Introduction
According to the 2009 annual report from the
National Cancer Institute (NCI), the rates of new diagnose and
death from all cancers combined have declined significantly for men
and women, but the incidence rate for lung cancer in women has
increased.
Anacardic acid (2-hydroxy-6-pentadecylbenzoic acid,
AA; Fig. 1) is a constituent of
the shell of the cashew-nut (Anacardium occidentale)
(1) and has been discovered in
many plants including Ginkgo biloba(2). AA has a number of roles including the
inhibition of lipid synthesis, enzyme activity such as
lipoxygenase, prostaglandin endoperoxide synthase and histone
acetyltransferase and the expression of nuclear factor-κB (NF-κB)
as well as the activation of aurora kinase A (2–6).
Additionally, AA has an antibacterial and anticancer effect
(7,8).
The name ‘apoptosis’ coined by Kerr et al is
classified as a type I programmed cell death (PCD) (9). Apoptosis is a physiological process
characterized by chromatin condensation, nuclear fragmentation, DNA
fragmentation and final removal by phagocytosis (10). Cell death is frequently thought to
be ‘caspase-independent’ when it is not suppressed by
broad-spectrum caspase inhibitors such as
N-benzyloxycarbonyl-Val-Ala-Asp-fluoromethylketone (Z-VAD-fmk).
However, the efficiency of Z-VAD-fmk is different from caspases and
it also inhibits calpains and cathepsins, especially at high
concentrations (10 μM) (10).
The main pathways of apoptosis are the death
receptor pathway (extrinsic) and the mitochondrial pathway
(intrinsic). When the apoptotic ligands bind to death receptors,
the extrinsic pathway occurs through the activation of initiators
such as caspases 8 and 10, and the activation of executioners,
caspases 3, 6, and 7 resulting in DNA fragmentation (11–13).
The mitochondrion is an essential mediator of the intrinsic
pathway. The arrival of signals leading to changes in permeability
of the outer mitochondrial membrane is the cause of the releasing
intermitochondrial apoptotic molecules into the cytosol (14,15).
Cytochrome c, one of the best characterized pro-apoptotic
molecules, leads to activation of caspases via the formation of the
apoptosome (16). AIF, a
phylogenetically conserved mitochondrial flavoprotein, also has a
key role in apoptosis. When apoptosis is induced, AIF relocates
from the mitochondria to the nucleus where it mediates chromatin
condensation and large-scale DNA fragmentation (17,18).
The experiments reported in this research were
designed to investigate the cytotoxic effect on cancer cells and to
find the potential cell signaling pathways leading to cell death on
AA treated human lung adenocarcinoma A549 cells.
Materials and methods
Cell culture and reagents
A549 (human lung adenocarcinoma), HEK293 (human
embryonic kidney cell), HepG2 and SK-Hep1 (human hepatocarcinoma)
were purchased from the American Tissue Culture Collection
(Manassas, VA, USA). Cells were cultured in RPMI-1640 (A549), DMEM
(HEK293), EMEM (HepG2 and SK-Hep1) (HyClone Laboratories, Logan,
UT, USA) medium supplemented with 10% heat inactivated fetal bovine
serum (FBS; HyClone Laboratories), 100 U/ml penicillin and 10
μg/ml streptomycin (PAA Laboratories GmbH, Pasching,
Austria) in a humidified atmosphere containing 5% CO2 at
37°C. Except the cells were used in the cell viability, A549 cells
were investigated, after incubation in the FBS-free RPMI-1640
medium for 24 h, anacardic acid (AA), Calbiochem (San Diego, CA,
USA) was added to the medium. Pan-caspase inhibitor,
N-benzyloxycarbonyl-Val-Ala-Asp-fluoromethylketone (Z-VAD-fmk) was
purchased from Sigma-Aldrich (St. Louis, MO, USA).
Cell cytoxicity
The exponential phase of cells were seeded into the
wells of 96-well plates at an initial density of
0.5×105–1×105 cells in medium containing 10%
FBS per well of 100 μl. Following 24 h of incubation, AA,
dissolved in 100% dimethyl sulfoxide (DMSO; Sigma-Aldrich), was
added to the culture medium at various concentrations. Cells were
incubated for 24 h and 10 μl of EZ-Cytox Cell Viability
Assay Solution (WST-1™; Daeil Lab Service, Seoul, Korea) was added
and incubated for 4 h at 37°C. The intensity was measured at 450 nm
using an ELISA reader (Molecular Devices, Sunnyvale, CA, USA).
Western blot analysis
A549 cell lysates were prepared in a
Radioimmunoprecipitation assay buffer (RIPA, Cell Signaling
Technology, Danvers, MA, USA) and proteins were visualized using
enhanced chemiluminescent (ECL) detection solution (Pierce
Biotechnology, Rockford, IL, USA). Antibodies include anti-cleaved
caspase 3, anti-cleaved caspase 7, anti-cytochrome c,
anti-Bax, anti-Bad, anti-Bak, anti-Bcl-XL, anti-cleaved PARP,
anti-FoxO1, anti-FoxO3a, anti-FoxO4, anti-β-actin, secondary rabbit
and mouse antibodies conjugated with HRP were purchased from Cell
Signaling Technology. Anti-AIF, anti-Hsp70, anti-polyclonal Hsp27,
anti-Noxa, anti-STRAP, anti-Bim and secondary antibodies conjugated
with HRP rabbit goat were purchased from Santa Cruz Biotechnology
(Santa Cruz, CA, USA).
DAPI staining
4′,6-diamidino-2-phenylindole (DAPI) staining was
used for the morphological observation of apoptosome. A549 cells
were seeded in plates, and the cells were treated with or without
AA (3 μg/ml) for 24 h, then the cells were washed with PBS.
Two-to-three milliliters of diluted DAPI was added to the cells and
incubated for 15 min at 37°C. The cells were rinsed once with
methanol and the result was analyzed by fluorescence microscopy
using an Eclipse 50i microscope.
TEM
For observation of more detailed morphological
features, transmission electron microscopy (TEM) was used. Cultured
A549 cells were pre-fixed in pellets at 4°C with 2.5%
glutaraldehyde in 0.1 M phosphate buffer and then post-fixed with
1% osmium tetraoxide (OsO4) in 0.1 M phosphate buffer,
pH 7.4. After fixing, cells were embedded in Epon 812 using routine
procedures. Approximately 70 μm ultrasected specimens by
Ultracut Reichert-jung were stained using uranyl acetate and lead
citrate, and examined with Hitachi H600 TEM (Tokyo, Japan). All
reagents used in the TEM experiment were purchased from Electron
Microscopy Science (EMS).
FACS analysis
A549 cells were harvested by trypsinization and
fixed with ice-cold ethanol (70%) for 5 h at 4°C. Followed
resuspending with PBS containing RNase A (0.2 μg/ml) and
incubation for 1 h at 37°C. Cells were stained with propidium
iodide (40 μg/ml) for 30 min. The distribution of sub-G1 DNA
content was analyzed using the FACS Calibur apparatus
(Becton-Dickinson, Mountain View, CA, USA).
Genomic DNA extraction
For analyzing DNA fragmentation, genomic DNA was
extracted using DNeasy Blood and Tissue kit purchased from Qiagen
Inc. (Valencia, CA, USA) to the manufacturer’s protocol.
Results
Cytotoxicity of AA
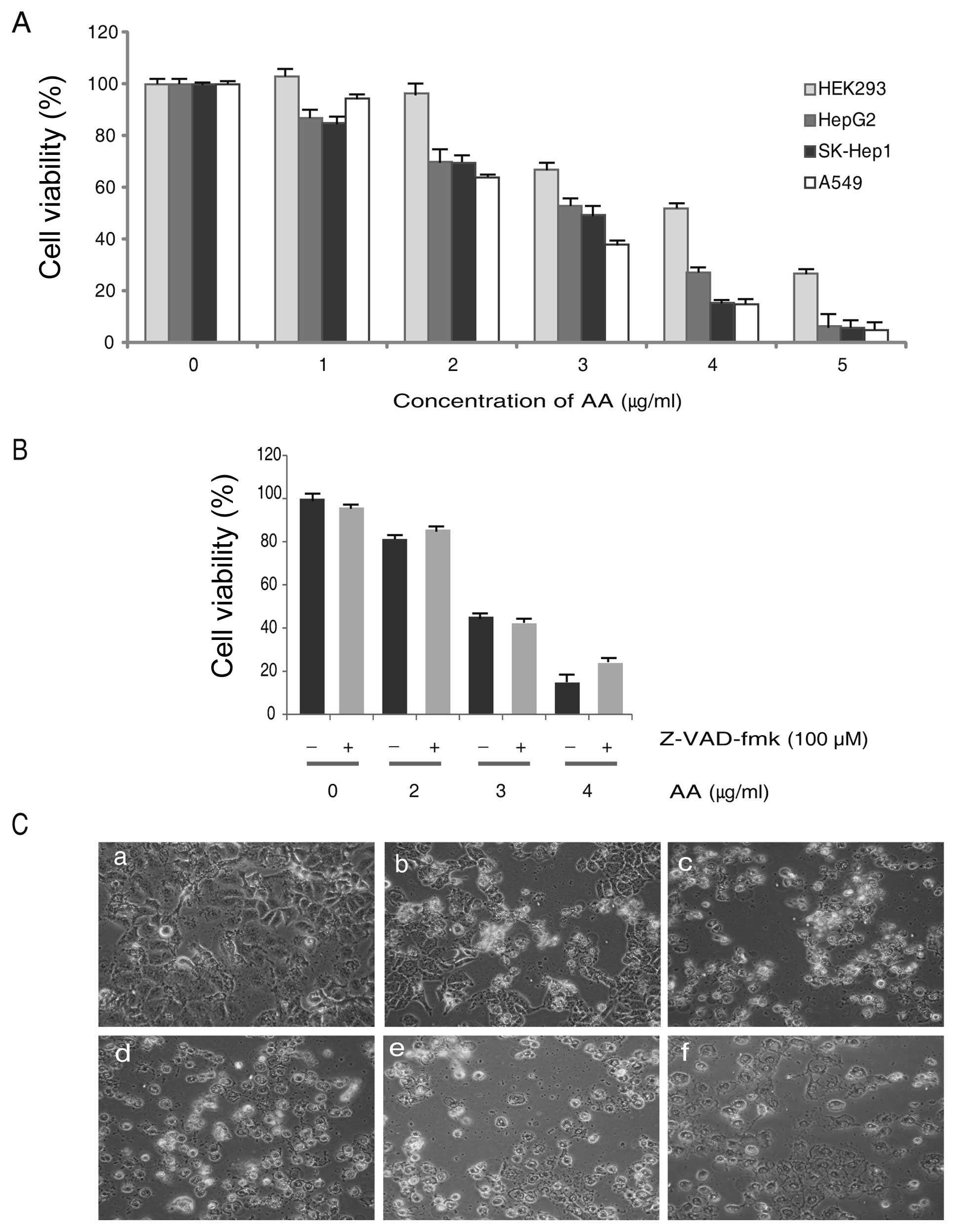
In order to investigate the cytotoxicity, we used a
normal cells (HEK293), and lung (A549) and liver (HepG2, SK-Hep1)
cancer cells were treated with AA in a dose-dependent manner. After
24 h of exposure, the results of cell viability assay showed that
AA inhibits the proliferation of all cells used. Although AA
inhibited cellular proliferation of HEK cells, the cytotoxicity was
less than cancer cells (Fig. 2A).
To study the effect of the caspase inhibitor, cells were pretreated
for 2 h with the cell-permeable and irreversible pan-caspase
inhibitor Z-VAD-fmk (100 μM) and then exposed to AA for 24 h
in the continued presence of Z-VAD-fmk. Viability was then assessed
by MTT assay using WST-1™ and the result showed
Z-VAD-fmk did not inhibit the cell viability in AA treated A549
cells (Fig. 2B). Considering our
interest in lung cancer, we used A549 cells for further research
and the IC50 value of AA treated A549 cells was
2.75±0.25 μg/ml. Therefore, we used 3.0 μg/ml of AA
for investigating the molecular mechanism of cell death in A549.
Cellular morphology of AA treated A549 cells were shown under a
microscope and cytotoxicity was increased dose-dependently
(Fig. 2C).
Induction of apoptosis in AA treated A549
cells
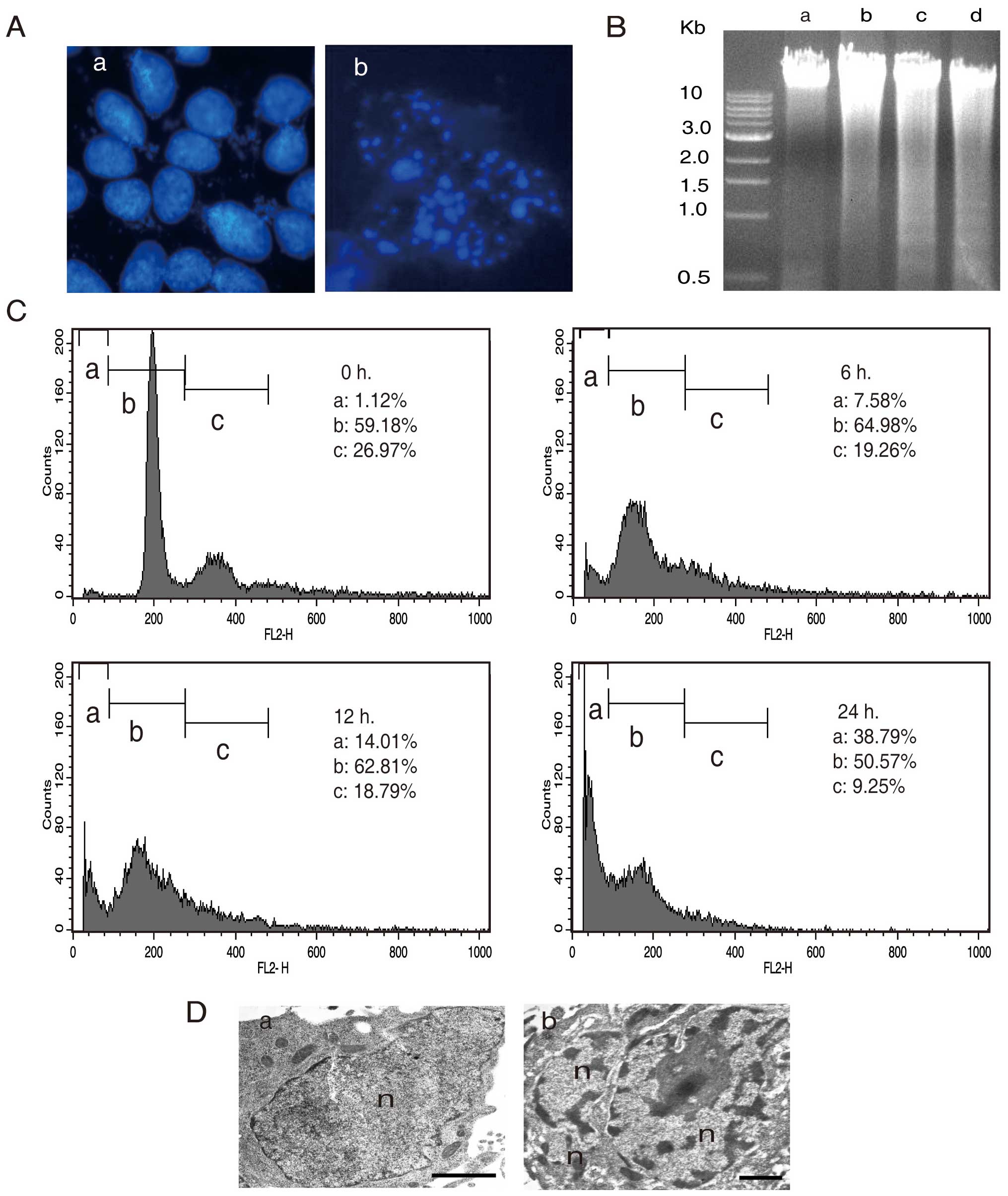
In order to further elucidate the nature of AA
induced cell death in A549, we investigated the nuclear morphology
of AA treated cells by using 4′,6-diamidino-2-phenylindole (DAPI)
staining. The results showed totally different patterns between the
AA-treated and -untreated cells. AA treated cells exhibited
formations of apoptosome (Fig.
3A–b), indicating that AA induces apoptosis in A549 cells. For
further evidence of apoptosis, we analyzed the fragmentation of
chromosomal DNA by using agarose gel electrophoresis (Fig. 3B). The fragmentation of chromosomal
DNA by AA increased to the exposure time (Fig. 3B-b, c and d), while it was not
observed in the untreated cells (Fig.
3B-a). In addition, to study the cell death induced by AA, we
quantified the sub-G1 DNA content by fluorescence activated cell
sorting (FACS) analysis. The sub-G1 genomic DNA content was
gradually increased after 6, 12 and 24 h to 7.58, 14.01 and 38.79%,
respectively (Fig. 3C). TEM also
showed apoptotic features such as chromatin condensation and
nuclear fragmentation (Fig. 3D-b).
These results support the view that AA plays a major role in
apoptosis induction in A549 cells.
Effect of AA on the intrinsic pathway of
apoptosis in A549 cells
To determine the apoptosis signaling pathway induced
by AA, we analyzed the expression of several genes using western
blot analysis. Cleaved caspase 3, cleaved caspase 7 and cytochrome
c, known as the executioners of the intrinsic pathway,
showed gradual increase in a time dependent manner on AA treated
A549 cells. Caspase 9 was also analyzed (Fig. 4A). In addition, the expression of
pro- and anti-apoptotic Bcl-2 family was determined. Pro-apoptotic
members of the Bcl-2 family, Bim, Bad, Bak and Noxa increased, and
the anti-apoptotic member Bcl-xL, decreased (Fig. 4B). The expression of Bak and Bad
showed gradual increase time-dependently with AA-treatment and the
expression of Bax and Noxa showed further increased level at 12 h
after AA exposure. Bim has three isoforms (BimS, BimL and BimEL)
with different intrinsic toxicities and promotes apoptosis
(19). The cleaved form of Bim
appeared at 6 h and the expression of Bcl-xl decreased gradually to
18 h after AA exposure.
Effects of AA on AIF related pathway
Because the inhibition of the pan-caspase inhibitor,
Z-VAD-fmk, failed to prevent cell death, we analyzed the
possibility of a caspase-independent pathway by apoptogenic
molecules, apoptosis-inducing factor (AIF). Poly (ADP-ribose)
polymerase-1 (PARP-1), the mediator of AIF release, was also
investigated. The expression of AIF showed gradual increase, as did
the cleaved PARP-1 after 18 h (Fig.
4C).
Decrease of the chaperone genes
We examined the expression of several genes that
encode proteins known as molecular chaperones by assisting the
correct folding of nascent and stress-accumulated misfolded
proteins (20). We investigated
the expression of Hsp70 and Strap, stress-responsive activator of
p300 and the results showed downregulation of Hsp70 and Strap by AA
(Fig. 4D).
Activation of forkhead transcription
factors by AA
Members of the non-phosphorylated mammalian forkhead
transcription factors (FoxOs) are involved in regulating the
expression of genes involved in apoptosis. FoxO1, FoxO4 and FoxO3a
are known as mammalian forkhead transcription factors that trigger
the up-regulation of proteins such as Bim and NOXA (21). The expression of FoxO1, FoxO4 and
FoxO3a in AA treated A549 cells increased with the optimal
expression time being slightly different depending on the subfamily
(Fig. 4E).
Discussion
The current study focused on the findings of a cell
signaling pathway leading to death in A549 cells by AA. Our
research provides strong evidence to support the view that AA
induces apoptosis in a caspase-independent manner with no
inhibition of cytotoxicity by pan-caspase inhibitor, Z-VAD-fmk, in
A549 cells. In addition to the morphological features by TEM and
microscopy and FACS analysis, the analysis of gene expression by
western blotting demonstrates the induction of apoptosis by AA.
Previous research reported that the release of
cytochrome c from mitochondria is an early event during
apoptosis, and pro-apoptotic Bcl-2 family members induce the
release of cytochrome c and anti-apoptotic Bcl-2 proteins
inhibit the release of cytochrome c(22,23).
The balance between the pro-apoptotic (Bid, Bad, Bim, Bax, Bak and
Noxa) and anti-apoptotic (Bcl-2, Bcl-xL, A1 and Bcl-w) Bcl-2
protein families is an important factor contributing to cytochrome
c release, and in determining cell fate (24,25).
Following cytochrome c release, caspases are activated and
the cell undergoes apoptosis through the formation of apoptosomes,
Apaf-1/caspase 9 complex (25).
Bax and Bak are also known to promote apoptosis by modulating ER
and mitochondrial Ca2+ stores (26). We are studying on the possibility
of apoptosis by ER stress. The pro-apoptotic BH3-only protein Bim,
induces cell death by binding the anti-apoptotic Bcl-2 family
protein and Noxa is known as a mediator of p53-induced apoptosis
(27). The expression of Bim and
Noxa is regulated by the transcription factor of Forkhead (FKHR) in
the rhabdomyosarcoma family including FoxO (21). FoxO transcription factors modulate
the expression of genes involved in apoptosis, cell cycle, cell
differentiation and other cellular functions (28). In this study, AA induced the
expression of the pro-apoptotic Bcl-2 family proteins, Bim, Bad,
Bak, Bax and Noxa and cytochrome c, and reduced the
expression of the anti-apoptotic Bcl-2 family protein, Bcl-XL, in
A549 cells. The expression of FoxOs increased on AA treated A549
cells. These results showed the possibility that the increase of
pro-apoptotic BH3-only protein, Bim and Noxa by FoxOs and decrease
of anti-apoptotic protein, Bcl-XL, induce the outer membrane
disruption of mitochondria in AA treated A549. The disruption of
mitochondria membrane may induce the release of proapoptotic
mediators such as AIF and cytochrome c from
mitochondria.
Mitochondrial intermembrane flavoprotein AIF was
originally characterized as a cell death mediator (17) and AIF has a potential role as a
prognostic marker and a target for radiochemotherapeutic
intervention in CH27 human lung carcinoma cells (29). AIF translocate from mitochondria to
the cytosol and then move to the nucleus to cause peripheral
chromatin condensation and large scale fragmentation of DNA
(17,18,30).
AIF is an important mitochondrial protein involved in
caspase-dependent and -independent pathways (31). One well-known mechanism to release
AIF from mitochondria is by the activation of poly (ADP-ribose)
polymerase-1 (PARP-1) which is a key molecule in AIF-induced cell
death and mediates the release and translocation of AIF (32).
While PARP-1 is involved in the release of AIF from
mitochondria (32), heat shock
protein 70 (Hsp70) negatively regulates the AIF function by
inhibiting translocation to the nucleus (33). Furthermore, Hsp70 inhibits
apoptosis through the inhibition of a downstream pathway of
cytochrome c release, upstream of caspase 3 activation and
Apaf-1 apoptosome formation (33–35).
Hsps also block caspase-dependent and -independent apoptosis in
Jurkat T cells (36) and a
depletion of Hsp70 produces apoptosis-like death in various tumor
cell types, including human oral carcinoma cells (37). Heat shock proteins are important
prognostic factors in malignant diseases due to their abundant
expression in many cancer cells. Strap is known as the Hsp70
transcription cofactor (38).
Current results show the possibility that the decrease of Hsp70 and
increase of proapoptotic protein AIF and PARP-1 promote chromatin
condensation and DNA fragmentation and induce apoptosis in AA
treated A549 cells.
In conclusion, for the first time our research
suggests that AA induces caspase-independent apoptosis, that the
pan-caspase inhibitor z-VAD-fmk does not inhibit cell death, and
the activation of a mitochondrial-mediated cell death signaling
pathway have a major role in apoptotic cell death in A549 cells.
The release of the intermembrane proapoptotic factors from the
mitochondria by imbalances between pro- and anti-apoptotic Bcl-2
family members, decrease of Hsp70 and increase of AIF could have a
key role in apoptosis in AA treated A549 cells. Based on our
results, we suggest (Fig. 5) the
possible signaling pathway leading to apoptosis in AA treated
A549.
Acknowledgements
This study was supported by a grant
from the Next-Generation BioGreen 21 Program (SSAC, grant no.
PJ008171), Rural Development Administration, Republic of Korea.
References
|
1
|
Kubo I, Kinst-Hori I and Yokokawa Y:
Tyrosinase inhibitors from Anacardium occidentale fruits. J Nat
Prod. 57:545–551. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Murata M, Irie J and Homma S: Inhibition
of lipid synthesis of bacteria, yeast and animal cells by anacardic
acids, glycerol-3-phosphate dehydrogenase inhibitors from Ginkgo.
Lebensm Wiss Technol. 30:458–463. 1997. View Article : Google Scholar
|
|
3
|
Grazzini R, Hesk D, Heininger E, et al:
Inhibition of lipoxygenase and prostaglandin endoperoxide synthase
by anacardic acids. Biochem Biophys Res Commun. 176:775–780. 1991.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kishore AH, Vedamurthy BM, Mantelingu K,
et al: Specific small-molecule activator of aurora kinase A induces
autophosphorylation in a cell-free system. J Med Chem. 28:792–797.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Sun Y, Jiang X, Chen S and Price BD:
Inhibition of histone acetyltransferase activity by anacardic acid
sensitizes tumor cells to ionizing radiation. FEBS Lett.
580:4353–4356. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Sung B, Pandey MK, Ahn KS, Yi T,
Chaturvedi MM, Liu M and Aggarwal BB: Anacardic acid (6-nonadecyl
salicylic acid), an inhibitor of histone acetyltransferase,
suppresses expression of nuclear factor-kappaB-regulated gene
products involved in cell survival, proliferation, invasion, and
inflammation through inhibition of the inhibitory subunit of
nuclear factor-kappaB alpha kinase, leading to potentiation of
apoptosis. Blood. 111:4880–4891. 2008.
|
|
7
|
Choi JG, Jeong SI, Ku CS, Sathishkumar M,
Lee JJ, Mun SP and Kim SM: Antibacterial activity of hydroxyalkenyl
salicylic acids from sarcotesta of Ginkgo biloba against
vancomycin-resistant Enterococcus. Fitoterapia. 80:18–20.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Sukumari-Ramesh S, Singh N, Jensen MA,
Dhandapani KM and Vender JR: Anacardic acid induces
caspase-independent apoptosis and radiosensitizes pituitary adenoma
cells. J Neurosurg. 114:1681–1690. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kerr JF, Wyllie AH and Currie AR:
Apoptosis: a basic biological phenomenon with wide-ranging
implications in tissue kinetics. Br J Cancer. 26:239–257. 1972.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kroemer G, Galluzzi L, Vandenabeele P, et
al: Classification of cell death: recommendations of the
Nomenclature Committee on Cell Death 2009. Cell Death Differ.
16:3–11. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Millan A and Huerta S: Apoptosis-inducing
factor and colon cancer. J Surg Res. 151:163–170. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Slee EA, Adrain C and Martin SJ:
Executioner caspase-3, -6, and -7 perform distinct, non-redundant
roles during the demolition phase of apoptosis. J Biol Chem.
276:7320–7326. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Walczak H and Krammer PH: The CD95
(APO-1/Fas) and the TRAIL (APO-2L) apoptosis systems. Exp Cell Res.
256:58–66. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zamzami N, Susin SA, Marchetti P, Hirsch
T, Gómez-Monterrey I, Castedo M and Kroemer G: Mitochondrial
control of nuclear apoptosis. J Exp Med. 183:1533–1544. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Schultz DR and Harrington WJ Jr:
Apoptosis: programmed cell death at a molecular level. Semin
Arthritis Rheum. 32:345–369. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Adrain C and Martin SJ: The mitochondrial
apoptosome: a killer unleashed by the cytochrome seas. Trends
Biochem Sci. 26:390–397. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Susin SA, Lorenzo HK, Zamzami N, et al:
Molecular characterization of mitochondrial apoptosis-inducing
factor. Nature. 397:441–446. 1999. View
Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ye H, Cande C, Stephanou NC, et al: DNA
binding is required for the apoptogenic action of apoptosis
inducing factor. Nat Struct Biol. 9:680–684. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
O’Connor L, Strasser A, O’Reilly LA,
Hausmann G, Adams JM, Cory S and Huang DC: Bim: a novel member of
the Bcl-2 family that promotes apoptosis. EMBO J. 17:384–395.
1998.
|
|
20
|
Beckmann RP, Mizzen LE and Welch WJ:
Interaction of Hsp70 with newly synthesized proteins: implications
for protein folding and assembly. Science. 248:850–854. 1990.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Obexer P, Geiger K, Ambros PF, Meister B
and Ausserlechner MJ: FKHRL1-mediated expression of Noxa and Bim
induces apoptosis via the mitochondria in neuroblastoma cells. Cell
Death Differ. 14:534–547. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kluck RM, Bossy-Wetzel E, Green DR and
Newmeyer DD: The release of cytochrome c from mitochondria: a
primary site for Bcl-2 regulation of apoptosis. Science.
275:1132–1136. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Yang J, Liu X, Bhalla K, et al: Prevention
of apoptosis by Bcl-2: release of cytochrome c from mitochondria
blocked. Science. 275:1129–1132. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Green DR and Amarante-Mendes GP: The point
of no return: mitochondria, caspases, and the commitment to cell
death. Results Probl Cell Differ. 24:45–61. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Li P, Nijhawan D, Budihardjo I,
Srinivasula SM, Ahmad M, Alnemri ES and Wang X: Cytochrome c and
dATP-dependent formation of Apaf-1/caspase-9 complex initiates an
apoptotic protease cascade. Cell. 91:479–489. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Nutt LK, Pataer A, Pahler J, Fang B, Roth
J, McConkey DJ and Swisher SG: Bax and Bak promote apoptosis by
modulating endoplasmic reticular and mitochondrial Ca2+
stores. J Biol Chem. 277:9219–9225. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Oda E, Ohki R, Murasawa H, et al: Noxa, a
BH3-only member of the Bcl-2 family and candidate mediator of
p53-induced apoptosis. Science. 288:1053–1058. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Huang H and Tindall DJ: Dynamic FoxO
transcription factors. J Cell Sci. 120:2479–2487. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Leung HW, Wu CH, Lin CH and Lee HZ:
Luteolin induced DNA damage leading to human lung squamous
carcinoma CH27 cell apoptosis. Eur J Pharmacol. 508:77–83. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Zhang X, Chen J, Graham SH, et al:
Intranuclear localization of apoptosis-inducing factor (AIF) and
large scale DNA fragmentation after traumatic brain injury in rats
and in neuronal cultures exposed to peroxynitrite. J Neurochem.
82:181–191. 2002. View Article : Google Scholar
|
|
31
|
Cregan SP, Dawson VL and Slack RS: Role of
AIF in caspase-dependent and caspase-independent cell death.
Oncogene. 23:2785–2796. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Yu SW, Wang H, Poitras MF, et al:
Mediation of poly (ADP-ribose) polymerase-1-dependent cell death by
apoptosis-inducing factor. Science. 297:259–263. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Gurbuxani S, Schmitt E, Cande C, et al:
Heat shock protein 70 binding inhibits the nuclear import of
apoptosis-inducing factor. Oncogene. 22:6669–6678. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Li CY, Lee JS, Ko YG, Kim JI and Seo JS:
Heat shock protein 70 inhibits apoptosis downstream of cytochrome c
release and upstream of caspase-3 activation. J Biol Chem.
275:25665–25671. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Saleh A, Srinivasula SM, Balkir L, Robbins
PD and Alnemri ES: Negative regulation of the Apaf-1 apoptosome by
Hsp70. Nat Cell Biol. 2:476–483. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
36
|
Creagh EM, Carmody RJ and Cotter TG: Heat
shock protein 70 inhibits caspase-dependent and -independent
apoptosis in Jurkat T cells. Exp Cell Res. 257:58–66. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Nylandsted J, Wick W, Hirt UA, et al:
Eradication of glioblastoma, and breast and colon carcinoma
xenografts by Hsp70 depletion. Cancer Res. 62:7139–7142.
2002.PubMed/NCBI
|
|
38
|
Xu D, Zalmas LP and La Thangue NB: A
transcription cofactor required for the heat-shock response. EMBO
Rep. 9:662–669. 2008. View Article : Google Scholar : PubMed/NCBI
|