Contents
Epidemiology
Etiology
Gastric polyps
Diagnosis
Screening and surveillance
Prevention and treatment
Conclusions
Epidemiology
Gastric cancer (GC) is the second leading cause of
cancer-related mortality worldwide, with an estimated 989,600 new
cases (male-to-female ratio 2:1) and 738,000 GC-related deaths in
2010, accounting for 8% of the total cancer cases and 10% of total
deaths from cancer. Moreover, >70% of these new cases and the
GC-related deaths occur in developing countries, particularly in
Eastern Asia (1).
The most common histopathological features of
gastric malignancies are adenocarcinoma and lymphoma of mucosa-
associated lymphoid tissue (MALT). Approximately 90% of gastric
tumors are adenocarcinomas, whereas gastric MALT lymphomas are
considerably less common (approximately 3% of all gastric tumors).
MALT lymphoma is a distinct subtype of marginal zone B-cell
non-Hodgkin’s lymphoma (NHL), accounting for approximately 7–8% of
all NHLs, with the gastrointestinal tract being the most common
site of the disease (2).
Helicobacter pylori (HP) infection plays an
important cancerogenic role in both gastric carcinoma and MALT
lymphoma. Indeed, HP is estimated to colonize the stomach in
approximately one-half of the world population, with a variable
prevalence in different countries (3). For example, in Eastern regions of
Asia and in some parts of Latin America, HP infection occurs at an
early age, usually in childhood, resulting in approximately 80% of
the population being infected by the age of 20. By contrast, in
developed countries such as France, the USA, the UK, or Australia,
the prevalence of HP infection is low in children under the age of
10 years but increases to approximately 40% in adults 30–40 years
of age. In the USA, the prevalence of HP infection varies among the
different subpopulations and is several-fold higher in the children
of ethnic groups, such as Afro-Americans and Asians, with lower
socioeconomic status. Moreover, migrant populations from high-risk
areas, including Japan, show a marked risk reduction when they move
to low-incidence regions, such as the USA, and subsequent
generations acquire risk levels comparable to those of the host
country. Thus, the difference in prevalence among ethnic groups of
similar socioeconomic status possibly reflects environmental causes
and host genetic alterations.
It has been calculated that the risk of gastric
adenocarcinoma and MALT lymphoma in HP-infected individuals is 3-
to 6-fold higher than in those who are uninfected (2). The association of HP infection with
gastric carcinoma mainly involves intestinal-type and distal forms
of the malignancy. There are 2 main sites of gastric
adenocarcinoma: proximal (cardia) and distal (non-cardia), with
different epidemiological and clinical features. In non-cardia GC,
other risk factors have been implicated, including low
socioeconomic status, smoking, salty and smoked food intake, low
consumption of fruits and vegetables and a family history of GC
(4). In contrast to the decline in
distal GCs, there has been a progressive increase in proximal
tumors since the 1970s, particularly among males in Western
countries. Gastric cardia cancers share certain molecular profiles
with adenocarcinomas of the distal esophagus and gastro-esophageal
junction, suggesting that they represent a similar disease entity.
In fact, the main risk factors for all of these tumors are obesity,
gastro-esophageal reflux disease and Barrett’s esophagus (5).
Histologically, GCs are subdivided into 2 main types
(Lauren classification): i) intestinal type; and ii)
undifferentiated or diffuse type. GC does not arise from a normal
mucosa. The intestinal type is related to corpus-dominant gastritis
with intestinal metaplasia, whereas the diffuse type usually
originates from superficial pangastritis without atrophy. In
addition, while the former is more common in males, blacks and
older age groups, the latter has a comparable male-to-female ratio
and is more frequent in younger individuals. Intestinal-type tumors
predominate in high-risk geographical areas (‘epidemic type’), such
as Eastern Asia, Eastern Europe and Central and South America.
Diffuse-type carcinomas of the stomach have a more uniform
geographical distribution (‘endemic type’). However, the incidence
of diffuse-type gastric carcinoma, in particular signet ring-type,
has been increasing (6).
Thus, GC incidence rates vary by up to 10-fold
worldwide. Regional variations in part reflect differences in
dietary patterns, particularly in European countries and in the
prevalence of HP infection. Gastric colonization by HP is usually
asymptomatic and, although in approximately 20% of infected
populations the bacterium is responsible for pre-neoplastic
changes, gastric neoplasms develop in <2%. Gastric lymphoma is
an even rarer consequence of HP infection, occurring in <1% of
infected individuals (7) (Fig. 1).
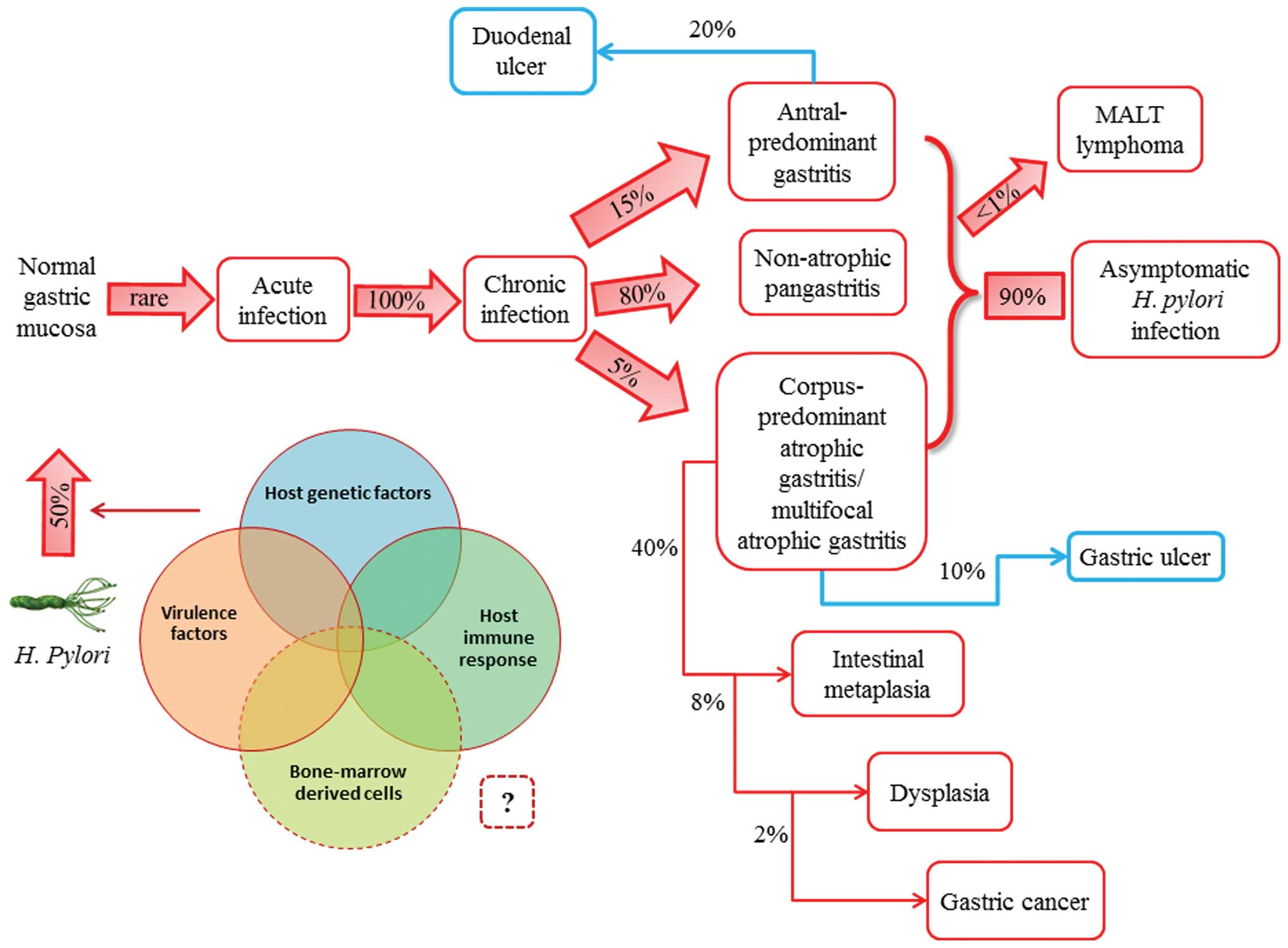 | Figure 1.Natural history of Helicobacter
pylori (H. pylori) infection. H. pylori is
usually acquired in childhood, whereas acute infection with the
bacterium is rarely diagnosed. Instead, chronic gastritis develops
in almost all persistently colonized individuals, 90% of whom will
remain asymptomatic. The clinical course of H. pylori
infection is highly variable depending on bacterial and host
(genetic and immune) factors. Recent studies have supported the
possible role of bone marrow-derived cells (i.e., gastric stem
cells) in tumor progression. Patients with increased acid secretion
are more likely to have antral-predominant gastritis, which
predisposes to duodenal ulcers. Patients with low acid secretion
will more likely develop gastritis in the body of the stomach and
are thus more likely to develop gastric ulcer, leading to gastric
atrophy, intestinal metaplasia, dysplasia and, finally, in rare
cases, gastric carcinoma. This sequence of events is more frequent
in people of advanced age. H. pylori infection induces the
formation of mucosa-associated lymphoid tissue (MALT) in the
gastric mucosa and MALT lymphoma is another rare complication of
H. pylori infection. |
GC rates have substantially decreased in most
geographical areas, possibly due to the increased use of
refrigeration, the availability of fresh fruits and vegetables and
the decreased consumption of salted and preserved foods. Despite
the potentially low malignant transformation of gastric
pre-cancerous lesions induced by HP, the eradication of this
bacterial infection may reduce the risk of gastric adenocarcinoma
and gastric MALT lymphoma.
In fact, the regression of gastric MALT lymphoma
occurs in 60–80% of HP-positive patients, suggesting HP eradication
as the first-choice treatment (8).
In a meta-analysis, the relative risk of GC following HP
eradication was calculated to be 0.65 overall (9). Nevertheless, GC still implies a poor
prognosis and high mortality. In general, patients living in
countries with a higher incidence of GC have better survival rates
than patients from countries with a lower incidence. This
association is mainly due to the location of the tumor within the
stomach. Patients with tumors located in the gastric cardia (which
are more frequent in areas with a lower incidence) have a much
poorer prognosis than those with tumors arising in the pyloric
antrum (more frequent in areas with a higher incidence), with lower
5-year survival rates and higher operative mortality. In addition,
accurate screening for the early detection in high-risk areas has
led to a reduction in mortality. In Japan, mortality rates for
males with GC have markedly decreased over the past several
decades, due to mass screening programs.
When the disease is confined to the inner lining of
the gastric wall, the 5-year survival is approximately 95%.
Unfortunately, the majority of GCs are not discovered at an early
stage, thus leading to 5-year relative survival rates of <20%.
Therefore, early detection is a crucial prognostic factor. Patients
with a family history of non-hereditary GC have a higher risk of
developing these tumors. Familial clusters of stomach cancer
reflect a shared exposure to environmental hazards and to inherited
factors. GC is associated with inherited syndromes in 1–3% of
cases. E-cadherin mutations are more frequent in hereditary
syndromes, occurring in approximately 25% of families with
autosomal dominant hereditary diffuse GC (HDGC), which arises as a
result of germline mutations in the E-cadherin/DCH1 gene (10).
Etiology
Gastric carcinogenesis is a multifactorial process
in which HP infection is the most important risk factor. Host
genetic features, such as a pro-inflammatory cytokine profile
and/or a positive family history, as well as bacterial virulence,
further increase the risk of GC development. Other environmental
issues, such as nutrition and socioeconomic conditions, are
additional important influences.
The correlation between HP infection and the
occurrence of GC represents a model of cancer development as a
consequence of a microbial infection and chronic inflammation. The
World Health Organization (WHO) has classified HP as a class I
carcinogen since 1994 (11). The
mechanisms whereby HP significantly increases the risk of gastric
adenocarcinoma are clearer for intestinal-type GC, which progresses
in a well-defined series of histological steps. The development of
this type is marked by a slow progression, beginning with HP
infection and subsequently progressing to chronic active gastritis,
which occurs in all infected individuals. Under the influence of
variable environmental and host factors, chronic active gastritis
may in turn evolve into atrophic gastritis and intestinal
metaplasia. In certain individuals, the metaplastic epithelium
undergoes further genomic and phenotypic changes, resulting in
gastric dysplasia and, finally, in adenocarcinoma. Variable
progression rates to GC ranging from 0 to 2% per year have been
reported in patients with atrophic gastritis. However, progression
rates from intestinal metaplasia and dysplasia to GC are widely
variable, ranging per year from 0 to 10% and from 0 to 73%,
respectively (12).
Prior to the discovery of HP, in 1988, Correa
proposed a multistep cascade leading to GC (13). This model has been clearly
reproduced in Mongolian gerbils infected with HP (14). This Gram-negative spiral-shaped
bacterium has unipolar flagella that helps it to colonize the
stomach. As noted above, infection is usually acquired during
infancy and it typically induces a life-long chronic gastritis. HP
is specifically adapted to survive in the hostile acidic gastric
environment, with gastric colonization resulting in the development
of gastritis in virtually all infected individuals. The adhesion of
the bacteria to epithelial cells induces an inflammatory response,
resulting in the recruitment of neutrophils, followed by B and T
lymphocytes, macrophages and plasma cells. Consequently, large
amounts of reactive oxygen or nitrogen species, involved in
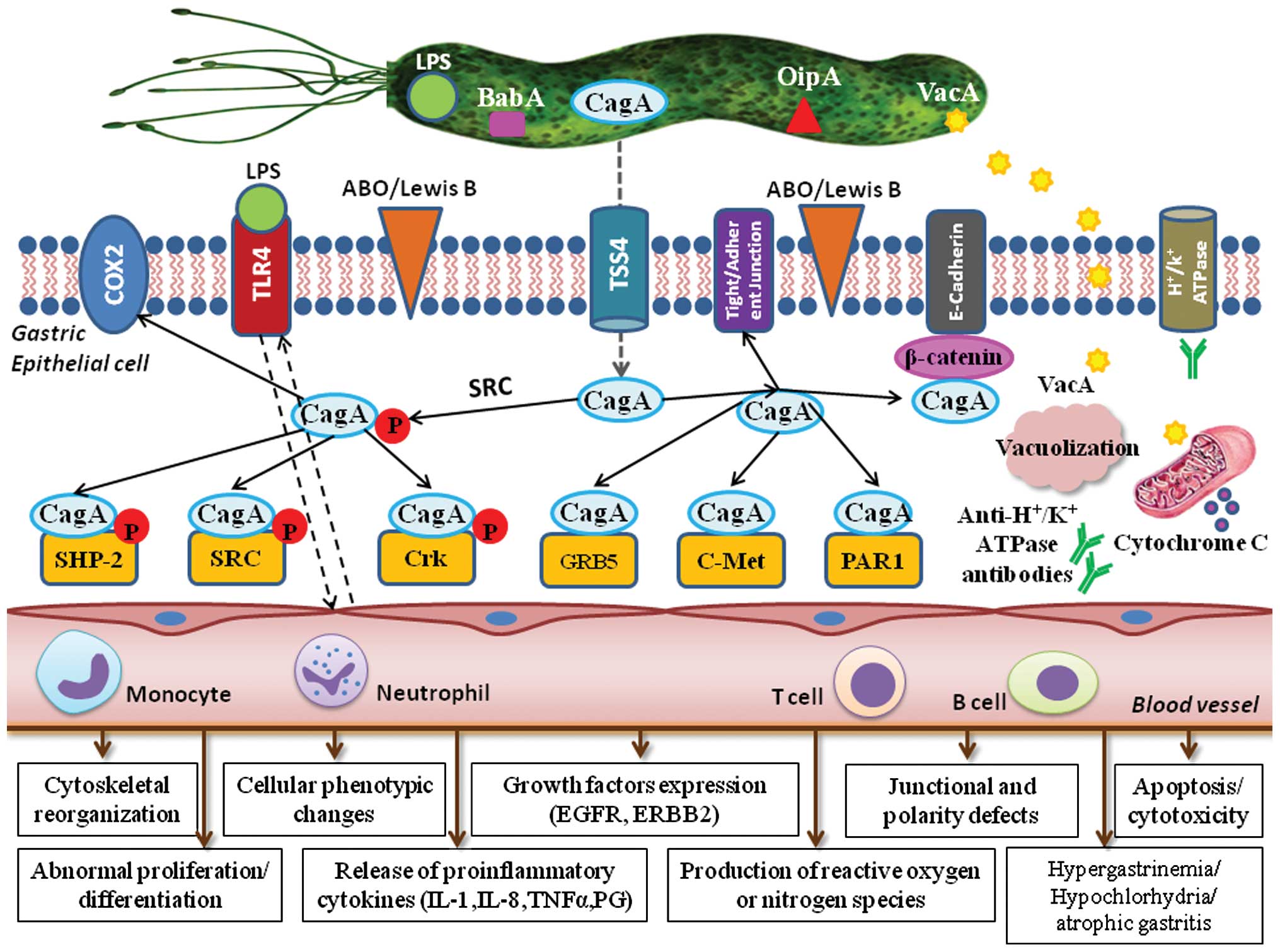
epithelial cell damage and carcinogenesis, are generated (15). The major virulence factors of HP
with a well-established role in the induction of mucosal
inflammation include the cytotoxin-associated gene (cag)
pathogenicity island (PAI)-encoded virulence factors, such as the
cytotoxin-associated antigen (CagA) protein, the vacuolating
toxin-A (VacA), the blood group antigen-binding adhesin (BabA) and
the outer inflammatory protein (OipA). These proteins are encoded
in a 40-kilobase segment of DNA that includes a group of
approximately 30 genes, including those for type IV secretion
system components.
CagA, encoded by cag PAI, is translocated
into the epithelial cytosol. This cytotoxin is a 121- to 145-kDa
immunodominant protein that is commonly considered a putative
bacterial oncoprotein. In fact, it has been used as a marker for
epidemiological studies of GC. Within Western populations,
CagA-positive strains are more commonly associated with peptic
ulceration, atrophic gastritis and gastric adenocarcinoma than
cag-negative strains. Conversely, in many populations with a
high incidence of GC, such as the Eastern regions of Asia, almost
all HP strains are cag-positive (16).
Epithelial cells recognize the translocated CagA as
a signaling molecule that is activated following tyrosine
phosphorylation by Src kinases. This form interacts with the
tyrosine phosphatase SHP-2, the C-terminal Src tyrosine kinase
(SCK) and the adaptor protein Crk, together resulting in
cytoskeletal reorganization and cell elongation. In turn, these
changes lead to cell scattering and so-called ‘hummingbird’
morphological changes. They also induce MAP kinase signaling,
resulting in abnormal cell proliferation by promoting cell cycle
progression. The latter finding, together with the phenotypic
changes, demonstrates that CagA-activated SHP-2 plays an important
role in cell transformation and GC promotion. Phosphorylated CagA
binds the adaptor protein Crk, leading to cytoskeletal
reorganization, the disruption of epithelial cell tight junctions
and tissue damage. Non-phosphorylated CagA also interacts with
certain host cell proteins, such as epithelial tight junctions, the
hepatocyte growth factor receptor C-Met, E-cadherin/β-catenin, the
adaptor protein GRB-5 and kinase PAR1. These CagA-host-protein
interactions disrupt tight and adherent junctions, leading to a
loss of cell polarity and inducing pro-inflammatory and mitogenic
effects that may be important in gastric carcinogenesis.
As a consequence of direct interactions between CagA
and E-cadherin, the formation of E-cadherin/β-catenin complexes is
impaired and cytoplasmic and nuclear accumulations of β-catenin
ensue. Downstream events include the transcription of genes
involved in intestinal differentiation, such as cdx1/cdx2 and the
muc2 mucin gene, causing trans-differentiation from gastric to
intestinal-type epithelial cells (17). HP stimulates gastric epithelial
cells to express and release excessive amounts of pro-inflammatory
cytokines, including interleukin-8 (IL-8) and IL-1.
Pro-inflammatory IL-1 gene cluster polymorphisms (IL-1B, encoding
IL-1B and IL-IRN, encoding its naturally occurring receptor
antagonist) increase the risk of both intestinal- and diffuse-types
of non-cardia GC, while causing a reduction in gastric acid
secretion, stimulating hypergastrinemia and promoting mucosal
damage in atrophic gastritis. Thus, a high-risk IL-1 genotype
increases the likelihood of non-cardia GC, a disease that is
characterized by hypochlorhydria, while it has no effect on cancers
associated with high-level acid exposure, such as esophageal
adenocarcinoma and certain gastric cardia cancers (18).
Another major HP virulence factor is VacA. After
entering the epithelial cell membrane, it induces vacuole formation
and exerts mitochondrial effects, leading to apoptosis. The
polymorphism is also found among the VacA alleles and results in
different levels of cytotoxicity (7). The detection and characterization of
HP CagA and VacA genotypes may be useful for the identification of
patients with gastric pre-neoplastic lesions who are at a high risk
of disease progression and therefore need more intensive
surveillance (19).
The HP OipA, if expressed together with CagA, is
associated with an enhanced inflammatory response in the gastric
mucosa (18,19). OipA is universally present in HP
strains in Eastern Asian populations but in <50% of all strains
in Western countries. Functional receptors for HP adhesion to the
gastric epithelial cell surface include fucosylated ABO blood group
and Lewis b antigens. BabA is an outer membrane protein encoded by
the BabA2 gene; it binds to the Lewis b antigen, ABO antigens and
the sialyl-Lewis x/a antigens.
The BabA-mediated adhesion of HP to the gastric
mucosal cells facilitates HP colonization, induces mucosal
inflammation and promotes the expression of sialyl-Lewis x/a
(17). Moreover, CagA-positive HP
infection upregulates cyclo-oxygenase-2 (COX-2) expression in
gastric mucosa and in cancer. COX-2 is usually undetectable in
normal tissue but becomes abundant at sites of inflammation and may
be overexpressed in gastric carcinomas. The overexpression of COX-2
leads to an increase in the synthesis and release of
prostaglandins, such as PGE2. This COX-2-induced prostaglandin
pathway promotes carcinogenesis by increasing cell proliferation,
inhibiting apoptosis and enhancing the invasiveness of malignant
cells. HP infection is thought to induce COX-2 expression in
pre-cancerous gastric lesions, in turn upregulating the expression
of vascular endothelial growth factor (VEGF) and promoting
angiogenesis. COX-2 expression significantly decreases following HP
eradication in patients with atrophic gastritis (15,20).
Functional polymorphisms of toll-like receptor 4
(TLR4), a cell-surface lipopolysaccharide (LPS) receptor involved
in HP recognition and host response, have been associated with an
increased grade of inflammation and severe tissue damage in
HP-infected individuals. Specifically, carriers of the
TLR4+896A>G polymorphism have more severe gastric atrophy and
inflammation as well as an increased risk of non-cardia GC
(21). Finally, polymorphisms and
the genetic diversity of essential pathogenic elements, such as the
CagA, TRL4 and SHP2 genes appear to influence the oncogenic
potential of HP strains (15).
The ability to stimulate gastrin production is an
important aspect of HP-associated cancerogenesis. In transgenic
mice, hypergastrinemia is associated with the overexpression of
certain growth factors, the COX-2-prostaglandin system and
anti-apoptotic proteins, such as survivin and Bcl-2, leading to the
proliferation of mutated atrophic cells, enhanced angiogenesis and
the development of gastric tumors (Fig. 2).
A recent study demonstrated the causal role of HP
infection as well as specific alterations of DNA methylation
patterns in the gastric mucosa of HP-infected patients and in GC
cell lines in vitro(22).
Genes specifically methylated during HP infection include
E-cadherin (CDH1), a member of the transmembrane glycoprotein
family expressed by epithelial tissues. The CDH1 protein not only
acts as a cell adhesion molecule, but also plays an important role
in cellular growth and carcinogenesis. The importance of the CDH1
gene in diffuse-type GC is suppported by the finding that germline
mutations in CDH1 are responsible for the HDGC syndrome and are
commonly acquired in sporadic GC associated with HP infection.
Inactivation of E-cadherin correlates with the infiltrative and
metastatic potential of GC. Patients with E-cadherin-positive GCs
have significantly longer 3- and 5-year survival rates than
patients with E-cadherin-negative tumors (4). The methylation of the E-cadherin
promoter can be reversed by the eradication of HP (3).
The methylation of the tumor suppressor,
Runt-related transcription factor 3 (RUNX3), by HP infection
likewise contributes to GC progression. Circulating RUNX3
methylation may therefore be a valuable biomarker for the detection
of early GC (23).
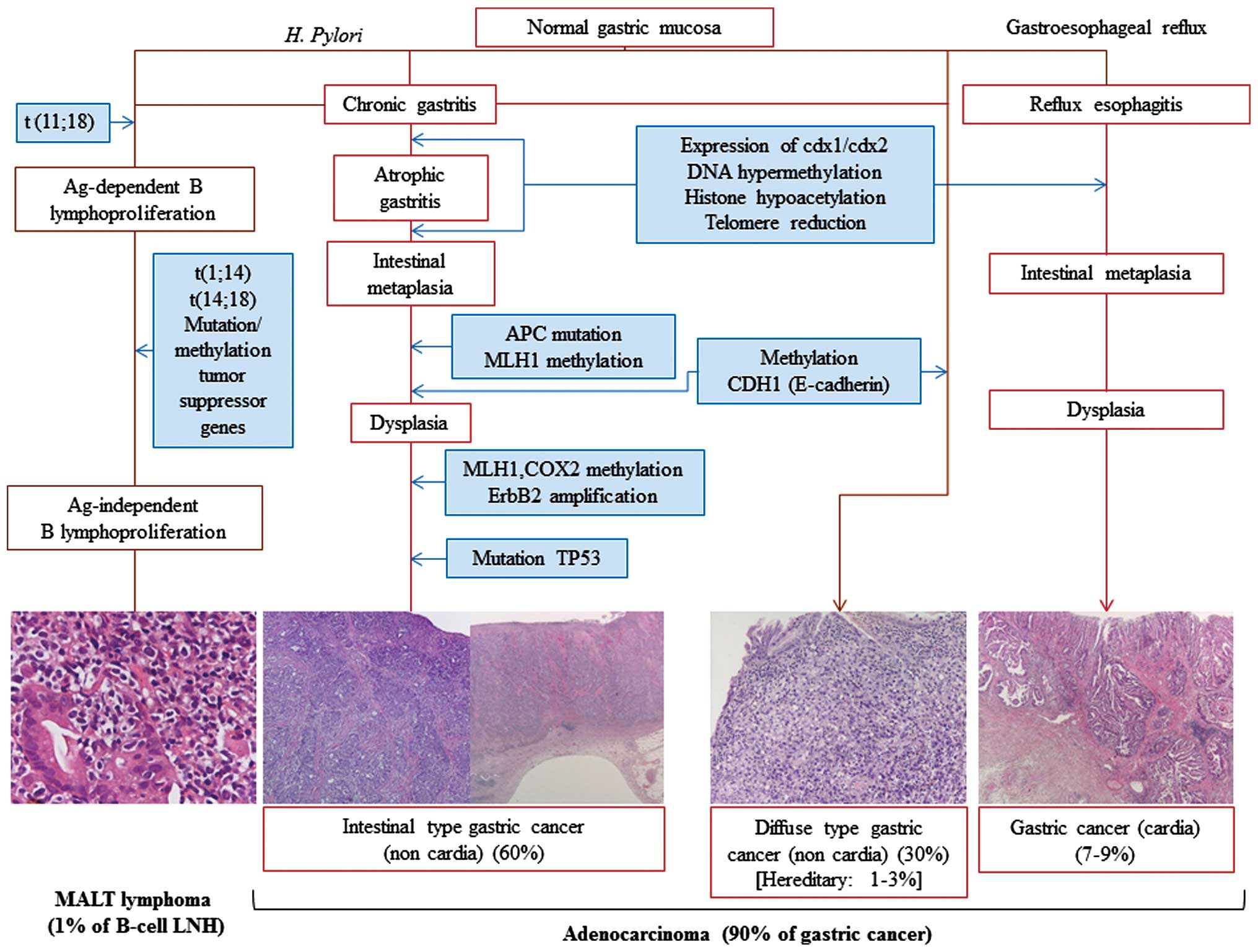
HP is considered the initiator of a chronic
inflammatory response that recruits bone marrow-derived cells to
the gastric mucosa, thereby contributing directly to GC (24). HP also plays an important role in
gastric MALT lymphomagenesis, favoring infection-associated
indirect lymphoid transformation. In addition to its chronic
persistence in host tissues, HP can trigger a sustained lymphoid
proliferation, which provides a selective advantage to lymphoid
clones that still remain dependent upon antigen stimulation. The
evolution into malignant NHL occurs in the presence of additional
oncogenic events, such as the constitutive activation of signaling
pathways following chromosomal translocations, including t(1;14)
and t(14;18), or the inactivation of tumor-suppressor genes by
hyper-methylation or mutations, causing the lymphoproliferation to
become independent of antigenic stimulation. Usually, mutations in
p53 are associated with the transformation of low-grade into
high-grade NHL (25) (Fig. 3).
Gastric polyps
Some gastric polyps may have malignant potential and
are associated with synchronous gastric adenocarcinomas.
Approximately 1–4% of patients who undergo gastric biopsy have
gastric epithelial polyps. Adenomatous polyps have a higher rate of
malignant transformation, whereas the risk of malignant
transformation is very low for hyperplastic gastric polyps and
virtually absent in sporadic fundic gland polyps. Therefore, the
diagnosis of an adenomatous gastric polyp may support the inclusion
of the patient in an endoscopic surveillance program, while only
limited follow-up is generally proposed for patients with diagnoses
of hyperplastic polyps, inflammatory polyps, or sporadic fundic
gland polyps (26).
Adenomatous polyps may occur sporadically or in
association with a familial adenomatous polyposis syndrome.
Endoscopically, these polyps are lobulated in appearance, usually
solitary (80%), located in the antrum and <2 cm in diameter.
They may form circumscribed lesions, or they may be pedunculated or
sessile. Histology will reveal a dysplastic epithelium without
detectable invasion of the lamina propria. The prevalence of
adenomatous polyps varies widely and is estimated to be 0.5–3.7% in
Western countries and 9–27% in areas with higher rates of GC, such
as China and Japan. The larger the adenomatous polyp, the greater
the probability of malignant evolution, particularly when foci of
adenocarcinoma are present.
Hyperplastic polyps often arise in patients with an
atrophic gastric mucosa and HP-associated gastritis (25% of cases).
They account for 18–70% of gastric polyps and are more frequently
detected in the antrum than in other parts of the stomach. Usually,
these polyps are multiple, smooth and small in diameter (measuring
0.5–1.5 cm), while dysplasia is rare (<3% of cases) (27).
Fundic gland polyps are the most common type of
polyps detected in patients undergoing esophagogastroduodenoscopy
in Western countries, with a prevalence of 13–77%. Endoscopically,
they appear as smooth, glassy, sessile, circumscribed elevations
(usually measuring <0.5 cm) in the oxyntic mucosa. Fundic gland
polyps may occur sporadically, in association with the use of
proton-pump inhibitors, or in patients with familial adenomatous
polyposis syndrome. They may or may not be associated with HP
infection. In patients who use proton-pump inhibitors, fundic gland
polyps may regress after the interruption of therapy.
Diagnosis
The symptoms of gastric pre-malignant and malignant
diseases are usually non-specific and vague, such that early
diagnosis is very difficult and GC is often diagnosed at an
advanced stage. However, dyspeptic symptoms may occur in
approximately 60–90% of patients with gastric pre-malignant
lesions. Dyspepsia is defined by the presence of one or more
symptoms of epigastric pain, burning, postprandial fullness, or
early satiation. Bloating and nausea often coexist with dyspepsia
but are non-specific and are thus not included in its definition.
Heartburn is also excluded from the diagnostic criteria of
dyspepsia since it is thought to primarily arise from the esophagus
and is suggestive of gastro-esophageal reflux disease, although it
too may occur concomitantly. These symptoms are generally
indistinguishable from gastric malignant disease. It is therefore
crucial to consider the diagnosis of gastric pre-malignant or
malignant cancer in symptomatic patients.
Upper endoscopy is performed as the initial
diagnostic test in patients with unexplained and persistent
dyspepsia who are considered to be at higher risk. They are over 45
years of age (this cut-off was set since GC below the age of 45 is
rare and its occurrence may vary between countries, depending on
the prevalence of GC) and/or complain of other alarming features
(anemia or evidence of acute/chronic bleeding, odynophagia,
dysphagia, recurrent or persistent vomiting, unintentional weight
loss, previous history of peptic ulcers).
Once the cause of any alarming features, typical
gastroesophageal reflux disease symptoms and possible offending
medications [such as non-steroidal anti-inflammatory drugs
(NSAIDs), COX-2 inhibitors, iron, bisphosphonates, erythromycin,
tetracycline, potassium supplements, acarbose, digitalis,
theophylline] has been excluded in dyspeptic patients under the age
of 45, evaluation for HP infection is warranted. Currently,
non-invasive testing for HP infection, followed by eradication
(‘test and treat’ strategy), is recommended for patients that are
persistently dyspeptic and are under the age of 45, without
alarming features. However, the effectiveness of HP ‘test and
treat’ is low in populations with a low HP prevalence and in this
situation empirical acid suppression is an equivalent option. Once
a patient has failed a 4- to 8-week trial of proton-pump inhibitor
therapy (in a geographical area with a low prevalence of HP) or
failed to respond to HP eradication (in a HP-endemic region), upper
endoscopy is indicated. In countries with a higher incidence of HP
infection, ‘test and treat’ leads to a reduced endoscopic workload
and is cost-effective (28). Thus,
screening for HP identification seems to be a suitable serological
test to non-invasively diagnose pre-malignant gastric lesions,
given the central role of this bacterium in gastric
carcinogenesis.
Non-invasive tests for the diagnosis of HP infection
include: the 13C urea breath test, stool antigen tests
(with polyclonal or monoclonal antibodies) and immunological tests
(laboratory-and office-based tests and tests on saliva and urine).
The urea breath procedure is an accurate, practical and readily
available test, with a sensitivity of 94% and a specificity of 95%.
The stool antigen test has a sensitivity and specificity of 91 and
93%, respectively; the sensitivity decreases to 69% after the
sample is left standing for 2–3 days at room temperature. Serology
is a widely available and low-cost non-invasive test; however, the
diagnostic accuracy is low (80–84%).
Prior treatment with proton-pump inhibitors can
result in false-negative, invasive and non-invasive diagnostic
tests. Therefore, these drugs should be stopped for at least 2
weeks prior to testing. However, this advice does not apply to
serology. Nonetheless, while serologic office-based tests are
extremely convenient, they are not accurate and are currently not
recommended for the detection of HP infection. Serology should be
considered as a diagnostic test only in the case of false-negative
results by other methods, such as in patients with bleeding ulcers,
gastric atrophy, MALT lymphoma, or in those being treated with
proton-pump inhibitors and antibiotics.
The detection of specific HP antibodies in urine and
saliva has no current role in patient management but may be helpful
for epidemiological studies. Since HP virulence factors differ and
host genetic factors may influence disease outcome, neither the
routine detection of HP pathogenic factors nor the assessment of
host genetic polymorphisms is currently recommended. The rapid
urease test can detect the presence of HP within 1 h, with a
satisfactory accuracy (90%). False-negative results can occur in
patients taking anti-secretory drugs. In patients presenting for
endoscopy without pre-treatment, a positive rapid urease test is
sufficient to initiate treatment. Non-invasive tests should be
employed for the confirmation of eradication, except in cases in
which repeat endoscopy is indicated, such as in patients with
gastric ulcers. When the urea breath test is not available, a stool
test, which is less accurate, may be used, preferably analyzed with
monoclonal antibodies. Confirmation of HP eradication should be
assessed at least 4 weeks following treatment (29).
Pepsinogen and gastrin levels, in combination with
HP serology, are useful to establish with high sensitivity and
specificity the presence of gastric pre-malignant lesions,
particularly atrophic gastritis. Serological tests for pepsinogens
I and II and for gastrin provide valuable information on the status
of the gastric mucosa. As is well known, pepsinogen I is produced
by mucosal neck cells in the fundic glands, whereas pepsinogen II
is produced throughout the entire stomach by mucosal neck cells and
by pyloric and Brunner’s glands. Gastric inflammation causes an
increased release of both pepsinogens into the bloodstream, with a
greater increase in pepsinogen II than in pepsinogen I production.
Atrophic gastritis causes a decrease in the production of both
pepsinogens which is more pronounced for pepsinogen I than for
pepsinogen II. As a result of these changes, chronic gastritis is
associated with a reduced pepsinogen I/II ratio, which decreases
even further when atrophic gastritis occurs. In addition, gastrin
is synthesized and secreted from antral G-cells. HP gastritis tends
to raise gastrin serum levels, possibly due to hyperplasia of the
antral G-cells and to an acid-suppressive effect of chronic
gastritis when the corpus mucosa is involved. The increased
production of gastrin also occurs in patients with atrophic
gastritis of the corpus, in response to reduced acid secretion. By
contrast, gastrin levels decrease in patients with
antral-predominant atrophic gastritis (30).
Pre-malignant gastric lesions are often diagnosed by
histological examination of random biopsy samples. At present, the
Sydney system is generally used, both in clinical practice and in
research, to grade gastritis. In this classification system,
several features of inflammation, atrophy and intestinal metaplasia
are separately assessed and then graded. Thus, atrophic gastritis
is defined as loss of glandular structures of the gastric mucosa
and intestinal metaplasia as the replacement of gastric columnar
epithelial cells by cells with an intestinal morphology. Intestinal
metaplasia possibly results from the differentiation of gastric
stem cells into cells with a small intestinal phenotype or colonic
components. It is characterized by the presence of intestinal-type,
mucin-containing goblet cells, Paneth cells and absorptive cells.
Gastric dysplasia (formerly non-invasive neoplasia; synonym,
intraepithelial neoplasia) is characterized by epithelial cells
that vary in size, shape and orientation, with nuclear enlargement
and atypia as well as the distortion of the normal glandular
arrangement (12).
It should be emphasized that differences exist
between Japanese and Western gastrointestinal pathologists with
respect to the classification of gastric dysplasia and cancer.
Japanese pathologists diagnose cancer based on cellular and
structural abnormalities, whereas Western pathologists focus on the
presence of tissue invasion as a prerequisite for a diagnosis of
cancer (31). In the year 2000,
the unified Padua classification was proposed, which divides
dysplasia and adenocarcinoma into 5 categories. The Vienna
classification further distinguishes the categories of low-and
high-grade dysplasia and was revised to improve the correlation
with clinical management (32,33).
Recently, an international group of
gastroenterologists and pathologists [the Operative Link for
Gastritis Assessment (OLGA)], proposed a system for reporting
gastritis in terms of stage (the OLGA staging system), which
arranges the histological phenotypes of gastritis along a scale of
progressively increasing GC risk, from the lowest (OLGA stage 0) to
the highest (OLGA stage IV). This staging framework is borrowed
from the oncology vocabulary and it applies to a gastritis
histology-based reporting format that was successfully adopted for
chronic hepatitis. Just as a given number of portal tracts is
required for the accurate staging of hepatitis, a well-defined
biopsy sampling protocol (as recommended by the Sydney system) is a
‘minimum requirement’ for the reliable staging of gastritis, which
is carried out by combining the extent of atrophy (scored
histologically) with the topographical location of the tumor (as
seen on the mapping protocol) (34).
Although the image quality of standard endoscopy has
improved over the last several decades, the findings at
conventional endoscopy often do not correlate with the histological
diagnoses of gastric pre-malignant lesions. This results from an
unsatisfactory visualization of structure, color and vascularity by
conventional techniques, as all these features play a role in the
adequate distinction of pre-malignant and early GC lesions.
Consequently, several alternative and supplementary strategies have
been developed to overcome the limitations of standard endoscopic
imaging, such as magnification endoscopy in patients with
pre-malignant gastric lesions or early GC.
Methylene blue staining is based on the absorptive
capability of cells and is used to demonstrate intestinal
metaplasia, whereas Indigo carmine enhances the architectural
changes in neoplastic lesions. Chromoendoscopy is an important
approach to the detection of pre-malignant gastric lesions and to
the identification of small foci of early gastric carcinoma not
visible with white light gastroscopy (35). A number of techniques that use the
specific spectral and absorptive features of light have been
developed, including narrow-band imaging, autofluorescence and
hemoglobin enhancement. Narrow-band imaging has been evaluated in
combination with magnification endoscopy. This technique uses
narrow filtered bands in the excitation light to improve imaging of
the superficial capillary network and of the surface contrast
within the mucosa. In autofluorescence endoscopy, the tissue is
exposed to light of shorter wavelengths, typically blue light, with
the subsequent emission of light by endogenous fluorophores.
Finally, the super-addition of a pseudocolor image based on the
mucosal hemoglobin content (hemoglobin enhancement) can be used to
facilitate the delineation of lesions (30).
Screening and surveillance
Since the incidence of GC is geographically highly
variable, the development of uniform worldwide screening strategies
seems inappropriate, given that, in screening, the burden for
patients as well as the costs and the restricted availability of
specific therapeutic interventions need to be taken into
consideration. A high individual risk justifies invasive
investigation by endoscopy, whereas a more conservative approach is
appropriate and ethically acceptable in individuals in low-risk
regions. The initial selection of subjects for screening in
low-incidence countries should possibly be based uniquely on
epidemiological factors, such as age, country of birth and
socioeconomic class, all of which are risk factors for HP
infection. Initial screening would consist of non-invasive tests
(36).
A serological diagnosis of atrophic gastritis should
be followed by endoscopy, with histological confirmation of the
diagnosis. However, in high-incidence countries, serologic and
endoscopic screening could be offered to the general population, as
is common practice in Japan. Indeed, in Japan, mortality rates have
declined, which may be ascribed to the introduction of
photofluorography. Screening may have also contributed to the
persistently high incidence rates reported by the Japanese
(37). Apart from screening,
endoscopic surveillance of patients with pre-malignant lesions is
an important approach to reduce GC morbidity and mortality.
Surprisingly, clear guidelines are not available for the
surveillance of patients with atrophic gastritis, intestinal
metaplasia, or gastric dysplasia, even more so since the guidelines
for surveillance of other gastrointestinal pre-malignant conditions
have been widely developed, for instance for Barrett’s esophagus or
colonic adenomas. Thus, the ‘screen and treat’ strategy for HP
infection should be considered an effective strategy for GC
prevention only in communities with a high incidence of GC
(38).
The diagnosis of gastric MALT lymphoma is based on
the histopathological evaluation of gastric biopsies and on
immunohistochemistry. Gastric MALT lymphoma is a B-cell, low-grade,
typically CD19+, CD20+, usually
CD5−, always CD10− and CD23− NHL.
Based on the close association between MALT lymphoma and HP, the
identification of this infection is highly recommended.
MALT lymphomas behave clinically as indolent NHLs,
with a long disease-free and overall survival. Their good prognosis
is likely related to their tendency to remain localized for long
periods of time and to the low frequency of transformation into
aggressive NHL. Often, only vague dyspeptic symptoms characterize
gastric MALT lymphoma and B symptoms are extremely rare, so that
the diagnosis is often incidental. In other cases, it may first be
detected as a complication of the gastric lesion, such as
gastrointestinal bleeding or perforation. Persistent vomiting and
weight loss are other possible presenting symptoms (39).
In approximately 50% of cases, gastric MALT
lymphomas disseminate within the gastrointestinal tract. The most
common non-gastric primary sites are the salivary glands and ocular
adnexa (25% of cases each), lung (14%) and skin (12%).
Extra-gastric MALT lymphomas are significantly more diffuse than
gastric MALT lymphomas (50 vs. 25%), thus reflecting a particular
homing pattern of lymphocytes generated within a MALT environment,
a possible difference between gastrointestinal and
non-gastrointestinal MALT.
In terms of genetic aberrations, 2 subgroups with a
higher risk of dissemination can be identified: i)
t(11;18)(q21;q21) gastric MALT lymphomas and ii) extra-gastric MALT
lymphomas with trisomy 18 (40).
HP-negative gastric MALT lymphoma accounts for 5–10% of all gastric
MALT lymphomas. Clinically, HP-negative tumors are located more
frequently in the proximal portion of the stomach and are less
frequently of the superficial type. HP-negative tumors may include
those related to autoimmune gastritis, which predominantly involves
the corpus, whereas antrum-predominant gastritis associated with
HP-negative gastric MALT lymphoma frequently displays
t(11;18)(q21;q21) and Bcl-10 nuclear expression. However,
HP-negative gastric MALT lymphoma has a favorable long-term
outcome, comparable to that of HP-positive lymphoma (41). The presence of genetic alterations
in cells of MALT lymphoma, such as trisomy 3, API2-MALT1
translocation, p53 mutation and p16 deletion, characterizes
neoplastic B cells with aggressive behavior, causing the so-called
lympho-epithelial lesions. These are a pathognomonic sign of
lymphoma, with the invasion and disruption of gastric glands.
At endoscopic observation, MALT lymphoma may present
with different macroscopic features, ranging from a
normal-appearing gastric mucosa to an ulcerative or vegetant mass,
clearly suggesting a malignancy. In addition to routine
histological analysis and immunohistochemistry, FISH analysis (or
PCR) for the detection of t(11;18) may be useful in identifying
patients unresponsive to antibiotic therapy. Rarely, patients may
have elevated lactate dehydrogenase or β2-microglobulin levels. A
majority of patients have no abnormal findings on physical
examination. The endoscopic appearance of gastric low-grade
lymphoma often mimics that of benign diseases, such as chronic
gastritis or a peptic ulcer. The histological evaluation of
subsequent biopsies remains an essential follow-up procedure in
MALT lymphoma. Unfortunately, the interpretation of a lymphoid
infiltrate in post-treatment gastric biopsies can be very difficult
and there are no uniform criteria for the definition of
histological remission. A preliminary breath test or stool antigen
test should be performed at least 4 weeks following antibiotic
treatment to establish whether or not HP has been eradicated.
Moreover, a strict endoscopic follow-up is recommended, with
multiple biopsies taken 2–3 months after treatment and subsequently
at least twice per year for 2 years, to monitor the histological
regression of the lymphoma. Gastric MALT lymphomas have a low
tendency to distant spreading and to histological transformation.
Long-term careful endoscopic control and systemic (blood counts and
minimal adequate radiological or ultrasound examinations) follow-up
once a year are recommended for all patients. Indeed, the risk of
gastric adenocarcinoma among patients diagnosed with gastric MALT
lymphoma is reportedly 6-fold higher than in the general population
(42,43).
Gastric polyps are associated with an elevated
frequency of pre-cancerous alterations of the gastric mucosa and,
consequently, with an elevated risk of synchronous or metachronous
cancer. They are often asymptomatic. Esophagogastroduodenoscopy is
the gold standard for their diagnosis. With the expanding
indications for this examination, polyps are more frequently
detected; currently, they are identified in approximately 5% of
upper gastrointestinal tract endoscopies, mostly as incidental
findings (44). One of the main
recommendations of the British Society of Gastroenterology is that
a biopsy sample should be obtained from all gastric polyps detected
at endoscopy. Histopathological assessment is required to establish
the diagnosis and to identify the dysplastic foci. All patients who
have hyperplastic or adenomatous polyps at
esophagogastroduodenoscopy should be evaluated for HP infection, in
that its eradication is associated with the regression of
approximately 80% of hyperplastic polyps whereas the advantage of
HP eradication in adenomas is less clear. In the surveillance of
gastric polyps, gastroscopy should be repeated after one year for
all polyps with dysplasia that have not been removed, after 6
months for adenomatous polyps with high-grade dysplasia and after
one year following complete polypectomy for all other high-risk
polyps (Table I).
 | Table I.Clinical features and management of
gastric polyps. |
Table I.
Clinical features and management of
gastric polyps.
| Polyp type | Prevalence
(frequency relative to other polyps) | Usual number, size
and site | Malignant
potential | Pathological
features of background gastric mucosa | Management |
|---|
| Fundic | 13–77% | Multiple; 1–5 mm;
upper and lower body | Low | Associated with PPI
use; may regress after interruption of PPI. No dysplasia. Consider
FAP | Biopsy to confirm
histological nature. No follow-up |
| Hyperplastic | 18–70% | Single or multiple;
0.5–15 mm; antrum or lower body | Low | HP associated
gastritis (25%); may regress after eradication therapy. Dysplasia
is rare (<3%) | Remove polyp if
dysplastic or symptomatic. Eradicate HP Repeat gastroscopy after 1
year |
| Adenomatous | 0.50–3.75% (in
Western countries); 9–27% (in Eastern countries) | Usually single;
<20 mm; antrum | High | Atrophic gastritis
with intestinal metaplasia. Malignant foci may coexist | Polypectomy. Repeat
gastroscopy after 1 year |
The correct management of hereditary cancer
syndromes requires genetic counseling, information on the family
pedigree going back 3 generations, histopathological confirmation
of gastric carcinoma and informed consent. The criteria for
screening GC families for HDGC require confirmed diffuse GC in a
minimum of 2 first- or second-degree relatives, provided one is
under 50 years of age, or 3 confirmed cases in family members of
any age. Approximately 30% of families meeting these original
criteria carry a predisposing CDH1 mutation (E-cadherin). This
autosomal dominant mutation is seen in at least one fourth of the
cases of HDGC, with a relatively high penetrance and a 70–80%
lifetime risk of GC occurrence (45).
Patients with HDGC typically present with
diffuse-type GC with signet ring cells and, at later stages,
linitis plastica. Advanced hereditary and sporadic diffuse forms of
GC are indistinguishable. Anatomical mapping of complete
gastrectomy specimens has shown that early-stage HDGC is
characterized by the presence of multiple foci of stage T1a, signet
ring cells confined to the superficial lamina propria and the
absence of nodal metastases. These foci are submucosal and not
readily identified by gastroscopy. The majority of foci appear
relatively indolent, with scanty mitoses, although they can rapidly
progress into advanced disease. Therefore, in early gastric lesions
it is important for pathologists to recognize a phenotype
consisting of patchy intramucosal signet ring cells often
associated with pagetoid spread. Even advanced-stage diffuse GC can
be missed at gastroscopy as it can infiltrate below an intact
epithelium. Improved endoscopic techniques that can detect
submucosal foci, coupled with histological or immunological
markers, are urgently required.
Despite these limitations, surveillance endoscopy
should be carried out annually using white light, high-definition
endoscopy: 1i) for carriers of mutation who decline prophylactic
surgery; ii) for patients younger than the age at which
prophylactic surgery is recommended (approximately 20 years); and
iii) prior to prophylactic surgery in newly diagnosed carriers
(46).
Prevention and treatment
Since symptoms are often absent or non-specific, GC
is frequently diagnosed at an advanced stage, when therapeutic
options are limited and prognosis is poor. Consequently, the
overall 5-year survival of these patients is <20% (1).
As previously stated, HP infection is considered an
important initial step in gastric carcinogenesis, conferring a
6-fold higher risk. The chronic inflammation of the gastric mucosa
associated with HP infection slowly progresses through the
aforementioned pre-malignant stages to gastric adenocarcinoma.
Since it has been estimated that 50% of the world population is
infected with the bacterium, its eradication seems a logical step
in the therapy of chronic gastritis and in the prevention of GC.
The aim of HP eradication therapy is either to restore the inflamed
mucosa to its normal healthy state or to prevent further
progression of advanced chronic lesions (atrophic gastritis and
intestinal metaplasia). Accordingly, it is better to administer
therapy prior to the onset of these pre-neoplastic mucosal
abnormalities (47). It should be
emphasized that HP eradication from the residual gastric mucosa
after the endoscopic treatment of early GC may also result in an
inhibitory effect on the occurrence of metachronous GC (48). Conversely, several meta-analyses
have shown that the progression of atrophic gastritis and
intestinal metaplasia to GC can indeed occur following HP
eradication, suggesting that other factors contribute to the
progression of pre-neoplastic lesions (48). For example, genetic and epigenetic
alterations may reach a point of no return, even after elimination
of the triggering carcinogen. Thus, the actual role of HP
eradication in the prevention of GC is still a matter of wide
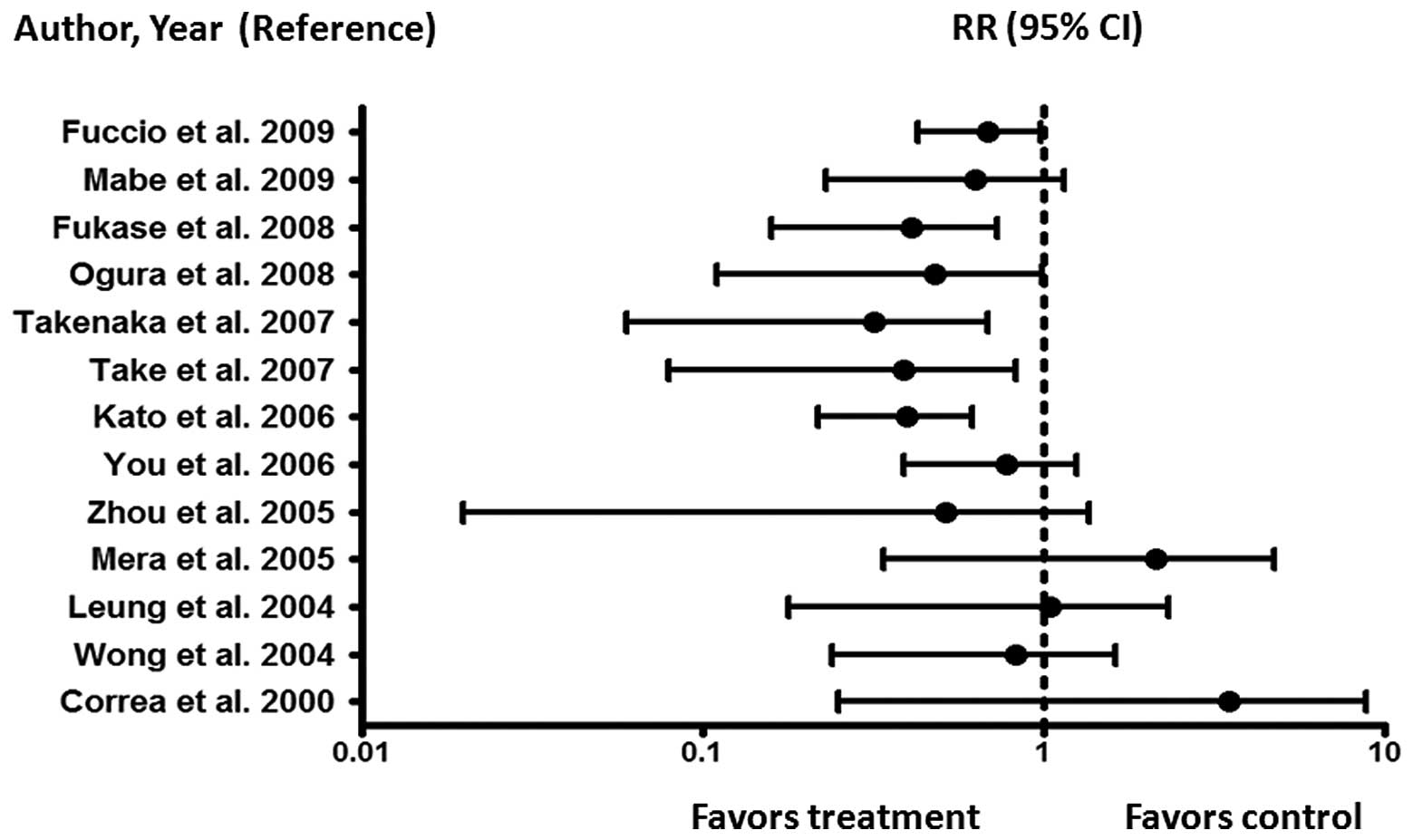
debate. Fig. 4 summarizes the
meta-analyses of randomized controlled trials and non-randomized
studies aimed at assessing whether HP eradication is indeed an
effective preventive strategy to reduce the risk of developing GC
(9,48,50–61).
For low-grade MALT lymphomas confined to the gastric
wall or perigastric lymph nodes (stages I or II), a meta-analysis
of more than 30 studies (8)
yielded an overall remission rate of 78% (95% confidence interval:
75–80%). According to this same report, the overall rate of
lymphoma recurrence was only 2% per year. Therefore, HP eradication
should be the first-choice treatment in patients with HP-positive
MALT lymphoma (42,62). Candidates for anti-HP therapy are
also patients with localized, mucosal, or submucosal, non-bulky,
flat disease who do not have metastases, lymphadenopathy, or frank
diffuse large B-cell lymphomas. It is estimated that <10% of
patients with gastric lymphoma are candidates for antibiotic
treatment as first-line therapy, although recent data suggested
that HP-negative patients will also benefit from this therapeutic
approach (63,64).
In patients who have not responded to eradication
therapy, surgical, radiation and systemic therapies (with
chemotherapeutic agents and/or anti-CD20 monoclonal antibodies)
should be administered, taking into account the stage of the
disease. Since the majority of patients are responsive to
eradication therapy, the timing of a new biopsy for response
assessment is crucial to avoid an inappropriate conclusion of
failure. Complete resolution of the lymphoid infiltrate usually
takes from several months to as long as 2 years. However, molecular
evidence of clonality, as determined by IgH gene rearrangement
studies, may persist for years after morphological remission but
without affecting patient outcome (42).
The t(11;18) translocation involves fusion of the
N-terminus of API2 (apoptosis inhibitor-2) on chromosome 11 and the
C-terminus of MALT1 on chromosome 18. It is the most common
cytogenetic abnormality found in MALT lymphomas of the
gastrointestinal tract, occurring in up to 25% of gastric MALT
lymphomas. This translocation correlates with resistance to
antibiotic therapy and with disseminated rather than stage I
disease. Although molecular studies are not routinely performed, a
search for t(11;18)(q21;q21) translocation should be considered in
order to determine prognosis and therapy for patients refractory to
conservative treatment (65).
Generally, the eradication of HP infection is
recommended in the following high-risk situations: i) patients with
gastric MALT lymphoma; ii) patients with atrophic gastritis; iii)
first-degree relatives of patients with gastric intestinal or
diffuse type of cancer; iv) patients with unexplained
iron-deficiency anemia; v) patients with gastroesophageal reflux
disease requiring long-term acid-suppression therapy, due to the
potential of proton-pump inhibitors to induce, in the presence of
HP, atrophic gastritis, with a subsequent risk of developing GC;
vi) patients with early GC treated by endoscopic mucosal resection,
as it can remove lesions posing a minimal mortality risk, but
likely to have higher rates of recurrence and incomplete
resections; and vii) patients with partial gastrectomy for GC
(28,29).
Triple therapy (proton-pump inhibitors + amoxicillin
+ clarithromycin) is the first-line regimen for the eradication of
HP. Metronidazole treatment is the recommended first-choice
treatment in association with proton-pump inhibitors and
amoxicillin in individuals with clarithromycin resistance (66), which account for <15–20% of the
general population (67).
Quadruple therapy (proton-pump inhibitors + bismuth subcitrate
potassium + metronidazole + tetracycline) remains the best
second-choice treatment. Recent studies have indicated that
quadruple therapy induces a higher eradication rate, whereas its
safety and tolerability are similar to those of triple therapy
(68). If second-line eradication
therapy fails or if there is resistance to metronidazole, an event
that occurs in approximately 40% of cases (67), a proton-pump inhibitor +
amoxicillin + levofloxacin is recommended. This combination is
expected to be effective, with a relatively low rate of adverse
reactions, although the more frequent use of the new quinolones
seems to result in increased resistance. Rifabutin-based triple
therapy has also been reported to be an effective salvage therapy
(66).
Proton-pump inhibitors are crucial for HP
eradication. They increase the stability of acid-labile antibiotics
and, by increasing the drug concentration in the gastric juice,
improve the sensitivity of HP. It is even possible that proton-pump
inhibitors have intrinsic, albeit modest, antimicrobial properties.
The metabolism of proton-pump inhibitors depends on hepatic
cytochrome P450 enzymes, particularly the CYP2C19 genotype, which
displays 3 polymorphisms that can interfere in drug metabolism and
thus the pharmacodynamics of proton-pump inhibitors. An increased
dose of esomeprazole (40 mg twice daily) in triple therapy may
therefore result in a better HP eradication rate than achieved with
omeprazole-based therapy for the wild-type, homozygous genotype of
CYP2C19. The choice of proton-pump inhibitors and their respective
dosage together with the evaluation of CYP2C19 genotyping may be a
more rational approach to obtain the highest HP eradication rates
in a clinical setting. Currently, however, this approach is hardly
applicable due to the elevated costs and the limited availability
of genotyping (69).
Sequential therapy has been proposed as an
alternative to standard triple therapy for the eradication of HP.
The primary goal of this regimen is to overcome clarithromycin
resistance. Hypothetically, during the first 5 days of therapy
amoxicillin would weaken the bacterial cell wall, thus preventing
the formation of the channels which block clarithromycin from
entering the bacterium and thereby causing resistance to this
antibiotic. In the second phase, clarithromycin and nitroimidazole
are added for 5 additional days. The proton-pump inhibitor is
continued throughout treatment. However, there are currently
insufficient data to recommend sequential therapy as a first-line
choice for HP therapy. A longer treatment duration, namely 14
instead of 7 days (66), may
increase eradication rates; however, this approach remains
controversial.
Non-invasive tests should be employed to confirm
eradication, except when a new endoscopy is indicated, such as in
patients with gastric ulcers. During follow-up after eradication
therapy, the urea breath test is the best option, based on its high
sensitivity and specificity. As noted above, the stool antigen test
is less accurate than the urea breath test; however, it is an
acceptable alternative if the latter is not available. Confirmation
of HP eradication should be performed at least 4 weeks after
treatment (70). Nonetheless, 1%
of patients still develop GC despite successful HP eradication;
furthermore, a tumor may develop several years following HP
eradication. Thus, it should be kept in mind that GC, similar to
other tumors, is a multifactorial disease and removing one factor
does not prevent all of these malignancies (49).
In consideration of the increased incidence of
treatment failure and antibiotic resistance, the development of a
prophylactic or therapeutic vaccine against HP seems a desirable
alternative, which may improve the rate of eradication success
obtained with standard regimens or reduce the bacterial density in
the gastric mucosa. Although vaccination studies in animal models
have yielded promising results, experiments in human volunteers
have revealed problems such as ‘post-immunization gastritis’.
Moreover, responses to vaccine antigens have been comparatively
poor, possibly since HP colonizes the gastric mucosa without
crossing the epithelium, thus making the bacterium inaccessible to
many immune effector mechanisms. While HP stimulates both innate
and acquired immune responses, it nonetheless persists by
exploiting a mechanism of reduced identification based on the
generation of different epitopes through point mutations and
recombinations. It also evades and manipulates the immune system by
sequestering itself in the gut lumen, where the immune system is
less efficient, while also mimicking host antigens and
downregulating the activation of immune cells (71).
New directions for active immunization include the
use of DNA, living vectors, microspheres, new vaccination schedules
and different routes of administration (oral, intranasal, rectal
and intramuscular). Many of these new approaches are currently
being evaluated (72). In a recent
study, a multi-epitope DNA-prime/peptide-boost immunization
strategy was proposed, using informatics tools in a mouse model of
GC induced by chronic HP infection. The multi-epitope vaccine was
administered intranasally and induced a broad immune response, as
determined by interferon-γ production in ELISpot assays. These
results suggest that the development of an epitope-based mucosal
vaccine may be beneficial in eradicating HP and in reducing the
burden of associated GC in humans (73).
As previously stated, hyperplastic polyp formation
is strongly associated with HP-positive gastritis. Up to 80% of
hyperplastic polyps regress after HP eradication and therefore do
not require endoscopic removal. Obviously, eradication therapy is
not indicated for the other types of gastric polyps as they are not
associated with HP infection. Conversely, all gastric adenomatous
polyps and all gastric polyps with dysplastic foci, as well as
symptomatic polyps, should be removed. Endoscopic mucosal resection
has become the standard of care for the removal of gastric large,
flat and sessile polyps and early cancer. Recently, endoscopic
submucosal dissection was introduced in Japan as an alternative
technique allowing the ‘en bloc’ resection of large lesions. The
indications for endoscopic mucosal resection are expanding and many
Western endoscopists are adopting the technique (74).
The most important hereditary GC is HDGC syndrome,
which is associated with CDH1 germline mutations. Prophylactic
gastrectomy should be considered only when these mutations occur,
in that they imply a >80% lifetime risk of developing GC. Other
inherited cancer predispositions are hereditary non-polyposis
colorectal cancer and Li-Fraumeni and Peutz-Jeghers syndromes; in
these cases, only endoscopic surveillance is recommended (46,75).
The overexpression of the COX-2 gene in GC suggests
its role in gastrointestinal carcinogenesis. The enzyme
participates in several key cellular activities, including cell
proliferation, apoptosis and angiogenesis. NSAIDs and aspirin are
therefore potential agents for the chemoprevention of GC. The
impact of HP infection on the relationship between the use of
NSAIDs and GC remains unclear. HP infection is known to be
associated with non-cardia GC. Thus, it is possible that NSAIDs
inhibit the replication and proliferation of HP, thus neutralizing
the increased COX-2 expression and elevated prostaglandin synthesis
associated with HP infection while reducing the risk of GC
(76).
A strong correlation can also be envisaged between
HP infection and dietary factors in gastric carcinogenesis. In
fact, HP gastritis enhances the growth of nitrose-producing
bacteria, which catalyze the production of carcinogenic N-nitrose
compounds. In addition, HP infection is known to inhibit the
gastric secretion of ascorbic acid, which is an important scavenger
of N-nitrose compounds and oxygen-free radicals. Moreover,
salt-preserved foods and the dietary nitrite found in preserved
meats are potentially carcinogenic. The intake of salted food may
increase the risk of HP infection and act synergistically to
promote the development of GC. Salt-induced mucosal damage may
increase the possibility of persistent infection with HP (4). Given that GC is often associated with
a poor prognosis, the main strategy for improving clinical outcome
remains prevention.
The widespread introduction of refrigeration has
further decreased the intake of chemically preserved foods and
increased the consumption of fresh fruits and vegetables. A decline
in the prevalence of HP infection may be due to improvements in
sanitary and housing conditions, as well as the use of eradication
therapy. In consideration of the multifactorial pathogenesis of GC,
all modifiable risk factors, such as high salt and nitrite
consumption, low fruit and vegetable intake, cigarette smoking and
HP infection may represent targets for prevention (77).
Conclusions
Over the past decades, HP infection has been clearly
correlated with gastric carcinogenesis. The strongest support for a
link between HP infection and GC development has come from the
preventive effect of HP eradication. At present and in the near
future, the most important challenge is to significantly reduce
mortality due to GC, as the prognosis of patients with GC remains
extremely poor. This can be achieved by the identification of
higher-risk patients, such as those with atrophic gastritis,
intestinal metaplasia, dysplasia of the stomach and, in rare cases,
in those with hyperplastic gastric polyps or germ-line mutations in
CDH1, responsible for HDGC. A number of recent studies on gastric
pre-cancerous lesions have offered the promise of novel biomarkers
allowing for the early detection of GC, while others have reported
improvements in invasive and non-invasive diagnostic tests for
pre-malignant stages of GC. In conclusion, recognition of the
pre-malignant condition at an early stage is a major advantage as
it allows endoscopic surveillance and, possibly, therapeutic
mucosal resection. In addition, preventive cancer strategies, such
as HP eradication with triple or quadruple treatment, the
development of prophylactic or therapeutic vaccination against HP
and perhaps chemoprevention in the form of NSAIDs and aspirin,
against a background of lifestyle and dietary modifications, may be
effective in ‘at risk’ populations.
Abbreviations:
|
BabA
|
blood group antigen-binding
adhesion
|
|
cag
|
cytotoxin-associated gene
|
|
COX-2
|
cyclo-oxygenase-2 (COX-2)
|
|
GC
|
gastric cancer
|
|
HP
|
Helicobacter pylori
|
|
IL
|
interleukin
|
|
MALT
|
mucosa-associated lymphoid tissue
|
|
NHL
|
non-Hodgkin’s lymphoma
|
|
OipA
|
outer inflammatory protein A
|
|
cag PAI
|
cag pathogenicity island
|
|
PGE2
|
prostaglandin E2
|
|
RUNX3
|
Runt-related transcription factor
3
|
|
SCK
|
Src tyrosine kinase
|
|
VacA
|
vacuolating toxin-A
|
|
VEGF
|
vascular endothelial growth factor
|
Acknowledgements
This study was supported in part by
grants from the Italian Association for Cancer Research (AIRC,
Milan, Italy), the Italian Foundation ‘Cassa di Risparmio di
Puglia’ (Bari, Italy) and the strategic project ‘Biotecnoter’ of
the Apulia Region (Bari, Italy).
References
|
1.
|
Jemal A, Bray F, Center MM, Ferlay J, Ward
E and Forman D: Global cancer statistics. CA Cancer J Clin.
61:69–90. 2011. View Article : Google Scholar
|
|
2.
|
Kim SS, Ruiz VE, Carroll JD and Moss SF:
Helicobacter pylori in the pathogenesis of gastric cancer
and gastric lymphoma. Cancer Lett. 305:228–238. 2011. View Article : Google Scholar
|
|
3.
|
Suerbaum S and Michetti P: Helicobacter
pylori infection. N Engl J Med. 347:1175–1186. 2002. View Article : Google Scholar
|
|
4.
|
Fuccio L, Eusebi LH and Bazzoli F: Gastric
cancer, Helicobacter pylori infection and other risk
factors. World J Gastrointest Oncol. 2:342–347. 2010.
|
|
5.
|
Conteduca V, Sansonno D, Ingravallo G, et
al: Barrett’s esophagus and esophageal cancer: an overview. Int J
Oncol. 41:414–424. 2012.
|
|
6.
|
Crew KD and Neugut AI: Epidemiology of
gastric cancer. World J Gastroenterol. 12:354–362. 2006.
|
|
7.
|
Polk DB and Peek RM Jr: Helicobacter
pylori: gastric cancer and beyond. Nat Rev Cancer. 10:403–414.
2010. View
Article : Google Scholar
|
|
8.
|
Zullo A, Hassan C, Cristofari F, et al:
Effects of Helicobacter pylori eradication on early stage
gastric mucosa-associated lymphoid tissue lymphoma. Clin
Gastroenterol Hepatol. 8:105–110. 2010.
|
|
9.
|
Fuccio L, Zagari RM, Eusebi LH, et al:
Meta-analysis: can Helicobacter pylori eradication treatment
reduce the risk for gastric cancer? Ann Intern Med. 151:121–128.
2009.
|
|
10.
|
Yaghoobi M, Bijarchi R and Narod SA:
Family history and the risk of gastric cancer. Br J Cancer.
102:237–242. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
IARC Working Group: Schistosomes, liver
flukes and Helicobacter pylori. IARC Working Group on the
Evaluation of Carcinogenic Risks to Humans Lyon, 7–14 June 1994.
IARC Monogr Eval Carcinog Risks Hum. 61:1–241. 1994.
|
|
12.
|
De Vries AC, Haringsma J and Kuipers EJ:
The detection, surveillance and treatment of premalignant gastric
lesions related to Helicobacter pylori infection.
Helicobacter. 12:1–15. 2007.PubMed/NCBI
|
|
13.
|
Correa P: A human model of gastric
carcinogenesis. Cancer Res. 48:3554–3560. 1998.
|
|
14.
|
Watanabe T, Tada M, Nagai H, Sasaki S and
Nakao M: Helicobacter pylori infection induces gastric
cancer in mongolian gerbils. Gastroenterology. 115:642–648. 1998.
View Article : Google Scholar
|
|
15.
|
Correa P and Houghton J: Carcinogenesis of
Helicobacter pylori. Gastroenterology. 133:659–672.
2007.
|
|
16.
|
Hatakeyama M: Helicobacter pylori
CagA-a bacterial intruder conspiring gastric carcinogenesis. Int J
Cancer. 119:1217–1223. 2006. View Article : Google Scholar
|
|
17.
|
Konturek PC, Konturek SJ and Brzozowski T:
Helicobacter pylori infection in gastric cancerogenesis. J
Physiol Pharmacol. 60:3–21. 2009.
|
|
18.
|
Gianfagna F, De Feo E, van Duijn CM,
Ricciardi G and Boccia S: A systematic review of meta-analyses on
gene polymorphisms and gastric cancer risk. Curr Genomics.
9:361–374. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
González CA, Figueiredo C, Lic CB, et al:
Helicobacter pylori cagA and vacA genotypes as predictors of
progression of gastric preneoplastic lesions: a long-term follow-up
in a high-risk area in Spain. Am J Gastroenterol. 106:867–874.
2011.
|
|
20.
|
Liu D, He Q and Liu C: Correlations among
Helicobacter pylori infection and the expression of
cyclooxygenase-2 and vascular endothelial growth factor in gastric
mucosa with intestinal metaplasia or dysplasia. J Gastroenterol
Hepatol. 25:795–799. 2010.
|
|
21.
|
Hold GL, Rabkin CS, Chow WH, et al: A
functional polymorphism of toll-like receptor 4 gene increases risk
of gastric carcinoma and its precursors. Gastroenterology.
132:905–912. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Nobili S, Bruno L, Landini I, et al:
Genomic and genetic alterations influence the progression of
gastric cancer. World J Gastroenterol. 17:290–299. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Lu XX, Yu JL, Ying LS, et al: Stepwise
cumulation of RUNX3 methylation mediated by Helicobacter
pylori infection contributes to gastric carcinoma progression.
Cancer. 118:5507–5517. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24.
|
Rocco A, Compare D and Nardone G: Cancer
stem cell hypothesis and gastric carcinogenesis: Experimental
evidence and unsolved questions. World J Gastrointest Oncol.
4:54–59. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
25.
|
Suarez F, Lortholary O, Hermine O, et al:
Infection-associated lymphomas derived from marginal zone B cells:
a model of antigen-driven lymphoproliferation. Blood.
107:3034–3044. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
26.
|
Carmack SW, Genta RM, Graham DY and
Lauwers GY: Management of gastric polyps: a pathology-based guide
for gastroenterologists. Nat Rev Gastroenterol Hepatol. 6:331–341.
2009. View Article : Google Scholar
|
|
27.
|
Kelly PJ and Lauwers GY: Clinical
guidelines: Consensus for the management of patients with gastric
polyps. Nat Rev Gastroenterol Hepatol. 8:7–8. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
28.
|
Malfertheiner P, Megraud F, O’Morain C, et
al: Current concepts in the management of Helicobacter
pylori infection: the Maastricht III Consensus Report. Gut.
56:772–781. 2007.PubMed/NCBI
|
|
29.
|
Malfertheiner P, Megraud F, O’Morain CA,
et al: Management of Helicobacter pylori infection - the
Maastricht IV/ Florence Consensus Report. Gut. 61:646–664.
2012.
|
|
30.
|
De Vries AC and Kuipers EJ: Epidemiology
of premalignant gastric lesions: implications for the development
of screening and surveillance strategies. Helicobacter. 12:22–31.
2007.PubMed/NCBI
|
|
31.
|
Schlemper RJ, Itabashi M, Kato Y, et al:
Differences in diagnostic criteria for gastric carcinoma between
Japanese and western pathologists. Lancet. 349:1725–1729. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
32.
|
Schlemper RJ, Riddell RH, Kato Y, et al:
The Vienna classification of gastrointestinal epithelial neoplasia.
Gut. 47:251–255. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
33.
|
Dixon MF: Gastrointestinal epithelial
neoplasia: Vienna revisited. Gut. 51:130–131. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
34.
|
Rugge M, Pennelli G, Pilozzi E, et al:
Gastritis: the histology report. Dig Liver Dis. 43(Suppl 4):
S373–S384. 2011. View Article : Google Scholar
|
|
35.
|
Ohnita K, Isomoto H, Shikuwa S, et al:
Magnifying chromoendoscopic findings of early gastric cancer and
gastric adenoma. Dig Dis Sci. 56:2715–2722. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
36.
|
Yeh JM, Hur C, Kuntz KM, Ezzati M and
Goldie SJ: Cost-effectiveness of treatment and endoscopic
surveillance of precancerous lesions to prevent gastric cancer.
Cancer. 116:2941–2953. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
37.
|
Hamashima C, Shibuya D, Yamazaki H, et al:
The Japanese guidelines for gastric cancer screening. Jpn J Clin
Oncol. 38:259–267. 2008. View Article : Google Scholar
|
|
38.
|
Fock KM, Katelaris P, Sugano K, et al:
Second Asia-Pacific consensus guidelines for Helicobacter
pylori infection. J Gastroenterol Hepatol. 24:1587–1600. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
39.
|
Zullo A, Hassan C, Cristofari F, Perri F
and Morini S: Gastric low-grade mucosal-associated lymphoid
tissue-lymphoma: Helicobacter pylori and beyond. World J
Gastrointest Oncol. 2:181–186. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
40.
|
Oh SY, Kim WS, Kim JS, et al: Multiple
mucosa-associated lymphoid tissue organs involving marginal zone B
cell lymphoma: organ-specific relationships and the prognostic
factors. Consortium for improving survival of lymphoma study. Int J
Hematol. 92:510–517. 2010. View Article : Google Scholar
|
|
41.
|
Shinagare AB, Ramaiya NH, O’Regan K,
Jagannathan JP, Hornick JL and LaCasce AS: Helicobacter
pylori-negative gastric mucosa-associated lymphoid tissue
lymphoma. J Clin Oncol. 29:297–300. 2011. View Article : Google Scholar
|
|
42.
|
Zucca E and Dreyling M; ESMO Guidelines
Working Group: Gastric marginal zone lymphoma of MALT type: ESMO
Clinical Practice Guidelines for diagnosis, treatment and
follow-up. Ann Oncol. 21(Suppl 5): v175–v176. 2010. View Article : Google Scholar
|
|
43.
|
Ruskoné-Fourmestraux A, Fischbach W,
Aleman BM, et al: EGILS consensus report. Gastric extranodal
marginal zone B-cell lymphoma of MALT. Gut. 60:747–758.
2011.PubMed/NCBI
|
|
44.
|
Goddard AF, Badreldin R, Pritchard DM, et
al: British Society of Gastroenterology: The management of gastric
polyps. Gut. 59:1270–1276. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
45.
|
Fitzgeral RC, Hardwick R, Huntsman D, et
al: Hereditary diffuse gastric cancer: updated consensus guidelines
for clinical management and directions for future research. J Med
Genet. 47:436–444. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
46.
|
Guilford P, Humar B and Blair V:
Hereditary diffuse gastric cancer: translation of CDH1 germline
mutations into clinical practice. Gastric cancer. 13:1–10. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
47.
|
Bornschein J, Rokkas T, Selgrad M and
Malfertheiner P: Helicobacter pylori and clinical aspects of
gastric cancer. Helicobacter. 14(Suppl 1): 41–45. 2009. View Article : Google Scholar
|
|
48.
|
Fukase K, Kato M, Kikuchi S, et al: Effect
of eradication of Helicobacter pylori on incidence of
metachronous gastric carcinoma after endoscopic resection of early
gastric cancer: an open-label, randomised controlled trial. Lancet.
372:392–397. 2008.PubMed/NCBI
|
|
49.
|
Take S, Mizuno M, Ishiki K, et al: The
long-term risk of gastric cancer after the successful eradication
of Helicobacter pylori. J Gastroenterol. 46:318–324. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
50.
|
Kodama M, Murakami K, Okimoto T, et al:
Helicobacter pylori eradication improves gastric atrophy and
intestinal metaplasia in long-term observation. Digestion.
85:126–130. 2012. View Article : Google Scholar
|
|
51.
|
Correa P, Fontham ET, Bravo JC, et al:
Chemoprevention of gastric dysplasia: randomized trial of
antioxidant supplements and anti-Helicobacter pylori
therapy. J Natl Cancer Inst. 92:1881–1888. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
52.
|
Wong BC, Lam SK, Wong WM, et al:
Helicobacter pylori eradication to prevent gastric cancer in
a high-risk region of China: a randomized controlled trial. JAMA.
291:187–194. 2004. View Article : Google Scholar
|
|
53.
|
Leung WK, Lin SR, Ching JY, et al: Factors
predicting progression of gastric intestinal metaplasia: results of
a randomised trial on Helicobacter pylori eradication. Gut.
53:1244–1249. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
54.
|
Mera R, Fontham ET, Bravo LE, et al: Long
term follow up of patients treated for Helicobacter pylori
infection. Gut. 54:1536–1540. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
55.
|
Zhou LY, Lin SR, Ding SG, et al: The
changing trends of the incidence of gastric cancer after
Helicobacter pylori eradication in Shandong area. Chin J Dig
Dis. 6:114–115. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
56.
|
You WC, Brown LM, Zhang L, et al:
Randomized double-blind factorial trial of three treatments to
reduce the prevalence of precancerous gastric lesions. J Natl
Cancer Inst. 98:974–983. 2006. View Article : Google Scholar
|
|
57.
|
Kato M, Asaka M, Nakamura T, et al:
Helicobacter pylori eradication prevents the development of
gastric cancer - results of a long-term retrospective study in
Japan. Aliment Pharmacol Ther. 24:203–206. 2006. View Article : Google Scholar
|
|
58.
|
Take S, Mizuno M, Ishiki K, et al:
Baseline gastric mucosal atrophy is a risk factor associated with
the development of gastric cancer after Helicobacter pylori
eradication therapy in patients with peptic ulcer diseases. J
Gastroenterol. 42(Suppl 17): 21–27. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
59.
|
Takenaka R, Okada H, Kato J, et al:
Helicobacter pylori eradication reduced the incidence of
gastric cancer, especially of the intestinal type. Aliment
Pharmacol Ther. 25:805–812. 2007. View Article : Google Scholar
|
|
60.
|
Ogura K, Hirata Y, Yanai A, et al: The
effect of Helicobacter pylori eradication on reducing the
incidence of gastric cancer. J Clin Gastroenterol. 42:279–283.
2008.
|
|
61.
|
Mabe K, Takahashi M, Oizumi H, et al: Does
Helicobacter pylori eradication therapy for peptic ulcer
prevent gastric cancer? World J Gastroenterol. 15:4290–4297.
2009.
|
|
62.
|
Kuo SH, Yeh KH, Wu MS, et al:
Helicobacter pylori eradication therapy is effective in the
treatment of early-stage H pylori-positive gastric diffuse large
B-cell lymphomas. Blood. 119:4838–4844. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
63.
|
Park HS, Kim YJ, Yang WI, Suh CO and Lee
YC: Treatment and outcome of localized Helicobacter
pylori-negative low-grade gastric MALT lymphoma. World J
Gastroenterol. 16:2158–2162. 2010.
|
|
64.
|
Gill H, Chim CS, Au WY, et al: Non-gastric
marginal zone B cell lymphoma: clinicopathologic features and
treatment results. Ann Hematol. 90:1399–1407. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
65.
|
Owens SR and Smith LB: Molecular aspects
of H. pylori-related MALT lymphoma. Patholog Res Int.
2011:1931492011.PubMed/NCBI
|
|
66.
|
O’Connor A, Gisbert JP, McNamara D and
O’Morain C: Treatment of Helicobacter pylori infection 2010.
Helicobacter. 15:46–52. 2010.
|
|
67.
|
Graham DY and Fischbach L: Helicobacter
pylori treatment in the era of increasing antibiotic
resistance. Gut. 59:1143–1153. 2010. View Article : Google Scholar
|
|
68.
|
Malfertheiner P, Bazzoli F, Delchier JC,
et al: Helicobacter pylori eradication with a capsule
containing bismuth subcitrate potassium, metronidazole and
tetracycline given with omeprazole versus clarithromycin-based
triple therapy: a randomised, open-label, non-inferiority, phase 3
trial. Lancet. 377:905–913. 2011. View Article : Google Scholar
|
|
69.
|
Kang JM, Kim N, Lee DH, et al: Effect of
the CYP2C19 polymorphism on the eradication rate of Helicobacter
pylori infection by 7-day triple therapy with regular proton
pump inhibitor dosage. J Gastroenterol Hepatol. 23:1287–1291.
2008.PubMed/NCBI
|
|
70.
|
Perri F, Manes G, Neri M, Vaira D and
Nardone G: Helicobacter pylori antigen stool test and
13C-Urea breath test in patients after eradication
treatments. Am J Gastroenterol. 97:2756–2762. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
71.
|
World Gastroenterology Organisation: World
Gastroenterology Organisation Global Guideline: Helicobacter
pylori in developing countries. J Clin Gastroenterol.
45:383–388. 2011. View Article : Google Scholar
|
|
72.
|
Czinn SJ and Blanchard T: Vaccinating
against Helicobacter pylori infection. Nat Rev Gastroenterol
Hepatol. 8:133–140. 2011. View Article : Google Scholar
|
|
73.
|
Moss SF, Moise L, Lee DS, et al:
HelicoVax: epitope-based therapeutic Helicobacter pylori
vaccination in a mouse model. Vaccine. 29:2085–2091. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
74.
|
Othman MO and Wallace MB: Endoscopic
mucosal resection (EMR) and endoscopic submucosal dissection (ESD)
in 2011, a Western perspective. Clin Res Hepatol Gastroenterol.
35:288–294. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
75.
|
Pandalai PK, Lauwers GY, Chung DC, Patel D
and Yoon SS: Prophylactic total gastrectomy for individuals with
germline CDH1 mutation. Surgery. 149:347–355. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
76.
|
Wu CY, Wu MS, Kuo KN, Wang CB, Chen YJ and
Lin JT: Effective reduction of gastric cancer risk with regular use
of nonsteroidal anti-inflammatory drugs in Helicobacter
pylori-infected patients. J Clin Oncol. 28:2952–2957. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
77.
|
Navarro Silvera SA, Mayne ST, Risch HA, et
al: Principal component analysis of dietary and lifestyle patterns
in relation to risk of subtypes of esophageal and gastric cancer.
Ann Epidemiol. 21:543–550. 2011.PubMed/NCBI
|