Introduction
Hepatocellular carcinoma (HCC) is one of the most
lethal malignancies worldwide (1,2). HCC
is caused mainly by chronic liver inflammation due to hepatitis B
virus, hepatitis C virus (HCV) or alcohol abuse (1). Despite curative resection and recent
advances in treatments, the clinical course of HCC is variable and
many patients suffer recurrence after surgery. Poor prognoses in
cases of HCC can be explained largely by the high rate of
intrahepatic recurrence (IHR), which results from metastatic spread
of cancer cells (3).
Previously, we identified a gene, inhibitor of
DNA binding 2 (ID2), that is significantly downregulated
in HCCs, especially in advanced HCCs, relative to surrounding liver
tissues (4,5). Moreover, we found that ID2 is
a portal vein invasion-related gene in HCV-related HCC (6) and that ID2 negatively
regulates the invasive potential of cancer cells (7). Therefore, HCC patients with low
ID2 expression have poor prognoses (7). ID2 belongs to a protein family
that comprises ID1 to ID4; these proteins have a helix-loop-helix
structure and form heterodimers with basic helix-loop-helix
transcription factors to act as dominant-negative inhibitors of
transcription (8–10). IDs are involved in proliferation
processes, differentiation, development, senescence and
angiogenesis (11–15), and are linked to various malignant
tumors (16–31).
In this study, we searched for antitumor drugs that
are effective against cells with low ID2 expression because
such antitumor drugs might be useful in the treatment of patients
who have HCC and a poor prognosis. We found that alteration of
ID2 expression affected the susceptibility of cells to
histone deacetylase (HDAC) inhibitors and that HDAC inhibitors were
the only antitumor drugs tested for which alteration of ID2
expression had an effect. HDAC inhibitors have emerged as a new
class of antitumor agents (32–34).
HDAC inhibitors can cause multiple epigenetic changes in aberrant
cells. Treatment with HDAC inhibitors most frequently induces
apoptosis (35–37). Although their precise mode of
action remains uncertain, HDAC inhibitors can modulate the cell
cycle, apoptosis, angiogenesis, invasion and metastases (32,33,38–40).
Here, we aimed to investigate how and whether ID2 affected
the anti-tumor activity of sodium butyrate (NaB), an HDAC
inhibitor.
Materials and methods
Hepatoma cell lines
Human hepatoma-derived cell lines, HLE and HuH-7,
were purchased from the Health Science Research Resources Bank
(Osaka, Japan). Cells were cultured in DMEM (Nissui Pharmaceutical,
Tokyo, Japan) containing 10% heat-inactivated fetal bovine serum
(Life Technologies, Tokyo, Japan) and supplemented with penicillin
(100 U/ml), streptomycin (100 μg/ml) and sodium bicarbonate
(1.5 g/l) at 37°C in 5% CO2 in air. As in our previous
report (7), ID2-knockdown
and ID2-overexpression were accomplished by transfection of
HuH-7 and HLE cells with ID2-specific small interfering RNAs
(siRNAs) or an ID2-expression plasmid vector, respectively.
HuH-7 and HLE cells transfected with control siRNA or empty
pcDNA3.1(-) plasmid DNA were used as the respective control.
Administration of histone deacetylase
inhibitors
NaB (Sigma-Aldrich, Tokyo, Japan), sodium
4-phenyl-butyrate (NaPB; Funakoshi, Tokyo, Japan), tricostatin A
(TSA; Sigma-Aldrich), suberoylanilide hydroxamic acid (SAHA; Cosmo
Bio, Tokyo, Japan), MS-275 (Sigma-Aldrich), apicidin
(Sigma-Aldrich) and HC-toxin (Sigma-Aldrich) were each used as an
HDAC inhibitor in this study. HDAC inhibitors were added to
cultures 24 h after cells had been seeded; cultures were then
further incubated with the inhibitor for defined periods at 37°C in
5% CO2 in air.
MTS assay
The CellTiter 96 AQueous One Solution Cell
Proliferation Assay (Promega, Tokyo, Japan) which includes
3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium,
inner salt (MTS) was used according to the manufacturer’s
instructions to evaluate cell survival. Cells (3×103)
were seeded into the wells of 96-well plates and cultivated. At the
appropriate time, MTS was added to the cells, which were then
incubated for 2 additional hours at 37°C. The optical density of
the culture medium at 492 and 650 nm were then measured by using an
EnVision plate reader (PerkinElmer, Waltham, MA). Triplicate wells
were analyzed in each assay.
Annexin V staining
The Annexin V-FLUOS Staining Kit (Roche Diagnostics,
Tokyo, Japan), which includes Annexin V/propidium iodide (PI), was
used according to the manufacturer’s instructions to detect
apoptosis. The cultures were observed under a fluorescent
microscope (IX71; Olympus, Tokyo, Japan). Simultaneously,
Hoechst33342-positive cells were counted as total number of cells
in cultures; Hoechst33342 was purchased from Sigma-Aldrich.
Semiquantitative real-time RT-PCR
Semiquantitative real-time RT-PCR (semi-qRT-PCR) was
performed as described previously (7,41)
with minor modifications. Real-time PCR amplification was performed
using the LightCycler 480 Probe Master and Universal ProbeLibrary
Probes in a LightCycler System Version 3 (all from Roche
Diagnostics). Primers and probes that were used are listed in
Table I. Amplification was
performed according to a two-step cycle procedure consisting of 45
cycles of denaturation at 95°C for 10 sec and annealing/elongation
at 60°C for 30 sec. We used the Δ/Δ threshold cycle method to
semiquantitatively measure mRNA levels;
glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and
β-actin (ACTB) were both used as reference genes. All
values of mRNA levels are expressed relative to control.
 | Table IThe primers and hydrolysis probes
used in this study. |
Table I
The primers and hydrolysis probes
used in this study.
| Primers and
probes | Sequence
(5′→3′) |
|---|
| ID2 | |
| 5′-primer |
ATATCAGCATCCTGTCCTTGC |
| 3′-primer |
AAAGAAATCATGAACACCGCTTA |
| Hydrolysis
probe | UPL Probe
#5a |
| BCL2 | |
| 5′-primer |
TTGACAGAGGATCATGCTGTACTT |
| 3′-primer |
ATCTTTATTTCATGAGGCACGTT |
| Hydrolysis
probe | UPL Probe
#6a |
| BCL2L1
(BCL-XL) | |
| 5′-primer |
GCTGAGTTACCGGCATCC |
| 3′-primer |
AGATTCTGAAGGGAGAGAAAGAGA |
| Hydrolysis
probe | UPL Probe
#83a |
| BAX | |
| 5′-primer |
ATGTTTTCTGACGGCAACTTC |
| 3′-primer |
ATCAGTTCCGGCACCTTG |
| Hydrolysis
probe | UPL Probe
#57a |
| CDKN1A
(P21) | |
| 5′-primer |
TCACTGTCTTGTACCCTTGTGC |
| 3′-primer |
GGCGTTTGGAGTGGTAGAAA |
| Hydrolysis
probe | UPL Probe
#32a |
| GAPDH | |
| 5′-primer |
AGCCACATCGCTCAGACAC |
| 3′-primer |
GCCCAATACGACCAAATCC |
| Hydrolysis
probe | UPL Probe
#60a |
| ACTB | |
| 5′-primer |
CCAACCGCGAGAAGATGA |
| 3′-primer |
CCAGAGGCGTACAGGGATAG |
| Hydrolysis
probe | UPL Probe
#64a |
Statistical analysis
Data are presented as mean ± standard deviation.
Dunnett’s test for multiple comparisons was used to evaluate the
differences between three groups. Calculations were performed using
SPSS Statics 17.0 software (IBM, Tokyo, Japan). P<0.05 was
considered statistically significant.
Results
Susceptibility to histone deacetylase
inhibitors in HCC-derived cell lines in which ID2 was knocked down
or overexpressed
We used the MTS assay and previously established
HCC-derived cell lines in which ID2 expression was
suppressed or enhanced (7) to
examine the susceptibility of HCC cells to antitumor drugs. Among
the tested antitumor drugs, the antitumor activity of an HDAC
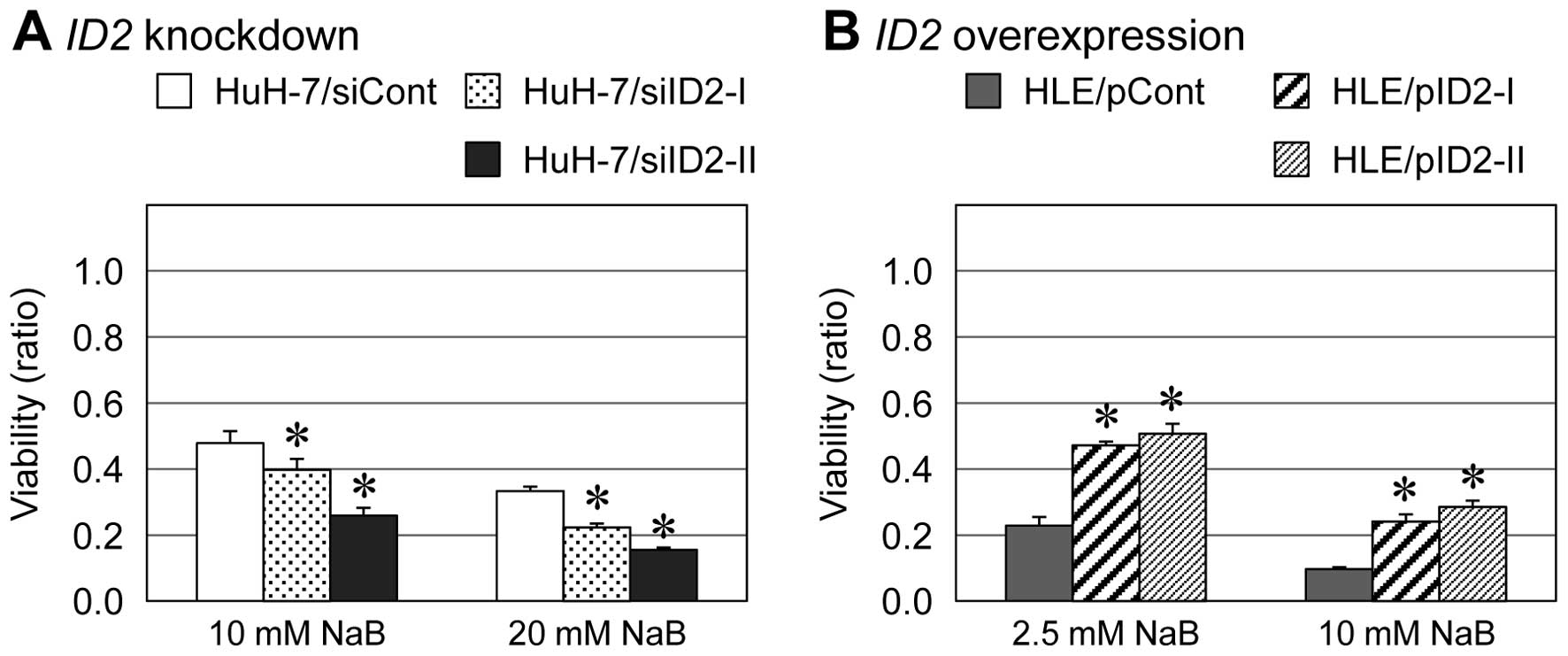
inhibitor, NaB, was increased in ID2 knockdown cells and
decreased in ID2-overexpressing cells (Fig. 1). Similar results were obtained
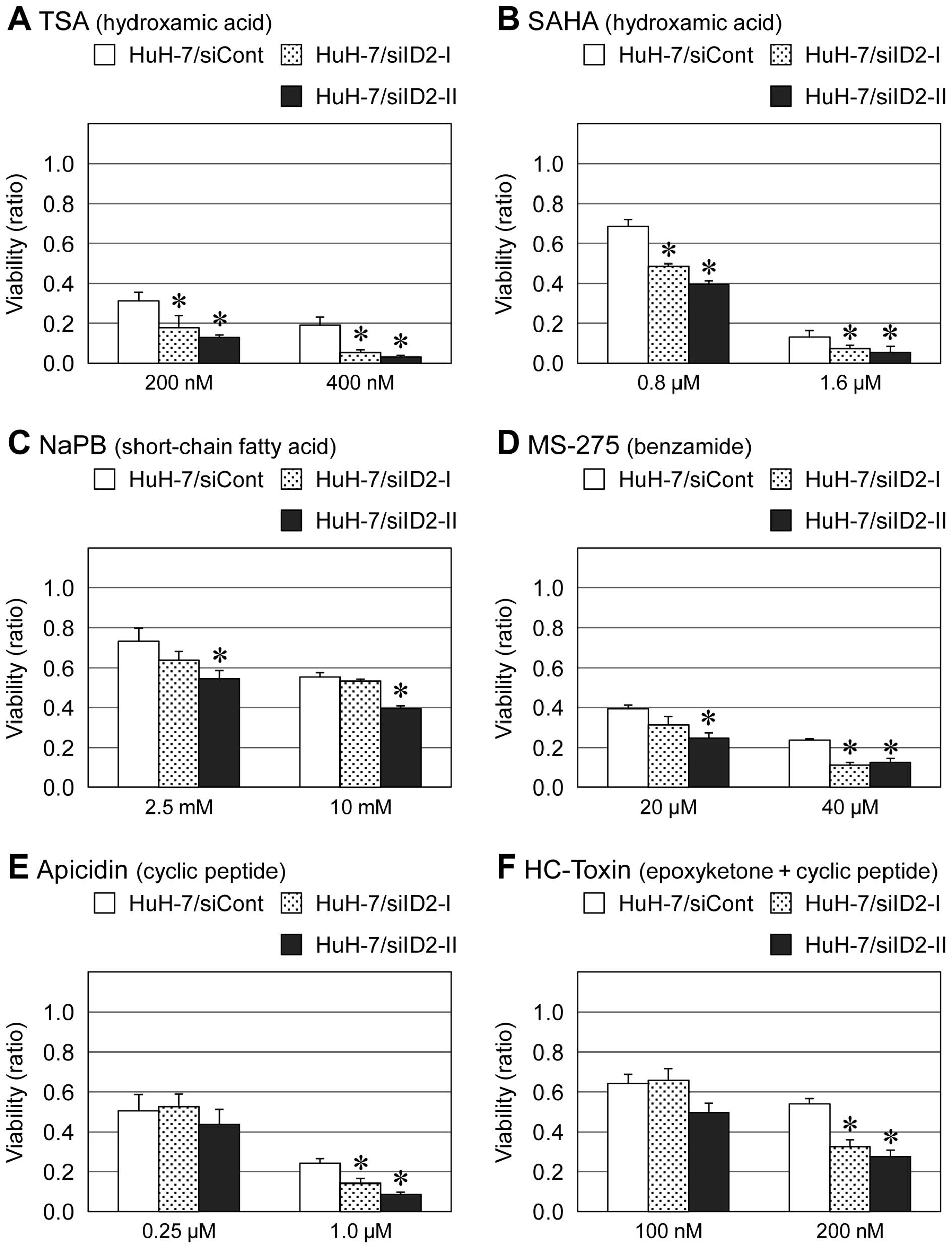
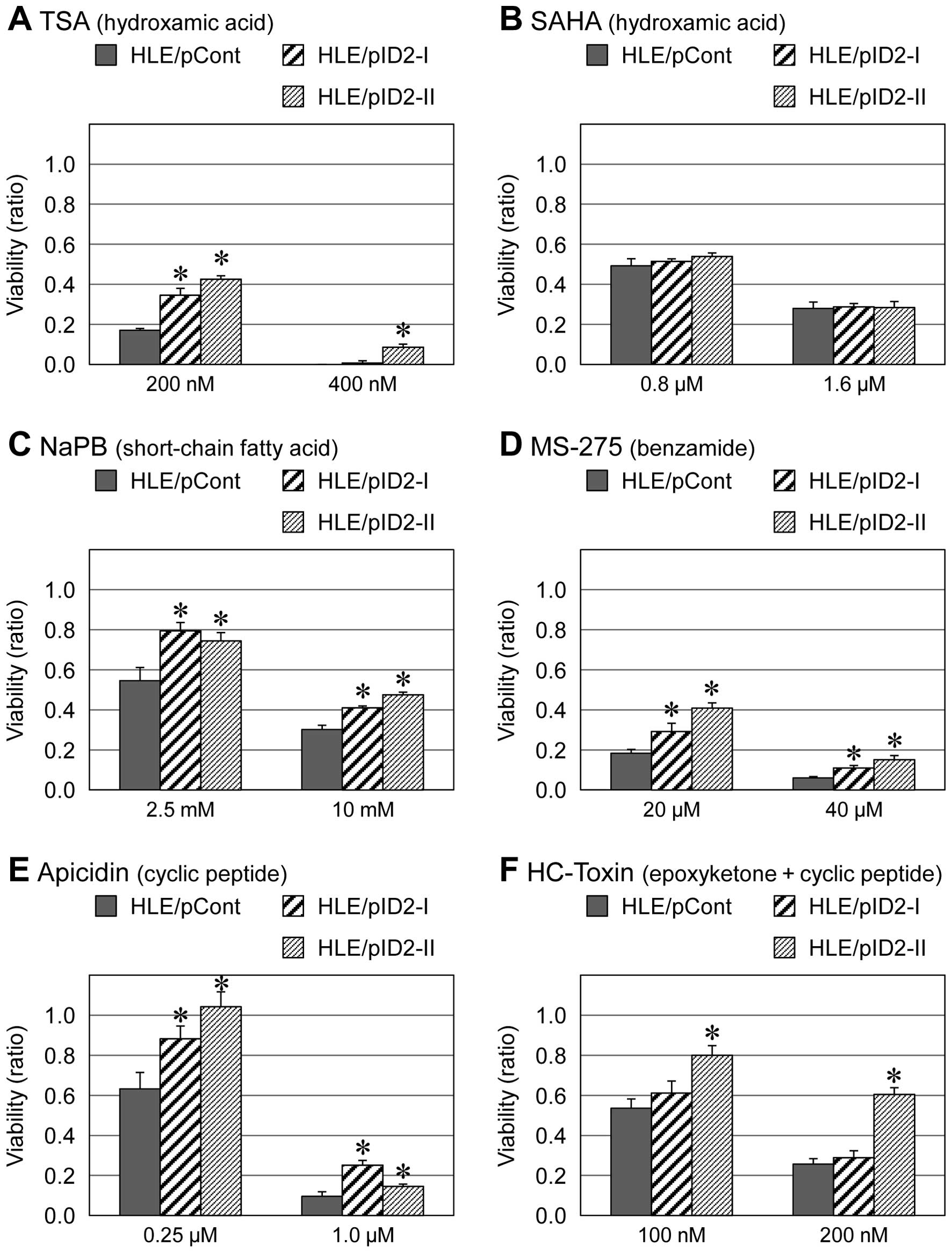
with other HDAC inhibitors including TSA, SAHA, PBA, MS-275,
apicidin and HC-toxin (Figs. 2 and
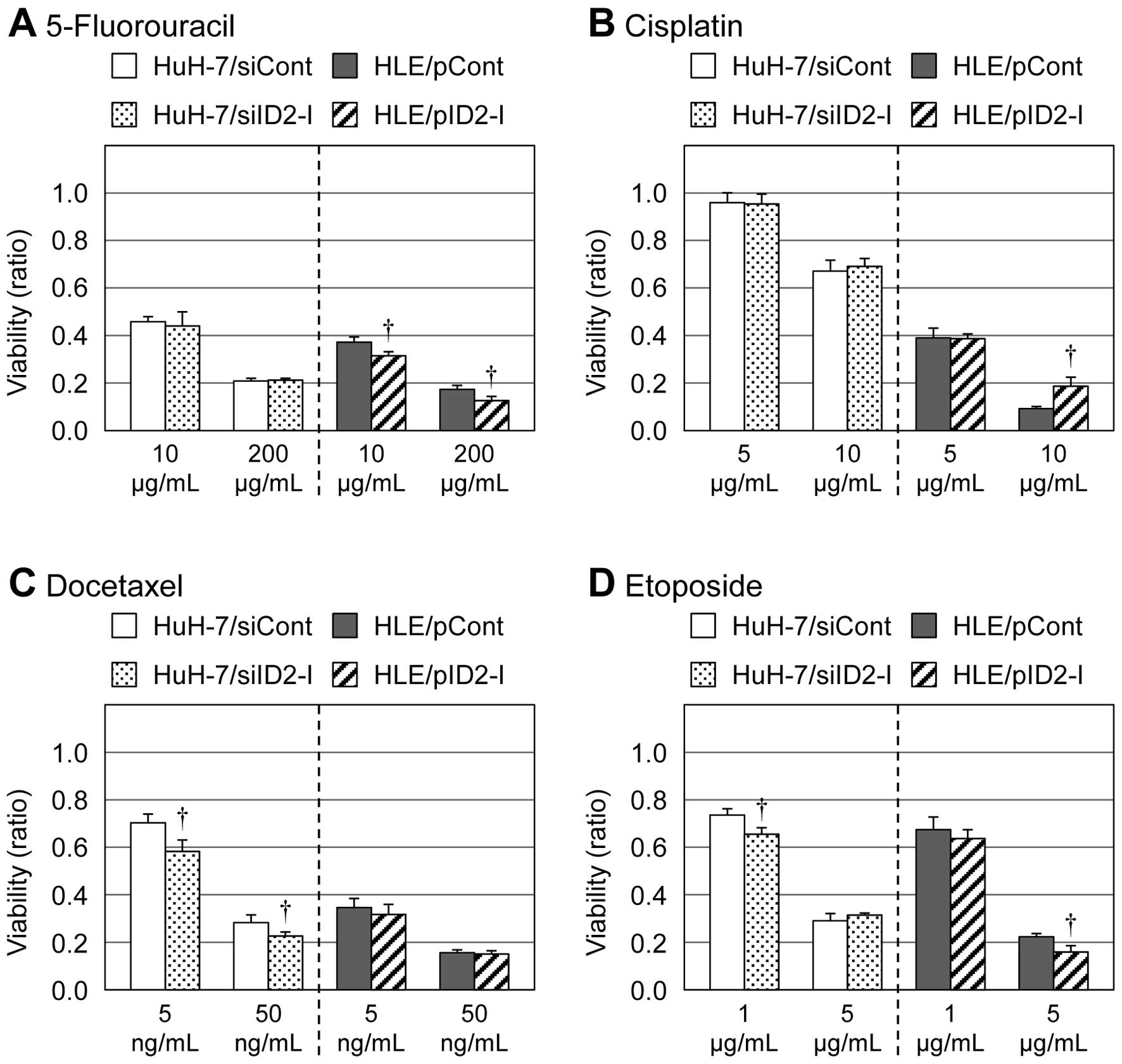
3). However, for other types of
antitumor agents than HDAC inhibitors (e.g., 5-fluorouracil,
cisplatin, docetaxel and etoposide), such results were not observed
(Fig. 4).
Influence of ID2 on NaB-induced
apoptosis
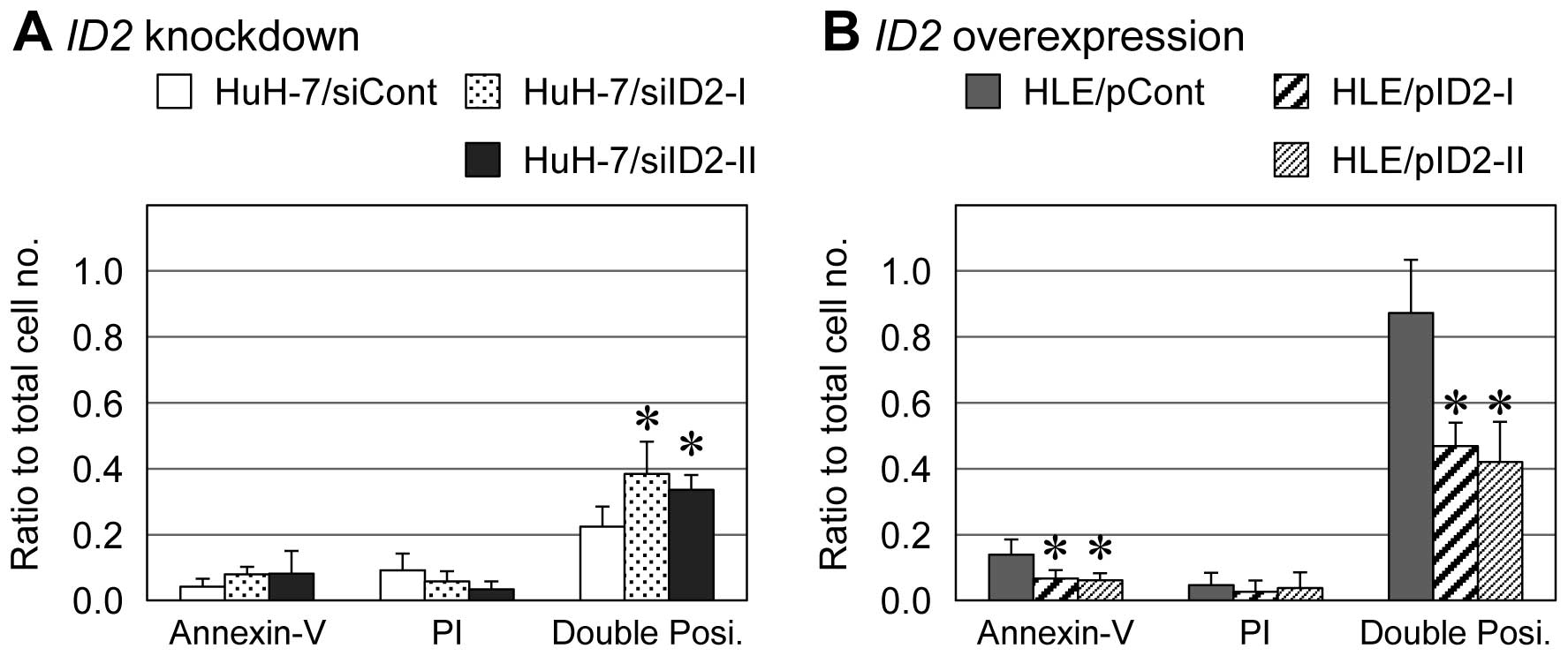
In HLE derivatives treated with 20 mM NaB for 72 h,
the number of cells positive for both Annexin V and PI (late
apoptosis) was significantly lower among ID2-overexpressing
cells than empty-vector control cells (approximately 44 vs. 87%,
respectively) (Fig. 5B). For HuH-7
derivatives treated with 20 mM NaB for 72 h, the percentage of
cells positive for both Annexin V and PI (late apoptosis) was 34%
among ID2-knockdown cells and 25% among siRNA-transfected
control cells (Fig. 5A). In both
HLE and HuH-7 derivatives, Annexin V single-positive cells (early
apoptosis) showed same tendency with the Annexin V and PI
double-positive cells, although Annexin V single-positive cells
were less than 10% of each cell type.
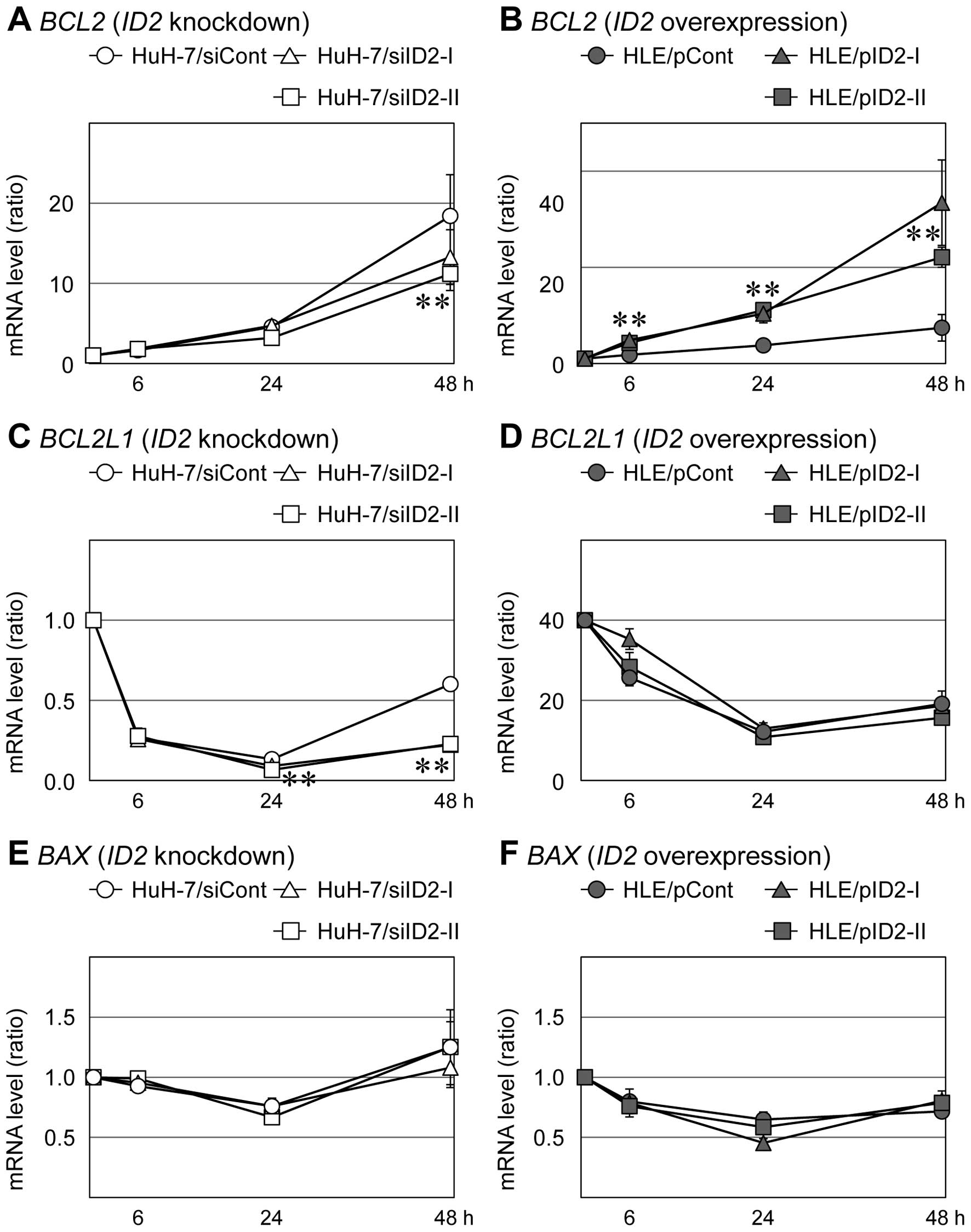
We examined expression of apoptosis-related genes in
HLE and HuH-7 cells that had been treated with NaB. Following
addition of 20 mM NaB, about half of the HLE cells had died within
24 h and about half of the HuH-7 cells had died within 48 h.
Treatment with NaB induce expression of BCL2 mRNA (an
anti-apoptotic mRNA) in HuH-7 cells transfected with control siRNA
and in HLE cells transfected with empty vector; however, this
NaB-dependent induction was suppressed in HuH-7 cells transfected
with ID2-specific siRNAs and enhanced in HLE cells that
overexpressed ID2 (Fig. 6A and
B). Levels of another anti-apoptotic mRNA, BCL2L1
(BCL-XL), decreased immediately after addition of NaB in
control HuH-7 cells and in ID2-knockdown HuH-7 cells; 48 h
after NaB addition, BCL2L1 levels had partially recovered in
control cells, but they had not recovered in ID2-knockdown
HuH-7 cells (Fig. 6C). HLE cells
that overexpressed ID2 and control HLE cells did not differ
significantly in BCL2L1 mRNA levels (Fig. 6D). The mRNA level of BAX, a
pro-apoptotic gene, was not influenced by ID2 expression
(Fig. 6E and F).
Discussion
In this study, ID2 negatively regulated the
susceptibility of HCC-derived cells to HDAC inhibitors (Figs. 1–3). Several types of antitumor drugs were
tested for their effects on HCC-derived cells with altered
ID2 levels; cells became more susceptible HDAC inhibitors
when ID2 was downregulated, but no other type of antitumor
drug had this effect (Fig. 4).
Previous reports showed that ID2 expression was linked to
poor prognosis of HCC (5–7). ID2 expression is low in HCC
samples that also exhibit other poor prognosis indicators such as
poor differentiation or portal vein invasion. Therefore, we
reasoned that HDAC inhibitors may be useful for treating HCC
characterized by indicators of poor prognosis.
HDAC inhibitors have emerged as a new class of
antitumor drugs that are intended to modulate epigenetic regulation
and several clinical trials have been conducted (42,43).
Although HDAC inhibitors act on HDACs specifically, genome-wide
acetylation of chromatin as a result of HDAC inhibition cause
changes in the expression of many genes (45–47).
Interestingly, addition of NaB to HCC-derived cell lines induced
expression of anti-apoptotic BCL2 and this induction of
BCL2 was positively regulated by ID2 expression
(Fig. 6). This finding indicates
that ID2 may exert an anti-apoptotic function via regulation
of anti-apoptotic genes in the presence of this HDAC inhibitor
(Figs. 5 and 6), although a pro-apoptotic gene,
BAX, was not influenced by ID2 expression (Fig. 6). Cyclin-dependent kinase
inhibitor 1A (CDKN1A; p21, Cip1) is one of the
genes activated by HDAC inhibitors and activated CDKN1A
inhibits the transition from G1 to S phase (48). The activation of CDKN1A
induced by HDAC inhibitors results in growth arrest and apoptosis
in several malignant cell types (47,49,50).
We also observed NaB-mediated induction of CDKN1A and this
induction was significantly suppressed by ID2 overexpression
(data not shown). In ID2 knockdown cells, however, that
NaB-mediated CDKN1A induction was not affected. The
expression of ID2 itself was gradualy induced following the
addition of NaB (data not shown). This increase in ID2
expression might be an endogenous defensive effect in response to
HDAC inhibitors. Because ID2 acts as a dominant-negative inhibitor
of basic helix-loop-helix transcription factors by forming
heterodimers (8–10), some of counter partners forming
heterodimers with ID2 may responsible for HDAC susceptibility.
We suggest that ID2 could serve as a
predictive marker of the response of HCC to HDAC inhibitors.
ID2 influences the susceptibility of HCC cells to the HDAC
inhibitor by regulating the expression of anti-apoptotic genes.
Further investigation of the mechanism by which ID2 affects
susceptibility to HDAC inhibitors, and of the influence of
ID2 on DNA methylation are needed because histone
acetylation and DNA methylation are each correlated with epigenetic
regulation. Biomarkers for clinical response are strongly needed
for improvement of patients’ quality of life and also medical
economics. ID2 may be useful as a biomarker of the likely
response of HCC to HDAC inhibitors; moreover, further research on
ID2 expression in HCC may contribute to the identification
of new molecular targets that can be altered to enhance the effects
of HDAC inhibitors. Such advances may lead to the improvement of
antitumor therapy that is based on HDAC inhibitors.
Acknowledgements
This study was partly supported by
JSPS KAKENHI Grant Number 18390366, 24659610, Research Fellowships
of the Japan Society for the Promotion of Science for Young
Scientists (JSPS KAKENHI Grant Number 187616) and the New Frontier
Project of Yamaguchi University School of Medicine.
References
|
1
|
Thorgeirsson SS and Grisham JW: Molecular
pathogenesis of human hepatocellular carcinoma. Nat Genet.
31:339–346. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Parkin DM, Bray F, Ferlay J and Pisani P:
Global cancer statistics. CA Cancer J Clin. 2005.55:74–108. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Bruix J, Boix L, Sala M and Llovet JM:
Focus on hepatocellular carcinoma. Cancer Cell. 5:215–219. 2004.
View Article : Google Scholar
|
|
4
|
Iizuka N, Oka M, Yamada-Okabe H, Mori N,
Tamesa T, Okada T, Takemoto N, Sakamoto K, Hamada K, Ishitsuka H,
Miyamoto T, Uchimura S and Hamamoto Y: Self-organizing-map-based
molecular signature representing the development of hepatocellular
carcinoma. FEBS Lett. 579:1089–1100. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Damdinsuren B, Nagano H, Kondo M, Yamamoto
H, Hiraoka N, Yamamoto T, Marubashi S, Miyamoto A, Umeshita K, Dono
K, Nakamori S, Wakasa K, Sakon M and Monden M: Expression of Id
proteins in human hepatocellular carcinoma: relevance to tumor
dedifferentiation. Int J Oncol. 26:319–327. 2005.PubMed/NCBI
|
|
6
|
Tsunedomi R, Iizuka N, Yamada-Okabe H,
Tamesa T, Okada T, Sakamoto K, Takashima M, Hamaguchi T, Miyamoto
T, Uchimura S, Hamamoto Y, Yamada M and Oka M: Identification of
ID2 associated with invasion of hepatitis C virus-related
hepatocellular carcinoma by gene expression profile. Int J Oncol.
29:1445–1451. 2006.PubMed/NCBI
|
|
7
|
Tsunedomi R, Iizuka N, Tamesa T, Sakamoto
K, Hamaguchi T, Somura H, Yamada M and Oka M: Decreased ID2
promotes metastatic potentials of hepatocellular carcinoma by
altering secretion of vascular endothelial growth factor. Clin
Cancer Res. 14:1025–1031. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Benezra R, Davis R, Lockshon D, Turner D
and Weintraub H: The protein ID: a negative regulator of
helix-loop-helix DNA binding proteins. Cell. 61:49–59. 1990.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kadesch T: Consequences of heteromeric
interactions among helix-loop-helix proteins. Cell Growth Differ.
4:49–55. 1993.PubMed/NCBI
|
|
10
|
Norton JD: ID helix-loop-helix proteins in
cell growth, differentiation and tumorigenesis. J Cell Sci.
113:3897–3905. 2000.PubMed/NCBI
|
|
11
|
Biggs J, Murphy EV and Israel MA: Id-like
helix-loop-helix protein expressed during early development. Proc
Natl Acad Sci USA. 89:1512–1516. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Hara E, Yamaguchi T, Nojima H, Ide T,
Campisi J, Okayama H and Oda K: Id-related genes encoding
helix-loop-helix proteins are required for G1 progression and are
repressed in senescent human fibroblasts. J Biol Chem.
269:2139–2145. 1994.PubMed/NCBI
|
|
13
|
Rivera R and Murre C: The regulation and
function of the Id proteins inlymphocyte development. Oncogene.
20:8308–8316. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zebedee Z and Hara E: Id proteins in cell
cycle control and cellular senescence. Oncogene. 20:8317–8325.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Benezra R, Rafii S and Lyden D: The Id
proteins and angiogenesis. Oncogene. 20:8334–8341. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ellmeier W, Aguzzi A, Kleiner E, Kurzbauer
R and Weith A: Mutually exclusive expression of a helix-loop-helix
gene and N-myc in human neuroblastomas and in normal development.
EMBO J. 11:2563–2571. 1992.PubMed/NCBI
|
|
17
|
Israel MA, Hernandez MC, Florio M,
Andres-Barquin PJ, Mantani A, Carter JH and Julin CM: Id gene
expression as a key mediator of tumor cell biology. Cancer Res.
59:1726s–1730s. 1999.PubMed/NCBI
|
|
18
|
Lyden D, Young AZ, Zagzag D, Yan W, Gerald
W, O’Reilly R, Bader BL, Hynes RO, Zhuang Y, Manova K and Benezra
R: Id1 and Id3 are required for neurogenesis, angiogenesis and
vascularization of tumour xenografts. Nature. 401:670–677. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Maruyama H, Kleeff J, Wildi S, Friess H,
Büchler MW, Israel MA and Korc M: Id-1 and Id-2 are overexpressed
in pancreatic cancer and in dysplastic lesions in chronic
pancreatitis. AmJ Pathol. 155:815–822. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lin CQ, Singh J, Murata K, Itahana Y,
Parrinello S, Liang SH, Gillett CE, Campisi J and Desprez PY: A
role for Id-1 in the aggressive phenotype and steroid hormone
response of human breast cancer cells. Cancer Res. 60:1332–1340.
2000.PubMed/NCBI
|
|
21
|
Langlands K, Down GA and Kealey T: Id
proteins are dynamically expressed in normal epidermis and
dysregulated in squamous cell carcinoma. Cancer Res. 60:5929–5933.
2000.PubMed/NCBI
|
|
22
|
Wilson JW, Deed RW, Inoue T, Balzi M,
Becciolini A, Faraoni P, Potten CS and Norton JD: Expression of Id
helix-loop-helix proteins in colorectal adenocarcinoma correlates
with p53 expression and mitotic index. Cancer Res. 61:8803–8810.
2001.PubMed/NCBI
|
|
23
|
Schindl M, Schoppmann SF, Ströbel T,
Heinzl H, Leisser C, Horvat R and Birner P: Level of Id-1protein
expression correlates with poor differentiation, enhanced malignant
potential and more aggressive clinical behavior of epithelial
ovarian tumors. Clin Cancer Res. 9:779–785. 2003.
|
|
24
|
Itahana Y, Singh J, Sumida T, Coppe JP,
Parrinello S, Bennington JL and Desprez PY: Role of Id-2 in the
maintenance of a differentiated and noninvasive phenotype in breast
cancer cells. Cancer Res. 63:7098–7105. 2003.PubMed/NCBI
|
|
25
|
Coppe JP, Itahana Y, Moore DH, Bennington
JL and Desprez PY: Id-1 and Id-2 proteins as molecular markers for
human prostate cancer progression. Clin Cancer Res. 10:2044–2051.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Umetani N, Takeuchi H, Fujimoto A,
Shinozaki M, Bilchik AJ and Hoon DS: Epigenetic inactivation of ID4
in colorectal carcinomas correlates with poor differentiation and
unfavorable prognosis. Clin Cancer Res. 10:7475–7483. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
de Candia P, Benera R and Solit DB: A role
for Id proteins in mammary gland physiology and tumorigenesis. Adv
Cancer Res. 92:81–94. 2004.PubMed/NCBI
|
|
28
|
Stighall M, Manetopoulos C, Axelson H and
Landberg G: High ID2 protein expression correlates with a
favourable prognosis in patients with primary breast cancer and
reduces cellular invasiveness of breast cancer cells. Int J Cancer.
115:403–411. 2005. View Article : Google Scholar
|
|
29
|
Umetani N, Mori T, Koyanagi K, Shinozaki
M, Kim J, Giuliano AE and Hoon DS: Aberrant hypermethylation of ID4
gene promoter region increases risk of lymph node metastasis in T1
breast cancer. Oncogene. 24:4721–4727. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Matsuda Y, Yamagiwa S, Takamura M, Honda
Y, Ishimoto Y, Ichida T and Aoyagi Y: Overexpressed Id-1 is
associated with a high risk of hepatocellular carcinoma development
in patients with cirrhosis without transcriptional repression of
p16. Cancer. 104:1037–1044. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Lee TK, Poon RT, Yuen AP, Ling MT, Wang
XH, Wong YC, Guan XY, Man K, Tang ZY and Fan ST: Regulation of
angiogenesis by Id-1 through hypoxia-inducible factor-1a-mediated
vascular endothelial growth factor up-regulation in hepatocellular
carcinoma. Clin Cancer Res. 12:6910–6919. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Drummond DC, Noble CO, Kirpotin DB, Guo Z,
Scott GK and Benz CC: Clinical development of histone deacetylase
inhibitors as anticancer agents. Annu Rev Pharmacol Toxicol.
45:495–528. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Liu T, Kuljaca S, Tee A and Marshall GM:
Histone deacetylase inhibitors: multifunctional anticancer agents.
Cancer Treat Rev. 32:157–165. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Khan O and La Thangue NB: HDAC inhibitors
in cancer biology: Emerging mechanisms and clinical applications.
Immunol Cell Biol. 90:85–94. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Rosato RR, Almenara JA, Dai Y and Grant S:
Simultaneous activation of the intrinsic and extrinsic pathways by
histone deacetylase (HDAC) inhibitors and tumor necrosis
factor-related apoptosis-inducing ligand (TRAIL) synergistically
induces mitochondrial damage and apoptosis in human leukemia cells.
Mol Cancer Ther. 2:1273–1284. 2003.
|
|
36
|
Emanuele S, Lauricella M and Tesoriere G:
Histone deacetylase inhibitors: Apoptotic effects and clinical
implications. Int J Oncol. 33:637–646. 2008.PubMed/NCBI
|
|
37
|
Zhang J, Kan S, Huang B, Hao Z, Mak TW and
Zhong Q: Mule determines the apoptotic response to HDAC inhibitors
by targeted ubiquitination and destruction of HDAC2. Genes Dev.
25:2610–2618. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Vigushin DM and Coombes RC: Histone
deacetylase inhibitors in cancer treatment. Anticancer Drugs.
13:1–13. 2002. View Article : Google Scholar
|
|
39
|
Lin HY and Chen CS, Lin SP, Weng JR and
Chen CS: Targeting histone deacetylase in cancer therapy. Med Res
Rev. 26:397–413. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Xu WS, Parmigiani RB and Marks PA: Histone
deacetylase inhibitors: molecular mechanisms of action. Oncogene.
26:5541–5552. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Tsunedomi R, Ogawa Y, Iizuka N, Sakamoto
K, Tamesa T, Moribe T and Oka M: The assessment of methylated BASP1
and SRD5A2 levels in the detection of early hepatocellular
carcinoma. Int J Oncol. 36:205–212. 2010.PubMed/NCBI
|
|
42
|
Kouzarides T: Histone acetylases and
deacetylases in cell proliferation. Curr Opin Genet Dev. 9:40–48.
1999. View Article : Google Scholar
|
|
43
|
Marks PA, Richon VM and Rifkind RA:
Histone deacetylase inhibitors:inducers of differentiation or
apotosis of transformed cells. J Natl Cancer Inst. 92:1210–1216.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Timmermann S, Lehrmann H, Polesskaya A and
Harel-Bellan A: Histone acetylation and disease. Cell Mol Life Sci.
58:728–736. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Ogawa K, Yasumura S, Atarashi Y, Minemura
M, Miyazaki T, Iwamoto M, Higuchi K and Watanabe A: Sodium butyrate
enhances Fas-mediated apoptosis of human hepatoma cells. J Hepatol.
40:278–284. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Joseph J, Mudduluru G, Antony S, Vashistha
S, Ajitkumar P and Somasundaram K: Expression profiling of sodium
butyrate (NaB)-treated cells: identification of regulation of genes
related to cytokine signaling and cancer metastasis by NaB.
Oncogene. 23:6304–6315. 2004. View Article : Google Scholar
|
|
47
|
Chiba T, Yokosuka O, Arai M, Tada M, Fukai
K, Imazeki F, Kato M, Seki N and Saisho H: Identification of genes
up-regulated by histone deacetylase inhibition with cDNA microarray
and exploration of epigenetic alterations on hepatoma cells. J
Hepatol. 41:436–445. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Chen J, Saha P, Kornbluth S, Dynlacht BD
and Dutta A: Cyclin-binding motifs are essential for the function
of p21CIP1. Mol Cell Biol. 16:4673–4682. 1996.PubMed/NCBI
|
|
49
|
Shin JY, Kim HS, Park J, Park JB and Lee
JY: Mechanism for inactivation of the KIP family cyclin-dependent
kinase inhibitor genes in gastric cancer cells. Cancer Res.
60:262–265. 2000.PubMed/NCBI
|
|
50
|
Han JW, Ahn SH, Kim YK, Bae GU, Yoon JW,
Hong S, Lee HY, Lee YW and Lee HW: Activation of
p21WAF1/Cip1 transcription through Sp1 sites by histone
deacetylase inhibitor apicidin. J Biol Chem. 276:42084–42090.
2001.PubMed/NCBI
|