal reagent M solution and total GSH detection
working solution were then added to the samples for total GSH or
GSSG detection, and were incubated at 25°C for 5 min, before the
addition of NADPH. The absorbance of the sample was detected at 512
nm with a microplate reader. The GSH content was calculated as
follows: GSH=total GSH-GSSG x2. The GSH content was normalized with
respect to the protein amount of the samples, which was determined
using the BCA method.
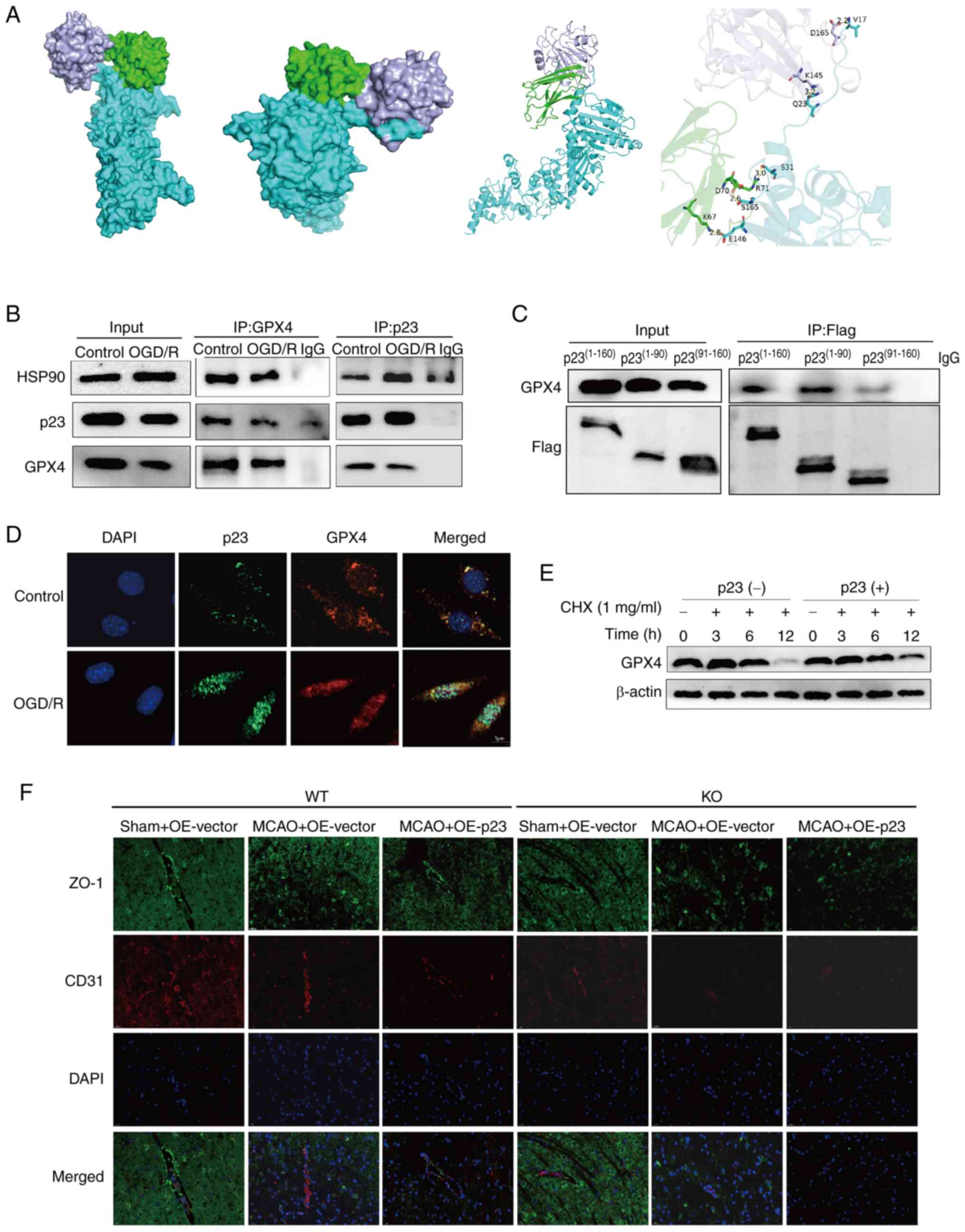
Molecular docking
The structures of HSP90, p23 and GPX4 were
downloaded from the Protein Data Bank (PDB) database (https://www.rcsb.org/); HSP90 and p23 are in the same
protein file (PDB ID: 7L7J), whereasGPX4 (PDB ID: 6HN3) has its own
protein structure file. The interaction modes between GPX4, HSP90
and p23 were investigated using the ZDOCK (https://zdock.wenglab.org/) server.
Transmission electron microscopy
Briefly, 5,000 cells/well were plated on a 96-well
slide. After OGD/R, the cells were washed with PBS and successively
fixed with 2.5% glutaraldehyde and 1% osmium acid at room
temperature for 6 h. Following dehydration using varying
concentrations of acetone, the samples were embedded in acetone and
embedding solution at room temperature for 12 h, and solidified in
an oven at 60°C for 24 h. Subsequently, the slides were
double-stained with 3% uranium acetate and lead nitrate at room
temperature for 2-3 min. Finally, the samples were observed and
images were captured using a Hitachi HT7700 transmission electron
microscope (Hitachi, Ltd.).
Co-immunoprecipitation assay
This assay was performed using agarose beads (Thermo
Fisher Scientific, Inc.). Proteins from cell samples were extracted
using radioimmunoprecipitation assay lysis buffer (Guangzhou
Dingguo Biotechnology Co., Ltd.). After extraction, 300 µg
cellular protein was incubated with the p23 (cat. no. 160150;
dilution 1:200; Cayman Chemical Company), GPX4 (cat. no. ab125066;
dilution 1:3,000; Abcam), and Flag (cat. no. 20543-1-AP; dilution
1:2,000; Proteintech Group, Inc.) antibodies at 4°C for 1 h with
agitation. Subsequently, 20 µl suspended agarose beads were
added and incubated at 4°C overnight with agitation. The
immunoprecipitation products were collected and centrifuged at
2,500 × g for 5 min at 4°C, and the supernatant was discarded. The
agarose beads were washed with PBST and centrifuged again; this was
repeated four times. Finally, the agarose beads were resuspended
with 40 µl electrophoretic loading buffer, boiled at 85°C
for 5 min, and 20 µl samples underwent SDS-PAGE and western
blotting.
Statistical analysis
All experiments were independently repeated at least
three times. The results, presented as the mean ± SD, were analyzed
using one-way or two-way ANOVA along with the multiple-comparison
Bonferroni correction test. Statistical analysis was performed
using GraphPad Prism 8 software (GraphPad Software Inc.). P<0.05
was considered to indicate a statistically significant
difference.
Results
Increased permeability of BMECs in
ischemic stroke
To investigate the effect of the MCAO model on BBB
dysfunction, the Evans Blue extravasation test was used to measure
cerebral microvascular permeability. As shown in Fig. 1A, the ischemia group had markedly
higher Evans Blue leakage than the sham group. TJ proteins are
transmembrane proteins located between endothelial cells of the
BBB, which serve critical roles in regulating the paracellular
permeability of the BBB (23).
The TJ protein ZO-1 was markedly decreased after 12-h I/R (Fig. 1B). To investigate the effects of
OGD/R on BBB permeability during hypoxia in vitro, a
Transwell assay was used to generate an in vitro BBB model,
and BBB permeability was measured in BMECs using Evans Blue
staining. The content of Evans Blue in the lower Transwell chamber
was higher in the 0 and 12 h OGD/R groups than in the normoxia
group (Fig. 1C). Subsequently,
western blotting was performed to determine whether OGD/R affected
the protein levels of ZO-1 and occludin. The expression levels of
ZO-1 and occludin were decreased in BMECs in the OGD/R group at 12
h after reoxygenation compared with those in the control group
(Fig. 1D-F). In addition, the
expression of the TJ ZO-1 was detected in OGD/R-treated BMECs using
immunofluorescence. The results showed that the expression of ZO-1
in BMECs in the OGD/R group was significantly decreased and
exhibited a discontinuous distribution (Fig. 1I and J).
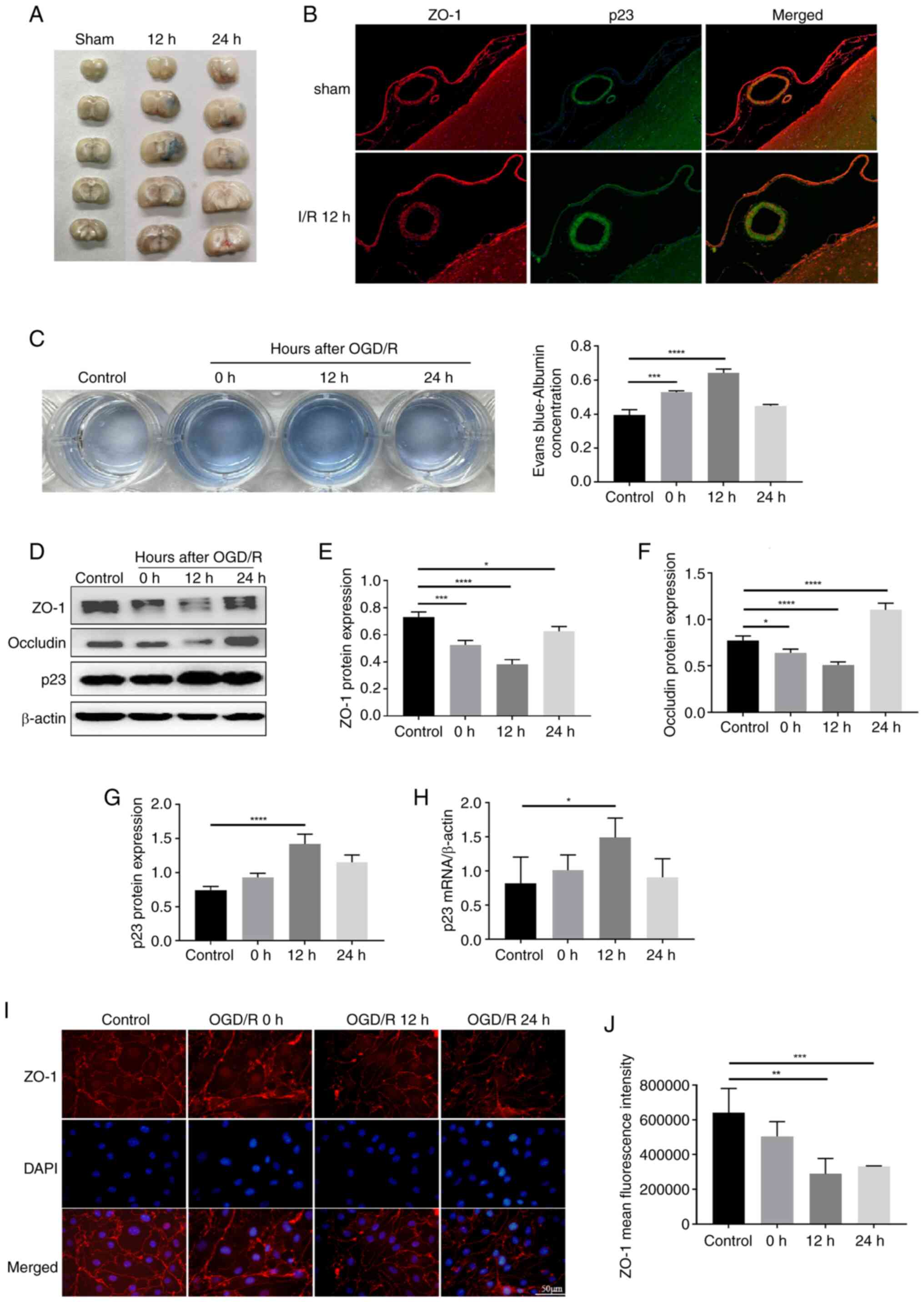 | Figure 1I/R-induced injury of BMECs. (A)
Evans Blue extravasation test was used to measure BBB permeability
in rats with different perfusion times. (B) Immunofluorescence
showed colocalization of p23 (green) and ZO-1 (red). (C) A
Transwell system was used to establish an in vitro BBB
model, and the permeability of BMECs was measured using Evans Blue
and underwent statistical analysis (n=3). (D) Protein expression
levels of (E) ZO-1 (n=3), (F) occludin (n=3) and (G) p23 (n=3) were
determined by western blotting in BMECs following OGD/R. (H)
Expression of p23 in BMECs after OGD/R was detected by quantitative
PCR (n=3). (I) Expression of ZO-1 in BMECs after OGD/R was detected
by immunofluorescence and (J) statistical analysis was performed
(n=3) (magnification, ×200). Data are presented as the mean ± SD.
*P<0.05, **P<0.01,
***P<0.001 and ****P<0.0001. I/R,
ischemia/reperfusion; BMECs, brain microvascular endothelial cells;
BBB, blood-brain barrier; ZO-1, zonula occludens-1; OGD/R,
oxygen-glucose deprivation/reoxygenation. |
Ferroptosis in BMECs is involved in BBB
damage in ischemic stroke
Increasing evidence has indicated that ferroptosis
in BMECs is the main cause of BBB injury induced by I/R (24,25). Pretreatment with Fer-1 was
performed via intraventricular injection for 2 h, followed by MCAO
modeling, and the results showed that, as compared with in the sham
+ vehicle (saline) group, ZO-1 expression in brain tissues was
decreased in the I/R + vehicle group, whereas it was increased in
the I/R + Fer-1 group compared with that in the I/R + vehicle group
(Fig. 2A). In addition, BMECs
were pretreated with Fer-1, and it was revealed that the reduction
in cell viability induced by OGD/R was significantly suppressed
(Fig. 2B). Following OGD/R,
BMECs exhibited mitochondrial swelling, and a decrease or
disappearance in cristae (Fig.
2C), as well as decreased levels of the antioxidant molecules
GSH and GPX4, and increased levels of MDA and ROS (Fig. 2D-J). This indicated that
intracellular lipid peroxidation levels were increased. In
addition, the expression levels of COX-2, another biomarker of
ferroptosis, were significantly increased in the 12 h OGD/R group
(Fig. 2K-M). These results
suggested that ferroptosis may be involved in OGD/R-induced BMECs
injury. Inhibiting ferroptosis in BMECs could protect against BBB
damage caused by cerebral I/R.
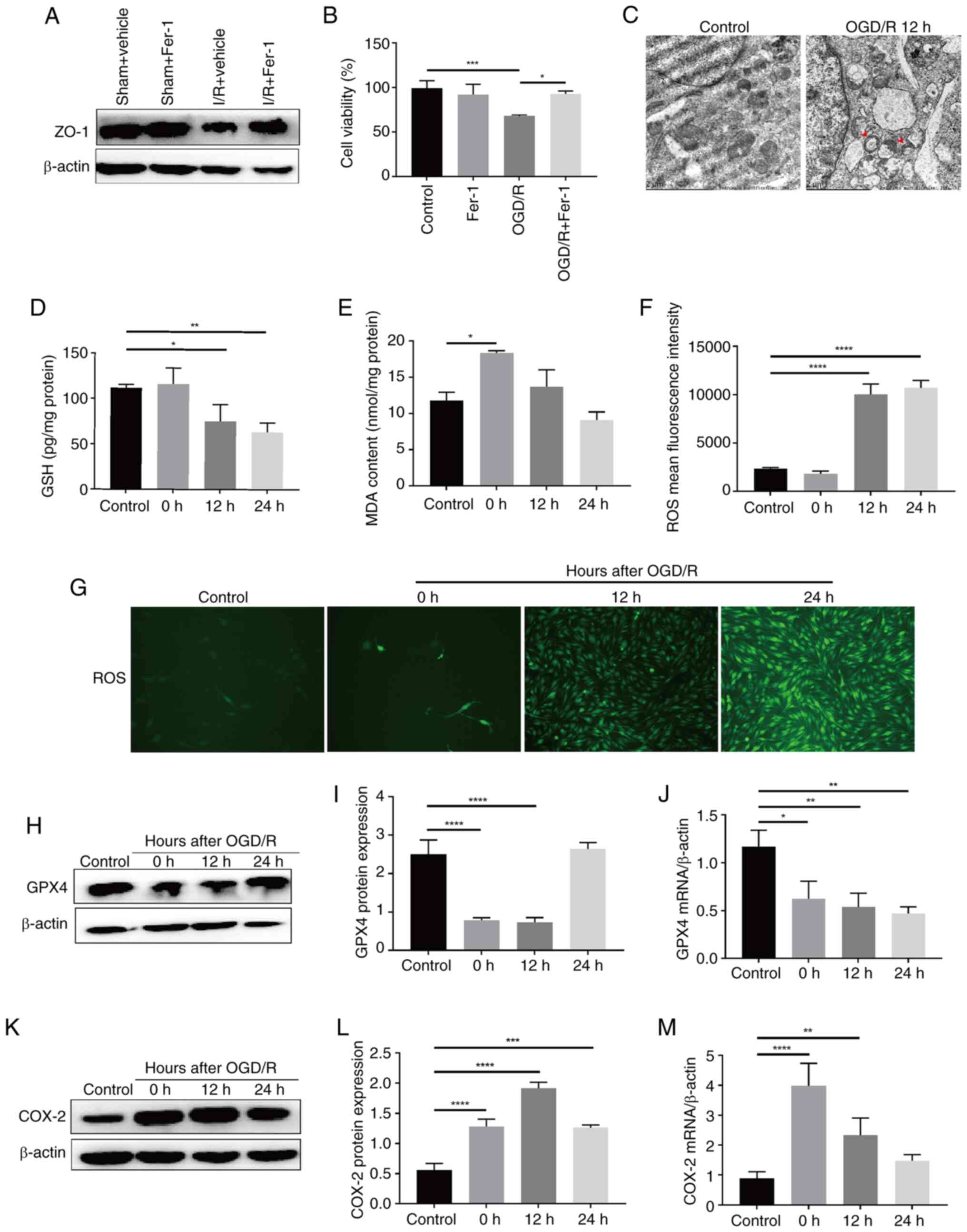 | Figure 2Ferroptosis is involved in
OGD/R-induced BMEC injury. (A) Expression levels of ZO-1 protein in
the rat brain tissue of a middle cerebral artery occlusion after 2
h of Fer-1 pretreatment were determined by western blotting. (B)
Viability of BMECs induced by OGD/R following pretreatment with
Fer-1 was determined using the Cell Counting Kit-8 assay (n=3). (C)
Representative transmission electron microscopy images displaying
the morphology of mitochondria in BMECs following OGD/R. The
mitochondria presented increased membrane density and a shrunken
form (indicated by red arrows) (magnification, ×20,000).
Intracellular (D) GSH (n=3), (E) MDA (n=3) and (F) ROS (n=3) levels
in BMECs following OGD/R. (G) Representative images of
intracellular ROS levels in BMECs following OGD/R (magnification,
×100). (H) Protein expression levels and (I) statistical analysis
(n=3), and (J) mRNA levels (n=3) of GPX4 in BMECs following OGD/R.
(K) Protein expression levels and (L) statistical analysis (n=3),
and (M) mRNA levels (n=3) of COX-2 in BMECs following OGD/R. Data
are presented as the mean ± SD. *P<0.05,
**P<0.01, ***P<0.001 and
****P<0.0001. OGD/R, oxygen-glucose
deprivation/reoxygenation; BMECs, brain microvascular endothelial
cells; ZO-1, zonula occludens-1; Fer-1, Ferrostatin-1; GSH,
glutathione; GPX4, glutathione peroxidase 4; MDA, malondialdehyde;
ROS, reactive oxygen species; COX-2, cyclooxygenase-2. |
p23 protects against BMEC injury in
ischemic stroke
p23 can bind to client proteins and serves a
regulatory role in numerous diseases by regulating their biological
functions. In the present study, the in vitro results showed
that p23 expression was significantly induced by OGD/R in BMECs
(Fig. 1D, G and H) Subsequently,
the permeability of BMECs was examined following OE or knockdown of
p23 in BMECs. First, in the OGD/R model, siRNA was used to
knockdown p23 expression (Fig.
3A), the results showed that p23 and ZO-1 expression levels
were inhibited by p23 knockdown (Fig. 3C and D), and the permeability of
BMECs was clearly increased (Fig.
3B), which indicated that the injury of BMECs induced by OGD/R
was aggravated by p23 knockdown. Transfection with the p23 OE
plasmid significantly increased the expression of p23 in the OGD/R
model (Fig. 4A-C). Compared with
in the vector + OGD/R group, the content of Evans Blue in the p23 +
OGD/R group was significantly decreased (Fig. 4D and E), and the expression of
ZO-1 was increased (Fig. 4F),
which indicated that the OE of p23 reversed the OGD/R-induced
damage of BMECs. In vivo, p23 knockdown by shRNA reduced the
expression of ZO-1 in the MCAO group compared with that in the MCAO
+ shNC group (Fig. 3E), while
p23 OE in the MCAO group could rescue the decrease in ZO-1 compared
with in the MCAO + vector group (Fig. 4G); CD31 was used to indicate the
location of blood vessels.
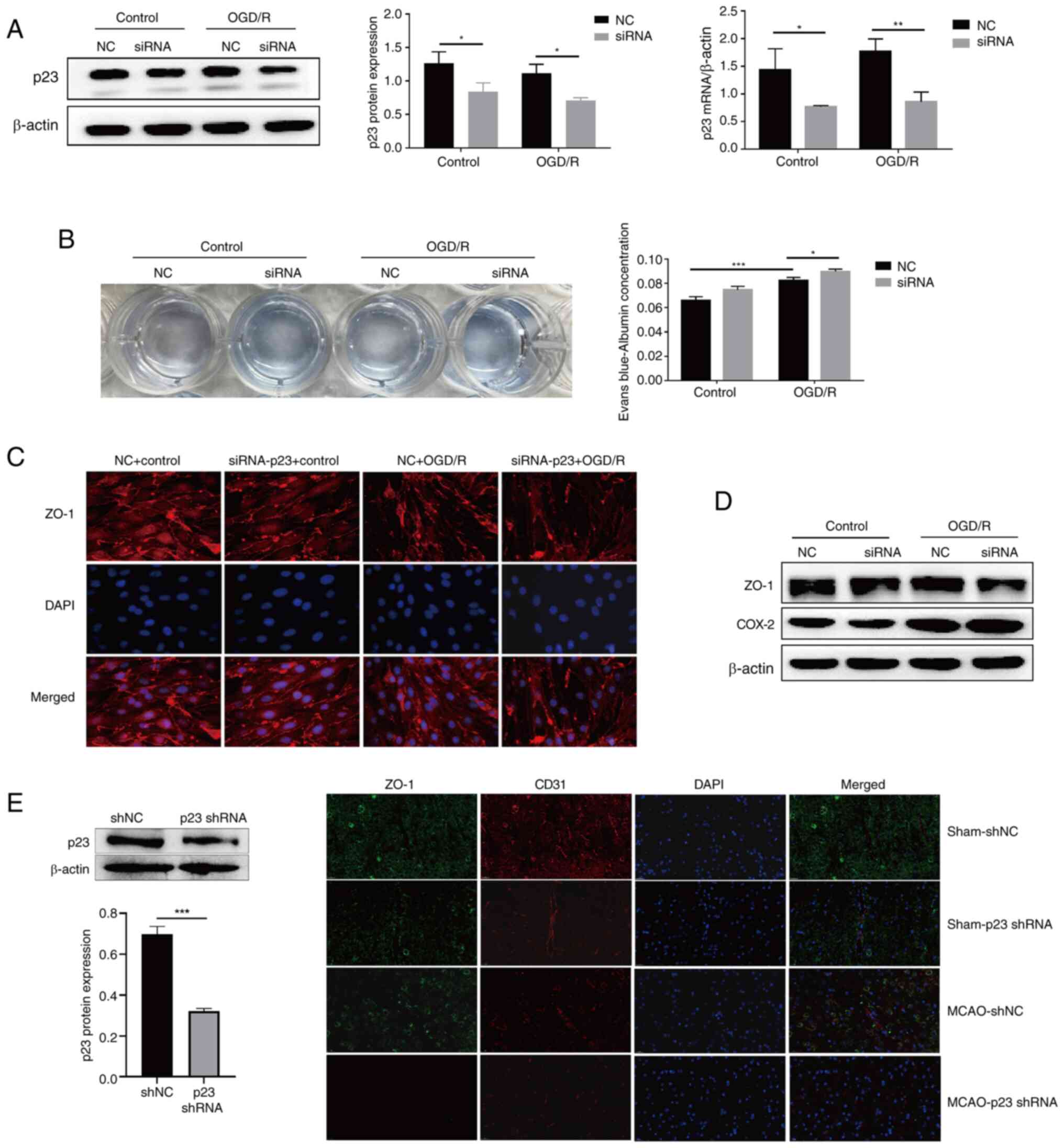 | Figure 3p23 protects against BMEC injury in
ischemic stroke. (A) p23 expression levels in BMECs with p23
siRNA-mediated knockdown (n=3). (B) Permeability of BMECs induced
by OGD/R following p23 siRNA transfection was detected using Evans
Blue (n=3). (C) Expression of ZO-1 in BMECs induced by OGD/R
following p23 siRNA transfection was detected using
immunofluorescence (magnification, ×200). (D) Expression of ZO-1
and COX-2 in rats following MCAO and p23 shRNA treatment was
detected using western blotting. (E) Expression levels of p23
(western blot repeats, n=3) and ZO-1 (magnification, ×100) were
detected in rats following MCAO and p23 shRNA treatment. Data are
presented as the mean ± SD. *P<0.05,
**P<0.01 and ***P<0.001. BMECs, brain
microvascular endothelial cells; siRNA, small interfering RNA; NC,
negative control; sh, short hairpin; OGD/R, oxygen-glucose
deprivation/reoxygenation; ZO-1, zonula occludens-1; COX-2,
cyclooxygenase-2; MCAO, middle cerebral artery occlusion. |
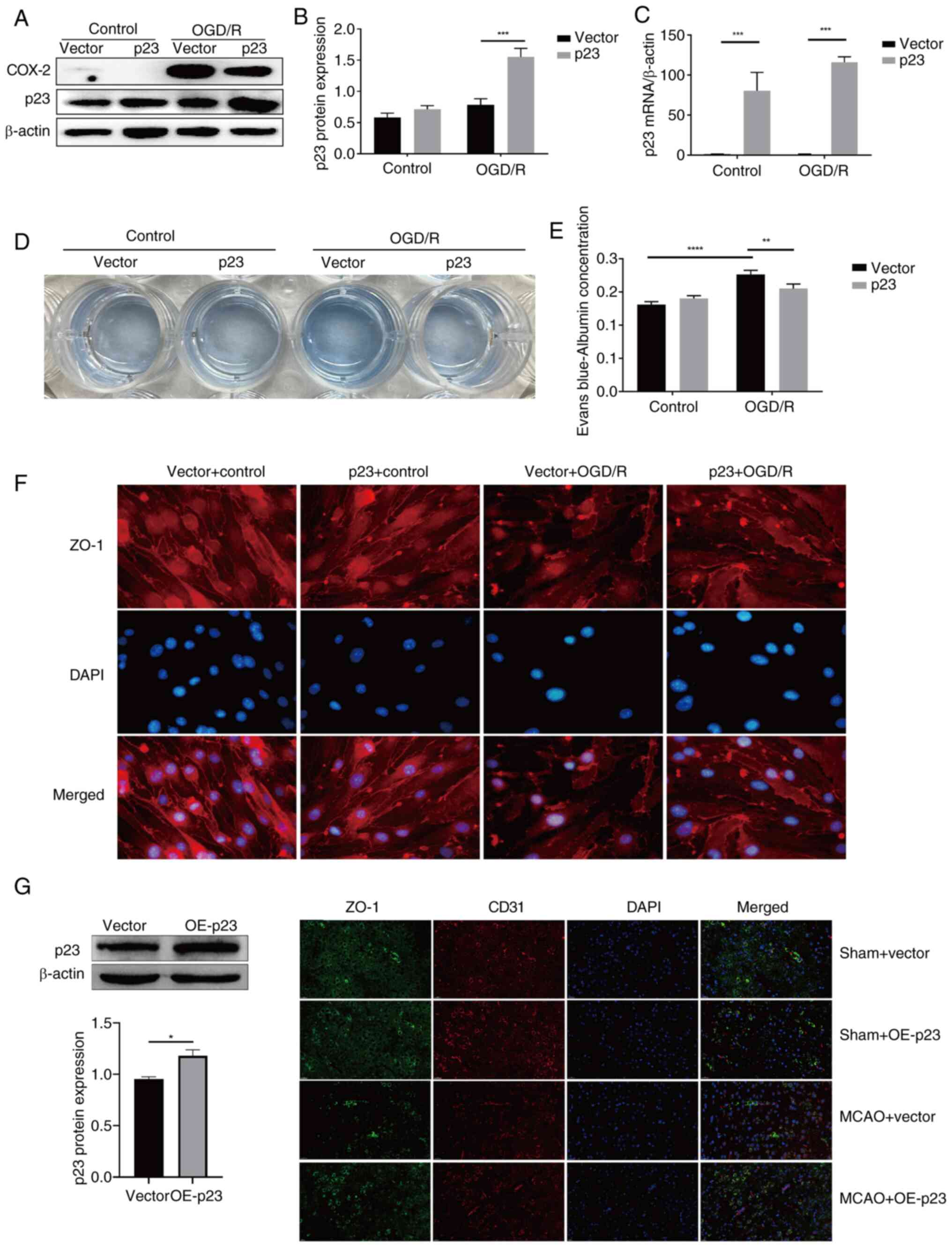 | Figure 4p23 protects against BMECs injury in
ischemic stroke. (A) Western blot analysis of p23 and COX-2
expression levels in BMECs with p23 OE. Statistical analysis of (B)
protein levels and (C) mRNA levels of p23 in BMECs with p23 OE
(n=3). (D) Permeability of BMECs in an OGD/R model following p23 OE
was detected using Evans Blue. (E) Statistical analysis of
permeability of BMECs in an OGD/R model following p23 OE (n=3). (F)
ZO-1 expression in BMECs in an OGD/R model following p23
overexpression was detected by immunofluorescence (magnification,
×200). (G) p23 (western blot repeats, n=3) and ZO-1 (magnification,
×100) expression were detected in rats following MCAO and p23 OE
treatment. Data are presented as the mean ± SD.
*P<0.05, **P<0.01,
***P<0.001 and ****P<0.0001. BMECs,
brain microvascular endothelial cells; OGD/R, oxygen-glucose
deprivation/reoxygenation; ZO-1, zonula occludens-1; MCAO, middle
cerebral artery occlusion; OE, overexpression. |
p23 protects against OGD/R-induced
ferroptosis in BMECs
Given that ferroptosis is involved in BBB damage in
ischemic stroke, the effect of p23 on OGD/R-induced ferroptosis in
BMECs was determined. Compared with in the control group, the
mortality of BMECs increased by p23 knockdown (Fig. 5A). In addition, p23 was
overexpressed at the cellular level, and p23 could rescue cell
death induced by OGD/R (Fig.
5B). In the OGD/R group, transfection with the p23 siRNA
further decreased the contents of GSH and GPX4, and increased the
levels of ROS, MDA, and acyl-CoA synthetase long-chain family
member 4 (ACSL4) and subunit solute carrier family 7 member 11
(SLC7A11; Fig. 5C-I) compared
with in the OGD/R + NC group, indicating that the increase of lipid
peroxidation in BMECs promoted OGD/R-induced ferroptosis. After
OGD/R, p23 OE decreased the levels of ROS, MDA, ACSL4 and SLC7A11,
and increased those of GSH and GPX4 compared with those in the
vector group (Fig. 5J-P),
indicating that the levels of antioxidants in BMECs were increased,
inhibiting OGD/R-induced ferroptosis. These results suggested that
the regulatory role of p23 in OGD/R-induced BMECs may be achieved
through ferroptosis.
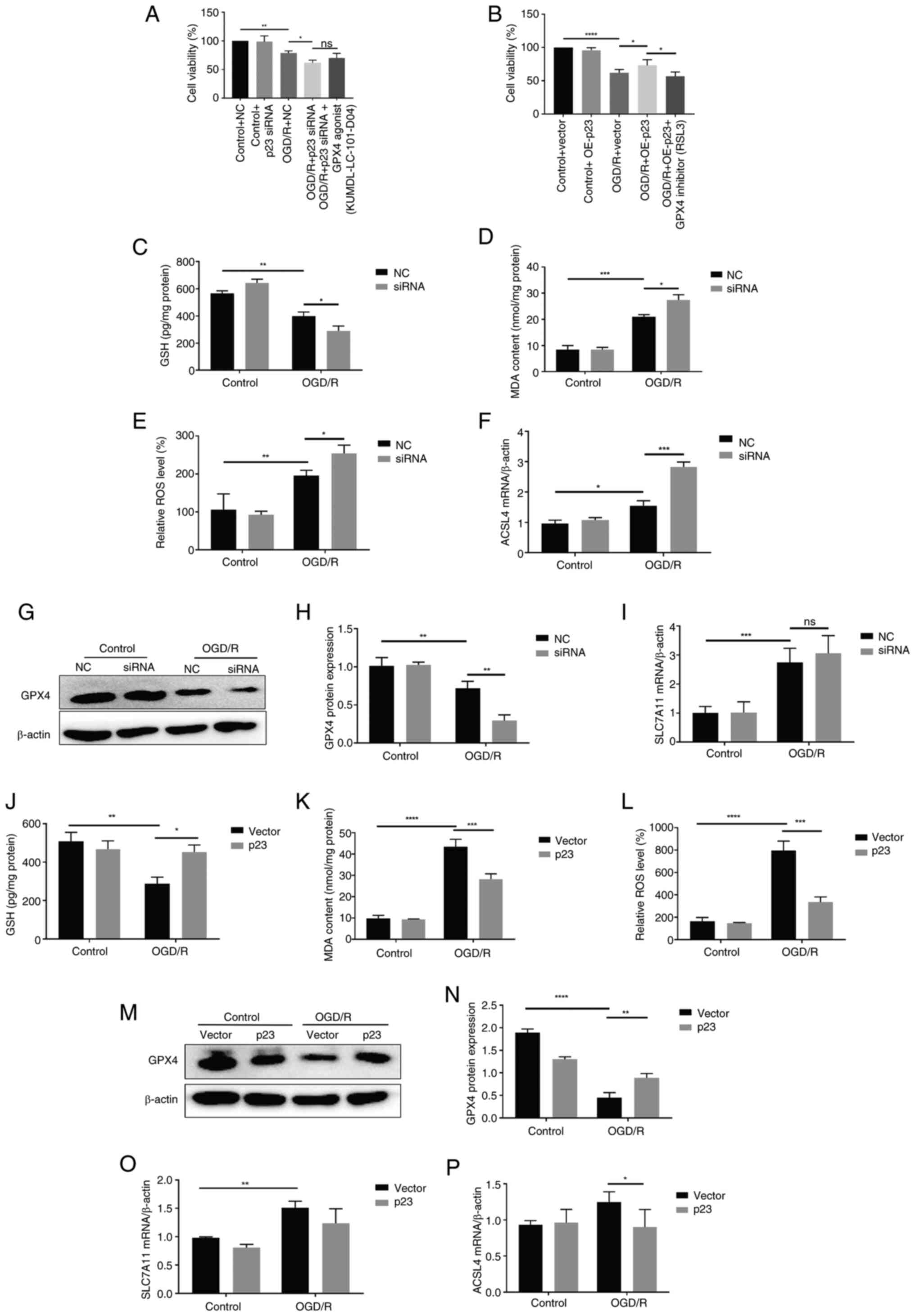 | Figure 5p23 protects against OGD/R-induced
ferroptosis of BMECs. (A) Cell viability induced by OGD/R following
transfection with p23 siRNA in BMECs was detected using the CCK-8
assay (n=3). (B) Cell viability induced by OGD/R following
transfection with p23 OE vector in BMECs was detected using the
CCK-8 assay (n=3). Intracellular (C) GSH (n=3), (D) MDA (n=3) and
(E) ROS (n=3) levels following the transfection of BMECs with p23
siRNA. (F) mRNA expression levels of ACSL4 in BMECs following p23
siRNA transfection (n=3). (G) Western blot analysis of GPX4 in
BMECs following p23 siRNA transfection and (H) statistical analysis
(n=3). (I) mRNA expression levels of SLC7A11 in BMECs following p23
siRNA transfection (n=3). Intracellular (J) GSH (n=3), (K) MDA
(n=3) and (L) ROS (n=3) levels in BMECs with p23 OE. (M) Western
blot analysis of GPX4 and (N) statistical analysis in BMECs with
p23 OE (n=3). mRNA expression levels of (O) SLC7A11 (n=3) and (P)
ACSL4 (n=3) in BMECs with p23 OE. Data are presented as the mean ±
SD. *P<0.05, **P<0.01,
***P<0.001 and ****P<0.0001. CCK, Cell
Counting Kit; OGD/R, oxygen-glucose deprivation/reoxygenation;
BMECs, brain microvascular endothelial cells; GSH, glutathione;
MDA, malondialdehyde; ROS, reactive oxygen species; ACSL4, acyl-CoA
synthetase long-chain family member 4; GPX4, glutathione peroxidase
4; SLC7A11, subunit solute carrier family 7 member 11; COX-2,
cyclooxygenase-2; OE, overexpression; siRNA, small interfering RNA;
NC, negative control; RSL3, RAS-selective lethal 3; ns, not
significant. |
p23 enhances GPX4 stability to suppress
ferroptosis
Notably, p23 can function as an independent
molecular chaperone or as a co-chaperone of HSP90 to exert its
biological functions (26). To
further explore the mechanism of p23-induced inhibition of
ferroptosis, protein docking showed that p23 formed a complex with
HSP90 and GPX4 through its N-terminal domain (1-90aa) (Fig. 6A). Co-IP experiments demonstrated
that anti-GPX4 antibodies could precipitate p23 and HSP90 proteins
in cells. Similarly, anti-p23 antibodies could precipitate GPX4 and
HSP90 proteins in cells. (Fig.
6B). Full-length and truncated p23 plasmids were then
constructed: Flag-p23(1-160), N-terminal Flag-p23(1-90) and
C-terminal Flag-p23(91-160) fragments. After transfecting the BMECs
with the truncated plasmids, immunoprecipitation was performed with
anti-Flag antibody. The results showed that GPX4 could be
precipitated by anti-Flag antibody, and the most important binding
region was p23(1-90), which indicated that p23 formed a complex
with HSP90 and GPX4 to inhibit GPX4 degradation and stabilize the
expression of GPX4 through the N-terminal domain (1-90aa) of p23,
thus inhibiting ferroptosis (Fig.
6C). In addition, immunofluorescence co-localization
verification showed that p23 and GPX4 were co-located in cells
(Fig. 6D). The effect of p23 on
the stability of GPX4 protein was then tested. The results showed
that the stability of GPX4 protein was markedly increased following
p23 OE (Fig. 6E). To further
verify the experimental results, GPX4 FosCreERT2 mice
were used to construct a model of MCAO. The results showed that the
reduction of ZO-1 in GPX4 FosCreERT2/− mice in the MCAO
model was partially reversed by p23 OE, while p23 OE did not have
this effect on GPX4 FosCreERT2/+ mice (Fig. 6F); CD31 was used to indicate the
location of blood vessels, indicating that p23 may have a
protective role by regulating ferroptosis through GPX4. The present
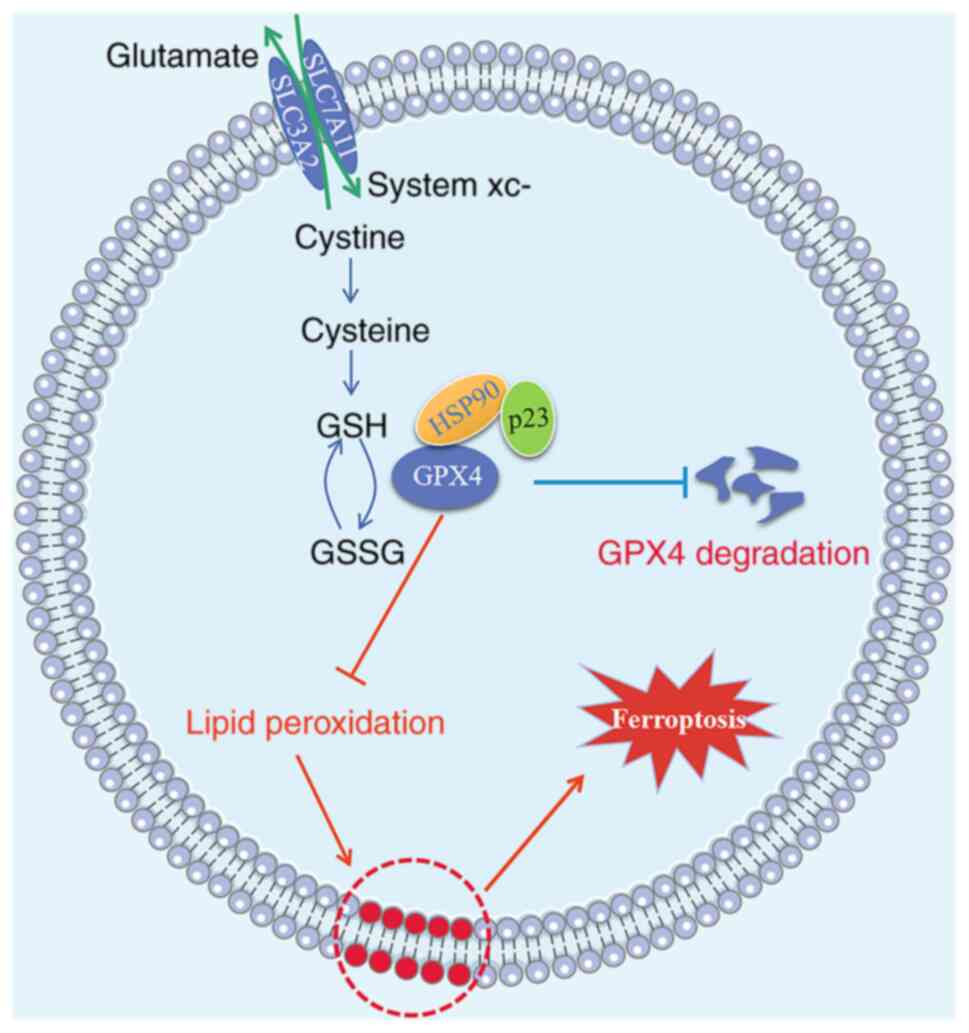
findings demonstrated that p23 may form complexes with HSP90 and
GPX4 through its N-terminal domain (1-90aa), thus enhancing the
stability of GPX4, inhibiting the degradation of GPX4 and
ultimately inhibiting BMEC ferroptosis, thereby protecting against
cerebral I/R-induced BBB damage.
Discussion
The present study illustrated the crucial role of
p23 as a key regulator of BBB injury following CIRI. First, p23 was
revealed to be markedly upregulated under stroke-like conditions in
both in vitro and in vivo models. p23 also formed a
complex with HSP90 and GPX4 through its N-terminal structure
(1-90aa), which improved their stability, inhibited GPX4
degradation, and ultimately inhibited ferroptosis in BMECs, thereby
protecting against I/R-induced BBB injury. The aforementioned
results are shown in Fig. 7.
These findings may have important implications for potential future
stroke treatment or prevention.
Ischemic stroke is the leading cause of death and
long-term disability worldwide, and reperfusion after ischemia can
trigger a series of harmful events, among which BBB disruption is
one of the most critical indicators (27). TJs are complexes formed between
microvascular endothelial cells, and their function is especially
crucial for maintaining the integrity of the BBB. When TJs open,
the integrity of the BBB is undermined. TJs are composed of a
complex network of proteins, such as occludin, claudin and ZO-1, -2
and -3. Among them, occludin and ZO-1 have been discovered to be
particularly significant in the assembly of endothelial cell
connections that sustain the BBB, and their loss is associated with
disruption of the BBB (28,29). Ferroptosis is a novel
iron-dependent form of regulated cell death that differs in
genetics, morphology and biochemistry from apoptosis, autophagy and
necrotic cell death (9).
Notably, it has been discovered to serve an important role in BBB
dysfunction (10,14,30). In line with previous reports, the
present results showed that cerebral I/R may lead to BBB injury and
increased BBB permeability. In vitro, OGD/R led to BMEC
damage and decreased the expression of TJ proteins ZO-1 and
occludin. In addition, rats were pretreated with an
intracerebroventricular injection of Fer-1 and a MCAO model was
established. The results indicated that Fer-1 markedly enhanced
ZO-1 expression. In vitro, the level of lipid peroxidation
in BMECs rose significantly, and the antioxidant content was
markedly decreased following OGD/R treatment, indicating that
ferroptosis may participate in OGD/R-induced BMEC injury.
Ferroptosis has become a global research focus. The
co-chaperone p23 is also known as cytosolic prostaglandin E (PGE)
synthase (15), and the
COX-2/PGE2 pathway has been reported to be closely associated with
CIRI (31). p23 is involved in
PGE2 synthesis, and there is evidence suggesting a connection
between the COX-2/PGE2 pathway and ferroptosis (32). Recent studies have shown that
PGE2 can prevent CIRI-induced ferroptosis (13,33), but the specific mechanism
requires further exploration. In the present study, in vitro
experiments showed that p23 knockdown promoted OGD/R-induced
ferroptosis in BMECs, and further aggravated I/R-induced BBB
damage. After OE of p23, the occurrence of ferroptosis in BMECs
could be inhibited and BBB damage could be improved. These results
indicated that p23 may exert a protective effect by regulating
GPX4.
The biochemical mechanism of ferroptosis involves
the iron-dependent formation of lipid-ROS, and consumption of GSH
or inactivation of GPX4 (34,35). Phospholipid hydroperoxide is an
enzyme that reduces phospholipid hydroperoxides to the
corresponding phospholipid alcohol, and its expression or activity
is regulated by selenium and GSH (36). In the course of the present
study, it was discovered that the expression levels of GPX4 were
regulated by p23, suggesting that p23 may regulate ferroptosis by
targeting GPX4. To further prove this, the effect of p23 on GPX4
protein stability was assessed, and it was demonstrated that GPX4
protein stability was markedly increased following p23 OE. In GPX4
FosCreERT2/+ mice, p23 almost lost the ability to
increase the expression of ZO-1, suggesting that GPX4 is an
important site for p23 to serve a protective role in BBB. p23 often
plays a unique biological function as a molecular chaperone and
serves a regulatory role in a variety of diseases by binding to a
various protein to regulate their biological function. First, it is
an important co-molecular chaperone of HSP90, assisting HSP90 in
completing its molecular chaperone function through its C-terminal
co-chaperone complex (37).
Genome and proteomics screening in yeast has revealed a network
showing that p23 may have an HSP90-independent effect in
intracellular receptor-regulated gene expression, and shows the
characteristics of ribosomal biogenesis and vesicle-mediated
transport (18,38). Through immunofluorescence
co-localization experiments, the present study revealed that p23
co-localizes with GPX4 in cells. Further verification by protein
docking and co-immunoprecipitation experiments showed that p23
forms a complex with HSP90 and GPX4 through its N-terminal domain
(1-90aa) and enhances the stability of GPX4, inhibits the
degradation of GPX4 and ultimately inhibits ferroptosis in BMECs,
protecting against I/R-induced BBB injury. To the best of our
knowledge, the present study was the first to explore the mechanism
of p23 regulating ferroptosis in BMECs to protect against BBB
dysfunction, providing hope for future clinical applications and
laying the foundation for more effective stroke treatment by
protecting against BBB damage.
There are some limitations in the current study.
Although the OE of p23 did not significantly change protein
expression levels in the control group, there was a significant
difference in the OGD/R group, as well as in the mRNA levels of p23
in the control group. It could be hypothesized that this may be due
to the fact that in the control group, after p23 is overexpressed,
cells may counteract functional activation by downregulating the
protein level of p23, thus achieving homeostasis of the body, while
in the OGD/R group, the destruction of homeostasis led to effective
OE.
In conclusion, p23 exerts a protective effect
against cerebral I/R-induced BBB injury by suppressing the
ferroptosis of BMECs through enhancing the stability of GPX4,
offering a potential therapeutic target for ischemic stroke.
Supplementary Data
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
YL and JZ designed and guided the project. YZ and YX
conducted the experiments. YZ, YX, QX and NH analyzed the data. All
authors discussed the results and contributed to the final version
of the manuscript. YZ and YX wrote the manuscript. YL and JZ
revised the manuscript. YL is the principal corresponding author
and is responsible for the integrity of the work in its entirety
from its initiation to the publication of the article. YL and JZ
confirm the authenticity of all the raw data. All authors read and
approved the final version of the manuscript.
Ethics approval and consent to
participate
All the procedures carried out in studies involving
animals were in line with the ethical standards of the institution
or practice where the studies were conducted, and were approved by
the Experimental Animal Center of Central South University
(Changsha, China; approval no. 2018sydw0222).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
Not applicable.
Funding
This work was funded by the National Natural Science Foundation
of China (grant nos. 82172147, 81571880, 81373147, 30901555 and
30972870) and the Natural Science Foundation of Hunan Province
(grant nos. 2021JJ30900 and 2016JJ2157). All these funding bodies
supported the study design, data collection, analysis,
interpretation and manuscript writing.
References
|
1
|
Miller JB, Merck LH, Wira CR, Meurer WJ,
Schrock JW, Nomura JT, Siket MS, Madsen TE, Wright DW, Panagos PD
and Lewandowski C: The advanced reperfusion Era: Implications for
emergency systems of ischemic stroke care. Ann Emerg Med.
69:192–201. 2017. View Article : Google Scholar
|
|
2
|
Eltzschig HK and Eckle T: Ischemia and
reperfusion-from mechanism to translation. Nat Med. 17:1391–1401.
2011. View
Article : Google Scholar : PubMed/NCBI
|
|
3
|
Leigh R, Christensen S, Campbell BC, Marks
MP, Albers GW and Lansberg MG: Pretreatment blood-brain barrier
disruption and post-endovascular intracranial hemorrhage.
Neurology. 87:263–269. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Jiang X, Andjelkovic AV, Zhu L, Yang T,
Bennett MVL, Chen J, Keep RF and Shi Y: Blood-brain barrier
dysfunction and recovery after ischemic stroke. Prog Neurobiol.
163-164:144–171. 2018. View Article : Google Scholar :
|
|
5
|
Leigh R, Jen SS, Hillis AE, Krakauer JW
and Barker PB: Pretreatment blood-brain barrier damage and
post-treatment intracranial hemorrhage in patients receiving
intravenous tissue-type plasminogen activator. Stroke.
45:2030–2035. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Desilles JP, Rouchaud A, Labreuche J,
Meseguer E, Laissy JP, Serfaty JM, Lapergue B, Klein IF, Guidoux C,
Cabrejo L, et al: Blood-brain barrier disruption is associated with
increased mortality after endovascular therapy. Neurology.
80:844–851. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Villringer K, Sanz Cuesta BE, Ostwaldt AC,
Grittner U, Brunecker P, Khalil AA, Schindler K, Eisenblätter O,
Audebert H and Fiebach JB: DCE-MRI blood-brain barrier assessment
in acute ischemic stroke. Neurology. 88:433–440. 2017. View Article : Google Scholar
|
|
8
|
Ji B, Zhou F, Han L, Yang J, Fan H, Li S,
Li J, Zhang X, Wang X, Chen X and Xu Y: Sodium Tanshinone IIA
sulfonate enhances effectiveness Rt-PA treatment in acute ischemic
stroke patients associated with ameliorating blood-brain barrier
damage. Transl Stroke Res. 8:334–340. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Dixon SJ, Lemberg KM, Lamprecht MR, Skouta
R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS,
et al: Ferroptosis: An iron-dependent form of nonapoptotic cell
death. Cell. 149:1060–1072. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zhao Y, Liu Y, Xu Y, Li K, Zhou L, Qiao H,
Xu Q and Zhao J: The role of ferroptosis in blood-brain barrier
injury. Cell Mol Neurobiol. 43:223–236. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Tuo QZ, Lei P, Jackman KA, Li XL, Xiong H,
Li XL, Liuyang ZY, Roisman L, Zhang ST, Ayton S, et al:
Tau-mediated iron export prevents ferroptotic damage after ischemic
stroke. Mol Psychiatry. 22:1520–1530. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Yan BC, Cao J, Liu J, Gu Y, Xu Z, Li D and
Gao L: Dietary Fe3O4 Nanozymes prevent the injury of neurons and
blood-brain barrier integrity from cerebral ischemic stroke. ACS
Biomater Sci Eng. 7:299–310. 2021. View Article : Google Scholar
|
|
13
|
Xu Y, Li K, Zhao Y, Zhou L, He N, Qiao H,
Xu Q, Zhang H, Liu Y and Zhao J: Inhibition of
15-hydroxyprostaglandin dehydrogenase protects neurons from
ferroptosis in ischemic stroke. MedComm. 5:e4522024. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Liu H, Hua Y, Keep RF and Xi G: Brain
ceruloplasmin expression after experimental intracerebral
hemorrhage and protection against Iron-induced brain injury. Transl
Stroke Res. 10:112–119. 2019. View Article : Google Scholar :
|
|
15
|
Tanioka T, Nakatani Y, Semmyo N, Murakami
M and Kudo I: Molecular identification of cytosolic prostaglandin
E2 synthase that is functionally coupled with cyclooxygenase-1 in
immediate prostaglandin E2 biosynthesis. J Biol Chem.
275:32775–32782. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Exner KS and Ivanova A: A
doxorubicin-peptide-gold nanoparticle conjugate as a functionalized
drug delivery system: Exploring the limits. Phys Chem Chem Phys.
24:14985–14992. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Martinez-Yamout MA, Venkitakrishnan RP,
Preece NE, Kroon G, Wright PE and Dyson HJ: Localization of sites
of interaction between p23 and Hsp90 in solution. J Biol Chem.
281:14457–14464. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Echtenkamp FJ, Zelin E, Oxelmark E, Woo
JI, Andrews BJ, Garabedian M and Freeman BC: Global functional map
of the p23 molecular chaperone reveals an extensive cellular
network. Mol Cell. 43:229–241. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Grad I, McKee TA, Ludwig SM, Hoyle GW,
Ruiz P, Wurst W, Floss T, Miller CA III and Picard D: The Hsp90
cochaperone p23 is essential for perinatal survival. Mol Cell Biol.
26:8976–8983. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lovgren AK, Kovarova M and Koller BH:
cPGES/p23 is required for glucocorticoid receptor function and
embryonic growth but not prostaglandin E2 synthesis. Mol Cell Biol.
27:4416–4430. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Patwardhan CA, Fauq A, Peterson LB, Miller
C, Blagg BS and Chadli A: Gedunin inactivates the co-chaperone p23
protein causing cancer cell death by apoptosis. J Biol Chem.
288:7313–7325. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
23
|
Liu WY, Wang ZB, Zhang LC, Wei X and Li L:
Tight junction in blood-brain barrier: An overview of structure,
regulation, and regulator substances. CNS Neurosci Ther.
18:609–615. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Liu H, Zhang TA, Zhang WY, Huang SR, Hu Y
and Sun J: Rhein attenuates cerebral ischemia-reperfusion injury
via inhibition of ferroptosis through NRF2/SLC7A11/GPX4 pathway.
Exp Neurol. 369:1145412023. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Xiao P, Huang H, Zhao H, Liu R, Sun Z, Liu
Y, Chen N and Zhang Z: Edaravone dexborneol protects against
cerebral ischemia/reperfusion-induced blood-brain barrier damage by
inhibiting ferroptosis via activation of nrf-2/HO-1/GPX4 signaling.
Free Radic Biol Med. 217:116–125. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Yu Z, Peng Y, Gao J, Zhou M, Shi L, Zhao
F, Wang C, Tian X, Feng L, Huo X, et al: The p23 co-chaperone is a
succinate-activated COX-2 transcription factor in lung
adenocarcinoma tumorigenesis. Sci Adv. 9:eade03872023. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Neuwelt EA, Bauer B, Fahlke C, Fricker G,
Iadecola C, Janigro D, Leybaert L, Molnár Z, O'Donnell ME,
Povlishock JT, et al: Engaging neuroscience to advance
translational research in brain barrier biology. Nat Rev Neurosci.
12:169–182. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Hawkins BT and Davis TP: The blood-brain
barrier/neurovascular unit in health and disease. Pharmacol Rev.
57:173–185. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zlokovic BV: The blood-brain barrier in
health and chronic neurodegenerative disorders. Neuron. 57:178–201.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Rand D, Ravid O, Atrakchi D, Israelov H,
Bresler Y, Shemesh C, Omesi L, Liraz-Zaltsman S, Gosselet F,
Maskrey TS, et al: Endothelial iron homeostasis regulates
Blood-brain barrier integrity via the HIF2α-Ve-cadherin pathway.
Pharmaceutics. 13:3112021. View Article : Google Scholar
|
|
31
|
Xu Y, Liu Y, Li K, Miao S, Lv C, Wang C
and Zhao J: Regulation of PGE2 pathway during cerebral ischemia
reperfusion injury in rat. Cell Mol Neurobiol. 41:1483–1496. 2021.
View Article : Google Scholar
|
|
32
|
Chen B, Chen Z, Liu M, Gao X, Cheng Y, Wei
Y, Wu Z, Cui D and Shang H: Inhibition of neuronal ferroptosis in
the acute phase of intracerebral hemorrhage shows long-term
cerebroprotective effects. Brain Res Bull. 153:122–132. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Xu Y, Liu Y, Li K, Yuan D, Yang S, Zhou L,
Zhao Y, Miao S, Lv C and Zhao J: COX-2/PGE2 pathway inhibits the
ferroptosis induced by cerebral ischemia reperfusion. Mol
Neurobiol. 59:1619–1631. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Zou Y, Li H, Graham ET, Deik AA, Eaton JK,
Wang W, Sandoval-Gomez G, Clish CB, Doench JG and Schreiber SL:
Cytochrome P450 oxidoreductase contributes to phospholipid
peroxidation in ferroptosis. Nat Chem Biol. 16:302–309. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Yang WS, Kim KJ, Gaschler MM, Patel M,
Shchepinov MS and Stockwell BR: Peroxidation of polyunsaturated
fatty acids by lipoxygenases drives ferroptosis. Proc Natl Acad Sci
USA. 113:E4966–E4975. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Friedmann Angeli JP and Conrad M: Selenium
and GPX4, a vital symbiosis. Free Radic Biol Med. 127:153–159.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Daneri-Becerra C, Valeiras B, Gallo LI,
Lagadari M and Galigniana MD: Cyclophilin A is a mitochondrial
factor that forms complexes with p23-correlative evidence for an
anti-apoptotic action. J Cell Sci. 134:2021. View Article : Google Scholar
|
|
38
|
Freeman BC, Felts SJ, Toft DO and Yamamoto
KR: The p23 molecular chaperones act at a late step in
intracellular receptor action to differentially affect ligand
efficacies. Genes Dev. 14:422–434. 2000. View Article : Google Scholar : PubMed/NCBI
|