Introduction
Despite the large number of studies in this field,
controversies regarding the definition of cell death and the
classification of cell death types have not yet been resolved.
Based on morphological criteria, three types of cell death have
been defined: apoptosis, autophagic cell death and necrosis.
Apoptosis (programmed cell death-type I, PCD-type I) and necrosis
are well-known mechanisms of cell death induced by several stimuli.
Emerging studies have demonstrated the existence of a nonapoptotic
form of programmed cell death called autophagic cell death
(programmed cell death-type II, PCD-type II). However, the
clear-cut distinctions among these three types of cell death are
still controversial, and a clear equivalence between the
ultrastructural alterations and biochemical cell death
characteristics has not been established (1).
All Bcl-2 family members contain at least one of the
four conserved α-helical motifs known as Bcl-2 homology (BH)
domains (BH1-BH4) (2). Bcl-2
family members are grouped into three categories: the
anti-apoptotic members, including Bcl-2, Bcl-xL and Mcl-1; the
multidomain proapoptotic members, such as Bax and Bak; and the BH3
domain only proteins, such as Bim, Bid, Bad and Bik (3). Bcl-2 family members are known to
regulate apoptosis. Although Bcl-2 family proteins have been found
to have diverse subcellular locations, the principal site in which
the action of apoptosis is regulated by Bcl-2 family proteins is
likely the mitochondrial membrane. The anti-apoptotic multidomain
proteins (Bcl-2, Bcl-xL, Bcl-w and Mcl-1) mainly reside in the
mitochondria. The anti-apoptotic Bcl-2 family members prevent the
translocation and/or activation of Bax-like proteins in the
mitochondria (4), inhibiting
cytochrome c release from the mitochondria or mitochondrial
membrane depolarization. Certain members of the Bcl-2 family are
present on the endoplasmic reticulum (ER), where they participate
in regulating ER-mediated apoptosis (5).
Previous studies have elucidated that Bcl-2 inhibits
autophagy. Antisense knockdown of Bcl-2 in HL-60-induced autophagy
(6), and Bcl-2 gene silencing
increase autophagy, and Bcl-2 transgenic expression reduces the
level of starvation-induced autophagy. Furthermore, the study using
Bcl-2 mutants restricting subcellular localization showed that
Bcl-2 functions at the ER and not at the mitochondria to inhibit
starvation-induced autophagy (7).
Thus, Bcl-2 not only functions as an anti-apoptotic protein but
also as an anti-autophagic factor. Increasing data suggest that
Bcl-2 plays a pivotal role in modulating the complex interaction
that exists between autophagy and the apoptotic cell death pathway.
Bcl-2 is known to interact with the evolutionarily conserved
autophagy protein, Beclin-1 protein, which is part of a Class III
PI3K complex that participates in autophagosome formation,
mediating the localization of other autophagy proteins to the
preautophagosomal membrane (8).
Bcl-2 overexpression interferes with the formation of the
autophagy-promoting Beclin-1/hVps34 complex (7).
The resistance of tumor cells to the current
chemotherapeutic drugs is a challenging hurdle to cancer treatment.
Upregulation of the anti-apoptotic molecules in tumors impairs
remission and cure with chemotherapy, protecting the tumor cells
from the apoptotic effects of various antineoplastic agents. High
expression of the anti-apoptotic protein Bcl-2 is found in numerous
human tumors (9). Anti-apoptotic
Bcl-2 proteins inhibit apoptosis induced by various stimuli
including chemotherapeutics (10).
The functional blockade of Bcl-2 or other anti-apoptotic proteins,
such as Bcl-xL, could either induce apoptosis in cancer cells or
sensitize these cells to chemotherapy (11). Because anti-apoptotic factors,
including Bcl-2, impair the ability to achieve remission and cure
with chemotherapy, protecting the tumor cells from the apoptotic
effects of various antineoplastic agents, anti-apoptotic members of
the Bcl-2 family have attracted interest in drug discovery to
develop a new class of anticancer agents.
Etoposide (VP-16) is a semi-synthetic derivative of
podophyllotoxin and acts as a topoisomerase II inhibitor by forming
a ternary complex. Etoposide, which is one of the most widely used
cancer chemotherapy agents (12),
has been used clinically both as a single agent and a constituent
of combination chemotherapy regimens and is known to improve the
treatment of various human cancers (13). Numerous studies have demonstrated
that etoposide induces apoptotic cell death. However, the exact
molecular mechanism leading to apoptotic cell death by etoposide
remains to be elucidated.
This study examined whether etoposide overcomes the
resistance conferred by Bcl-2 in Hep3B hepatoma cells. We found
that etoposide overcomes the resistance conferred by Bcl-2 in Hep3B
hepatoma cells via the induction of autophagic cell death.
Materials and methods
Reagents and antibodies
The following reagents were obtained commercially:
rabbit polyclonal anti-human Bcl-2, Beclin-1, retinoblastoma
protein (pRB), p130, E2F1, E2F4, cyclin D1, cyclin E,
cyclin-dependent kinases (Cdk2, Cdk4 and Cdk6) and goat polyclonal
anti-human MAP LC3β and mouse monoclonal anti-human cyclin A
antibodies from Santa Cruz Biotechnology (Santa Cruz, CA, USA);
rabbit polyclonal anti-human caspase-3, caspase-6, caspase-7 and
SQSTM1/p62, mouse monoclonal anti-human caspase-9 antibodies,
HRP-conjugated goat anti-rabbit and horse anti-mouse IgGs from Cell
Signaling (Danvers, MA, USA); mouse monoclonal anti-human β-actin
antibody, Hoechst 33342, dimethyl sulfoxide (DMSO), propidium
iodide (PI), staurosporine, acridine orange, monodansylcadaverine,
3-methyladenine (3MA), and Annexin V-FITC apoptosis detection kit
from Sigma-Aldrich (Irvine, CA, USA); caspase inhibitor I zVAD-fmk
and neomycin sulfate (G418), mouse monoclonal anti-human p27, p21,
p16 antibodies and necrostatin-1 from Calbiochem (San Diego, CA,
USA); 3,3′-dihexyloxacarbocyanine iodide (DiOC6) from
Molecular Probes (Eugene, OR, USA); SuperSignal West Pico enhanced
chemiluminescence western blot detection reagent from Pierce
(Rockford, IL, USA); RNase A and proteinase K from Biosesang
(Sungnam, Korea); etoposide (VP-16) from Nippon Kayaku (Tokyo,
Japan); siPORT Amine was from Ambion (Austin, TX, USA);
Lipofectamine was from Invitrogen (Calsbad, CA, USA).
Cells
Hep3B cells were obtained from the American Type
Culture Collection (ATCC HB-8064; Rockville, MD, USA).
Cell culture and establishment of
Hep3B/Bcl-2 cells
Hep3B cells were cultured in complete Dulbecco’s
modified Eagle’s medium (DMEM; Gibco, Gaithersburg, MD)
supplemented with 10% heat-inactivated fetal bovine serum (FBS;
Gibco) and 100 U/ml penicillin in 5% CO2 at 37°C.
Mammalian expression vector encoding Flag-tagged Bcl-2 was kindly
provided by Professor A. Strasser (The Walter and Eliza Hall
Institute of Medical Research, Melbourne, Australia). Hep3B cells
were transfected with the expression vector encoding Flag-tagged
Bcl-2 using FuGENE6 reagent (Roche, Mannheim, Germany) according to
the manufacturer’s instructions. The transfected cells were
incubated for two days, and stable cells were then selected with
changes of fresh medium containing puromycin (4 μg/ml) for
four weeks. Single-cell clones were isolated by limiting dilutions
and subsequently analyzed for an increase of Bcl-2 expression
relative to identically cloned empty vector controls.
Treatment with etoposide and other
pharmacological agents
Forty-eight hours after Hep3B/vec and Hep3B/Bcl-2
cells were cultured, the original medium was removed. Cells were
washed with PBS and then incubated in the same fresh medium.
Etoposide and staurosporine were stocked in DMSO. Etoposide from a
stock solution was added to the medium to obtain indicated
dilutions (0–70 μg/ml) of the drug for 0–48 h. Staurosporine
from a stock solution was added to the medium to obtain indicated
dilutions (0–500 nM) of the drug for 0–48 h. The concentration of
DMSO used in this study had no effect on Hep3B/vec and Hep3B/Bcl-2
cells proliferation in our preliminary studies. To examine the
effect of caspase or autophagy inhibitor, cells were incubated in
the presence or absence of 100 μM zVAD-fmk or 1 mM 3MA for 3
h. Cells then exposed to 40 μg/ml etoposide or 300 nM
staurosporine for 48 h.
Cell viability assay
Cell viability was determined by the Vi-Cell cell
counter (Beckman Coulter, Miami, FL, USA), which performs an
automated trypan blue exclusion assay.
Nuclear morphology
Cell suspensions were cytospun onto clean fat-free
glass slides using a cytocentrifuge. Centrifuged samples were fixed
in 4% paraformaldehyde for 10 min and stained in 4 μg/ml
Hoechst 33342 for 30 min at 37°C. Cells were observed and
photographed under an epifluorescence microscope by an observer who
was blinded to the experimental group.
Quantification of DNA hypoploidy and cell
cycle phase analysis by flow cytometry
Ice-cold 95% ethanol with 0.5% Tween-20 was added to
cell suspensions to a final concentration of 70% ethanol. Fixed
cells were pelleted and washed with PBS containing 1% bovine serum
albumin (BSA). Cells were resuspended in 1 ml PBS containing 11
Kunitz U/ml RNase, incubated at 4°C for 30 min, washed once with
BSA-PBS and resuspended in PI solution (50 μg/ml). After the
cells had been incubated at 4°C for 30 min in the dark and washed
with PBS, DNA content was measured using Epics XL (Beckman
Coulter), and the data were analyzed using the Multicycle software
which allowed a simultaneous estimation of cell cycle parameters
and apoptosis.
Western blot analysis
Cells (2×106) were washed twice with
ice-cold PBS, resuspended in RIPA buffer (Elpis Biotech, Daejeon,
Korea) and incubated at 4°C for 30 min. The lysates were
centrifuged at 14,000 rpm for 20 min at 4°C. Protein concentrations
of cell lysates were determined with Bradford protein assay reagent
(Bio-Rad) and 30 μg of proteins were loaded onto 7.5–15%
SDS-PAGE. The gels were transferred to nitrocellulose membrane
(Amersham Pharmacia Biotech, Piscataway, NJ, USA) and reacted with
each antibody. Immunostaining with antibodies was performed using
the Super Signal West Pico enhanced chemiluminescence substrate and
detected with LAS-4000PLUS (Fuji Photo Film Company, Kanagawa,
Japan).
Transfection of GFP-LC3 in Hep3B/vec and
Hep3B/Bcl-2 cells and confocal microscopy
Hep3B/vec and Hep3B/Bcl-2 cells plated in six-well
dishes were transfected with 4 μg of LC3 cDNA using
Lipofectamine reagent (Invitrogen, Calsbad, CA, USA). The mammalian
expression construct of human LC3 cloned into pEGFP was a gift from
Dr N. Mizushima (Tokyo Medical and Dental University, Tokyo,
Japan). An empty pEGFP vector was used as a control for the stable
expression of LC3. Stable transfectants of Hep3B/vec and
Hep3B/Bcl-2 cells were selected with changes in medium containing
400 μg/ml of G418. Cells grown in coverslides were treated
as indicated and fixed with 4% paraformaldehyde for 10 min.
Fluorescent images were observed and analyzed under Zeiss LSM 700
laser-scanning confocal microscope (Göettingen, Germany).
Transmission electron microscopy
Forty-eight hours after treatment, cells were
harvested, pelleted and fixed in 2.5% glutaraldehyde
(Sigma-Aldrich) in phosphate buffer. After rinsing with phosphate
buffer, the samples were postfixed in 1% osmium tetroxide
(Sigma-Aldrich) for 1 h, rinsed with water, dehydrated in a graded
series of ethanol followed by propylene oxide (Sigma-Aldrich) and
kept overnight in Epon812 (Sigma-Aldrich). The samples were
embedded in Epon812 and cured in an oven at 60°C. Ultrathin
sections were obtained with a Reichert Ultracut E microtome. The
sections were stained with uranyl acetate and lead citrate and
observed using an H7650 transmission electron microscope (Hitachi,
Tokyo, Japan).
Detection of autophagosome formation with
acridine orange and monodansylcadaverine (MDC) by flow
cytometry
Cells were seeded on 6-well plates (1×105
cells/ml). After 24 h of incubation, cells were treated with
etoposide and staurosporine, and cultured for 48 h. Cells were
trypsinized, collected, and stained with acridine orange (final
concentration of 1 μg/ml) or MDC (final concentration of 50
μM) for 10 min at 37°C. After centrifugation, cells were
resuspended in PBS and analyzed with Epics XL.
Flow cytometric analysis of Annexin
V-FITC binding for apoptosis
Apoptosis was determined by flow cytometry with the
Annexin V-FITC apoptosis detection kit according to the
manufacturer’s instruction. Cells were incubated with etoposide and
staurosporine for 48 h. The cells were collected, centrifuged, and
resuspended in binding buffer, and incubated with Annexin V-FITC
and PI for 20 min at room temperature in the dark. After double
staining, the cells were analyzed with Epics XL.
DNA electrophoresis
Cells (0.6×106) were resuspended in 1.5
ml of lysis buffer (10 mM Tris-HCl, 10 mM EDTA, 10 mM NaCl, 0.5%
SDS, pH 7.5) into which proteinase K (200 μg/ml) was added.
After samples were incubated overnight at 48°C, 200 μl of
ice cold 5 M NaCl was added and the supernatant containing
fragmented DNA was collected after centrifugation. The DNA was then
precipitated overnight at −20°C in 50% isopropanol and RNase
A-treated for 1 h at 37°C. The DNA from 1×106 or
2×106 cells (15 ml) was equally loaded on each lane of
2% agarose gels in Tris-acetic acid/EDTA buffer containing 0.5
μg/ml ethidium bromide at 50 mA for 1.5 h.
Pulsed-field gel electrophoresis
(PFGE)
For PFGE, cells (2×106) were suspended in
50 μl of PBS and mixed with 50 μl of PBS containing
1% low melting temperature agarose. The cell suspension was poured
into a template (5×2×10 mm), plugged, and cooled on ice. The
hardened agarose gel blocks were incubated with 250 μl of a
mixture of proteinase K (1 mg/ml), N-lauroyl sarcosine sodium (1%
w/v), and 0.5 M EDTA (pH 9.2) at 50°C for 48 h. After incubation,
half the volume of the digested agarose gel block was loaded into a
sample well of a 1% (w/v) agarose gel (Sigma type II, 150×150×4.4
mm) in 0.5X TBE buffer (89 mM Tris-boric acid, 2 mM EDTA, pH 8.0).
The PFGE apparatus used was the Gene Path System from Bio-Rad. PFGE
was carried out in 0.5X TBE maintained at 14°C by circulating cool
water for 16 h (constant, 6 V; switch times are initial 60 sec and
final 90 sec). DNA in the gel was stained with ethidium bromide and
detected with LAS-4000Plus (Fuji Photo Film Company, Kanagawa,
Japan). Chromosomal DNA from Saccharomyces cerevisiae (from
Bio-Rad), a mixture of λ DNA, its concatemers, and HindIII-digested
λ DNA (from Sigma-Aldrich) were used as DNA size markers.
Beclin-1 small interfering RNA (siRNA)
transfection and combination treatment with etoposide or
staurosporine
Transfection of siRNA against Beclin-1 transcripts
was performed by using siPORT Amine (Ambion, Austin, TX, USA) in
Opti-MEM (GibcoBRL) media according to manufacturer’s
recommendations. Beclin-1 siRNA was purchased from Cell signaling.
Cells were treated with etoposide and staurosporine for 48 h.
Co-immunoprecipitation (Co-IP)
Cell extracts that were incubated with antibodies
were precipitated with protein A-Sepharose beads.
Immunoprecipitated proteins were separated on SDS-PAGE, and western
blot analysis was performed as described.
Statistical analysis
Four independent experiments were carried out in
vitro. The results are expressed as means ± SD from four
experiments, each performed in triplicate. The results of the
experimental and control groups were tested for statistical
significance by the nonparametric Kruskall-Wallis test. In all
cases, a p-value <0.05 was considered significant.
Results
Etoposide, but not staurosporine,
bypasses the chemoresistance conferred by Bcl-2 in Hep3B cells
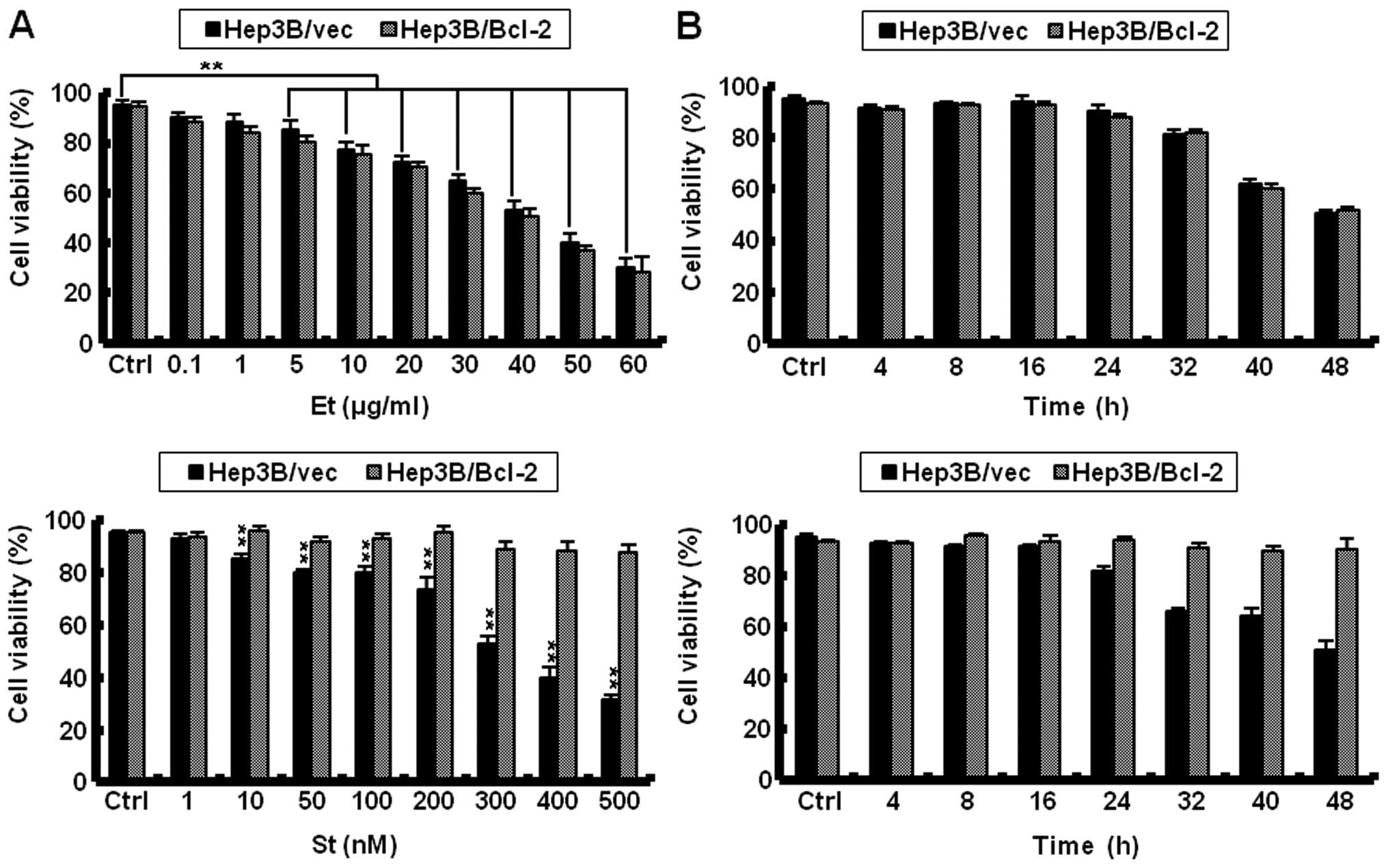
Etoposide at 5-60 μg/ml significantly reduced
the viability of Hep3B cells compared to the control Hep3B cells.
Noticeably, etoposide at the same doses also significantly reduced
the viability in Bcl-2-overexpressing Hep3B cells, indicating that
etoposide bypasses the chemoresistance conferred by Bcl-2 in Hep3B
cells. Another representative apoptosis inducer staurosporine at
10-500 nM also significantly reduced the viability of Hep3B cells
in a dose-dependent manner. However, staurosporine at the same
doses did not significantly reduce viability in
Bcl-2-overexpressing Hep3B cells, indicating that staurosporine
does not bypass the chemoresistance conferred by Bcl-2 in Hep3B
cells (Fig. 1A). Because the
viability of Hep3B/vec cells treated with 40 μg/ml etoposide
or 300 nM staurosporine for 48 h was approximately 50%, this single
concentration was utilized for further study. Etoposide at 40
μg/ml reduced the viability of Hep3B/Bcl-2 and Hep3B/vec
cells in a time-dependent manner. Staurosporine at 300 nM for 48 h
reduced the viability of Hep3B/vec cells in a time-dependent
manner. However, ectopic expression of Bcl-2 prevented the
reduction in viability by staurosporine in Hep3B cells (Fig. 1B).
Etoposide induces G1/S phase
arrest in Hep3B/vec and Hep3B/Bcl-2 cells
Analysis via a flow cytometric method, which allows
the simultaneous estimation of cell cycle parameters and apoptosis,
elucidated not only the rate at which etoposide altered cell cycle
progression but also the correlation between cell cycle progression
and cell death. Time-course analysis of cell cycle distribution
after etoposide treatment revealed an increase in the percentage of
S-phase cells and also a sustained increase in the S-phase
population, while producing a concomitant fall in the percentage of
G0/G1 phase cells. The increase in the
S-phase cell percentage and decrease in the
G0/G1 phase populations paralleled an
increase in the subG1 portion. In contrast to etoposide,
staurosporine treatment revealed an increase in the percentage of
G2/M-phase cells, while producing a concomitant fall in
the percentage of G0/G1 phase cells. The
increase in the G2/M phase cell percentage and
concomitant fall in the percentage of G0/G1
phase cells paralleled an increase in the subG1 portion
(Fig. 2A). We next examined the
expression levels of checkpoint proteins in the G1/S
transition and cell cycle regulating factors at the G1/S
boundary by western blot analysis. The total level of pRB
expression decreased remarkably and changed from the
hyperphosphorylated form to the hypophosphorylated form after
etoposide treatment; however, staurosporine-treated cells remained
unchanged. Moreover, the protein level of E2F-4 was completely
inhibited after 48 h of exposure to etoposide; however, the level
of E2F-1 was not. These results suggest that etoposide inhibits the
release of E2F family proteins from pRB. Furthermore, while the
protein levels of cyclin A, cyclin E and Cdk6 were significantly
inhibited by etoposide treatment in a time-dependent manner, the
levels of cyclin D1, Cdk2 and Cdk4 remained unchanged in the
etoposide-treated cells. These results suggest that the suppressive
effects of etoposide at the G1/S phase in Hep3B cells
are partly caused by downregulating the levels of cyclin A, cyclin
E and Cdk6. Because Cdk activity is highly regulated by an
association with Cdk inhibitors, such as p16, p21 and p27, we next
examined the possible upregulation of these proteins in cells
treated with etoposide or staurosporine. Noticeably, etoposide
treatment resulted in a time-dependent increase in p27 protein,
which was not undetectable in untreated control cells and
staurosporine-treated cells. The expression levels of other Cdk
inhibitors, such as p16 and p21, did not change after etoposide
treatment (Fig. 2B). These data
suggested that the etoposide-induced G1/S phase arrest
in human hepatocarcinoma Hep3B/vec and Hep3B/Bcl-2 cells requires
increased p27 expression but not p16 and p21 expression. Taken
together, etoposide, but not staurosporine, induces G1/S
phase arrest in Hep3B/vec and Hep3B/Bcl-2 cells.
Hep3B cells undergoing cell death after
treatment with etoposide show apoptotic manifestations
To examine whether etoposide reduces the viability
of Hep3B/vec and Hep3B/Bcl-2 cells by inducing apoptosis, various
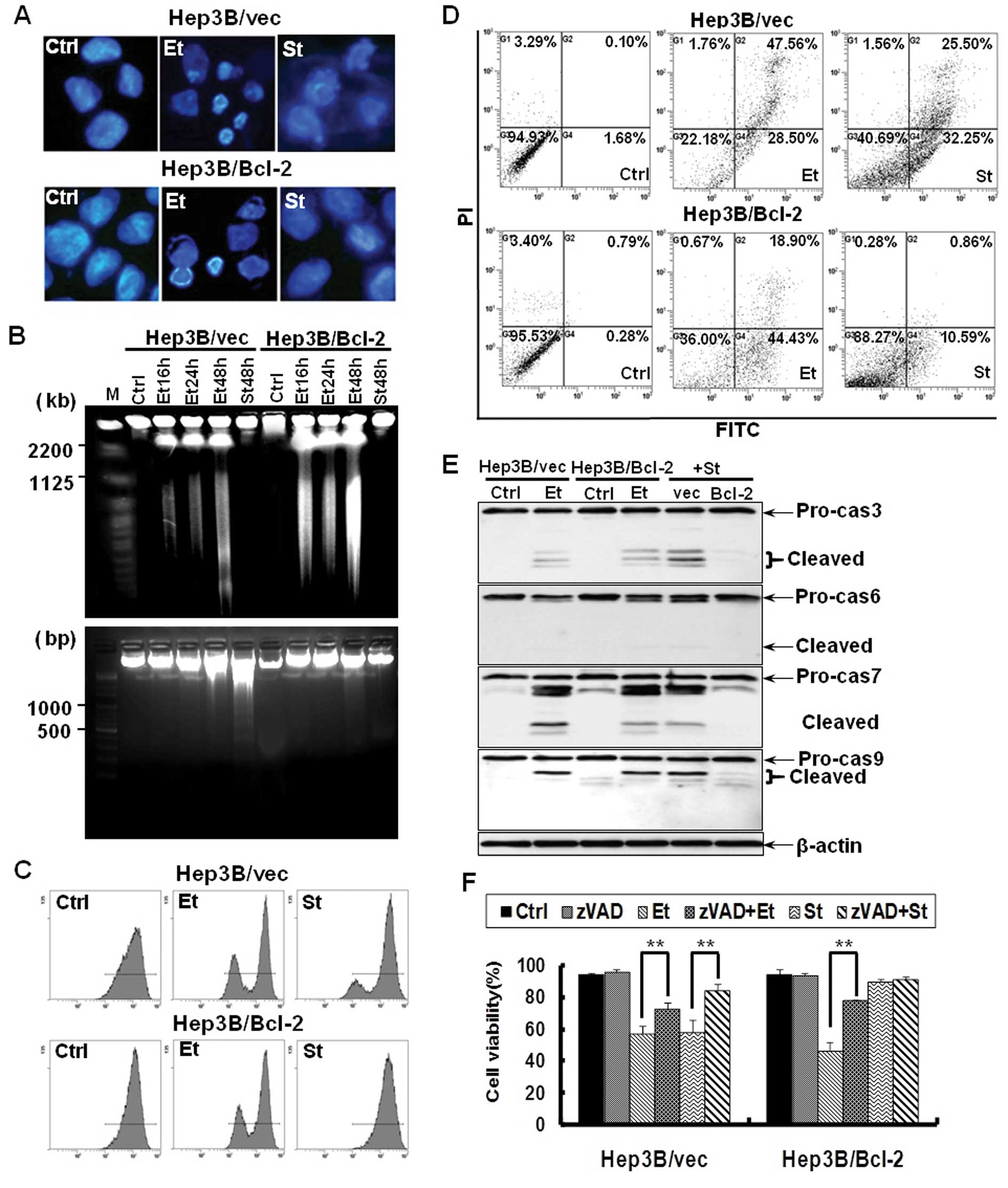
assays were performed. Hoechst staining showed nuclear condensation
in Hep3B cells treated with etoposide or staurosporine (Fig. 3A). While etoposide did not
demonstrate ladder-like DNA fragments on agarose gel,
disintegration of nuclear DNA into giant fragments of 1–2 Mbp and
high molecular weight fragments of 200–800 kbp were recognized by
PFGE. Conversely, staurosporine demonstrated DNA ladder on an
agarose gel (Fig. 3B). Flow
cytometry indicated a reduction in the mitochondrial membrane
potential (Fig. 3C). Flow
cytometry using Annexin V-FITC/PI double staining also demonstrated
the induction of apoptosis in Hep3B cells treated with etoposide or
staurosproine (Fig. 3D). Etoposide
or staurosporine produced the cleaved products procaspase-3, -6, -7
and -9 in Hep3B cells (Fig. 3E).
Etoposide treatment caused similar apoptotic phenotypes in
Hep3B/Bcl-2 cells. However, Bcl-2 overexpression protected Hep3B
cells against apoptosis induced by staurosporine (Fig. 3A-E). The pan-caspase inhibitor
zVAD-fmk protected Hep3B cells against apoptosis induced by
etoposide or staurosporine (Fig.
3F). Taken together, etoposide bypasses the chemoresistance
conferred by Bcl-2 in Hep3B cells via the induction of
apoptosis.
Hep3B cells treated with etoposide or
staurosporine show autophagic phenotypes
We next observed the fine structure of Hep3B/vec and
Hep3B/Bcl-2 cells treated with etoposide. The control Hep3B/vec
cell has a normal appearing nucleus and a normal distribution of
organelles with numerous microvilli. Mitochondria have a round to
tubular shape with intact outer and inner membranes and cristae.
The lumen of the ERs is not dilated. The control Bcl-2
overexpressing Hep3B cell also shows a normal appearance of the
nucleus and a normal distribution of organelles with numerous
microvilli. Most mitochondria have a round shape with intact outer
and inner membranes and cristae. The lumen of the ERs is not
dilated (Fig. 4Aa and 4Ab). The
etoposide-treated Hep3B/vec cells have mitochondria that are
fragmented. Cellular organelles are merged with lysosomes,
indicating the formation of an autophagolysosome. Numerous vesicles
with a double membrane are observed in the cytoplasm. The Bcl-2
overexpressing Hep3B cells treated with etoposide also had numerous
vesicles with a double membrane in the cytoplasm. Cup-shaped
membrane structures were found, reflecting preautophagosomal
structure phagopore formation. Autophagosomes contained
mitochondria and membranous structures in different stages of
degradation. Numerous autophagosomes were fused with lysosomes.
Autophagosomes contained mitochondria and lamellar structures with
residual digested materials (Fig.
4Ac–Af). Staurosporine-treated Hep3B cell also showed
autophagic phenotypes (Fig. 4Ag).
Bcl-2 overexpressing Hep3B cells showed the normal appearance
(Fig. 4Ah) or autophagic
phenotypes (Fig. 4Ai). Confocal
microscopy demonstrated that etoposide or staurosporine induced LC3
puncta in Hep3B cells transfected with GFP-LC3 (Fig. 4B). These findings indicate that
etoposide or staurosporine treatment induces autophagy in Hep3B and
Hep3B/Bcl-2 cells.
Autophagy in Hep3B cells induced by
etoposide and staurosproine is cell death-inducing and
cyto-protective, respectively
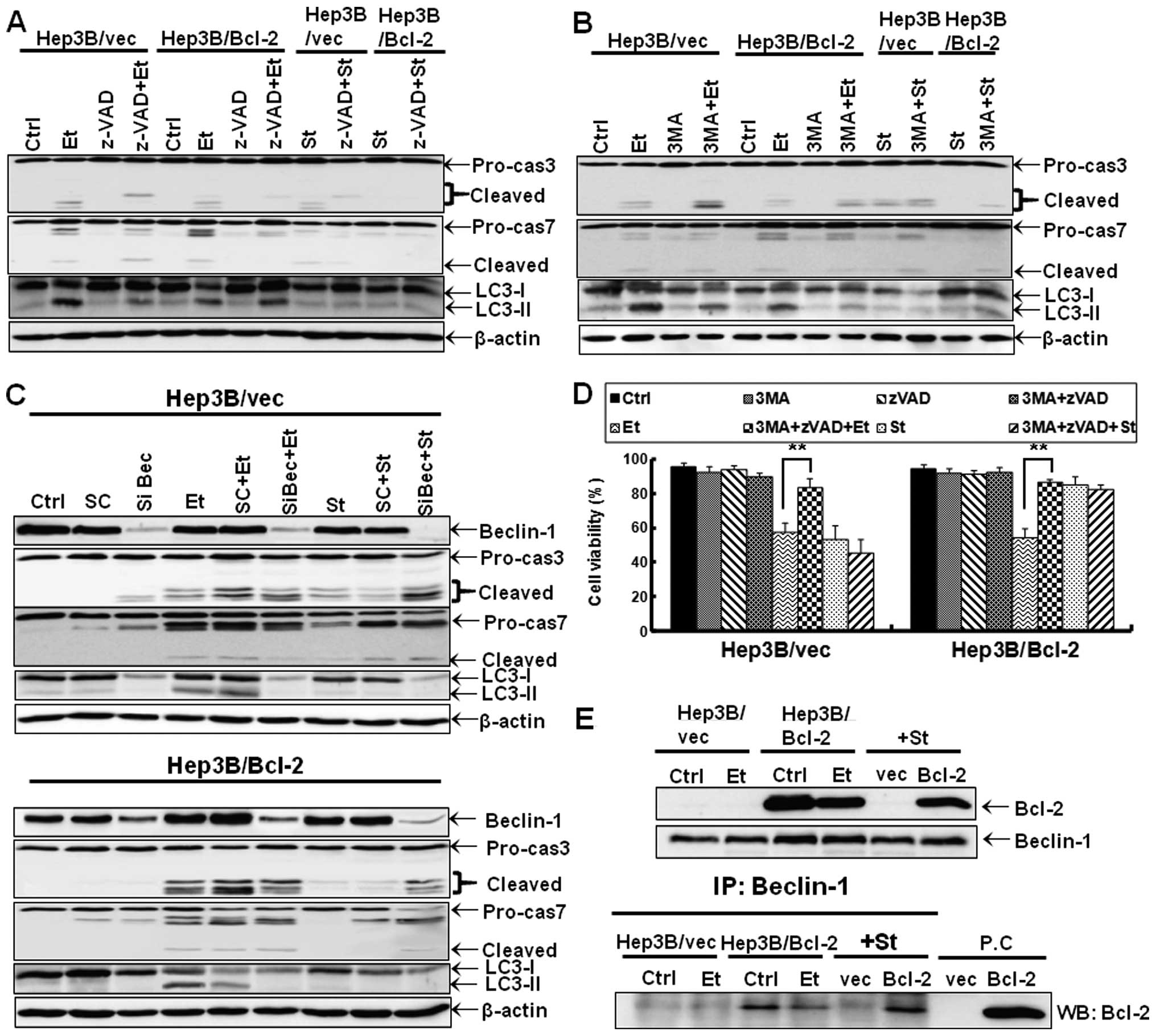
We next examined autophagic flux. Flow cytometry
revealed increased acridine orange intensity or MDC recruitment to
autophagosomes in Hep3B/vec and Hep3B/Bcl-2 cells treated with
etoposide or staurosporine. These data indicate that both etoposide
and staurosporine increase the percent of cells with acidic
vesicular organelles. Importantly, the percent of both acridine
orange- and MDC-positive cells was substantially higher in
staurosporine-treated cells than in etoposide-treated cells
(Fig. 5A and B). We further
conducted an autophagy flux assay based on the turnover of LC3-II
by western blot analysis in the presence and absence lysosomal
degradation. To prevent lysosomal degradation, bafilomycin A1 was
used. A western blot assay showed that an increase in LC3-II levels
in cells treated with etoposide was markedly enhanced by
bafilomycin A1 treatment (Fig.
5C). Although LC3-II is not observed in Hep3B cells treated
with staurosporine alone, bafilomycin A1 treatment evidently
increased LC3-II level in staurosporine-treated Hep3B cells. These
data indicate that autophagic flux, which refers to the complete
process of autophagy, is more active in staurosporine-treated cells
compared to etoposide-treated cells. We next examined the effect of
the inhibition of autophagy on the reduction of cell viability by
etoposide or staurosporine. We observed that the inhibition of
autophagy by 3MA or siRNA against Beclin-1 prevented the reduction
in viability by etoposide in Hep3B/vec and Hep3B/Bcl-2 cells.
Conversely, the inhibition of autophagy by 3MA or siRNA against
Beclin-1 augmented the reduction in viability by staurosporine in
Hep3B/vec and Hep3B/Bcl-2 cells (Fig.
5D and E). These findings indicate that autophagy in Hep3B
cells induced by etoposide and staurosproine is cell death-inducing
and cyto-protective in Hep3B cells, respectively.
 | Figure 5Autophagy in Hep3B cells induced by
etoposide and staurosporine is cell death-inducing and
cyto-protective in Hep3B cells, respectively. Cells with positive
staining were monitored using flow cytometry 48 h after treatment
with etoposide or staurosporine. (A) Flow cytometric histograms
showing the intensity of red fluorescence in acridine
orange-stained cells (left panel). Flow cytometric analysis
indicates the percent of acridine-orange positive cells, which is
represented as the mean ± SD (right panel). The percentage of
acridine orange-positive cells is larger in cells treated with
etoposide or staurosporine compared to untreated control cells. The
percentage of acridine orange-positive cells is larger in cells
treated with etoposide than in cells treated with staurosporine.
(B) Flow cytometric histograms showing increased MDC recruitment to
autophagosomes in Hep3B/vec and Hep3B/Bcl-2 cells treated with
etoposide or staurosporine (left panel). Flow cytometric analysis
indicates the percent of MDC positive cells, which is represented
as the mean ± SD (right panel). The percentage of MDC-positive
cells is larger in cells treated with etoposide or staurosporine
compared to untreated control cells. The percentage of MDC-positive
cells is larger in cells treated with etoposide compared to cells
treated with staurosporine. (C) Autophagy flux assay based on the
turnover of LC3-II by western blot analysis in the presence and
absence lysosomal degradation. The cells were pretreated with
bafilomycin A1 for 3 h and were further exposed to etoposide or
staurosporine for 48 h. BfA1, bafilomycin A1. Western blot analysis
shows that an increase in LC3-II in cells treated with etoposide
was markedly enhanced by bafilomycin A1 treatment. Although LC3-II
is not observed in Hep3B cells treated with staurosporine alone,
bafilomycin A1 treatment evidently increased LC3-II in
staurosporine-treated Hep3B cells. (D) Viability assay showing the
effect of the inhibition of autophagy by 3MA, which also
significantly prevents the reduction in viability by etoposide, in
Hep3B/ vec and Hep3B/Bcl-2 cells. **p<0.01.
Conversely, 3MA significantly augmented the reduction in viability
by staurosporine in Hep3B/vec and Hep3B/Bcl-2 cells.
**p<0.01. (E) Viability assay showing the effect of
the inhibition of autophagy by siRNA against Beclin 1.
Additionally, siRNA against Beclin-1 significantly prevents the
reduction in viability by etoposide in Hep3B/vec and Hep3B/Bcl-2
cells. **p<0.01. Conversely, siRNA against Beclin-1
significantly augments the reduction in viability by staurosporine
in Hep3B/vec and Hep3B/Bcl-2 cells. **p<0.01. SC,
scrambled siRNA; SiBec, siRNA Beclin-1. See Fig. 1 for other definitions. |
Apoptotic and autophagic cell death occur
independently in etoposide-treated Hep3B cells
Because our data support that etoposide induces a
mixed type of programmed cell death, we further examined whether
necroptosis, another type of programmed cell death, plays a role in
the cell death of etoposide-treated Hep3B cells. However,
necrostatin-1 did not alter the amount of the etoposide-mediated
cell death (negative data not shown). These data suggest that
etoposide induces a mixed type of programmed cell death via
apoptosis and autophagic cell death and overcomes the resistance
conferred by Bcl-2 in Hep3B hepatoma cells. We next asked whether
autophagic cell death induced by etoposide interplays with
apoptosis in this type of cell death. We observed that a
pan-caspase inhibitor zVAD-fmk did not prevent the increase in
LC3-II, although it efficiently prevented the activation of
caspase-3 and -7 (Fig. 6A). We
further observed that 3MA did not prevent the activation of
caspase-3 or caspase-7, although it efficiently prevented the
increase in LC3-II (Fig. 6B).
Beclin-1 siRNA also showed a similar effect with 3MA (Fig. 6C). The viability assay showed that
co-treatment with 3MA and zVAD-fmk almost completely abolished the
reduction in viability by etoposide (Fig. 6D). Although we did not completely
exclude the possibility that autophagic cell death leads to
apoptosis or vice versa in Hep3B cells treated with
etoposide, our data suggest that autophagy and apoptosis may
contribute to cell death independently, bypassing the resistance
conferred by Bcl-2. Conversely, co-treatment with 3MA and zVAD-fmk
did not prevent the reduction in viability induced by staurosporine
(Fig. 6D), although zVAD-fmk and
3MA or Beclin-1 siRNA protected Hep3B cells against apoptotic and
autophagic phenotypes in Hep3B cells treated with staurosporine,
respectively. In concert with the data showing that zVAD-fmk almost
completely abolished the reduction in viability by staurosporine
(Fig. 3F), these data indicate
that autophagy does not contribute to cell death in Hep3B cells
treated with staurosporine.
Etoposide decreases the Bcl-2 to Beclin-1
interaction
To this end, we examined whether etoposide has the
ability to dissociate the interaction between Bcl-2 and Beclin-1.
According to western blot data, both etoposide and staurosporine
slightly downregulated the expression of Bcl-2 protein in
Hep3B/Bcl-2 cells. Importantly, etoposide but not staurosporine
dissociated the interaction between Bcl-2 and Beclin-1 (Fig. 6E).
Discussion
Although surgery is the most effective treatment for
hepatocellular carcinoma (HCC), the numbers of patients with HCC
who are eligible for surgical intervention are limited. Thus,
numerous approaches have been conducted to search for other
options, such as efficient chemotherapeutic agents. However, HCC is
weakly chemosensitive (14).
One of the most important problems that hamper the
efficiency of cancer chemotherapeutic drugs, such as cisplatin and
etoposide, is chemoresistance (15,16).
Accumulated data strongly suggest a causal relationship between
defects in apoptosis and drug resistance (17). The expression of genes that
regulate apoptotic cell death plays an important role in
determining the sensitivity of tumor cells to chemotherapy.
Although most of the anticancer drugs, regardless of their targets
and mechanisms, ultimately induce apoptosis, cancer cells are
genetically predisposed to apoptotic resistance, blocking the
action of anticancer drugs. For example, the efficacy of
anthracycline antibiotics can be significantly reduced after
cancers develop apoptotic resistance (18). Similarly, the efficacy of other
anticancer agents, such as vinca alkaloids, taxanes,
epipodophylotoxins, and imatinib, is reduced with the cancer
resistance to apoptosis (19).
Autophagy has been used to describe the catabolic
pathways of the degradation of intracellular macromolecules.
Autophagy begins with the sequestration of intracellular cargo,
such as protein aggregates, organelles and ribosomes, in a membrane
vacuole called an autophagosome. Next, the loaded autophagosome
fuses with lysosomes, where the cellular materials are degraded by
lysosomal acid proteases, and lysosomal permeases and transporters
export amino acids and other by-products of degradation back out to
the cytoplasm, where they can be re-used for building
macromolecules and for metabolism (20). In theory, autophagy may help
promote cell survival either by promoting energy efficiency through
ATP generation or by mediating damage control by removing
non-functional proteins and organelles (21). Numerous studies have demonstrated
that autophagy protects cells by preventing them from undergoing
apoptosis. Autophagy sequesters damaged mitochondria, preventing
cytochrome c from being able to form a functional
apoptosome, and protects cells from the caspase-independent death
that occurs after mitochondrial outer membrane permeabilization
(MOMP) (22,23). Thus, autophagy may contribute to
chemoresistance by preventing apoptosis.
Paradoxically, autophagy may also represent a form
of nonapoptotic cell death. Numerous recent studies have shown that
increased autophagic activity is associated with cell death
(24,25). Although it is unclear whether
autophagy directly contributes to cell death or is a failed effort
to preserve cell viability, autophagy appears to promote cell death
through the excessive self-digestion and degradation of essential
cellular constituents. Unlike apoptosis, autophagic cell death is
usually considered caspase-independent (25). Furthermore, autophagic cell death
is known to occur in cells with profound defects in the apoptotic
machinery (26). However,
autophagic cell death undergoes cross-talk with apoptosis via
classical apoptotic mediators and involves the caspase-dependent
mechanism in some circumstances (27). In this context, autophagic cell
death defects may also contribute to chemoresistance. Thus, the
induction of autophagic cell death could be a strategy in
circumventing cancer drug resistance.
Etoposide acts as a topoisomerase II inhibitor.
Topoisomerase II modulates DNA topology during synthesis by passing
an intact helix through a transient double stranded break before
closing up for further extension, thereby regulating over- and
under-winding of the backbone and resolving knots and tangles
(28). Etoposide induces covalent
protein DNA complex formation, impairing this mode of progression
(29). Although topoisomerase
inhibitor-induced DNA damage is known to induce apoptosis, the
signaling pathway has not been fully defined (18). The mechanisms involved in the
induction of apoptosis by topoisomerase inhibitors are believed to
be largely mediated by the mitochondrial apoptotic pathway
(30). Because Bcl-2 plays a
central role in regulating changes in mitochondrial outer membrane
permeability (3), Bcl-2 inhibits
the mitochondria-dependent apoptosis induced by etoposide. Because
increased expression of Bcl-2 has been associated with poor
response to chemotherapy in various malignancies, overexpression of
Bcl-2 justly appears to inhibit or attenuate the pathway to
apoptosis (31).
However, previous studies have demonstrated that
chemotherapeutic drugs can bypass Bcl-2-mediated protection against
apoptosis in Bcl-2-overexpressing cells (32). Although the molecular mechanism of
cell death in Bcl-2-overexpressing cells remains unclear, the
involvement of lipid peroxidation, the modulation of p53 target
genes, such as Puma and Noxa, and the formation of
PML (promyelocytic leukemia) nuclear bodies have been suggested to
underlie the induction of apoptosis in Bcl-2-overexpressing
cells.
Arguably, the most intriguing finding of our study
is that etoposide bypasses the resistance by inducing a mixed type
of programmed cell death in Hep3B hepatoma cells. On the basis of
previous studies showing that blocked apoptosis contributes to
cancer drug resistance, most pharmacological approaches have been
aimed at restoring the efficacy of chemotherapeutics by activating
apoptosis. However, cancer drug resistance is complex in nature,
and many different types of cell death appear to contribute to
cancer drug resistance. Previous studies have shown that the
induction of necroptosis circumvents apoptotic resistance (33). Therefore, simultaneous activation
of multiple death pathways seems to be an effective strategy to
bypass cancer cell resistance. To date, few chemicals have been
demonstrated to induce multiple types of programmed cell death. The
concomitant occurrence of autophagy and necroptosis (34) or apoptosis and autophagic cell
death has been reported (35).
According to their data, two PCDs independently contribute to cell
death execution or the one PCD is the consequence of the other PCD.
Our data suggest that etoposide induces PCD I and PCD II
concomitantly, both of which contribute to the cytotoxic effects in
Hep3B /Bcl-2 and Hep3B cells.
Another interesting finding is that the concomitant
occurrence of apoptosis and autophagy after etoposide treatment is
mediated with cell cycle arrest in the G1/S phase. The
progression of eukaryotic cells through the cell cycle is
orchestrated by the sequential activation and inactivation of the
Cdks, which is associated with their respective cyclin subunits.
G1 progression and G1/S transition are
regulated by Cdk4/Cdk6, which assemble with D-type cyclins during
the mid-G1 phase and with Cdk2, which later combines
with cyclin E. Although Cdk2 controls the S-phase when it is
associated with cyclin A, the G2/M transition is
regulated by Cdk2 in combination with cyclins A and B (36). Moreover, the relative balance
between the cellular concentrations of Cdk inhibitors also
regulates cell cycle progression. Cdk inhibitors are divided into
two families according to substrate specificity. In mammalian
cells, these are the CIP/KIP family, which consists of p21, p27 and
p57, and the INK4a family, including p15, p16 and p18 (37). Cdk inhibitors mediate cell cycle
arrest in response to several anti-proliferative signals. The
activity of Cdks is also negatively regulated by binding to Cdk
inhibitors in response to a variety of antiproliferative signals
and thus modulates retinoblastoma protein (pRB) phosphorylation
events, which are essential for various cell cycle transitions
(38). These observations suggest
new approaches that could alter uncontrolled human cancer cell
growth by modulating cell cycle regulators causing cell cycle
arrest and could be useful in prevention and/or intervention in
human cancer (39). In line with
several previous studies reporting that etoposide could induce
S-phase arrest (40), etoposide
induced S-phase arrest in Hep3B cells. Numerous previous studies
have reported that cell cycle arrest is associated with the
induction of apoptosis. Furthermore, recent studies have suggested
that cell cycle arrest is associated with autophagy. Among those
studies, only a few studies have shown that autophagy is associated
with S-phase arrest (41).
According to recent studies, the functional networks of cell cycle,
apoptosis and autophagy appear to be intermingled. Cell cycle
machineries are involved in the progress of apoptosis and
autophagy. In light of our results, etoposide targets many proteins
in Hep3B cells, leading to cell cycle arrest, apoptosis and
autophagic cell death, which determine cell fate; however, the
entire molecular mechanism is not fully understood.
Because autophagy and apoptosis can be triggered by
overlapping signaling mechanisms, and because Bcl-2 plays important
roles in inhibiting not only apoptosis but also autophagy (42), Bcl-2 is an important potential
therapeutic target for overcoming cancer cell resistance by a
simultaneous activation of apoptosis and autophagy. However, to
date, only a few chemicals have been reported to simultaneously
activate apoptosis and autophagic cell death. In contrast to a
previous study showing Bcl-2-mediated etoposide resistance in 697
leukemia cells, we observed that etoposide concomitantly activates
apoptosis and autophagic cell death in Bcl-2-overexpressing Hep3B
cells.
This study revealed that etoposide dissociates the
interaction between Bcl-2 and Beclin-1 in Hep3B cells, efficiently
inducing autophagic cell death in Hep3B cells. Bcl-2 helps maintain
mitochondrial integrity, protects cells from apoptosis, and when
bound to Beclin-1, can inhibit autophagy as well (43). Autophagy is initially induced to
prolong cell survival, but when taken to extremes, it causes cell
death. Bcl-2 suppresses autophagy by binding to the protein
Beclin-1. Thus, Bcl-2 can help cells evade autophagic cell death.
We also observed the induction of autophagy in Hep3B cells treated
with staurosporine, which is a strong inducer of apoptosis in many
different cell types. However, autophagy induced by staurosproine
in Hep3B cells is associated with cell survival but not with
autophagic cell death. Importantly, staurosporine neither
dissociated the interaction between Bcl-2 and Beclin-1 nor bypassed
the resistance conferred by Bcl-2 in Hep3B cells. These data
suggest that bypassing resistance with etoposide in Hep3B cells can
be associated with the efficacy of etoposide in dissociating the
interaction between Bcl-2 and Beclin-1.
In conclusion, etoposide bypasses the resistance by
inducing a mixed type of programmed cell death in Hep3B hepatoma
cells. Because multiple mechanisms may be involved in
etoposide-cell death in Bcl-2-overexpressing cancer cells and may
vary between different cell types, further studies are
required.
Acknowledgements
This study was supported by the
National Research Foundation of Korea grant funded by the Korean
government (2010-0001942).
References
|
1
|
Kroemer G, Galluzzi L, Vandenabeele P,
Abrams J, Alnemri ES, Baehrecke EH, Blagosklonny MV, El-Deiry WS,
Golstein P, Green DR, Hengartner M, Knight RA, Kumar S, Lipton SA,
Malorni W, Nunez G, Peter ME, Tschopp J, Yuan J, Piacentini M,
Zhivotovsky B and Melino G: Classification of cell death:
recommendations of the Nomenclature Committee on Cell Death. Cell
Death Differ. 16:3–11. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Oltvai ZN, Milliman CL and Korsmeyer SJ:
Bcl-2 heterodimerizes in vivo with a conserved homolog, Bax, that
accelerates programmed cell death. Cell. 74:609–619. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Adams JM and Cory S: The Bcl-2 protein
family: arbiters of cell survival. Science. 281:1322–1326. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Murphy KM, Streips UN and Lock RB: Bcl-2
inhibits a Fas-induced conformational change in the Bax N terminus
and Bax mitochondrial translocation. J Biol Chem. 27:17225–17228.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Foyouzi-Youssefi R, Arnaudeau S, Borner C,
Kelley WL, Tschopp J, Lew DP, Demaurex N and Krause KH: Bcl-2
decreases the free Ca2+ concentration within the
endoplasmic reticulum. Proc Natl Acad Sci USA. 97:5723–5728. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Saeki K, You A, Okuma E, Yazaki Y, Susin
SA, Demaurex N and Krause KH: Bcl-2 down-regulation causes
autophagy in a caspase-independent manner in human leukemic HL60
cells. Cell Death Differ. 7:1263–1269. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Pattingre S, Tassa A, Qu X, Garuti R,
Liang XH, Mizushima N, Packer M, Schneider MD and Levine B: Bcl-2
antiapoptotic proteins inhibit Beclin-1-dependent autophagy. Cell.
122:927–939. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Liang XH, Kleeman LK, Jiang HH, Gordon G,
Goldman JE, Berry G, Herman B and Levine B: Protection against
fatal Sindbis virus encephalitis by Beclin, a novel
Bcl-2-interacting protein. J Virol. 72:8586–8596. 1998.PubMed/NCBI
|
|
9
|
Reed JC, Miyashita T, Takayama S, Wang HG,
Sato T, Krajewski S, Aime-Sempe C, Bodrug S, Kitada S and Hanada M:
BCL-2 family proteins: regulators of cell death involved in the
pathogenesis of cancer and resistance to therapy. J Cell Biochem.
60:23–32. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Letai AG: Diagnosing and exploiting
cancer’s addiction to blocks in apoptosis. Nat Rev Cancer.
8:121–132. 2008.
|
|
11
|
Real PJ, Cao Y, Wang R, Nikolovska-Coleska
Z, Sanz-Ortiz J, Wang S and Fernandez-Luna JL: Breast cancer cells
can evade apoptosis-mediated selective killing by a novel small
molecule inhibitor of Bcl-2. Cancer Res. 64:7947–7953. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Chen Ay and Liu LF: DNA topoisomerases:
Essential enzymes and lethal targets. Annu Rev Pharmacol Toxicol.
3:191–218. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Dubrez L, Goldwasser F, Genne P, Pommier Y
and Solary E: The role of cell cycle regulation and apoptosis
triggering in determining the sensitivity of leukemic cells to
topoisomerase I and II inhibitors. Leukemia. 9:1013–1024.
1995.PubMed/NCBI
|
|
14
|
Cohn AL, Myers JW, Mamus S, Deur C, Nicol
S, Hood K, Khan MM, Ilegbodu D and Asmar L: A phase II study of
pemetrexed in patients with advanced hepatocellular carcinoma.
Invest New Drugs. 26:381–386. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Lee JO, Lee KW, Oh DY, Kim JH, Im SA, Kim
TY and Bang YJ: Combination chemotherapy with capecitabine and
cisplatin for patients with metastatic hepatocellular carcinoma.
Ann Oncol. 20:1402–1407. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yuan JN, Chao Y, Lee WP, Li CP, Lee RC,
Chang FY, Yen SH, Lee SD and Whang-Peng J: Chemotherapy with
etoposide, doxorubicin, cisplatin, 5-fluorouracil, and leucovorin
for patients with advanced hepatocellular carcinoma. Med Oncol.
25:201–206. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Johnstone RW, Ruefli AA and Lowe SW:
Apoptosis: a link between cancer genetics and chemotherapy. Cell.
108:153–164. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Gewirtz DA: A critical evaluation of the
mechanisms of action proposed for the antitumor effects of the
anthracycline antibiotics Adriamycin and daunorubicin. Biochem
Pharmacol. 57:727–741. 1999. View Article : Google Scholar
|
|
19
|
Jordan MA and Wilson L: Microtubules as a
target for anticancer drugs. Nat Rev Cancer. 4:253–265. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Mizushima N: Autophagy: process and
function. Genes Dev. 21:2861–2873. 2007. View Article : Google Scholar
|
|
21
|
Glick D, Barth S and Macleod KF:
Autophagy: cellular and molecular mechanisms. J Pathol. 221:3–12.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ravikumar B, Berger Z, Vacher C, O’Kane CJ
and Rubinsztein DC: Rapamycin pre-treatment protects against
apoptosis. Hum Mol Genet. 15:1209–1216. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Colell A, Ricci JE, Tait S, Milasta S,
Maurer U, Bouchier-Hayes L, Fitzgerald P, Guio-Carrion A,
Waterhouse NJ, Li CW, Mari B, Barbry P, Newmeyer DD, Beere HM and
Green DR: GAPDH and autophagy preserve survival after apoptotic
cytochrome c release in the absence of caspase activation. Cell.
129:983–997. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Tsujimoto Y and Shimizu S: Another way to
die: autophagic programmed cell death. Cell Death Differ. 12(Suppl
2): 1528–1534. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Gozuacik D and Kimchi A: Autophagy as a
cell death and tumor suppressor mechanism. Oncogene. 23:2891–2906.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Shimizu S, Kanaseki T, Mizushima N, Mizuta
T, Arakawa-Kobayashi S, Thompson CB and Tsujimoto Y: Role of Bcl-2
family proteins in a non-apoptotic programmed cell death dependent
on autophagy genes. Nat Cell Biol. 6:1221–1228. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Lee SW, Song YS, Lee SY, Yoon YG, Lee SH,
Park BS, Yun I, Choi H, Kim K, Chung WT and Yoo YH: Downregulation
of protein kinase CK2 activity facilitates tumor necrosis
factor-α-mediated chondrocyte death through apoptosis and
autophagy. PLoS One. 6:e191632011.PubMed/NCBI
|
|
28
|
Hande KR: Etoposide: Four decades of
development of a topoisomeraseII inhibitor. Eur J Cancer.
34:1514–1521. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Pommier Y, Pourquier P, Fan Y and
Strumberg D: Mechanism of action of eukaryotic DNA topoisomerase I
and drugs targeted to the enzyme. Biochim Biophys Acta.
1400:83–105. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Debatin KM, Poncet D and Kroemer G:
Chemotherapy: targeting the mitochondrial cell death pathway.
Oncogene. 21:8786–8803. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Park JW, Choi YJ, Suh SI, Baek WK, Suh MH,
Jin IN, Min DS, Woo JH, Chang JS, Passaniti A, Lee YH and Kwon TK:
Bcl-2 overexpression attenuates resveratrol-induced apoptosis in
U937 cells by inhibition of caspase-3 activity. Carcinogenesis.
22:1633–1639. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Lee JS, Jeong SH, Soung YH, Kim TH, Choi
HJ, Park BS, Kwon TK and Yoo YH: SAHA treatment overcomes the
anti-apoptotic effects of Bcl-2 and is associated with the
formation of mature PML nuclear bodies in human leukemic U937
cells. Chem Biol Interact. 18:61–70. 2009.
|
|
33
|
Han W, Li S, Qiu S, Lu Q, Pan Q, Gu Y, Luo
J and Hu X: Shikonin circumvents cancer drug resistance by
induction of a necroptotic death. Mol Cancer Ther. 6:641–649.
2007.PubMed/NCBI
|
|
34
|
Stendel R, Biefer HR, Dékány GM, Kubota H,
Münz C, Wang S, Mohler H, Yonekawa Y and Frei K: The antibacterial
substance taurolidine exhibits anti-neoplastic action based on a
mixed type of programmed cell death. Autophagy. 5:194–210. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Tomic T, Botton T, Cerezo M, Robert G,
Luciano F, Puissant A, Gounon P, Allegra M, Bertolotto C, Bereder
JM, Tartare-Deckert S, Bahadoran P, Auberger P, Ballotti R and
Rocchi S: Metformin inhibits melanoma development through autophagy
and apoptosis mechanisms. Cell Death Dis. 2:e1992011. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Koff A, Giordano A, Desai D, Yamashita K,
Harper JW, Elledge S, Nishimoto T, Morgan DO, Franza BR and Roberts
JM: Formation and activation of a cyclin E-cdk2 complex during the
G1 phase of the human cell cycle. Science. 257:1689–1694. 1992.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Elledge SJ and Harper JW: Cdk inhibitors:
on the threshold of checkpoints and development. Curr Opin Cell
Biol. 6:847–852. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Sidle A, Palaty C, Dirks P, Wiggan O,
Kiess M, Gill RM, Wong AK and Hamel PA: Activity of the
retinoblastoma family proteins, pRB, p107, and p130, during
cellular proliferation and differentiation. Crit Rev Biochem Mol
Biol. 31:237–271. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Sandal T: Molecular aspects of the
mammalian cell cycle and cancer. Oncologist. 7:73–81. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Knudsen KE, Booth D, Naderi S,
Sever-Chroneos Z, Fribourg AF, Hunton IC, Feramisco JR, Wang JY and
Knudsen ES: RB-dependent S-phase response to DNA damage. Mol Cell
Biol. 20:7751–776. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Li M, Jiang X, Liu D, Na Y, Gao GF and Xi
Z: Autophagy protects LNCaP cells under androgen deprivation
conditions. Autophagy. 4:54–60. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Zhou F, Yang Y and Xing D: Bcl-2 and
Bcl-xL play important roles in the crosstalk between autophagy and
apoptosis. FEBS J. 278:403–413. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Ciechomska IA, Goemans GC, Skepper JN and
Tolkovsky AM: Bcl-2 complexed with beclin-1 maintains full
anti-apoptotic function. Oncogene. 28:2128–214. 2009. View Article : Google Scholar : PubMed/NCBI
|