Introduction
Hepatocellular carcinoma (HCC) is the fifth most
common cancer and the third leading cause of cancer related
mortality worldwide (1).
Clinically, surgical resection remains the primary mode of therapy
for advanced HCC. In the absence of an effective adjuvant therapy,
liver cancer has a poor prognosis (2). HCC is a highly malignant tumor with a
potent capacity to invade locally and metastasize distantly
(2,3); therefore, an approach that decreases
its ability to invade and metasta-size may facilitate the
development of an effective adjuvant therapy.
Focal adhesion kinase (FAK) is a non-receptor
cytoplasmic tyrosine kinase that plays a key role in the regulation
of cell proliferation and migration in both inflammation and
neoplasia (4). FAK appears to be
essential for the regulation of cell migration and invasion
properties (5–7). It is associated with integrin
receptors and integrates with other molecules to form a complex
that transmits signals from the extracellular matrix to the cell
cytoskeleton (8,9). In 2010, the expression and prognostic
significance of FAK were reported in liver cancer and FAK was found
to play an important role in invasion and metastasis of HCC
(10). Therefore, FAK may be a
promising therapeutic target for HCC (11). Our previous studies showed that
prostaglandin E2 (PGE2) induces cell adhesion
and migration by activating FAK in HCC cells (12). However, the specific mechanism of
this activation was not investigated.
PGE2 is one of the predominant metabolic
products of arachidonic acid. In previous studies, PGE2
was found to play a major role in promoting tumor cell growth,
migration and invasion in many cancer types (13–16).
PGE2 has been shown to regulate tumor development and
progression by combining with E prostanoid receptors (EP receptors)
on the surface of the cell membrane and activating their
predominant signal transduction pathways (17).
There are four types of EP receptors expressed on
the membrane surface of HCC cells (1). Among them, the EP1 receptor is
accepted to be involved in cell growth, invasion and metastasis in
many cancers, such as colon cancer (18), skin cancer (19) and cervical cancer (20). Recently, the EP1 receptor was
reported to enhance cell invasion in HCC cells (21), although it is unclear whether FAK
activation is involved. In the present study, PGE2 was
found to upregulate FAK phosphorylation via the EP1 receptor in HCC
cells. Protein kinase C (PKC), c-Src and epidermal growth factor
receptor (EGFR) were all involved in the EP1 receptor-mediated FAK
phosphorylation and cell adhesion and migration. Our data generated
from the present studies may provide significant insights into the
mechanisms of EP1 receptor-mediated FAK activation and ultimately
lead to the development of novel management strategies and
therapeutics for cell invasion and metastasis in hepatocellular
carcinoma.
Materials and methods
Materials
The human HCC cell line Huh-7 and human embryonic
kidney (HEK) 293 cells were obtained from American Type Culture
Collection (ATCC, Manassas, VA, USA). Dulbecco’s modified Eagle’s
medium (DMEM) was from Invitrogen (Carlsbad, CA, USA).
PGE2, 17-phenyl trinor-PGE2
(17-PT-PGE2) and sc-19220 were from Cayman Chemical Co.
(Ann Arbor, MI, USA). AG1478 was from Sigma-Aldrich (St. Louis, MO,
USA). Bisindolymaleimide I (#203290),
phorbol-12-myristate-13-acetate (#524400) and PP2 (#529573) were
obtained from Merck (Darmstadt, Germany). Recombinant human
epidermal growth factor (EGF; #AF-100-15) was from PeproTech (Rocky
Hill, NJ, USA). WST reagent from the Cell Counting Kit-8 (CCK-8)
was from Dojindo Laboratories (Kumamoto, Japan). The protein assay
was from Bio-Rad (Hercules, CA, USA). Electrochemiluminescence
(ECL) reagents were from Amersham Biosciences (Piscataway, NJ,
USA). The PKC assay kit was from Millipore (Billerica, MA, USA).
[γ-32P]ATP (#BLU002A) was from Perkin-Elmer (Waltham,
MA, USA). G418 sulfate was from Amresco (Solon, OH, USA). The
following were commerically obtained antibodies: anti-FAK antibody
(sc-558; Santa Cruz Biotechnology, Santa Cruz, CA, USA);
anti-phosphorylated FAK Y397 antibody (#611806, BD Biosciences,
Franklin Lakes, NJ, USA); phosphorylated Src antibody (#2101, Cell
Signaling Technology Danvers, MA, USA); total Src antibody (#21168,
SAB, College Park, MD, USA); anti-β-actin antibody (Sigma).
Cell lines and culture
Huh-7 cells and HEK293 cells were cultured in DMEM
with 10% fetal calf serum, 100 IU/ml penicillin and 100
μg/ml streptomycin at 37°C with 5% CO2.
Cell adhesion assays
Cell adhesion assays were performed in 96-well
culture cell plates. Cells (5×104) were added to each
well of the plates and pharmacological agents were added at the
indicated time. The plates were incubated at 37°C for 3 h and then
washed three times with phosphate-buffered saline (PBS) to remove
unattached cells. The attached cells were stained with WST at 37°C
for 2 h. Adherent cells were quantified by measuring the absorbance
at 450 nm.
Cell migration assays
Cell migration assays were performed in 12-well
transwell units. Before the experiment, the lower surfaces of the
membranes were coated with gelatin (1%) diluted in PBS. Huh-7 cells
(5×104) were added to the upper chamber and 1 ml
complete DMEM to the lower chamber of the transwell.
Pharmacological agents were added at the indicated time. After
incubation at 37°C for 12 h, the cells were fixed with ethanol and
then stained with 0.1% crystal violet for 30 min at room
temperature. After washing the wells with PBS, the cells were
removed from the upper surface of the membrane by wiping with a
moist cotton swab. The cells migrated to the lower surface of the
membrane were solubilized with 300 μl of 10% acetic acid and
quantified by measuring the absorbance at 570 nm.
PKC measurements
Cells were treated with pharmacological agents at
37°C for various times, as indicated in the experiments. The cells
were collected into lysis buffer (50 mM Tris-HCl, pH 8.0, 150 mM
NaCl, 0.5% sodium deoxycholate, 1% Nonidet P-40, 0.1% SDS, 100
μg/ml PMSF and aprotinin) and placed on ice for 30 min. Cell
lysates were sonicated on ice for at least 30 sec and then cleared
by centrifugation at 120,000 × g for 30 min at 4°C. Total protein
(30 μg) (10 μl) for each sample was added to a
microcentrifuge tube and assayed for PKC levels using a direct
human PKC enzyme activity assay kit according to the manufacturer’s
instructions. Briefly, 10 μl of substrate cocktail, PKA
inhibitor cocktail, Assay Dilution Buffer II (ADBII), lipid
activator and diluted [γ-32P]-ATP mixture were added to
a microcentrifuge tube and then the mixture was incubated for 10
min at 30°C with constant agitation. A 25-μl aliquot from
each sample was transferred onto the center of a P81
phosphocellulose paper. The assay squares were washed with 0.75%
phosphoric acid three times, followed by one wash with acetone. The
assay squares were transferred to vials with a scintillation
cocktail and read in a scintillation counter. The counts per minute
(cpm) of the enzyme samples were compared to those of the control
samples containing no enzyme.
Plasmid transfections
The pcDNA3-based plasmid encoding the human EP1
receptor (EP1R-pcDNA3) was a generous gift of Kathy Mccusker in
2007 (Merck Frosst Centre for Therapeutic Research, Canada). HEK293
cells (2×105) were seeded and grown in 6-well culture
plates for 24 h before transfection with the EP1R-pcDNA3 plasmid or
pcDNA3 empty vector control (2 μg) using Lipofectamine
2000TM (5 μl). After incubation for 5 h, the
medium was changed to fresh normal growth DMEM. The efficiency of
transfection was assayed by flow cytometry. The G418 antibiotic was
used to select for HEK293 cells stably expressing the EP1
receptor.
Western blotting
Cells were treated with pharmacological agents at
37°C for various times, as indicated in the experiments. The cells
were collected into lysis buffer (50 mM Tris-HCl, pH 8.0, 150 mM
NaCl, 0.5% sodium deoxycholate, 1% Nonidet P-40, 0.1% SDS, 100
μg/ml PMSF and aprotinin) and placed on ice for 30 min. Cell
lysates were sonicated on ice for ≥30 sec and then cleared by
centrifugation at 120,000 × g for 30 min at 4°C. Equal amounts of
total proteins (40 μg) were separated by SDS-PAGE and
transferred onto a nitrocellulose membrane. The membranes were
probed with the appropriate antibodies at 4°C overnight with gentle
shaking. The immunoreactivity was detected by ECL and analyzed
using Image lab 4.0 analysis software from Bio-Rad.
Statistical analysis
Data are presented as means ± SD. P-values were
calculated using the Student’s t-test for unpaired samples with MS
Excel software. The results were considered significantly different
at P<0.05.
Results
EP1 receptor promotes FAK
phosphorylation, cell adhesion and migration in Huh-7 cells
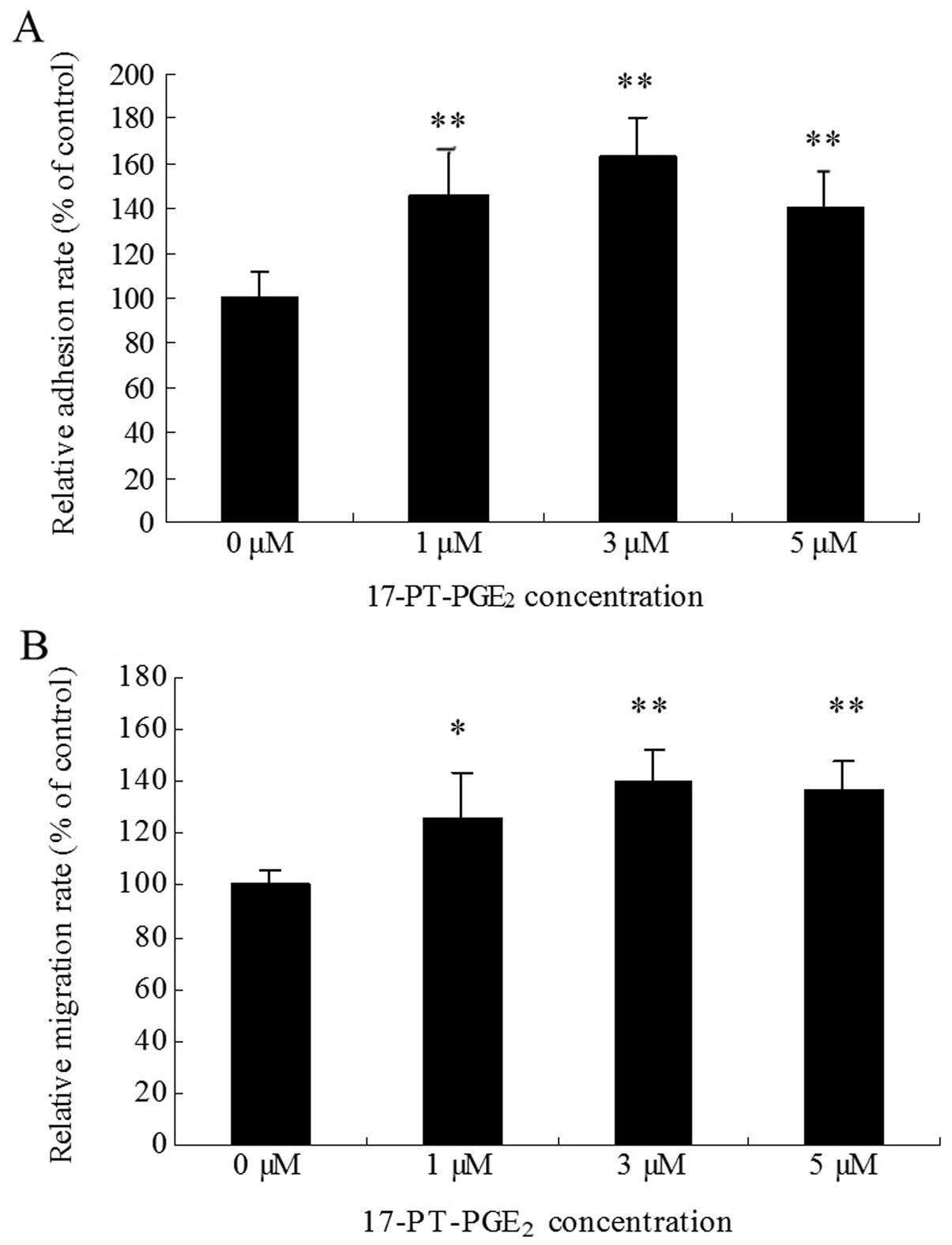
Huh-7 cells were treated with various concentrations
of the EP1 receptor agonist 17-PTPGE2 for 3 h. The WST
assay was used to detect the cell adhesion rate. The results showed
that 17-PT-PGE2 produced a 40–60% increase in cell
adhesion in Huh-7 cells (Fig. 1A).
In the transwell assay, cell migration was found to increase by
20–40% when the cells were treated with 17-PT-PGE2 for
12 h (Fig. 1B). At the
concentration of 3 μM, 17-PT-PGE2 caused maximal
responses both in cell adhesion and migration assays.
We previously found that PGE2 stimulates
cell adhesion and migration in HCC cells by increasing FAK Y397
phosphorylation (12). To
determine whether the EP1 receptor is involved in
PGE2-mediated FAK phosphorylation, Huh-7 cells were
exposed to different concentrations of exogenous
17-PT-PGE2 for different periods of time. As shown in
Fig. 2A, an increase in FAK
phosphorylation at the Y397 site was detected 15 min after
17-PT-PGE2 treatment and the maximal response (∼2-fold
induction) was reached at 30 min post-treatment with 3 μM
17-PT-PGE2 (Fig. 2B).
Based on these findings, treatment with 3 μM
17-PT-PGE2 for 30 min was used for subsequent
experiments in Huh-7 cells. As shown in Fig. 2C, pre-treatment with sc-19220, a
specific antagonist of the EP1 receptor, suppressed
PGE2-mediated upregulation of FAK Y397
phosphorylation.
 | Figure 2Effects of EP1 receptor on FAK Y397
phosphorylation in Huh-7 cells. (A) Effects of EP1 agonist on FAK
phosphorylation at Y397 in Huh-7 cells cultured and treated with 3
μM exogenous 17-PT-PGE2 for 0, 15, 30 and 60 min.
(B) Effects of EP1 agonist 17-PT-PGE2 treatment (0, 1, 3
or 5 μM for 30 min) on FAK Y397 phosphorylation in Huh-7
cells. FAK phosphorylation increased after stimulation with
different concentrations of 17-PT-PGE2 and the maximal
response was reached at 30 min post-treatment with 3 μM
17-PT-PGE2. (C) Effects of an EP1 antagonist on
17-PT-PGE2-mediated FAK phosphorylation in Huh-7 cells.
Cells were pre-treated with 10 μM sc-19220 for 30 min,
followed by treatment with 3 μM 17-PT-PGE2 for 30
min. (D) Effects of the EP1 receptor expression on
17-PT-PGE2-mediated FAK phosphorylation in HEK293 cells.
HEK293 cells (3×105 cells) were seeded in 6-well plates
and cultured for 24 h before transfection with 2 μg of the
EP1R-pcDNA3 plasmid or empty pcDNA3 plasmid as a control. After
transfection, cells expressing the EP1 receptor were selected by
300 μg/ml G418. EP1 receptor-transfected HEK293 cells were
exposed to 2 μM PGE2 for 30 min, with or without
sc-19220 pre-treatment. Equal amounts of total proteins were
separated by SDS-PAGE and relative levels of phosphorylated FAK and
total FAK expression were determined using anti-phospho-FAK and
anti-FAK antibodies. β-actin was detected as a loading control.
Densitometric quantitation of the above blots is shown. Results are
presented as means ± SD from three different experiments.
*P<0.05, compared to control cells;
**P<0.01, compared to control cells;
#P<0.05, compared to PGE2-treated
cells. |
To confirm the role of the EP1 receptor in the
induction of FAK phosporylation, HEK293 cells were transfected with
the EP1 receptor expression plasmid (EP1R-pcDNA3) and selected with
G418 to obtain a stable expression cell culture. As illustrated in
Fig. 2D, expression of the EP1
receptor did not alter the basal phosphorylation level of FAK.
However, FAK phosphorylation was significantly upregulated in the
EP1R-transfected cells when treated with PGE2, compared
with control cells. At the same time, pre-treatment with sc-19220
significantly decreased PGE2-mediated FAK Y397
phosphorylation in EP1R-transfected cells.
PKC/c-Src signaling is involved in
EP1-mediated FAK phosphorylation, cell adhesion and migration in
Huh-7 cells
The PKC/c-Src pathway is reportedly involved in
EP1-mediated cell migration (22).
Therefore, the relationship between PKC/c-Src activation and FAK
phosphorylation was examined in the present study. PKC activity and
c-Src phosphorylation in response to 17-PT-PGE2 were
directly measured in Huh-7 cells. Treatment of Huh-7 cells with
17-PT-PGE2 upregulated PKC activity >2-fold after 15
min and then decreased after 30 min (Fig. 3A). Pre-treatment of cells with the
PKC inhibitor bisindolymaleimide I (Bis) significantly reduced the
17-PT-PGE2-mediated FAK phosphorylation (Fig. 3B). At the same time,
17-PT-PGE2 induced the phosphorylation of c-Src at Y416,
which could be detected at 15 min after treatment and then
decreased after 60 min (Fig. 3C).
In addition, pre-treatment of cells with a c-Src inhibitor (PP2)
diminished 17-PT-PGE2-increased FAK phosphorylation
significantly (Fig. 3D). In
addition, pre-treatment of Huh-7 cells with Bis and PP2 completely
blocked 17-PT-PGE2-mediated cell adhesion and migration
(Fig. 3E and F).
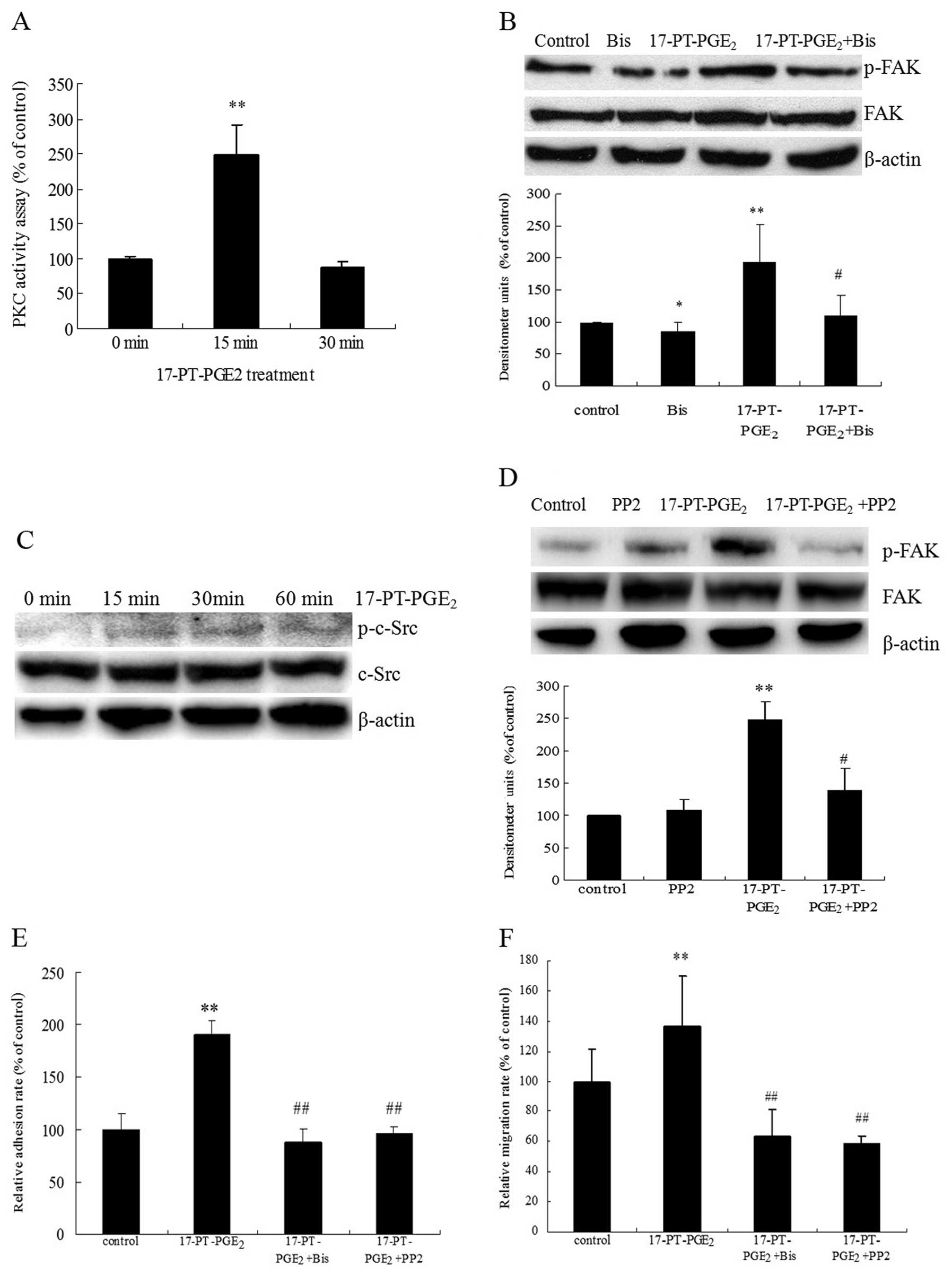 | Figure 3Roles of PKC and c-Src pathways in
17-PT-PGE2-mediated FAK phosphorylation in Huh-7 cells.
(A) PKC activity assay. Huh-7 cells were treated with 3 μM
17-P-T-PGE2 for 0, 15 or 30 min. Equal amounts of total
proteins (30 μg) were added to microcentrifuge tubes and
assayed for PKC levels using a direct human PKC enzyme activity
assay kit. (B) Effects of PKC inhibitor on
17-PT-PGE2-mediated FAK phosphorylation in Huh-7 cells
treated with 3 μM 17-PT-PGE2 for 30 min, with or
without pre-treatment of 5 μM Bis for 1 h. Equal amounts of
total proteins were separated by SDS-PAGE. Relative levels of
phosphorylated FAK and total FAK expression were determined using
anti-phospho-FAK and anti-FAK antibodies. β-actin was detected as a
loading control. Densitometric quantitation of the above blots is
shown. (C) Effects of 17-PT-PGE2 on c-Src
phosphorylation at Y416 in Huh-7 cells treated with 3 μM
exogenous 17-PT-PGE2 for 0, 15, 30 or 60 min. Equal
amounts of total proteins were separated by SDS-PAGE. Relative
levels of phosphorylated c-Src and total c-Src expression were
determined using anti-phospho-c-Src and anti-c-Src antibodies.
β-actin was detected as a loading control. These experiments were
performed three times with similar results. (D) Effects of c-Src
inhibitor on 17-PT-PGE2-mediated FAK phosphorylation in
Huh-7 cells treated with 3 μM 17-P-T-PGE2 for 30
min, with or without pre-treatment of 10 μM PP2 for 1 h.
Equal amounts of total proteins were separated by SDS-PAGE.
Relative levels of phosphorylated FAK and total FAK expression were
determined using anti-phospho-FAK and anti-FAK antibodies. β-actin
was detected as a loading control. Densitometric quantitation of
the above blots is shown. (E) Effects of PKC or c-Src inhibitors on
17-PT-PGE2-mediated cell adhesion in Huh-7 cells. The
cell adhesion assay was performed in 96-well plates. Huh-7 cells
were treated with 3 μM 17-P-T-PGE2 for 3 h, with
or without pre-treatment with 5 μM Bis or 10 μM PP2
for 1 h. The attached cells were stained with WST and quantified by
reading absorbance at 450 nm. (F) Effects of PKC or c-Src
inhibitors on 17-PT-PGE2-mediated cell migration in
Huh-7 cells. The cell migration assay was performed in 12-well
transwells. Huh-7 cells were treated with 3 μM
17-PT-PGE2 for 12 h, with or without pre-treatment of 5
μM Bis or 10 μM PP2 for 1 h. Cells on the lower
surface were stained with 0.1% crystal violet, solubilized with
acetic acid solution and quantified by measuring absorbance at 570
nm. Results are presented as means ± SD from three different
experiments. *P<0.05, compared to control cells;
**P<0.01, compared to control cells;
#P<0.05, compared to cells with 17-PT-PGE2
treatment; ##P<0.01, compared to 17-PT-PGE2-treated
cells. |
In order to clarify whether c-Src functions
downstream of the 17-PT-PGE2-induced PKC pathway,
effects of the PKC activator phorbol-12-myristate-13-acetate (PMA)
and inhibitor Bis on c-Src phosphorylation were examined. PMA
treatment increased c-Src phosphorylation, as well as the FAK
phosphorylation (Fig. 4A), while
Bis significantly suppressed the 17-PT-PGE2-induced
c-Src phosphorylation (Fig.
4B).
EGFR is involved in EP1-mediated FAK
phosphorylation
In our previous studies, we found that the
phosphorylation level of EGFR increases significantly after
17-PT-PGE2 treatment in HCC cells (23). In the present study, FAK Y397
phosphorylation was found to increase after exposure of Huh-7 cells
to exogenous EGF for 30 min (Fig.
5A). In addition, pre-treatment of cells with the EGFR
inhibitor AG1478 significantly suppressed the
17-PT-PGE2-induced FAK phosphorylation (Fig. 5B). As shown in Fig. 5C and D, pre-treatment with AG1478
mildly suppressed the 17-PT-PGE2-mediated cell adhesion
and completely blocked the 17-PT-PGE2-mediated migration
in Huh-7 cells. In order to clarify whether c-Src is involved in
the 17-PT-PGE2-induced EGFR/FAK pathway, the effect of
AG1478 on c-Src phosphorylation was detected, but surprisingly it
had no effect on 17-PT-PGE2-mediated c-Src
phosphorylation (Fig. 5E).
Discussion
Prostaglandin E2 is one of the major
products of cyclooxygenase-2 (COX-2), which has been shown to drive
cancer cell growth and invasion in many cancer cells. Previous
studies have indicated that COX-2 is overexpressed in many cancer
tissues and that the level of PGE2 is increased in COX-2
over-expressing cells (24–26).
Endogenous and exogenous PGE2 induce angiogenesis
(27,28) and promote tumor cell growth,
migration and invasion in colon cancer (13), renal cancer (16) and lung cancer (15). Selective COX-2 inhibitors were
shown to suppress PGE2 production (29,30)
and reduce cell growth, migration and invasion (29–32).
PGE2 exerts its functions through four
subtypes of receptors expressed on the surface of tumor cells: EP1,
EP2, EP3 and EP4 receptors (17).
It is well accepted that the EP1 receptor promotes development and
progression of many cancers (20).
For example, expression of the EP1 receptor is frequently observed
both in the cytoplasm and/or in the nuclear membrane in many cancer
cells (21,33,34).
BK5.EP1 transgenic mice produce more skin tumors than wild-type
(WT) mice (35). Furthermore, the
EP1 receptor level is associated with cell migration or invasion in
colon cancer cells (18),
chondrosarcoma cells (22) and
oral cancer cells (36).
FAK is a non-receptor, cytoplasmic protein tyrosine
kinase that appears to play a central role in integrin-mediated
signal transduction. Overexpression of FAK enhances cell
proliferation (8,37); while FAK depletion impairs cell
adhesion or migration in colon cancer (38), breast cancer cells (7) and melanoma cells (39). Recently, FAK mRNA and protein, as
well as phosphorylated FAK Tyr397, were found to be overexpressed
both in HCC clinical samples and HCC cell lines (10,11).
Increased FAK and phosphorylated FAK Tyr397 expression levels have
been correlated with tumor stage, vascular invasion and
intrahepatic metastasis in HCC (11).
In our previous study, we found that PGE2
improves cell adhesion, migration and invasion in HCC cells. FAK
plays an important role in PGE2-mediated cell adhesion
and migration, as exogenous PGE2 was shown to greatly
increase phosphorylation of FAK at the Y397 site in a
PGE2 concentration-dependent manner. RNA interference
targeting FAK can suppress PGE2-mediated cell adhesion
and migration significantly (12).
In the present study, 17-PT-PGE2 treatment mimicked the
effects of PGE2 and promoted cell adhesion and migration
in HCC cells. Therefore, we hypothesize that PGE2
upregulates FAK phosphorylation via the EP1 receptor in HCC
cells.
Autophosphorylation at Y397 is required for FAK
activation (40). To date, very
little is known about the effects of EP1 receptor on FAK Y397
activation and molecular mechanisms mediating these effects. In our
previous study, FAK phosphorylation and expression were found to be
upregulated by PGE2 in HCC cells (12). In the present study,
17-PT-PGE2 treatment caused upregulation of FAK Y397
phosphorylation in Huh-7 cells. Expression of the EP1 receptor in
HEK293 cells mimicked the effect of 17-PT-PGE2
treatment. In addition, pre-treatment with sc-19220, the specific
antagonist of the EP1 receptor, suppressed PGE2-mediated
upregulation of FAK Y397 phosphorylation. Our data suggest that the
EP1 receptor is involved in PGE2-mediated activation of
FAK. Because FAK is increasingly recognized as a critical factor in
the development of HCC (11), the
present findings suggest that the EP1 receptor may possibly be
explored as a drug target to prevent the phosphorylation of FAK
Y397 in the prevention and treatment of liver cancer.
PKC has been reported to be involved in the EP1
receptor signaling pathway (17).
The PKC family was first identified as intracellular receptors for
the tumor promoting phorbol esters (41). Activation of PKC by calcium ions
and the second messenger diacylglycerol is thought to play a
central role in the induction of cellular responses to a variety of
ligand-receptor systems and in the regulation of cellular
responsiveness to external stimuli (41,42).
PKC was found to be associated with the development of HCC. For
example, mRNA levels of several PKC isoforms were shown to be
significantly increased in HCC samples, as compared to the
corresponding non-cancerous liver tissues. PKCα expression is also
significantly correlated with tumor size and the tumor, node and
metastasis (TNM) stage (43).
Meanwhile, reduction of PKC expression by RNA interference or
selective inhibitors greatly decreases cell proliferation,
migration and invasion in HCC cells (44).
Our data show that PKC activities were enhanced
after 17-PT-PGE2 treatment for 15 min, while the
selective PKC inhibitor, Bis, completely blocked
17-PT-PGE2-mediated cell adhesion and migration. These
results support that PKC is involved in EP1 receptor-mediated cell
adhesion and migration in HCC cells. The involvement of PKC in EP1
receptor-mediated FAK phosphorylation was further confirmed by use
of PMA, a selective PKC activator, which promoted the
phosphorylation of FAK at Y397 in Huh-7 cells. In addition, Bis
pre-treatment diminished the 17-PT-PGE2-mediated FAK
phosphorylation.
Phosphorylation of FAK Y397 was determined to create
a high-affinity binding site for SH2 domains of Src family kinases
to form a FAK/Src complex. A major function of FAK is to recruit
and activate Src at cell-extracellular matrix adhesion sites
(40). However, FAK Y397 is not
strictly an autophosphorylation site and signal amplification can
also result from phosphorylation of this site by Src (45). Therefore, the effect of c-Src on
EP1 receptor-induced FAK Y397 phosphorylation in Huh-7 cells was
further evaluated in this study.
C-Src activity is regulated by tyrosine
phosphorylation at two distinct sites, Y416 and Y527 and the active
state of c-Src is p-Y416-c-Src (46). Our results showed that c-Src Y416
phosphorylation was enhanced after 17-PT-PGE2 treatment
for 15 min. The selective Src inhibitor PP2, caused a significant
reduction in 17-PT-PGE2-mediated FAK Y397
phosphorylation. PP2 pre-treatment completely blocked
17-PT-PGE2-mediated cell adhesion and migration.
Furthermore, PMA treatment increased c-Src phosphorylation, while
Bis partly suppressed the 17-PT-PGE2-induced c-Src
phosphorylation. These data suggest that the PKC/c-Src signal
pathway is involved in EP1 receptor-mediated FAK activation, cell
adhesion and migration in HCC cells.
EGFR has been widely accepted to improve cell growth
and invasion in many cancer cell types (47). Several studies in the last decade
have shown that the EP receptor pathway also modulates activation
of the EGFR (48). In 2006, Han
et al revealed a novel crosstalk between the EP1 and EGFR
signaling pathways that synergistically promote cancer cell growth
and invasion. The association of EP1 with EGFR was found by
immunoprecipitation after HCC cells were treated with
PGE2 or EP1 agonist (21). Of interest, EGFR activation was
found to be sufficient to induce activation of the Src-FAK pathway
in tumor cells (49,50). As the EGFR/Src/FAK pathway has been
implicated in cell growth, migration and invasion in various cancer
cells (49–51), the involvement of this signaling
pathway in EP1 receptor-mediated cell adhesion and migration was
explored in this study.
In our previous study, we found that the
phosphorylation level of EGFR increases significantly after
17-PT-PGE2 treatment in HCC cells (23). In the present study, exogenous EGF
was observed to increase FAK Y397 phosphorylation. The selective
EGFR inhibitor, AG1478, greatly suppressed
17-PT-PGE2-induced FAK Y397 phosphorylation. At the same
time, AG1478 pre-treatment mildly suppressed
17-PT-PGE2-mediated cell adhesion and completely blocked
17-PT-PGE2-mediated cell migration. Surprisingly, AG1478
had no effect on 17-PT-PGE2-mediated c-Src Y416
phosphorylation. Our results suggest that EGFR is also associated
with EP1 receptor-mediated FAK activation, while c-Src may be not
involved in EGFR-induced FAK activation in HCC cells.
In conclusion, this study demonstrated that the
PGE2 EP1 receptor upregulates FAK phosphorylation at
Y397 to improve cell adhesion and migration. PKC/c-Src and EGFR
signaling pathways are both involved in EP1 receptor-mediated FAK
phosphorylation. The PGE2/EP1/PKC/c-Src and
PGE2/EP1/EGFR signaling pathway may coordinately
regulate FAK activation, cell adhesion and migration in HCC
(Fig. 6). In this regard, our
present findings provide important new information regarding the
putative role of the EP1 receptor in FAK phosphorylation in HCC
cells and suggest that targeting the PGE2/EP1/FAK signal
pathway may provide new therapeutic strategies for both the
prevention and treatment of this malignant disease.
Acknowledgments
We thank Dr Kathy Mccusker (Merck Frosst Centre for
Therapeutic Research, Canada) for providing the pcDNA3 plasmid
construct encoding the EP1 receptor. This study was supported in
part by the National Natural Science Foundation of China (81101496,
81172003), Research Fund for the Doctoral Program of Higher
Education of China (20113234120009) and a Project Funded by the
Priority Academic Program Development of Jiangsu Higher Education
Institutions (PAPD).
References
|
1.
|
Wu T: Cyclooxygenase-2 in hepatocellular
carcinoma. Cancer Treat Rev. 32:28–44. 2006. View Article : Google Scholar
|
|
2.
|
Uchino K, Tateishi R, Shiina S, Kanda M,
Masuzaki R, Kondo Y, Goto T, Omata M, Yoshida H and Koike K:
Hepatocellular carcinoma with extrahepatic metastasis: clinical
features and prognostic factors. Cancer. 117:4475–4483. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Pang RW, Joh JW, Johnson PJ, Monden M,
Pawlik TM and Poon RT: Biology of hepatocellular carcinoma. Ann
Surg Oncol. 15:962–971. 2008. View Article : Google Scholar
|
|
4.
|
Mon NN, Ito S, Senga T and Hamaguchi M:
FAK signaling in neoplastic disorders: a linkage between
inflammation and cancer. Ann NY Acad Sci. 1086:199–212. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Meng XN, Jin Y, Yu Y, Bai J, Liu GY, Zhu
J, Zhao YZ, Wang Z, Chen F, Lee KY and Fu SB: Characterisation of
fibronectin-mediated FAK signalling pathways in lung cancer cell
migration and invasion. Br J Cancer. 101:327–334. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Kwiatkowska A, Kijewska M, Lipko M, Hibner
U and Kaminska B: Downregulation of Akt and FAK phosphorylation
reduces invasion of glioblastoma cells by impairment of MT1-MMP
shuttling to lamellipodia and downregulates mMPs expression.
Biochim Biophys Acta. 1813:655–667. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Chan KT, Cortesio CL and Huttenlocher A:
FAK alters invadopodia and focal adhesion composition and dynamics
to regulate breast cancer invasion. J Cell Biol. 185:357–370. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Guan JL: Integrin signaling through FAK in
the regulation of mammary stem cells and breast cancer. IUBMB Life.
62:268–276. 2010.PubMed/NCBI
|
|
9.
|
Mitra SK and Schlaepfer DD:
Integrin-regulated FAK-Src signaling in normal and cancer cells.
Curr Opin Cell Biol. 18:516–523. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Yuan Z, Zheng Q, Fan J, Ai KX, Chen J and
Huang XY: Expression and prognostic significance of focal adhesion
kinase in hepatocellular carcinoma. J Cancer Res Clin Oncol.
136:1489–1496. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Chen JS, Huang XH, Wang Q, Chen XL, Fu XH,
Tan HX, Zhang LJ, Li W and Bi J: FAK is involved in invasion and
metastasis of hepatocellular carcinoma. Clin Exp Metastasis.
27:71–82. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Bai XM, Zhang W, Liu NB, Jiang H, Lou KX,
Peng T, Ma J, Zhang L, Zhang H and Leng J: Focal adhesion kinase:
important to prostaglandin E2-mediated adhesion, migration and
invasion in hepatocellular carcinoma cells. Oncol Rep. 21:129–136.
2009.PubMed/NCBI
|
|
13.
|
Xia D, Wang D, Kim SH, Katoh H and DuBois
RN: Prostaglandin E2 promotes intestinal tumor growth via DNA
methylation. Nat Med. 18:224–226. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Harding P and LaPointe MC: Prostaglandin
E2 increases cardiac fibroblast proliferation and increases cyclin
D expression via EP1 receptor. Prostaglandins Leukot Essent Fatty
Acids. 84:147–152. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Kim JI, Lakshmikanthan V, Frilot N and
Daaka Y: Prostaglandin E2 promotes lung cancer cell migration via
EP4-betaArrestin1-c-Src signalsome. Mol Cancer Res. 8:569–577.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Wu J, Zhang Y, Frilot N, Kim JI, Kim WJ
and Daaka Y: Prostaglandin E2 regulates renal cell carcinoma
invasion through the EP4 receptor-Rap GTPase signal transduction
pathway. J Biol Chem. 286:33954–33962. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Boie Y, Stocco R, Sawyer N, Slipetz DM,
Ungrin MD, Neuschäfer-Rube F, Püschel GP, Metters KM and Abramovitz
M: Molecular cloning and characterization of the four rat
prostaglandin E2 prostanoid receptor subtypes. Eur J Pharmacol.
340:227–241. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
O’Callaghan G, Kelly J, Shanahan F and
Houston A: Prostaglandin E2 stimulates Fas ligand expression via
the EP1 receptor in colon cancer cells. Br J Cancer. 99:502–512.
2008.PubMed/NCBI
|
|
19.
|
Rundhaug JE, Simper MS, Surh I and Fischer
SM: The role of the EP receptors for prostaglandin E2 in skin and
skin cancer. Cancer Metastasis Rev. 30:465–480. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Liu H, Xiao J, Yang Y, Liu Y, Ma R, Li Y,
Deng F and Zhang Y: COX-2 expression is correlated with VEGF-C,
lymphangiogenesis and lymph node metastasis in human cervical
cancer. Microvasc Res. 82:131–140. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21.
|
Han C, Michalopoulos GK and Wu T:
Prostaglandin E2 receptor EP1 transactivates EGFR/MET receptor
tyrosine kinases and enhances invasiveness in human hepatocellular
carcinoma cells. J Cell Physiol. 207:261–270. 2006. View Article : Google Scholar
|
|
22.
|
Liu JF, Fong YC, Chang CS, Huang CY, Chen
HT, Yang WH, Hsu CJ, Jeng LB, Chen CY and Tang CH: Cyclooxygenase-2
enhances alpha2beta1 integrin expression and cell migration via EP1
dependent signaling pathway in human chondrosarcoma cells. Mol
Cancer. 9:432010. View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Bai XM, Jiang H, Ding JX, Peng T, Ma J,
Wang YH, Zhang L, Zhang H and Leng J: Prostaglandin E2 upregulates
survivin expression via the EP1 receptor in hepatocellular
carcinoma cells. Life Sci. 86:214–223. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24.
|
Leng J, Han C, Demetris AJ, Michalopoulos
GK and Wu T: Cyclooxygenase-2 promotes hepatocellular carcinoma
cell growth through Akt activation: evidence for Akt inhibition in
celecoxib-induced apoptosis. Hepatology. 38:756–768. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
25.
|
Wendum D, Masliah J, Trugnan G and Fléjou
JF: Cyclooxygenase-2 and its role in colorectal cancer development.
Virchows Arch. 445:327–333. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
26.
|
Zha S, Yegnasubramanian V, Nelson WG,
Isaacs WB and De Marzo AM: Cyclooxygenases in cancer: progress and
perspective. Cancer Lett. 215:1–20. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
27.
|
Alfranca A, López-Oliva JM, Genís L,
López-Maderuelo D, Mirones I, Salvado D, Quesada AJ, Arroyo AG and
Redondo JM: PGE2 induces angiogenesis via MT1-MMP-mediated
activation of the TGFbeta/Alk5 signaling pathway. Blood.
112:1120–1128. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
28.
|
Zhang Y and Daaka Y: PGE2 promotes
angiogenesis through EP4 and PKA Cγ pathway. Blood. 118:5355–5364.
2011.PubMed/NCBI
|
|
29.
|
Cui W, Yu CH and Hu KQ: In vitro and in
vivo effects and mechanisms of celecoxib-induced growth inhibition
of human hepatocellular carcinoma cells. Clin Cancer Res.
11:8213–8221. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
30.
|
Wu T, Leng J, Han C and Demetris AJ: The
cyclooxygenase-2 inhibitor celecoxib blocks phosphorylation of Akt
and induces apoptosis in human cholangiocarcinoma cells. Mol Cancer
Ther. 3:299–307. 2004.PubMed/NCBI
|
|
31.
|
Boonmasawai S, Akarasereenont P,
Techatraisak K, Thaworn A, Chotewuttakorn S and Palo T: Effects of
selective COX-inhibitors and classical NSAIDs on endothelial cell
proliferation and migration induced by human cholangiocarcinoma
cell culture. J Med Assoc Thai. 92:1508–1515. 2009.PubMed/NCBI
|
|
32.
|
Tsai WC, Hsu CC, Chou SW, Chung CY, Chen J
and Pang JH: Effects of celecoxib on migration, proliferation and
collagen expression of tendon cells. Connect Tissue Res. 48:46–51.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
33.
|
Thorat MA, Morimiya A, Mehrotra S, Konger
R and Badve SS: Prostanoid receptor EP1 expression in breast
cancer. Mod Pathol. 21:15–21. 2008. View Article : Google Scholar
|
|
34.
|
Han C and Wu T: Cyclooxygenase-2-derived
prostaglandin E2 promotes human cholangiocarcinoma cell growth and
invasion through EP1 receptor-mediated activation of the epidermal
growth factor receptor and Akt. J Biol Chem. 280:24053–24063. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
35.
|
Surh I, Rundhaug JE, Pavone A, Mikulec C,
Abel E, Simper M and Fischer SM: The EP1 receptor for prostaglandin
E2 promotes the development and progression of malignant murine
skin tumors. Mol Carcinog. 51:553–564. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
36.
|
Yang SF, Chen MK, Hsieh YS, Chung TT,
Hsieh YH, Lin CW, Su JL, Tsai MH and Tang CH: Prostaglandin E2/EP1
signaling pathway enhances intercellular adhesion molecule 1
(ICAM-1) expression and cell motility in oral cancer cells. J Biol
Chem. 285:29808–29816. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
37.
|
Yamamoto D, Sonoda Y, Hasegawa M,
Funakoshi-Tago M, Aizu-Yokota E and Kasahara T: FAK overexpression
upregulates cyclin D3 and enhances cell proliferation via the PKC
and PI3-kinase-Akt pathways. Cell Signal. 15:575–583. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
38.
|
von Sengbusch A, Gassmann P, Fisch KM,
Enns A, Nicolson GL and Haier J: Focal adhesion kinase regulates
metastatic adhesion of carcinoma cells within liver sinusoids. Am J
Pathol. 166:585–596. 2005.PubMed/NCBI
|
|
39.
|
Hess AR and Hendrix MJ: Focal adhesion
kinase signaling and the aggressive melanoma phenotype. Cell Cycle.
5:478–480. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
40.
|
Cox BD, Natarajan M, Stettner MR and
Gladson CL: New concepts regarding focal adhesion kinase promotion
of cell migration and proliferation. J Cell Biochem. 99:36–52.
2006.PubMed/NCBI
|
|
41.
|
Bosco R, Melloni E, Celeghini C, Rimondi
E, Vaccarezza M and Zauli G: Fine tuning of protein kinase C (PKC)
isoforms in cancer: shortening the distance from the laboratory to
the bedside. Mini Rev Med Chem. 11:185–199. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
42.
|
Rosse C, Linch M, Kermorgant S, Cameron
AJ, Boeckeler K and Parker PJ: PKC and the control of localized
signal dynamics. Nat Rev Mol Cell Biol. 11:103–112. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
43.
|
Wu TT, Hsieh YH, Wu CC, Hsieh YS, Huang CY
and Liu JY: Overexpression of protein kinase C alpha mRNA in human
hepatocellular carcinoma: a potential marker of disease prognosis.
Clin Chim Acta. 382:54–58. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
44.
|
Wu TT, Hsieh YH, Hsieh YS and Liu JY:
Reduction of PKC alpha decreases cell proliferation, migration, and
invasion of human malignant hepatocellular carcinoma. J Cell
Biochem. 103:9–20. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
45.
|
Siesser PM and Hanks SK: The signaling and
biological implications of FAK overexpression in cancer. Clin
Cancer Res. 12:3233–3237. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
46.
|
Lau GM, Lau GM, Yu GL, Gelman IH, Gutowski
A, Hangauer D and Fang JW: Expression of Src and FAK in
hepatocellular carcinoma and the effect of Src inhibitors on
hepatocellular carcinoma in vitro. Dig Dis Sci. 54:1465–1474. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
47.
|
Fischer OM, Hart S, Gschwind A and Ullrich
A: EGFR signal transactivation in cancer cells. Biochem Soc Trans.
31:1203–1208. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
48.
|
Buchanan FG, Wang D, Bargiacchi F and
DuBois RN: Prostaglandin E2 regulates cell migration via the
intracellular activation of the epidermal growth factor receptor. J
Biol Chem. 278:35451–35457. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
49.
|
Wang SE, Xiang B, Zent R, Quaranta V,
Pozzi A and Arteaga CL: Transforming growth factor β induces
clustering of HER2 and integrins by activating Src-FAK and receptor
association to the cytoskeleton. Cancer Res. 69:475–482. 2009.
|
|
50.
|
Aponte M, Jiang W, Lakkis M, Li MJ,
Edwards D, Albitar L, Vitonis A, Mok SC, Cramer DW and Ye B:
Activation of PAF-receptor and pleiotropic effects on tyrosine
phospho-EGFR/Src/FAK/Paxillin in ovarian cancer. Cancer Res.
68:5839–5848. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
51.
|
Calandrella SO, Barrett KE and Keely SJ:
Transactivation of the epidermal growth factor receptor mediates
muscarinic stimulation of focal adhesion kinase in intestinal
epithelial cells. J Cell Physiol. 203:103–110. 2005. View Article : Google Scholar
|