Introduction
Lung cancer is the most lethal cancer in the world.
Non-small cell lung cancer (NSCLC), the majority form of lung
cancer, is often in advanced stage when diagnosed and patients do
not have the opportunity for surgical resection. Patients did not
benefit much from traditional chemotherapy and radiotherapy;
therefore the 5-year survival rate remained poor (1). In recent years, along with the
invention of targeted therapy, more people benefit from the
treatment and survive longer.
Gefitinib is a first generation epidermal growth
factor receptor tyrosine kinase inhibitor (EGFR-TKI) which prevent
the phosphorylation of EGFR as well as the signaling transduction
(2). Clinically, only a subtype of
patients is suitable to take EGFR-TKI as the major therapy. They
were mostly Asians, females, non-smokers, adenocarcinomas and had
sensitive mutation in exons encoding the tyrosine kinase domain of
EGFR gene (3,4). However, to our disappointment, most
patients could not escape a relapse when they took the drug for a
median time of 9 months (5). A
wide array of research has been done to explore the exact mechanism
of acquired EGFR-TKI resistance. In 2005, the first case report on
T790M mutation found that patient relapsed after receiving TKI
drugs, Pao et al found that 50% of the cases progressed
because they had the T790M mutation (6,7).
Another 20% of secondary resistance was discovered in 2007 because
of the amplification of the c-Met gene (8). Still, the remaining 30% are
extensively investigated, for loss of PTEN gene, loss of
IGF-binding protein, expression of epithelial membrane protein-1
(EMP-1), and activation of downstream EGFR signaling (9–13).
Shaw et al reported in 2009 that in NSCLC patients with
metastatic disease, who harbored EML4-ALK mutation, would become
resistant to EGFR-TKI (14).
However, the majority of extensive pre-clinical studies involving
in vitro and in vivo experiments have not discovered
new drug candidate having scientific merit for further development.
Thus, we explored the untapped mechanism of acquired gefitinib
resistance so as to provide a combinatorial strategy in H1975
gefitinib-resistant cell line using gefitinib as a main
compound.
P-Akt was found elevated in many tumor tissue
samples and cancer cell lines, including esophageal cancer, lung
cancer and its expression is often related to poor clinical outcome
and indicate the resistance of therapy (15–17).
Since phosphoinositide 3-kinases (PI3K)/Akt pathway is vital in
tumor development and progression, once activated, it would affect
several downstream signaling pathways, such as proliferation,
survival, anti-apoptotic, cell cycle and angiogenesis (18,19).
Targeting the constitutively active Akt kinase and its key
downstream molecules might provide a possible anticancer effect.
Synergistic use of chemotherapy drugs with mTOR inhibitor exhibited
great inhibition influence on renal cell carcinoma and was approved
by FDA for use in patients with metastatic renal cell carcinoma
(20). However, due to complicated
interactions or compensatory upregulation of activated Akt,
combining EGFR-TKI with mTOR inhibitor in lung cancer failed to
reach the ideal result either in laboratory or in the latest
clinical trials (21,22). It is, therefore, important to
discover novel alternative Akt downstream targets which do not
cause a negative feedback loop, allowing to reverse therapeutic
resistance to TKI drugs. In 2004, Brown et al had found
reduced expression of p27 was a novel mechanism of docetaxel
resistance in breast cancer cells (23). Similarly, the downstream molecule
Bim of Akt signaling is also involved in mediating EGFR-TKI-induced
apoptosis in lung cancer cells (24). Knockdown of Bim was able to
attenuate apoptosis induced by EGFR-TKI (24).
It is well known that gefitinib induces apoptosis in
NSCLC cell line H3255 (2).
Consequently, the development of resistance of cancer cells to TKI
drugs may be a result of resistance to apoptosis. Bcl-2 is an Akt
downstream anti-apoptotic protein belonging to the Bcl-2 family. In
many tumor cells, such as breast cancer, and chronic lymphocytic
leukemia, the knockdown Bcl-2 expression can induce cell apoptosis
and even overcome drug resistance (25,26).
We wondered whether gefitinib resistance was associated with the
dysregulation of apoptosis in H1975 cell line. RNA interference is
a more effective method to silence gene expression from RNA level
to protein level compared with antisense method (27). We examined the influence of Bcl-2
small interfering RNA (siRNA) on the drug sensitization in H1975
cells. Our results revealed that the knockdown Bcl-2 gene
expression by siRNA induced cell apoptosis in H1975 lung cancer
cell line. Furthermore, gefitinib enhanced pro-apoptotic effect and
reversed acquired EGFR-TKI drug resistance. Our data predicted a
potential of a combined therapeutic approach with gefitinib and
Bcl-2 siRNA for the treatment of EGFR-TKI-resistant H1975 cell line
containing T790M mutation.
Materials and methods
Materials and reagents
The following monoclonal antibodies were used: Akt
(Epitomics, CA, USA), p-Akt (Cell Signaling Technology, Danvers,
MA, USA), Bcl-2 (Epitomics), Bax (Epitomics), caspase-3
(Epitomics), PARP-p110/85 (Epitomics), GAPDH (Epitomics). Goat
anti-rabbit peroxidase-conjugated secondary antibody (Boster,
Wuhan, China). The sequence of siRNA targeting Bcl-2 were:
si-h-BCL2_001 sense, 5′-CGG AGGCUGGGAUGCCUUUdTdT-3′; antisense,
3′-dTdTGC CUCCGACCCUACGGAAA-5′; si-h-BCL2_002 sense, 5′-GG
AUUGUGGCCUUCUUUGA dTdT-3′; antisense, 3′-dTdTCC
UAACACCGGAAGAAACU-5′; si-h-BCL2_003 sense, 5′-GG AUGACUGAGUACCUGAA
dTdT-3′; antisense, 3′-dTdT CCUACUGACUCAUGGACUU-5′. SiRNA against
Bcl-2 and a negative control siRNA were purchased from Guangzhou
RiboBio Co. Ltd. (Guangzhou, China). Lipofectamine 2000 was
obtained from Invitrogen (Carlsbad, CA, USA). Gefitinib was
generously provided by AstraZeneca and was dissolved in DMSO in
20-mM concentration stored at −20°C. Drugs were diluted in fresh
media immediately prior to use and the final DMSO concentration was
<0.1%. MTT cell viability assay kit was obtained from
Sigma-Aldrich (St. Louis, MO, USA). DAPI was purchased from
Beyotime Institute of Biotechnology (Shanghai, China). Annexin
V-FITC apoptosis detection kit was obtained from KeyGen Biotech Co.
(Nanjing, China).
Cell culture
The human lung cancer H1975 and HCC827 cell lines
were obtained from the Cell Bank of the Chinese Academy of Sciences
(Shanghai, China). H1975 cell line was cultivated in DMEM
high-glucose medium (Gibco, Grand Island, NY, USA) supplemented
with 10% fetal bovine serum (FBS) (Gibco), HCC827 cell line was
grown in RPMI-1640 medium (Gibco) supplemented with 10% fetal
bovine serum (Gibco). Cells were incubated in a humidified
incubator at 37°C with 5% CO2 atmosphere and maintained
in a logarithmic growth phase for all the experiments.
Transient transfection
To study the uptake of cy3-conjugated scrambled
negative control siRNA, after plating 2.5×104 cells/well
in 24-well plates, transfection efficiency of siRNA at various
concentrations was observed by fluorescence microscopy. We chose 50
nM which shows >80% transfection efficiency in the following
experiments. For confirmation of downregulation of Bcl-2 protein,
three siRNA oligonucleotides directed against Bcl-2 and a negative
control siRNA were tested for effectiveness of protein knockdown.
In brief, 1×105 cells were plated in 6-well plates,
incubated for 24 h and transfected with siRNA at 50 nM using
Lipofectamine 2000 reagent and OPTI-MEM reduced serum media (Gibco)
according to the manufacturer’s instructions. Later (4–6 h),
OPTI-MEM media was aspirated and DMEM fresh media containing 10%
FBS was added. At 48 h after transfection, cellular expression of
Bcl-2 was determined by western blot analysis.
MTT assay
Cell viability assay was performed with MTT cell
viability assay kit. Cells were seeded into 5 replicate wells of
each group, at a density of 6×103 viable cell per well
in 96-well plates. Allowed to attach for 24 h, transfection was
done. Forty-eight hours later, various concentrations of gefitinib
were added and cells were incubated for another 24 h. At the
indicated time, 3-(4,5-dimethylthiazol-2-yl)-2,
5-diphenyltetrazolium bromide (MTT) was added to incubate for 4 h.
Then media were aspirated, 100 μl DMSO was added. Cells were
incubated for 10 min at 37°C with gentle shaking. The absorbance
was then read at 490 nm with a 96-well micro-plate reader
(Bio-Rad). The values in the siRNA treated cells were normalized to
the values of control as to determine the percentage of viability.
Each assay was performed in triplicate. The inhibition rate (IR)
was calculated according to equation: IR = [A490 (control) − A490
(treatment)]/[A490 (control) −A490 (zero)] × 100%.
A490 (control) stands for the absorbance in control
group. A490 (treatment) was the absorbance in the drug-treated or
siRNA groups. A490 (zero) means the absorbance in the group with no
cells.
DAPI staining
Cells were seeded into 24-well plates, at a density
of 2×104 viable cells per well. After 48 h of
transfection, cells were treated with 5 μmol/l gefitinib.
Drug dose was based on the IC50 data in the cell
viability assay and available data from other studies (28). Following culture for further 24 h,
the cells were washed with phosphate buffer saline (PBS) and fixed
with 4% paraformaldehyde, then washed with PBS 3 times. Cells were
stained with 4′,6-diamidino-2-phenylindole (DAPI) for 10 min and
washed another 3 times. Cell nuclear morphology was examined by UV
fluorescent microscopy. Apoptotic cells were identified by
condensation and fragmentation of nuclear chromosome. At least 200
cells were counted and performed in a blinded manner.
Apoptosis detection
Cells (1×105) were plated in 6-well
plates in 2 ml of fresh media and cultured for 24 h. After H1975
cells had been transfected with Bcl-2 siRNA or negative siRNA for
48 h, cells were left untreated or exposed to gefitinib at a
concentration of 5 μmol/l. Twenty-four hours later, cells
attached to the growth surface were removed by trypsin treatment
[trypsin (0.05%); 37°C, 1 min]. Attached and detached cells were
collected for analysis, washed twice in cold PBS and resuspended in
500 μl of binding buffer containing 5 μl
Annexin-V-FITC and 5 μl propidium iodide mixtures according
to the manufacturer’s instructions. FACS analysis was performed on
FACScan (Becton-Dickinson Co., USA) using CellQuest software
(Becton-Dickinson), 10,000 events were collected for each
sample.
Protein extraction and
immunoblotting
Cells were collected, washed twice in ice-cold PBS
and lysed in ice-cold lysis buffer containing 20 mM Tris (pH 7.5),
150 mM NaCl, 1% Triton X-100, 2.5 mM sodium pyrophosphate, 1 mM
β-glycerophosphate, 1 mM EDTA, 1 mM Na3VO4, 1
μg/ml leupeptin with added 1 mM PMSF and 1 mM phosphatase
inhibitor. Centrifuged at 13,000 g for 5 min at 4°C and the
supernatant was saved and boiled with loading buffer for 5 min then
stored at −80°C for subsequent analysis. Protein concentration was
determined using BCA protein assay kit (Beyotime, Shanghai, China).
Equal amounts of proteins (40 μg) were loaded onto 10–12%
SDS-PAGE, separated by electrophoresis and transferred to PVDF
membrane (Millipore, USA). Membranes were then blocked with 5%
non-fat milk containing 0.1% Tween-20 at room temperature for 1 h,
incubated with primary antibodies [p-Akt (1:2,000); Akt (1:10,000);
Bcl-2 (1:500); Bax (1:1,000); caspase-3 (1:1,000); PARP p110/85
(1:1,000); GAPDH (1:10,000); 4°C, overnight] and subsequently with
goat anti-rabbit peroxidase-conjugated secondary antibody (1:2,000;
room temperature, 1 h). Peroxidase activity was visualized with
Pierce Super Signal West Pico Chemiluminescent Substrate (Pierce,
USA). Signal intensity was determined densitometrically and
normalized against those of total proteins present in the
corresponding lane on the membrane using Quality one software,
version 1.5 (Bio-Rad, CA, USA).
Statistical analysis
Statistical analysis was carried out using SPSS
software, version 17.0 (Chicago, IL, USA). Data are presented as
mean ± SD from at least three experiments. Results were analyzed
using Student’s t-test and p-values were indicated where
appropriate in the figures and in their legends. A p<0.05 was
considered statistically significant.
Results
Gefitinib-induced cytotoxicity is
substantially reduced in gefitinib-resistant H1975 cell line
compared with gefitinib-sensitive HCC827 cell line
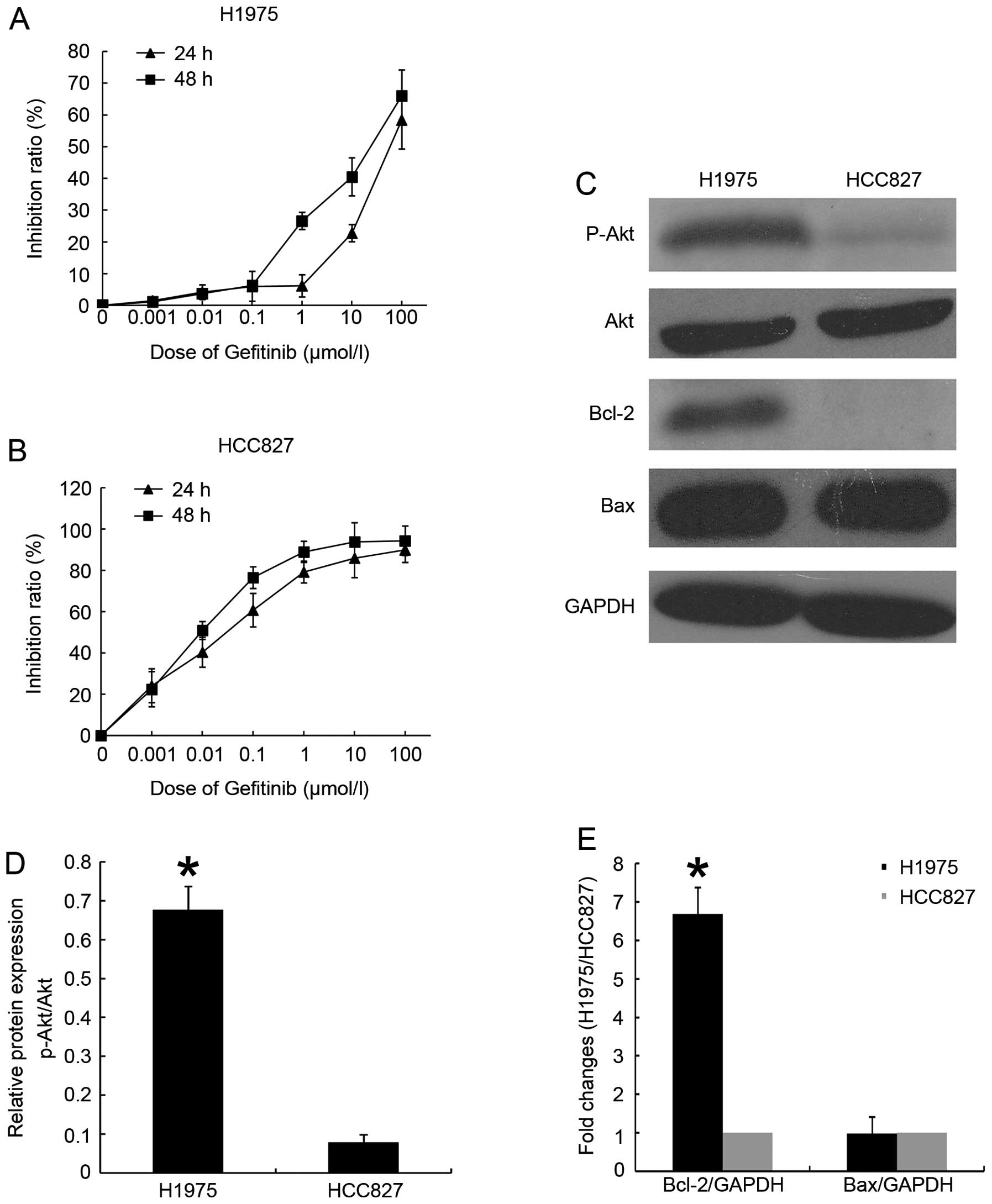
Lung cancer H1975 cell line is an established cell
line bearing T790M mutation besides L858R mutation and is being
used as a model for acquired resistance to EGFR-TKI. In this
experiment, we chose HCC827 lung cancer cell line containing exon
19 deletion as a sensitive control. To measure the growth
inhibitory action of geifitinib and to confirm each cell line as
either sensitive or resistant, we performed MTT assays. For each
cell line, cells were incubated for ≤48 h at indicated doses of
gefitinib (0, 0.001, 0.01, 0.1, 1, 10 or 100 μmol/l). The
results (Fig. 1A and B)
demonstrated that gefitinib exhibited time- and dose-dependent
inhibition effect on HCC827 cell line, and slightly inhibited the
proliferation of H1975 cells. After 24-h exposure to gefitinib,
H1975 gefitinib-resistant subtype exhibited greater resistance to
gefitinib than did HCC827 cells (IC50 35.75±0.77
μmol/l vs 0.003±0.18 μmol/l, respectively).
Akt activation and downstream protein
expression in different cell lines
To test whether PI3K/Akt signaling cascade
activation exists in cells with acquired resistance to gefitinib,
we used immunoblotting to test expression of several proteins
between gefitinib-resistant and gefitinib-sensitive cell lines. As
expected, phosphorylated Akt was elevated in H1975 cell line
compared with HCC827 cell line (Fig.
1C). Since we and others had found that gefitinib induce
apoptosis and down-regulate phosphorylated Akt expression in HCC827
cell line, we presumed that acquired gefitinib resistance might be
due to the overexpression of certain inhibitor proteins of
apoptosis which belong to the Akt downstream of signaling cascade
(2). We compared the basic level
of Bcl-2 family apoptosis-related proteins in two cell lines.
Indeed, we found abundant anti-apoptotic Bcl-2 protein in H1975
cells, whereas no evidence was detected in HCC827 cells (Fig. 1C). There was no significant
difference in the expression of pro-apoptotic Bcl-2 associated X
protein (Bax) (Fig. 1C).
Knockdown of the Bcl-2 gene in H1975 lung
cancer cell line
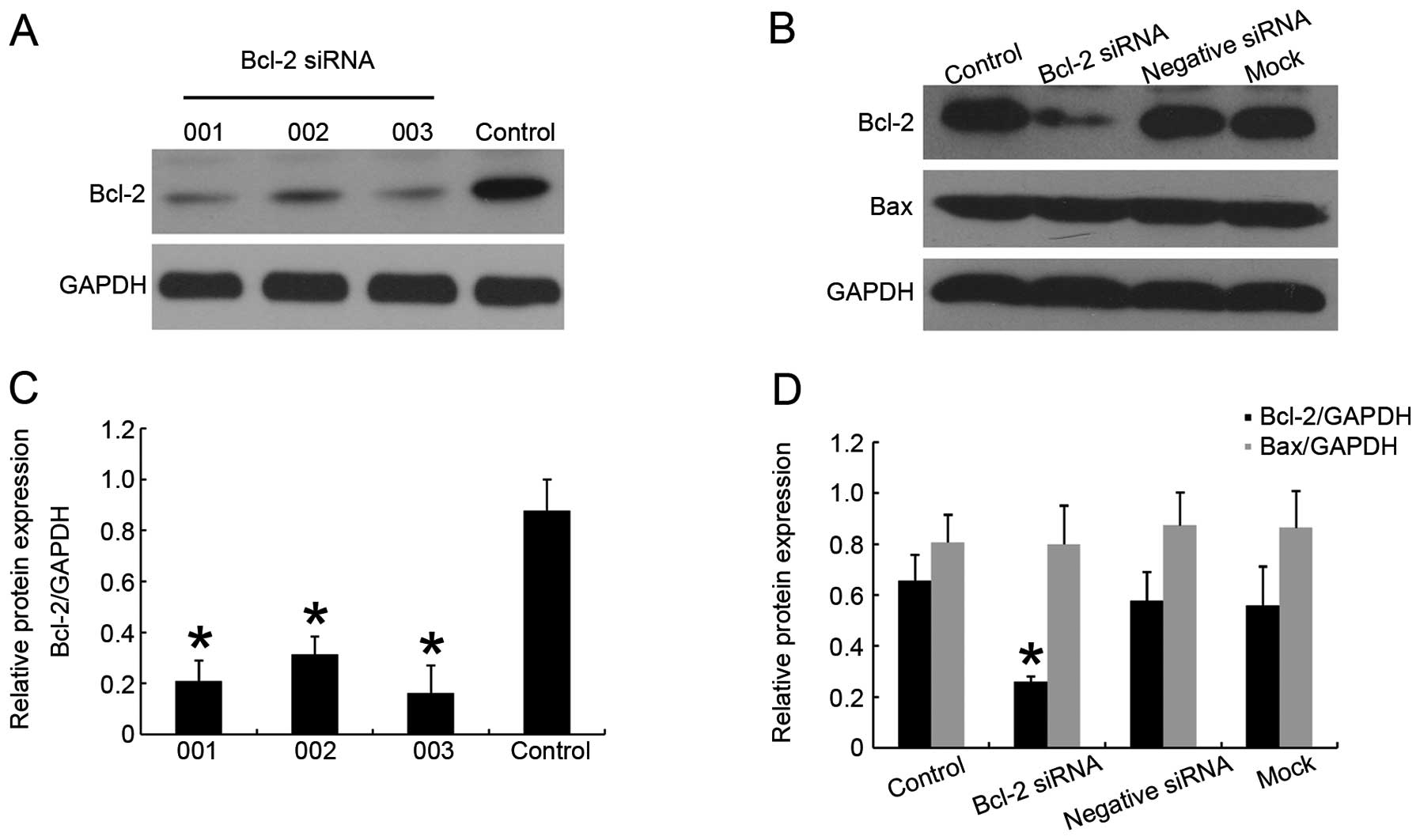
Based on the above results, we performed RNA
interference methodology to downregulate Bcl-2 protein expression
in H1975 cell line. We used cy3-conjugated siRNA to verify the
transfection efficiency though fluorescence microscopy, the
transfection efficiency in this cell line can reach >80% when
siRNA concentration is 50 nM. After 48 h of transfection, the
effect of siRNA on the protein level was assessed (Fig. 2A). A small amount of Bcl-2 in
transfected group was detected from the immunoblotting. The
suppression lasted for 72 h post-transfection. We chose
siRNA-Bcl-2_003 as the most efficacious one for the following
experiments (Fig. 2A). No Bcl-2
downregulation was detected in the negative siRNA group or in mock
transfected cells (Fig. 2B), and,
expression of protein Bax had not changed (Fig. 2B), indicating a specific and
effective function, thus excluding that the effects were due to the
upregulated Bax.
Bcl-2 siRNA increases gefitinib
sensitivity
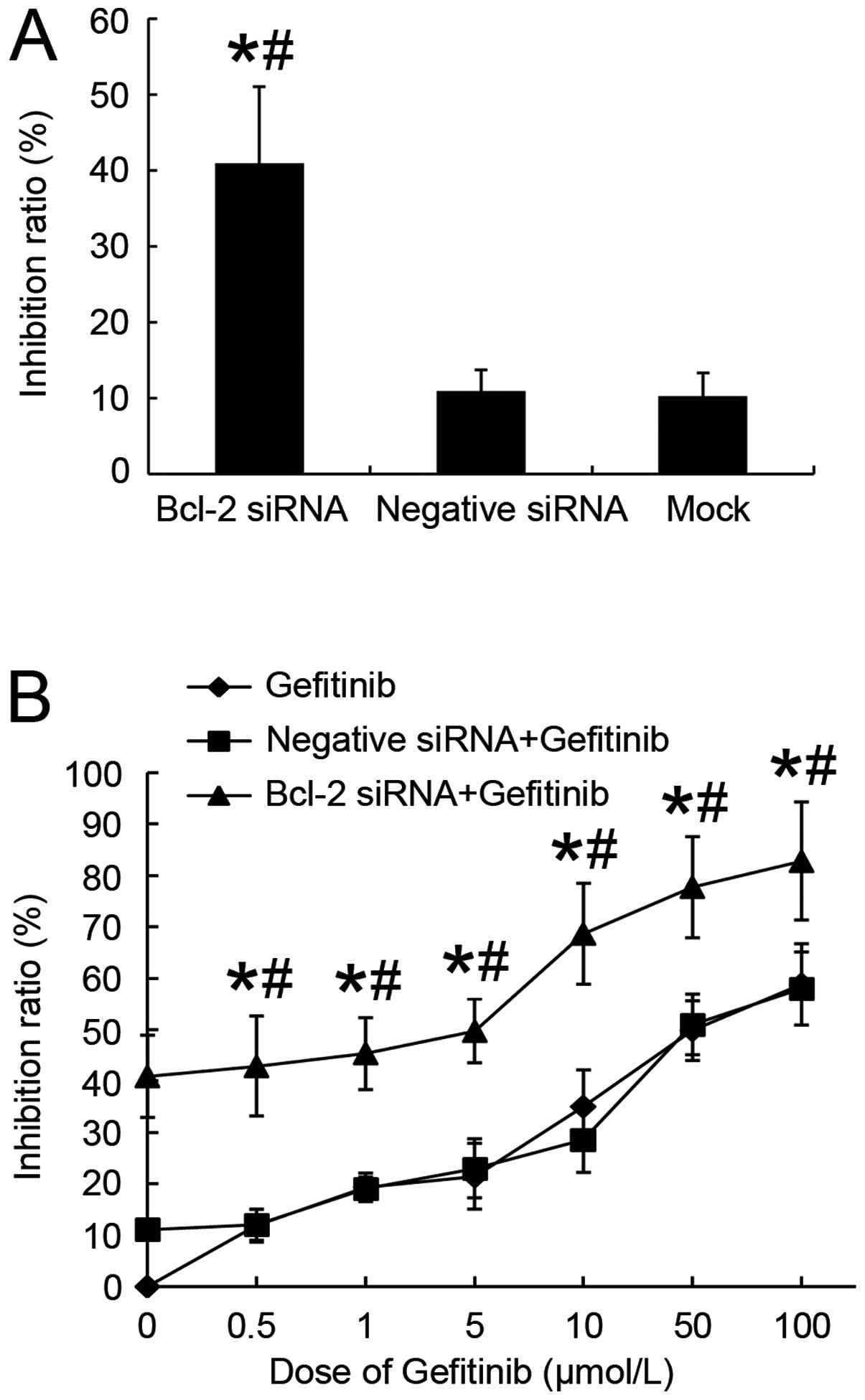
We examined the effects of siRNA mediated silencing
of Bcl-2 protein expression on the proliferation of H1975 lung
adenocarcinoma cells. After being transfected with Bcl-2 siRNA for
48 h, viable cells were reduced to 58.1% compared with the
untreated control group (Fig. 3A).
Although negative siRNA group and mock transfected cells exhibited
minor killing effect, there was no statistical significance
(p>0.05). The results showed that downregulating Bcl-2
expression was able to restore the effect of gefitinib in H1975
cells, Bcl-2 siRNA transfectants had higher IR than negative siRNA
transfectants or mock cells (p<0.05). After a 48-h period
allowing silencing of Bcl-2, H1975 cells were further exposed to a
series concentrations of gefitinib (0, 0.5, 1, 5, 10, 50 or 100
μmol/l) for 24 h. As shown in Fig. 3B, we observed that combination
treatment significantly augmented IR ∼4.6-fold compared with
control group with gefitinib (IC50 7.07±0.38 vs
32.47±0.71 μmol/l, respectively) and 4.3-fold compared with
negative siRNA plus gefitinib (IC50 7.07±0.38 vs
30.27±0.61 μmol/l, respectively). Results indicated that
combining Bcl-2 siRNA and gefitinib induced synergistic inhibitory
effect on H1975 cell proliferation and viability.
Effect of Bcl-2 siRNA on the nuclear
morphology change
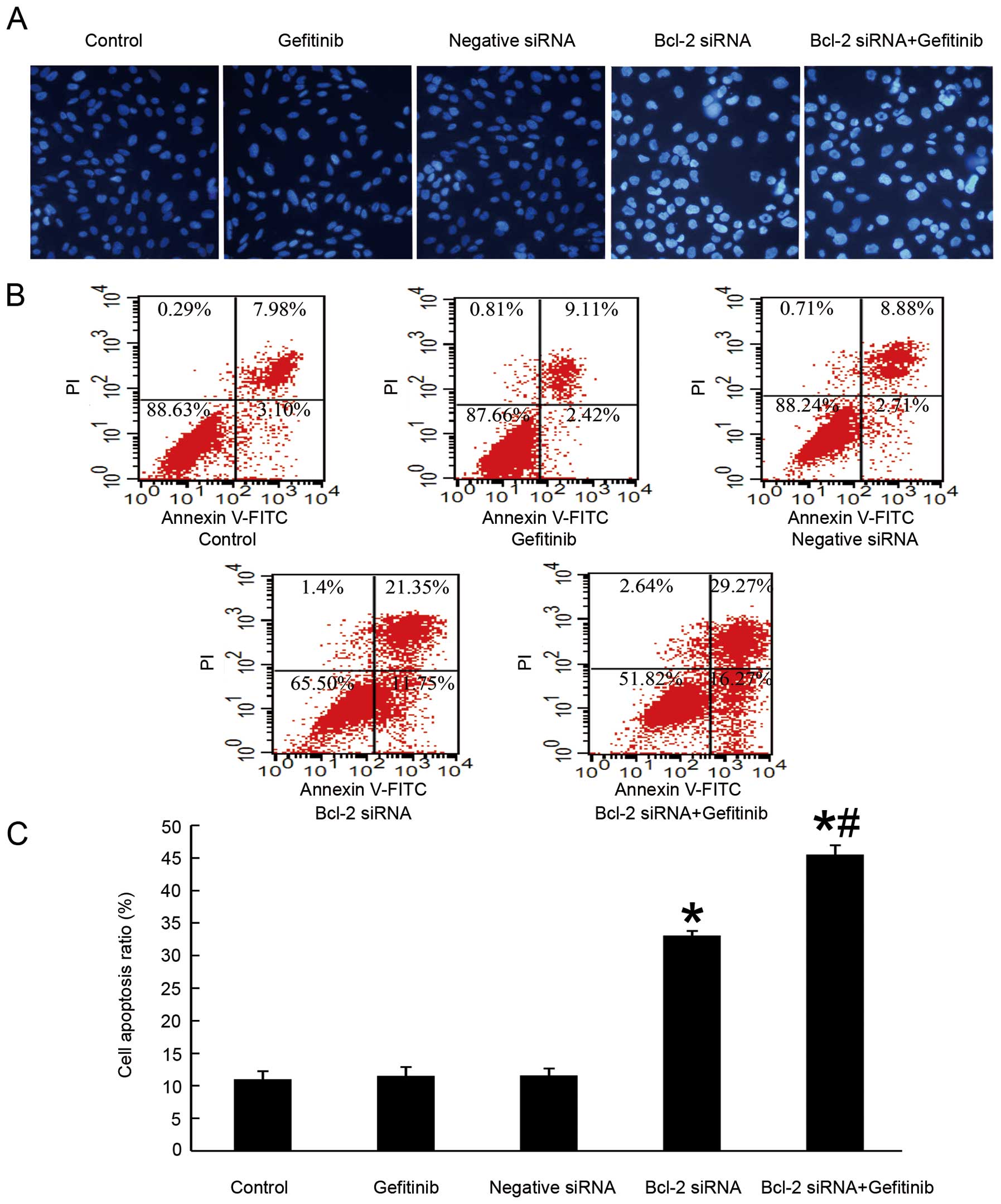
We investigated whether the anti-proliferation
effect of Bcl-2 siRNA in H1975 cells was due to the induction of
apoptosis, and whether the knockdown of Bcl-2 might sensitize the
cells to undergo apoptosis in response to gefitinib. To provide
confirmatory evidence, DAPI staining was performed to detect
nuclear morphological changes of the H1975 cells being transiently
transfected in the presence or absence of gefitinib. We chose 5
μmol/l according to the IC50 and also because
this is a concentration that can be achieved in serum of patients
being treated with gefitinib for short period (29). As shown in Fig. 4A, cells transfected with siRNA
targeting Bcl-2 for 48 h underwent characteristics of apoptosis,
including chromatin condensation and nuclear DNA fragmentation.
When followed by 5 μmol/l gefitinib administration for 24 h,
the nuclear change became more obvious. Inversely, untreated
control cells, merely suffering higher dose of gefitinib (20
μmol/l) and cells transfected with negative siRNA all showed
only slight signs of apoptosis.
Effect of Bcl-2 siRNA on cellular
apoptosis
To further investigate the exactly apoptosis rate
among different groups, we performed flow cytometry. The
Annexin-V-FITC assay was aimed to distinguish whether the reduction
in cell viability was due to apoptosis or necrosis. As seen in
Fig. 4B, after Bcl-2 siRNA
treatment, the rate of early apoptotic cells increased from 7.98 in
control group to 21.35%. This difference is statistically
significant (p<0.05). The percentage of late apoptotic cells was
also augmented. To determine whether the time to add gefitinib
impacted on the synergistic effect, we evaluated different groups
according to the time-points that gefitinib was added. Cells were
treated with 5 μmol/l gefitinib at the time of transfection
end, 24 h later or 48 h after transfection. When followed by 5
μmol/l gefitinib addition 48 h after transfection, the early
apoptotic fraction increased to a greater extent, from 21.35 in the
absence to 29.27% in the presence. Our results showed only the
group that added gefitinib ≥48 h after transfection had synergistic
function. This synergistic effect could not be seen in other groups
when gefitinib was added at an earlier time either immediately
after transfection or 24 h later. Results above suggested that
knockdown of Bcl-2 protein expression in H1975 lung cancer cell
line can suppress cell proliferation via apoptosis induction. It
also demonstrated that synergy between Bcl-2 siRNA and gefitinib
was augmented by optimizing drug scheduling with superior effects
when gefitinib was administered 48 h after transfection at the time
that Bcl-2 had been successfully knocked down. The data coincided
with the hypothesis that Bcl-2 expression level is associated to
the sensitivity of EGFR-TKI drugs.
Effect of Bcl-2 siRNA activated caspase-3
and increased PARP cleavage through regulating intrinsic
mitochondrial pathways of apoptosis
The pro-apoptotic effect of Bcl-2 siRNA was further
confirmed by examining the total and cleavage protein of caspase-3
and its downstream substrate PARP. As seen in Fig. 5A once cells were treated with Bcl-2
siRNA will activate procaspase-3 then induce 17-kDa active
caspase-3, PARP was cleaved from its 116-kDa intact form into
85-kDa cleaved fragment. Cleaved PARP which is a marker of
caspase-mediated early apoptosis was increased in Bcl-2 siRNA
treated cells and those followed by gefitinib treatment after
transfection (30). Inversely, not
much cleaved PARP could be seen in the other groups. From the
experiment, we observed onset of the Bcl-2 siRNA process of
intrinsic mitochondrial way of caspase-dependent apoptosis which
was consistent with the finding in other cell lines (31,32).
We found that gefitinib alone and Bcl-2 siRNA alone did not inhibit
or stimulate Akt phosphorylation in H1975 cell line which indicated
that no direct feedback loop exists between Bcl-2 and p-Akt;
however, when siRNA targeting Bcl-2 followed by addition of 5
μmol/l gefitinib, the phosphorylation of Akt was reduced
while the amount of total Akt expressed was consistent with the
treatment results of Bcl-2 siRNA or gefitinib.
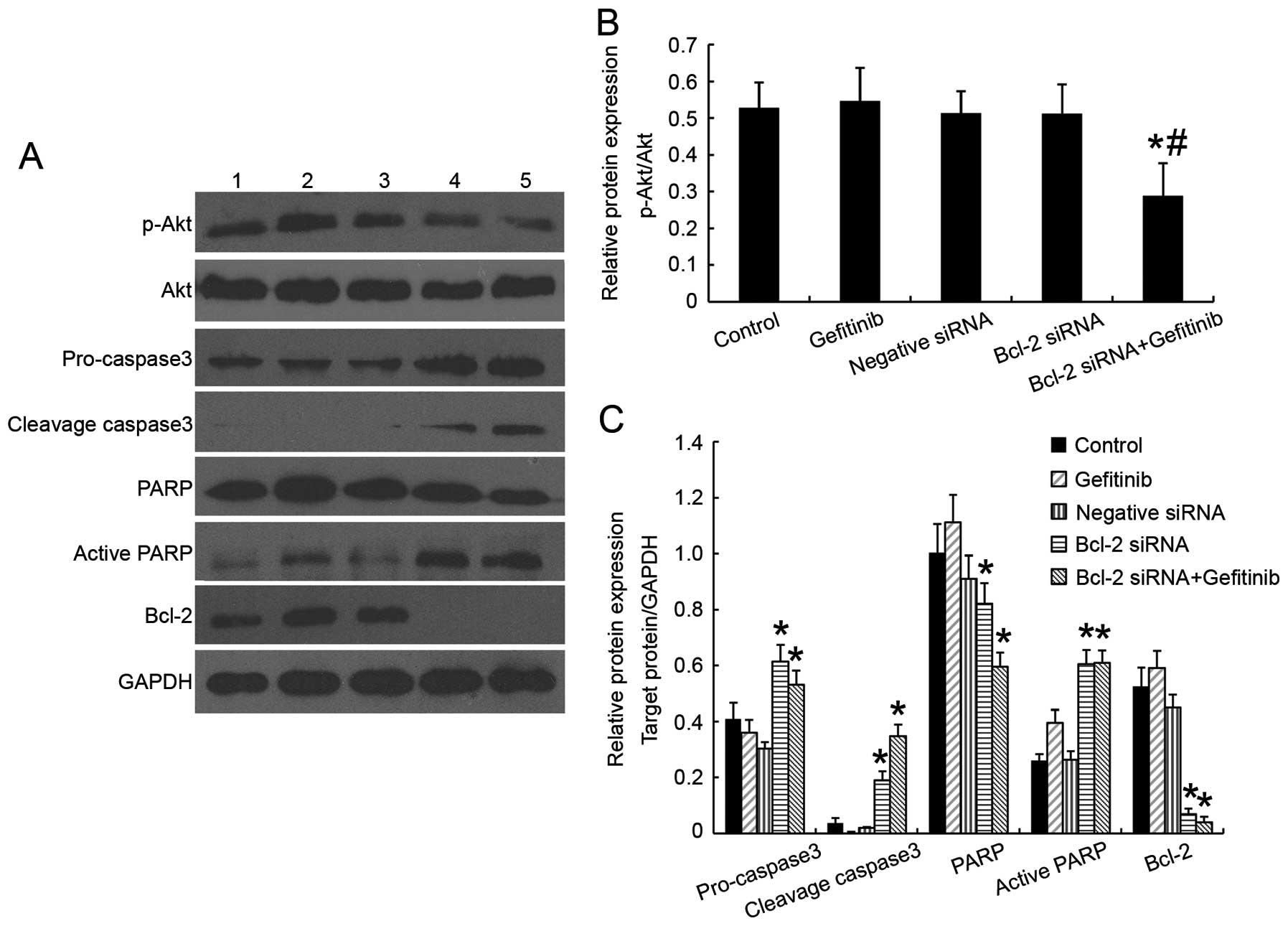 | Figure 5Effects of Bcl-2 siRNA with or
without gefitinib on Akt activation and apoptosis protein
expression in H1975 cell line. (A) Western blotting was used to
detect p-Akt, Akt, caspase-3, PARP and Bcl-2 protein expression.
Lane 1, cells without treatment (control); lane 2, cells with 20
μmol/l gefitinib for 24 h (gefitinib); lane 3, negative
siRNA transfectants (negative siRNA); lane 4, Bcl-2 siRNA
transfectants (Bcl-2 siRNA); lane 5, Bcl-2 siRNA transfected
followed by 5 μmol/l gefitinib for 24 h (Bcl-2
siRNA+gefitinib). Bcl-2 protein expression was remarkably reduced
in Bcl-2 siRNA transfected groups. Active caspase-3 and active PARP
was increased in Bcl-2 siRNA transfected groups. Phosphorylation of
Akt decreased only when Bcl-2 siRNA was combined with incubation of
5 μmol/l gefitinib for 24 h. (B) Relative protein expression
level of p-Akt compared with total Akt. *p<0.05 vs.
control group while #p<0.05 vs. Bcl-2 siRNA group.
(C) Relative protein expression level of pro-caspase-3, cleaved
caspase-3, PARP, cleaved PARP and Bcl-2 compared with GAPDH.
*p<0.05 vs. control group. The experiments were
carried out in triplicate and representative data are shown. |
Discussion
Activated Akt expression manifests poor clinical
outcome and confer chemotherapy and radiotherapy resistance in many
cancer types including NSCLC, and neuronal cells (16,33).
Consistent with previous literature, our results show that compared
with TKI-sensitive HCC827 lung cancer cell line, the level of p-Akt
in TKI-resistant H1975 lung cancer cell line was elevated. In the
former studies, it was found that combining EGFR-TKI with specific
inhibitors of the Ras or PI3K pathways exerted different effects in
several lung cancer cell types, which might correlate with the
diverse expression of downstream proteins (34). Compared with LY294002 which is a
potent inhibitor of PI3Ks, bortezomib, a proteasome inhibitor used
in an established EGFR-TKI-resistant cell line, induced a
significant inhibition of cancer cell growth and increased
apoptosis suggesting that besides interfering with Akt signaling,
other molecular mechanisms were involved in overcoming resistance
to anti-EGFR therapies (35).
Deregulation of the Akt-dependent pathway has been
well documented in a variety of human tumors (23,24).
We were eager to know whether the activation of Akt and its
downstream cascade differs in NSCLC cell lines with different
response to EGFR-TKI. We focused on apoptosis pathway downstream of
Akt aiming to avoid the negative feedback induced by mTOR
inhibition (36). We postulated
that tyrosine kinase inhibitor resistance in lung cancer H1975 cell
line bearing T790M mutation might result from elevated p-Akt and
the deregulation of apoptosis cascade. The reasons for apoptosis
evation can be divided as follows: i) overexpression of
anti-apoptotic protein, such as Bcl-2, Bcl-xl, ii) lack of
pro-apoptotic protein, such as Bax, Bad, iii) higher expression of
surviving genes, for instance, survivin (37). In our experiments, we examined
protein level of apoptosis-related proteins and found there was no
distinction in Bax protein expression but a distinguishable
difference in Bcl-2 protein expression between two cell lines of
NSCLC on the basis of obvious discrepancy in sensitivity to
gefitinib. Our results were in line with previous findings that
Bcl-2 protein expression level would increase when cells were
treated with certain chemotherapy drugs and became resistant
(38,39). The finding was unexpected since
Bcl-2 is rarely expressed in NSCLC, unlike the level of Bcl-2 that
is abundant in small cell lung cancer (SCLC) (40). We thus hypothesized that
substantial Bcl-2 might be related to gefitinib resistance. Therapy
targeted at anti-apoptotic protein from Bcl-2 family might
reactivate the apoptosis signaling pathway.
The function of Bcl-2 in Bcl-2 overexpressing tumors
such as lymphomas and SCLC have been explored. Clinically, chronic
lymphocytic leukemia (CLL) is suitable for targeting by imatinib. A
great body of basic experiments investigated the possible mechanism
of acquired drug-resistance to find new approaches to resensitize
the malignant cells to the treatment. Bcl-2 overexpression is a
hallmark in CLL refractory patients (41). Phase I study of Navitoclax
(ABT-263) or pan Bcl-2 family inhibitor Obatoclax in CLL patients
with relapsed disease showed that Bcl-2 could be a valid
therapeutic target (41,42). Similar phase I clinical studies had
been done in SCLC, the Bcl-2 family genes were greatly amplified
and had achieved good results with acceptable adverse effect
(43,44). Similar results were confirmed in
hepatocarcinoma cells resistant to LCL161 with a combination of
Bcl-2 siRNA and LCL161 (45). In
our study, data confirmed that the cytotoxic and pro-apoptosis
effects in H1975 cell line induced by Bcl-2 siRNA alone could be
enhanced when administered together with gefitinib thus reversing
acquired EGFR-TKI resistance.
Our data agree with that reported by Fan and
co-workers who demonstrated that the established early TKI
resistant cell lines exhibited dependence on activation of
Bcl-2/Bcl-xl signaling (46). They
also found that when using Bcl-2 homology domain 3 mimetic agents
ABT-737 could eradicate the tumor cells evading TKI drugs. Contrary
to us, they found that mere Bcl-2 siRNA could not induce dramatic
reduction in the early TKI-resistant tumor cells. Dual knockdown of
Bcl-2/Bcl-xl manifested more advantageously in cytotoxicity
comparing to single intervention. We propose the disparity might
lie in the different cell models, the very ‘early’ molecular events
found in their study were probably not the same with the protein
changes discovered by us in established cell lines.
However, the precise mechanism for increased Bcl-2
expression in H1975 cell line is still unclear. One potential
mechanism may be that micro-RNAs involved in regulating target
genes include Bcl-2 (47–49). A panel of miRNAs (miR-16, miR-143)
suppressed by estradiol (E2) dramatically induce Bcl-2 expression
in breast cancer cells (47). Wang
et al determined that miR-181d may act as a glioma
suppressor by targeting K-ras and Bcl-2 (48). Similar observation was reported
where miR-136 was downregulated in human glioma and lost the
capacity to repress anti-apoptotic genes, AEG-1 and Bcl-2 (49). In addition, hepatitis B virus
pre-S2 large mutant surface antigen (HBV pre-S2D) increased Bcl-2
expression in hepatocellular carcinoma cells (50). Therefore, further examination of
the specific mechanism that Bcl-2 overexpressed in H1975 lung
cancer cells are undertaken in our laboratory.
Only a few studies have focused on combining Bcl-2
siRNA with targeted therapy except in CLL. In this study, we
confirmed the effect of Bcl-2 siRNA on EGFR-TKI acquired resistant
H1975 lung cancer cell line. Although we have verified that Bcl-2
was vital for cell proliferation, anti-apoptotic, inducing acquired
drug resistance in H1975 cell line, there are still certain
limitations which need further clarification. However,
notwithstanding the limitation, this study does suggest the effect
of Bcl-2 siRNA on sensitizing TKI acquired resistant cell line to
gefitinib.
In conclusion, endogenous level of Bcl-2 can predict
the cell sensitivity to EGFR-TKI gefitinib and is an important
determinant in gefitinib resistance. Our in vitro study
suggested that knockdown the expression of Bcl-2 by siRNA can
reverse drug resistance to gefitinib in H1975 lung cancer cell line
harboring T790M mutation. Thus, these findings provide a new
concept for the development of novel therapeutic approaches in the
treatment of refractory and relapsed patients who are no longer
sensitive to EGFR-TKI drugs. Combination of common-used therapy
with one targeted to downstream blocking may raise new hope for
cure.
Abbreviations:
|
NSCLC
|
non-small cell lung cancer
|
|
SCLC
|
small cell lung cancer
|
|
EGFR-TKI
|
epidermal growth factor receptor
tyrosine kinase inhibitor
|
|
PI3K
|
phosphoinositide 3-kinases
|
|
siRNA
|
small interfering RNA
|
|
FBS
|
fetal bovine serum
|
|
MTT
|
3-(4,5-dimethylthiazol-2-yl)-2,
5-diphenyltetrazolium bromide
|
|
IR
|
inhibition rate
|
|
PBS
|
phosphate buffer saline
|
|
DAPI
|
4′,6-diamidino-2-phenylindole
|
|
CLL
|
chronic lymphocytic leukemia
|
Acknowledgements
This study was supported by National
Natural Science Foundation of China (no. 81172187) and in part by
grant from the Wujieping Foundation, China (no.
320.6720.10010).
References
|
1
|
Siegel R, Naishadham D and Jemal A: Cancer
statistics, 2012. CA Cancer J Clin. 62:10–29. 2012. View Article : Google Scholar
|
|
2
|
Tracy S, Mukohara T, Hansen M, Meyerson M,
Johnson BE and Janne PA: Gefitinib induces apoptosis in the
EGFRL858R non-small-cell lung cancer cell line H3255. Cancer Res.
64:7241–7244. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Lynch TJ, Bell DW, Sordella R, et al:
Activating mutations in the epidermal growth factor receptor
underlying responsiveness of non-small-cell lung cancer to
gefitinib. N Engl J Med. 350:2129–2139. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Pao W, Miller V, Zakowski M, et al: EGF
receptor gene mutations are common in lung cancers from ‘never
smokers’ and are associated with sensitivity of tumors to gefitinib
and erlotinib. Proc Natl Acad Sci USA. 101:13306–13311. 2004.
|
|
5
|
Sequist LV, Martins RG, Spigel D, et al:
First-line gefitinib in patients with advanced non-small-cell lung
cancer harboring somatic EGFR mutations. J Clin Oncol.
26:2442–2449. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kobayashi S, Boggon TJ, Dayaram T, et al:
EGFR mutation and resistance of non-small-cell lung cancer to
gefitinib. N Engl J Med. 352:786–792. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Pao W, Miller VA, Politi KA, et al:
Acquired resistance of lung adenocarcinomas to gefitinib or
erlotinib is associated with a second mutation in the EGFR kinase
domain. PLoS Med. 2:e732005. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Engelman JA, Zejnullahu K, Mitsudomi T, et
al: MET amplification leads to gefitinib resistance in lung cancer
by activating ERBB3 signaling. Science. 316:1039–1043. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Bianco R, Shin I, Ritter CA, et al: Loss
of PTEN/MMAC1/TEP in EGF receptor-expressing tumor cells
counteracts the antitumor action of EGFR tyrosine kinase
inhibitors. Oncogene. 22:2812–2822. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Sos ML, Koker M, Weir BA, et al: PTEN loss
contributes to erlotinib resistance in EGFR-mutant lung cancer by
activation of Akt and EGFR. Cancer Res. 69:3256–3261. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Guix M, Faber AC, Wang SE, et al: Acquired
resistance to EGFR tyrosine kinase inhibitors in cancer cells is
mediated by loss of IGF-binding proteins. J Clin Invest.
118:2609–2619. 2008.PubMed/NCBI
|
|
12
|
Jain A, Tindell CA, Laux I, et al:
Epithelial membrane protein-1 is a biomarker of gefitinib
resistance. Proc Natl Acad Sci USA. 102:11858–11863. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Uchida A, Hirano S, Kitao H, et al:
Activation of downstream epidermal growth factor receptor (EGFR)
signaling provides gefitinib-resistance in cells carrying EGFR
mutation. Cancer Sci. 98:357–363. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Shaw AT, Yeap BY, Mino-Kenudson M, et al:
Clinical features and outcome of patients with non-small-cell lung
cancer who harbor EML4-ALK. J Clin Oncol. 27:4247–4253. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Tang JM, He QY, Guo RX and Chang XJ:
Phosphorylated Akt overexpression and loss of PTEN expression in
non-small cell lung cancer confers poor prognosis. Lung Cancer J
Iaslc. 51:181–191. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Scrima M, De Marco C, Fabiani F, et al:
Signaling networks associated with AKT activation in non-small cell
lung cancer (NSCLC): new insights on the role of
phosphatydil-inositol-3 kinase. PLoS One. 7:e304272012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Brognard J, Clark AS, Ni Y and Dennis PA:
Akt/protein kinase B is constitutively active in non-small cell
lung cancer cells and promotes cellular survival and resistance to
chemotherapy and radiation. Cancer Res. 61:3986–3997.
2001.PubMed/NCBI
|
|
18
|
Janmaat ML, Kruyt FA, Rodriguez JA and
Giaccone G: Response to epidermal growth factor receptor inhibitors
in non-small cell lung cancer cells: limited antiproliferative
effects and absence of apoptosis associated with persistent
activity of extracellular signal-regulated kinase or Akt kinase
pathways. Clin Cancer Res. 9:2316–2326. 2003.
|
|
19
|
Yao M, Zhang W, Zhang Q, et al:
Overexpression of MUC1 enhances proangiogenic activity of
non-small-cell lung cancer cells through activation of Akt and
extracellular signal-regulated kinase pathways. Lung. 189:453–460.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Motzer RJ, Escudier B, Oudard S, et al:
Efficacy of everolimus in advanced renal cell carcinoma: a
double-blind, randomised, placebo-controlled phase III trial.
Lancet. 372:449–456. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Nakachi I, Naoki K, Soejima K, et al: The
combination of multiple receptor tyrosine kinase inhibitor and
mammalian target of rapamycin inhibitor overcomes erlotinib
resistance in lung cancer cell lines through c-Met inhibition. Mol
Cancer Res. 8:1142–1151. 2010. View Article : Google Scholar
|
|
22
|
Price KA, Azzoli CG, Krug LM, et al: Phase
II trial of gefitinib and everolimus in advanced non-small cell
lung cancer. J Thorac Oncol. 5:1623–1629. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Brown I, Shalli K, McDonald SL, et al:
Reduced expression of p27 is a novel mechanism of docetaxel
resistance in breast cancer cells. Breast Cancer Res. 6:R601–R607.
2004. View
Article : Google Scholar : PubMed/NCBI
|
|
24
|
Costa DB, Halmos B, Kumar A, et al: BIM
mediates EGFR tyrosine kinase inhibitor-induced apoptosis in lung
cancers with oncogenic EGFR mutations. PLoS Med. 4:1669–1679.
16802007.PubMed/NCBI
|
|
25
|
Lima RT, Martins LM, Guimaraes JE, Sambade
C and Vasconcelos MH: Specific downregulation of bcl-2 and xIAP by
RNAi enhances the effects of chemotherapeutic agents in MCF-7 human
breast cancer cells. Cancer Gene Ther. 11:309–316. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Tumilasci VF, Oliere S, Nguyen TL, Shamy
A, Bell J and Hiscott J: Targeting the apoptotic pathway with BCL-2
inhibitors sensitizes primary chronic lymphocytic leukemia cells to
vesicular stomatitis virus-induced oncolysis. J Virol.
82:8487–8499. 2008. View Article : Google Scholar
|
|
27
|
Aoki Y, Cioca DP, Oidaira H, Kamiya J and
Kiyosawa K: RNA interference may be more potent than antisense RNA
in human cancer cell lines. Clin Exp Pharmacol Physiol. 30:96–102.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Witta SE, Gemmill RM, Hirsch FR, et al:
Restoring E-cadherin expression increases sensitivity to epidermal
growth factor receptor inhibitors in lung cancer cell lines. Cancer
Res. 66:944–950. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Baselga J, Rischin D, Ranson M, et al:
Phase I safety, pharmacokinetic, and pharmacodynamic trial of
ZD1839, a selective oral epidermal growth factor receptor tyrosine
kinase inhibitor, in patients with five selected solid tumor types.
J Clin Oncol. 20:4292–4302. 2002. View Article : Google Scholar
|
|
30
|
Bursztajn S, Feng JJ, Berman SA and Nanda
A: Poly (ADP-ribose) polymerase induction is an early signal of
apoptosis in human neuroblastoma. Brain Res Mol Brain Res.
76:363–376. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wu XH, Lu Y, Fang YW and Jiang YX: The
polyamidoamine-mediated inhibition of bcl-2 by small hairpin RNA to
induce apoptosis in human lens epithelial cells. Mol Vis. 18:74–80.
2012.PubMed/NCBI
|
|
32
|
George J, Banik NL and Ray SK: Combination
of taxol and Bcl-2 siRNA induces apoptosis in human glioblastoma
cells and inhibits invasion, angiogenesis and tumour growth. J Cell
Mol Med. 13:4205–4218. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Dudek H, Datta SR, Franke TF, et al:
Regulation of neuronal survival by the serine-threonine protein
kinase Akt. Science. 275:661–665. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Janmaat ML, Rodriguez JA, Gallegos-Ruiz M,
Kruyt FA and Giaccone G: Enhanced cytotoxicity induced by gefitinib
and specific inhibitors of the Ras or phosphatidyl inositol-3
kinase pathways in non-small cell lung cancer cells. Int J Cancer.
118:209–214. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Morgillo F, D’Aiuto E, Troiani T, et al:
Antitumor activity of bortezomib in human cancer cells with
acquired resistance to anti-epidermal growth factor receptor
tyrosine kinase inhibitors. Lung Cancer J Iaslc. 71:283–290. 2011.
View Article : Google Scholar
|
|
36
|
Zitzmann K, Ruden J, Brand S, et al:
Compensatory activation of Akt in response to mTOR and Raf
inhibitors - a rationale for dual-targeted therapy approaches in
neuroendocrine tumor disease. Cancer Lett. 295:100–109. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Han SW and Roman J: Targeting apoptotic
signaling pathways in human lung cancer. Curr Cancer Drug Targets.
10:566–574. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Losert D, Pratscher B, Soutschek J, et al:
Bcl-2 downregulation sensitizes nonsmall cell lung cancer cells to
cisplatin, but not to docetaxel. Anticancer Drugs. 18:755–761.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Yoo SH, Yoon YG, Lee JS, et al: Etoposide
induces a mixed type of programmed cell death and overcomes the
resistance conferred by Bcl-2 in Hep3B hepatoma cells. Int J Oncol.
41:1443–1454. 2012.PubMed/NCBI
|
|
40
|
Lawson MH, Cummings NM, Rassl DM, et al:
Bcl-2 and beta1-integrin predict survival in a tissue microarray of
small cell lung cancer. Br J Cancer. 103:1710–1715. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Roberts AW, Seymour JF, Brown JR, et al:
Substantial susceptibility of chronic lymphocytic leukemia to BCL2
inhibition: results of a phase I study of navitoclax in patients
with relapsed or refractory disease. J Clin Oncol. 30:488–496.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Schimmer AD, O’Brien S, Kantarjian H, et
al: A phase I study of the pan bcl-2 family inhibitor obatoclax
mesylate in patients with advanced hematologic malignancies. Clin
Cancer Res. 14:8295–8301. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Gandhi L, Camidge DR, Ribeiro DOM, et al:
Phase I study of Navitoclax (ABT-263), a novel Bcl-2 family
inhibitor, in patients with small-cell lung cancer and other solid
tumors. J Clin Oncol. 29:909–916. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Chiappori AA, Schreeder MT, Moezi MM, et
al: A phase I trial of pan-Bcl-2 antagonist obatoclax administered
as a 3-h or a 24-h infusion in combination with carboplatin and
etoposide in patients with extensive-stage small cell lung cancer.
Br J Cancer. 106:839–845. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Chen KF, Lin JP, Shiau CW, et al:
Inhibition of Bcl-2 improves effect of LCL161, a SMAC mimetic, in
hepatocellular carcinoma cells. Biochem Pharmacol. 84:268–277.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Fan W, Tang Z, Yin L, et al:
MET-independent lung cancer cells evading EGFR kinase inhibitors
are therapeutically susceptible to BH3 mimetic agents. Cancer Res.
71:4494–4505. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Yu X, Zhang X, Dhakal IB, Beggs M,
Kadlubar S and Luo D: Induction of cell proliferation and survival
genes by estradiol-repressed microRNAs in breast cancer cells. BMC
Cancer. 12:292012. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Wang XF, Shi ZM, Wang XR, et al: MiR-181d
acts as a tumor suppressor in glioma by targeting K-ras and Bcl-2.
J Cancer Res Clin Oncol. 138:573–584. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Yang Y, Wu J, Guan H, et al: MiR-136
promotes apoptosis of glioma cells by targeting AEG-1 and Bcl-2.
FEBS Lett. 586:3608–3612. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Hung JH, Teng YN, Wang LH, et al:
Induction of Bcl-2 expression by hepatitis B virus pre-S2 mutant
large surface protein resistance to 5-fluorouracil treatment in
Huh-7 cells. PLoS One. 6:e289772011. View Article : Google Scholar : PubMed/NCBI
|