Introduction
Chemical-analytical studies have led to the
identification of ~3,000 compounds in tobacco. These include
carcinogens in processed tobacco as well as tumor initiators, tumor
promoters, cocarcinogens and organ-specific carcinogens in tobacco
smoke. Some of these compounds such as 4-(methylnitro
samino)-1-(3-pyridyl)-1-butanone and N′-nitrosonornicotine are
nicotine derivatives and highly carcinogenic (1). Nicotine is the major addictive
component of tobacco smoke. Although nicotine is generally thought
to have limited ability to initiate cancer, it can induce cell
proliferation and angiogenesis in a variety of systems. A recent
finding suggested that nicotine may at least be partially involved
in the initiation, promotion and even progression of tumors
(2). However, the effect of
nicotine on cancer cell invasion is still not clear.
Epidemiological studies have shown that the
consumption of green tea decreases the risk of developing human
cancers (3). The anti-carcinogenic
and anti-proliferative effects of green tea have been attributed to
the biological activities of its polyphenol components. Green tea
extract contains (−)-epicatechin (EC), (−)-epicatechin gallate
(ECG), (−)-epigallocatechin (EGC) and
(−)-epigallocatechin-3-gallate (EGCG) (4). EGCG, the most abundant polyphenol in
green tea, has been considered to be the major chemopreventive
constituent of tea and has been the focus of a great deal of
attention. EGCG inhibits proliferation and induces apoptosis of
tumor cells (5,6). The exact mechanisms underlying the
anticarcinogenic activity of tea are not well defined and warrant
further study. In addition to having a cancer chemopreventive
activity, polyphenols have been shown to inhibit tumor invasion,
which is a crucial step for the metastasis of all solid tumors. It
was proposed recently that the anticancer activity of EGCG is
associated with the inhibition of invasion by inhibiting the matrix
metalloproteinases (MMPs) (7),
activity of urokinase (8) and
removal of oxygen radicals (9),
all of which play key roles in cancer invasion and metastasis.
Previously, we reported that mice treated with EGCG resulted in
marked inhibition of vascularity and proliferation of human colon
cancer xenografts model (10).
Cancer invasion and metastasis are multistep
processes and require the coordinated action of cell-secreted
proteolytic enzymes and their inhibitors. Matrix metalloproteinases
(MMPs) are a family of zinc containing enzymes which are involved
in the degradation of different components of the extracellular
matrix and there is considerable evidence indicating that
individual MMPs have important roles in tumor invasion and spread
(11). Of the MMPs, MMP-9
(gelatinase B) cleaves matrix substrates including gelatin and
collagen types IV, V and VII (12). In addition, several extracellular
stimuli have been reported to regulate the activities of MMPs in
various cell types (13,14). In cancer, it has recently been
reported that MMP-9 is related to the initial step of cancer cell
invasion and that the plasma level of MMP-9 in patients correlates
with the tumor metastatic potential. As reported in related studies
(15,16), it is clear that MMP-mediated
degradation of extracellular matrix is a hallmark in several
pathologic conditions such as arthritis, inflammation, cancer,
angiogenesis, cardiovascular, pulmonary, ocular, gastrointestinal
and oral diseases. Invasion and metastasis, both fundamental
properties of malignant cancer cells, are the end result of a
complex series of steps involving multiple tumor-host interactions
(17,18). As a result, cancer cell migration
and invasion of surrounding tissues are mediated in part by MMPs,
especially MMP-9 (19,20).
In this study, the inhibitory effects of EGCG and
its molecular mechanism on nicotine-induced MMP-9 and cell
invasiveness in human ECV304 endothelial cells were examined.
Materials and methods
Cell culture and culture conditions
Human endothelial ECV304 cells obtained from the
American Type Culture Collection (Manassas, VA, USA) were cultured
in Dulbecco’s modified Eagle’s medium, supplemented with 10% fetal
bovine serum (FBS) and 1% penicillin-streptomycin at 37°C in an
atmosphere containing 5% CO2. To determine the effects
of nicotine on MMP-9 expression, cells treated with nicotine at
various concentrations were harvested and the level of MMP-9
messenger RNA (mRNA), protein and promoter activity were
determined. The role of EGCG (Sigma-Aldrich, St. Louis, MO, USA) in
the nicotine-induced expression of MMP-9 was examined by
pretreating the ECV304 cells with EGCG for 1 h before exposure to
nicotine.
Reverse transcription-PCR
Total RNA was extracted from the ECV304 cells using
TRIzol reagent (Invitrogen, Carlsbad, CA, USA). One microgram of
total RNA was used for first-strand complementary DNA synthesis
using random primers and the SuperScript reverse transcriptase
(Invitrogen). The complementary DNA was subjected to PCR
amplification with the primer sets for glyceraldehyde 3-phosphate
dehydrogenase (GAPDH), MMP-9, c-fos and c-jun. The specific primer
sequences were as follows: GAPDH sense, 5′-TTG TTG CCA TCA ATG ACC
CC-3′; GAPDH antisense, 5′-TGA CAA AGT GGT CGT TGA GG-3′ (836 bp);
MMP-9 sense, 5′-AAG TGG CAC CAC CAC AAC AT-3′; MMP-9 antisense,
5′-TTT CCC ATC AGC ATT GCC GT-3′ (516 bp); c-fos sense, 5′-AAC CGG
AGG AGG GAG CTG ACT GAT-3′; c-fos antisense, 5′-GGG CCT GGA TGA TGC
TGG GAA CA-3′ (375 bp); c-jun sense, 5′-ATG GAG TCC CAG GAG CGG ATC
AA-3′; and c-jun antisense, 5′-GTT TGC AAC TGC TGC GTT AG-3′ (251
bp). The PCR conditions were as follows: denaturation at 94°C for
30 sec, annealing at 58°C for 30 sec and extension at 72°C for 45
sec. The products were electrophoresed in 1.5% agarose gel
containing ethidium bromide.
Western blot analysis
The cells were washed in phosphate-buffered saline
(PBS), detached using trypsin-EDTA buffer and stored at −70°C until
needed. The protein was extracted with RIPA buffer [1% NP-40, 0.5%
sodium deoxycholate, 0.1% sodium dodecyl sulfate (SDS)], protease
inhibitors [aprotinin, leupeptin, phenylmethanesulfonylfluoride
(PMSF), benzamidine, trypsin inhibitor and sodium orthovanadate].
Fifty micrograms of the protein was then separated by 10% SDS-PAGE
and transferred to polyvinylidene fluoride membranes. The membranes
were blocked in a PBS solution containing 5% non-fat dry milk,
incubated with the primary antibodies in a blocking solution
overnight at 4°C and washed three times with TTBS (0.1% Tween-20 in
TBS) at 10-min intervals. Horseradish peroxidase-conjugated
secondary antibodies (Amersham, Arlington Heights, IL, USA) were
used to detect the immunoreactive proteins by chemiluminescence.
The following antibodies were used: anti-MMP-9 (R&D Systems,
Inc, Minneapolis, MN, USA) and anti-phospho-IκB (Santa Cruz
Biotechnology, Santa Cruz, CA, USA). The total protein levels were
assayed by washing the blotted membrane with a stripping solution
composed of 100 mM 2-mercaptoethanol, 2% SDS and 62.5 mM Tris-HCl
(pH 6.7) for 30 min at 50°C and the membrane was then reprobed with
anti-β-actin (Sigma-Aldrich) monoclonal antibodies.
Gelatin zymography
The quantity of protein in the conditioned media was
determined with a BSA protein assay kit (Pierce, USA).
Subsequently, the conditioned media was mixed with an equal volume
of 2X sample loading buffer [62.5 mM Tris-HCl (pH 6.8), 25%
glycerol, 4% sodium dodecyl sulfate (SDS) and 0.01% bromophenol
blue; Bio-Rad, USA] and loaded onto a 7.5% acrylamide:bisacrylamide
(29:1; Bio-Rad) gel containing 625 μg/ml gelatin (Sigma). Upon
electrophoresis at 100 V for 2 h, the gel was soaked in 1X zymogram
renaturation buffer (Bio-Rad) on a rocker for 1.5 h at room
temperature to remove residual SDS, rinsed in distilled water,
incubated at 37°C for 18 h in 1X zymogram development buffer
(Bio-Rad), stained with 0.25% (w/v) coomassie brilliant blue R-250
(Bio-Rad) and then destained in destaining buffer (10% acetic acid
and 20% methanol).
Quantitation of MMP-9
The amounts of MMP-9 in conditioned media from
ECV304 cells were determined using commercially available
enzyme-linked immunosorbent assay (ELISA) kits (R&D Systems)
according to the manufacturer’s instructions.
Measurement of MMP-9 promoter
activity
The transcriptional regulation of MMP-9 was examined
by the transient transfection of an MMP-9 promoter luciferase
reporter construct (pGL4-MMP-9). The plasmid pGL4-MMP-9 promoter
(21) was kindly provided by Dr
Young Han Lee (Konkuk University, Korea). ECV304 cells
(5×105) were seeded and grown until they reached 60–70%
confluence, then pRL-TK (an internal control plasmid containing the
herpes simplex thymidine kinase promoter linked to the
constitutively active Renilla luciferase reporter gene) and
pGL4-MMP-9 were cotransfected into the cells using FuGene
(Boehringer-Mannheim, Mannheim, Germany), according to the
manufacturer’s protocol. pRL-TK was transfected as an internal
control. Cells were incubated in the transfection medium for 20 h
and treated with nicotine for 5 h. The effects of signaling
inhibitors on MMP-9 promoter activity were determined by
pretreating cells with EGCG for 1 h prior to addition of nicotine.
The cotransfection studies were performed in the presence or
absence of NF-κB-inducting kinase (NIK), I-κBα or I-κBβ and AP-1
decoy oligodeoxynucleotides (ODNs). The dominant negative mutants
of I-κBα, I-κBβ and NIK were kindly provided by Dr D.W. Ballard
(22) and Dr W.C. Greene (23), respectively. The phosphorothioate
double-stranded ODNs with the sequences against the AP-1 binding
site (5′-CAC TCA GAA GTC ACT TC-3′ and 3′-GAA GTG ACT TCT GAG
CTG-5′) were prepared (Genotech, St. Louis, MO, USA) and annealed
(AP-1 decoy ODNs).
Measurement of intracellular hydrogen
peroxide (H2O2)
The level of intracellular
H2O2 was measured using 5- and 6-carboxyl-2′,
7′-dichlorodihydrofluorescein diacetate (DCFDA, Molecular Probes,
Eugene, OR, USA) according to a previously-described procedure
(24). Briefly, the cells were
grown in serum-starved DMEM medium supplemented with 0.5% FBS for
an additional 2 days. The cells were then stabilized in a
serum-free DMEM medium without phenol red for ≥30 min before being
exposed to nicotine for 15 min. The effect of EGCG was assessed by
pretreating the cells with EGCG for 1 h and incubating them with
nicotine sensitive fluorophore DCFDA (5 ng/ml) for 10 min. The
cells were observed immediately under a laser-scanning confocal
microscope. The DCF fluorescence was excited at 488 nm using an
argon laser and the emission evoked was filtered with a 515-nm long
pass filter.
Transient transfection of NF-κB and AP-1
reporter
The NF-κB and AP-1 reporter construct was purchased
from Clontech (Palo Alto, CA, USA). Once the cells had reached
60–70% confluence, they were washed with Dulbecco’s modified
Eagle’s medium and incubated in the medium without serum and
antibiotics for 18 h. The cells were then transfected with NF-κB
and AP-1 reporter containing the pGL3 vector using Lipofectamine
2000 (Invitrogen). Reporter transfected cells were pretreated with
EGCG for 1 h and incubated with 200 μg/ml nicotine for 5 h. The
luciferase activity was measured using a luminometer.
Matrigel invasion assay
ECV304 cells (5×104) in 250 μl of
complete DMEM were seeded in the upper chamber of a 10-well
chemotaxis chamber (Neuro Probe, USA) and serum-free DMEM was
placed in the lower chamber. A Matrigel-coated membrane was
inserted between the two chambers. Following overnight incubation
at 37°C, the media in the upper chamber was replaced with
serum-free DMEM and ECV304 cells were preincubated with the
indicated concentration of EGCG and anti-MMP-9 antibody for 1 h and
added to 200 μg/ml nicotine for 24 h. Upon additional 24 h
incubation at 37°C in 5% CO2 air, the membrane was fixed
and stained with a Hemacolor rapid staining kit (Merck, Germany) as
per the manufacturer’s instructions.
Statistics
Data are shown as the mean ± SD and represent the
mean of at least three separate experiments performed in
triplicate. Differences between data sets were determined by
Student’s t-test. Differences described as significant in the text
correspond to P<0.05.
Results
MMP-9 expression induced by nicotine in
human ECV304 endothelial cells
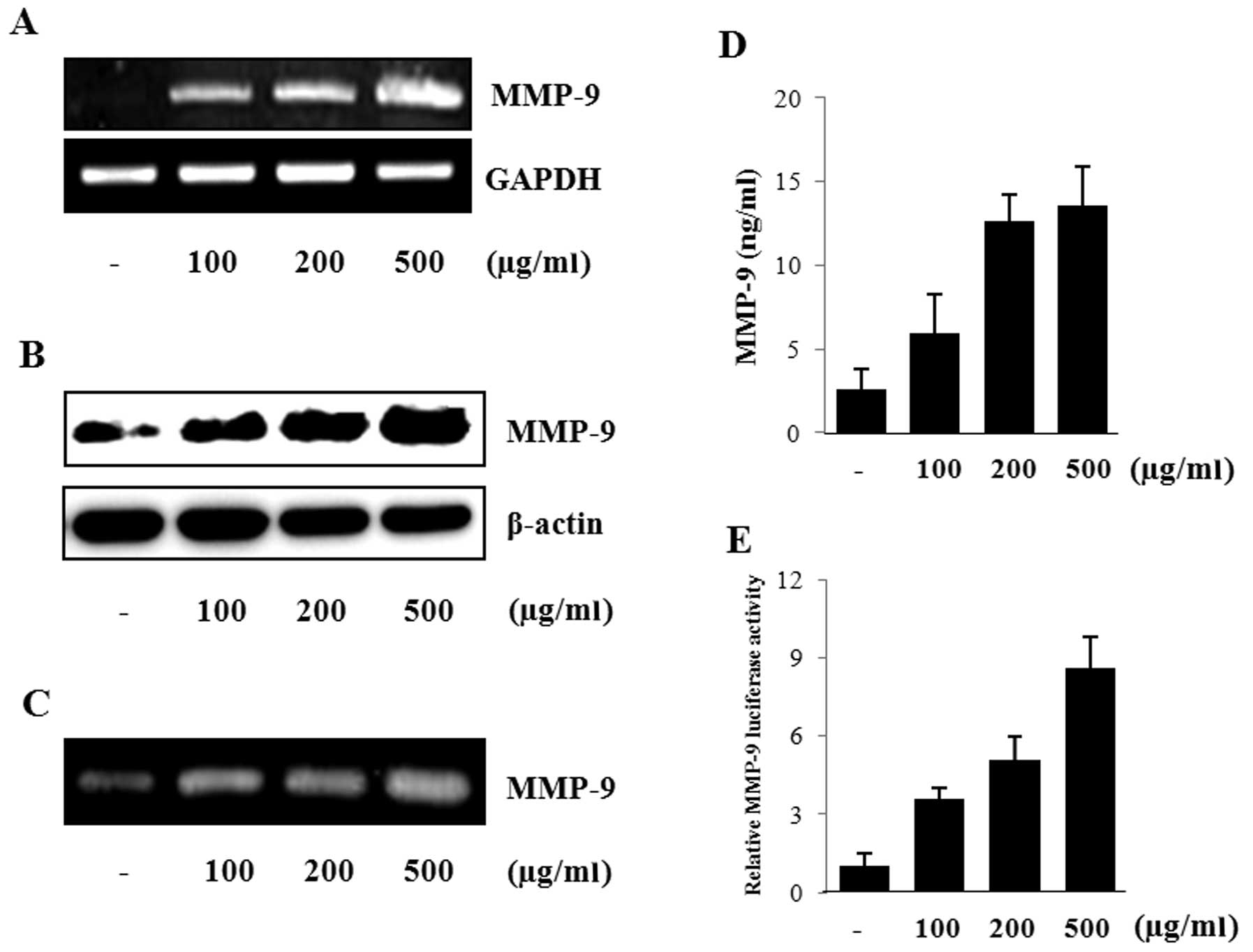
To determine the induction of MMP-9 expression by
nicotine in human ECV304 endothelial cells, the cells were
incubated with nicotine at 0–500 μg/ml. The level of MMP-9 mRNA and
protein in the cells were determined by reverse transcription-PCR
and western blot analysis, respectively. As shown in Fig. 1A and B, nicotine induced MMP-9 mRNA
and protein expression in a dose-dependent manner. Similarly to
other members of the metalloproteinase family, MMP-9 is secreted as
a pro-enzyme and then activated by proteolytic cleavage
extracellularly (25). Since MMP-9
plays a pivotal role in tumor cell invasiveness, we examined the
effect of nicotine on MMP-9 enzyme activities. For this goal, we
performed gelatin zymography and ELISA with conditioned media
harvested from nicotine-treated ECV304 cells. The gelatinolytic
activity of MMP-9 was upregulated with increasing concentrations of
nicotine (Fig. 1C). The level of
MMP-9 was also increased by treating with nicotine determined by
ELISA (Fig. 1D). Next, the effect
of nicotine on transcriptional regulation of the MMP-9 gene was
examined. To this end, ECV304 cells were transiently transfected
with the MMP-9 promoter luciferase reporter construct (pGL4-MMP-9)
and the MMP-9 promoter activity was determined. The cells treated
with nicotine displayed an increase in MMP-9 promoter activity in a
dose-dependent manner (Fig.
1E).
Suppressive effect of EGCG on
nicotine-induced MMP-9 upregulation in human ECV304 endothelial
cells
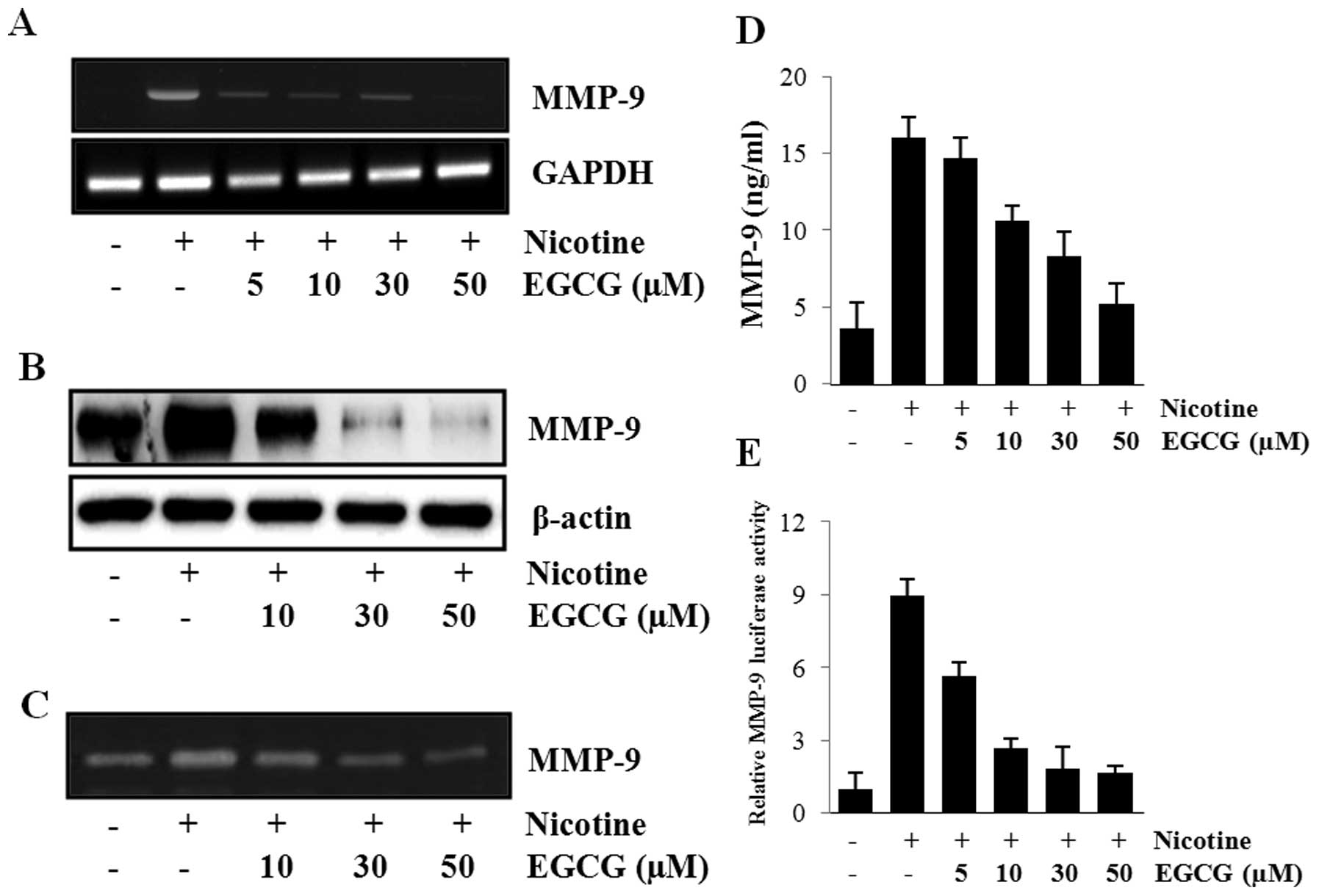
To examine the inhibitory effect of EGCG against
MMP-9 enzymatic activity, ECV304 endothelial cells were subjected
to RT-PCR and western blot analysis. In the presence of increased
concentrations of EGCG (0, 5, 10, 30 and 50 μM) following nicotine
stimulation, MMP-9 expressions were shown to be reduced in a
dose-dependent manner (Fig. 2A and
B). The inhibitory effects of EGCG on MMP-9 induction could
also be confirmed by zymographic analysis and ELSIA assay,
respectively (Fig. 2C and D).
Next, we investigated whether the EGCG treatment inhibited the
MMP-9 transcriptional activity induced by nicotine. As shown in
Fig. 2E, cells pretreated with
10–50 μM EGCG lost most of the nicotine-induced MMP-9
transcriptional activity. EGCG at the concentrations used did not
affect cell viability (data not shown).
Inhibition of nicotine-induced ROS
production by EGCG
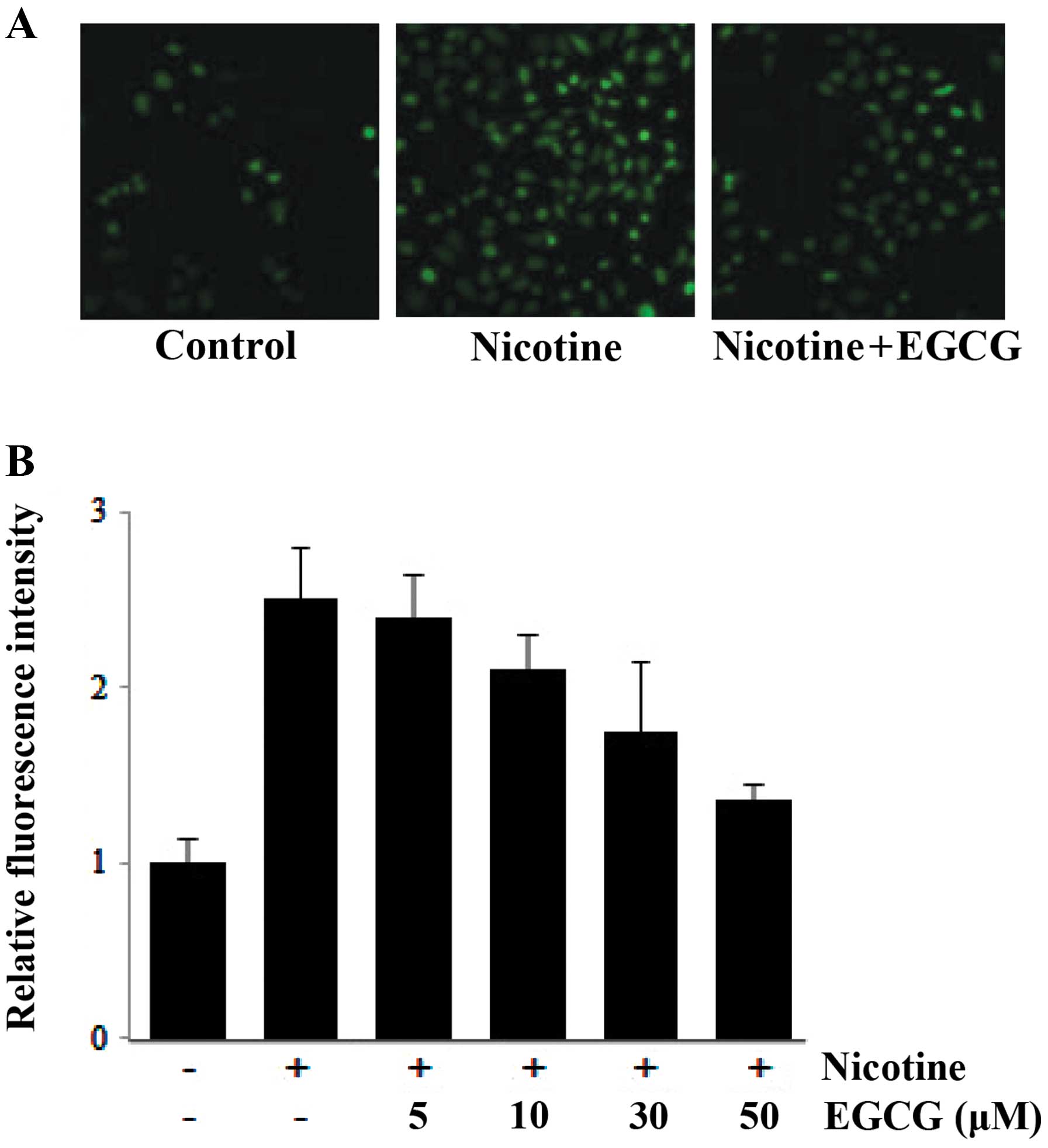
Since the ROS was known to be intracellular
molecules to regulate various genes (26), we determined the role of ROS in
which EGCG suppressed nicotine-induced MMP-9. The changes in the
level of ROS were assayed using the H2O2
sensitive fluorophore DCFDA in human ECV304 endothelial cells
treated with nicotine. Nicotine induced the production of ROS in
the cells and pretreating the cells with 0–50 μM EGCG inhibited the
production of ROS in a dose-dependent manner (Fig. 3).
Effect of EGCG on the activation of
transcription factor NF-κB during nicotine-induced MMP-9
expression
Considering that nicotine can generate ROS and that
NF-κB is a redox sensitive transcription factor (27,28),
the roles of NF-κB in the inhibitory effect of EGCG on MMP-9
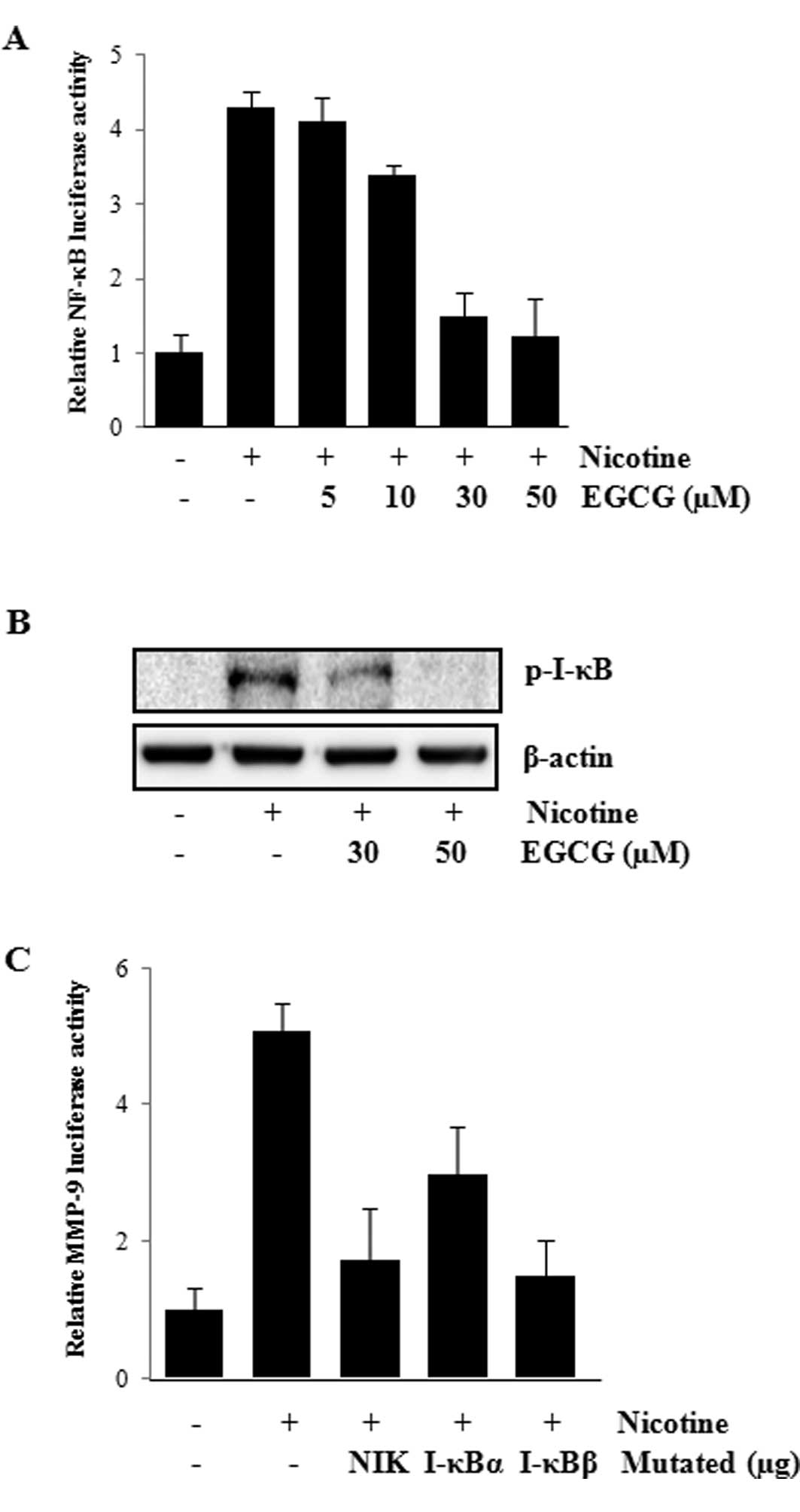
expression were investigated. NF-κB-dependent transcription study
showed that EGCG inhibited nicotine activated NF-κB in a
dose-dependent manner (Fig. 4A).
To gain further insight into the mechanism of EGCG-mediated
downregulation of NF-κB, the effects of EGCG on the I-κB
phosphorylation were examined. Activation of NF-κB is usually
associated with the induction of I-κB phosphorylation, which is
followed by its degradation by proteasome, NF-κB nuclear
translocation and subsequent activation of target gene expression.
The change in the amount of I-κB phosphorylation in ECV304 cells
was determined by western blotting using an antibody to I-κB
phosphorylation. As shown in Fig.
4B, EGCG inhibited the nicotine-induced phosphorylation of
I-κB. The involvement of NF-κB in the induction of MMP-9 by
nicotine was confirmed by cotransfecting ECV304 cells with the
MMP-9 promoter reporter and the dominant negative mutant forms of
NF-κB-related molecules. As shown in Fig. 4C, the expression of dominant
negative mutant forms of NIK, I-κBα, or I-κBβ resulted in a
decrease of the nicotine-induced MMP-9 promoter activity. These
data indicated that NF-κB may be a crucial molecule in the
inhibitory effect of EGCG on MMP-9 expression induced by
nicotine.
Role of AP-1 in EGCG-mediated MMP-9
regulation
The above results indicated that EGCG could inhibit
the nicotine induced MMP-9 expression at the transcriptional level.
Several tentative transcription factors, including NF-κB and AP-1,
have been suggested to control the MMP-9 expression (29). AP-1, which consists of subunits
c-fos and c-jun, is the downstream transcriptional target of Erk1/2
and JNK (30,31). To determine if EGCG regulates c-fos
and c-jun activation induced by nicotine, the expression of c-fos
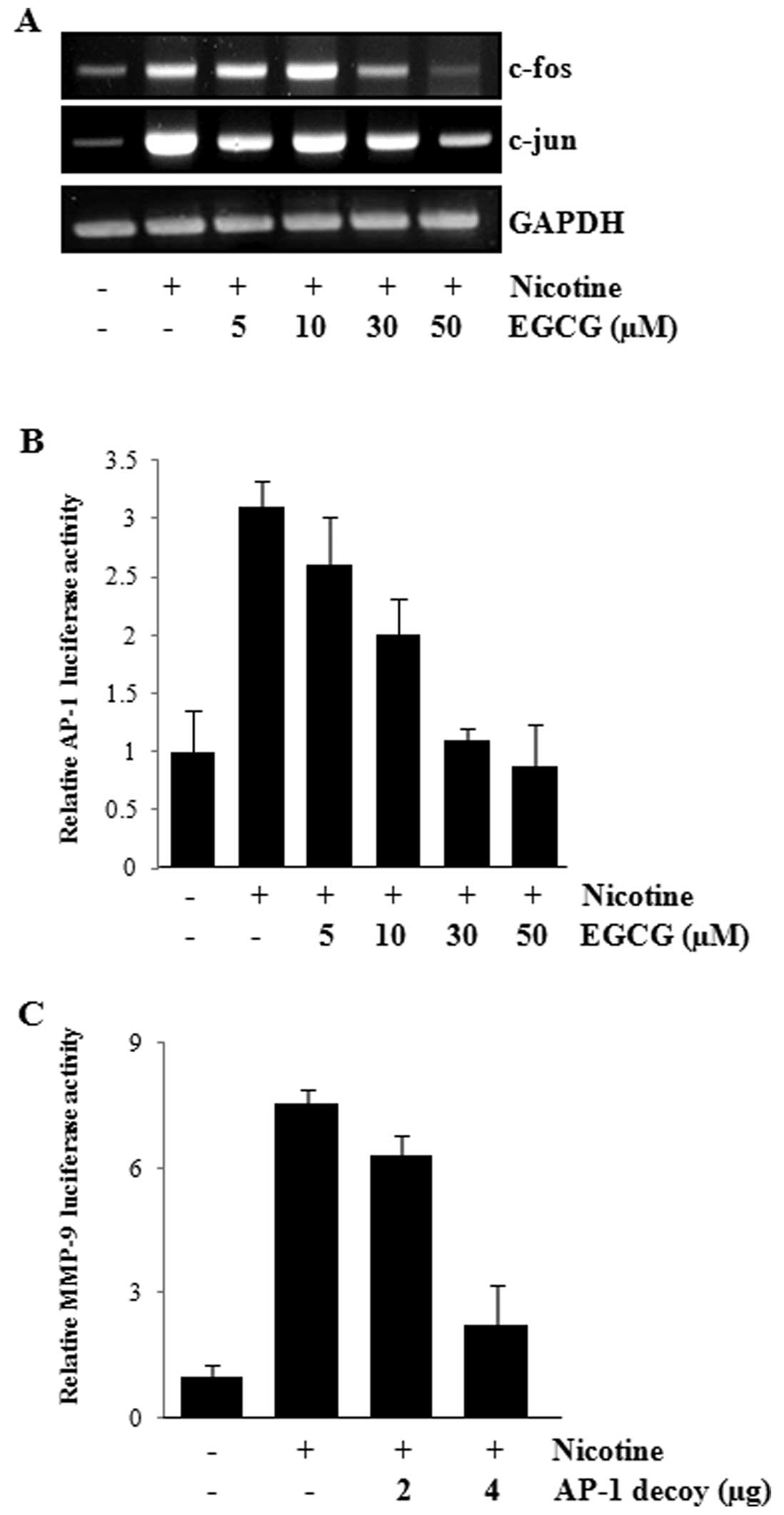
and c-jun were determined by RT-PCR. As shown in Fig. 5A, EGCG inhibited the
nicotine-induced c-fos and c-jun expression in a dose-dependent
manner. Furthermore, EGCG treatment caused a decrease in the
AP-1-dependent transcriptional activity, as revealed by the
transient transfection study using pAP-1-luciferase reporter
construct (Fig. 5B). To confirm
that AP-1 plays an important role in the MMP-9 expression in
endothelial cells, the cells were transiently transfected with AP-1
decoy oligonucleotides and change in the MMP-9 promoter activity
was examined. As shown in Fig. 5C,
the MMP-9 activity was significantly decreased by AP-1 decoy
transfection. The above results suggest that the transcription
factor AP-1 is also involved in nicotine-induced MMP-9 expression
and be a molecular target in EGCG-mediated MMP-9 regulation.
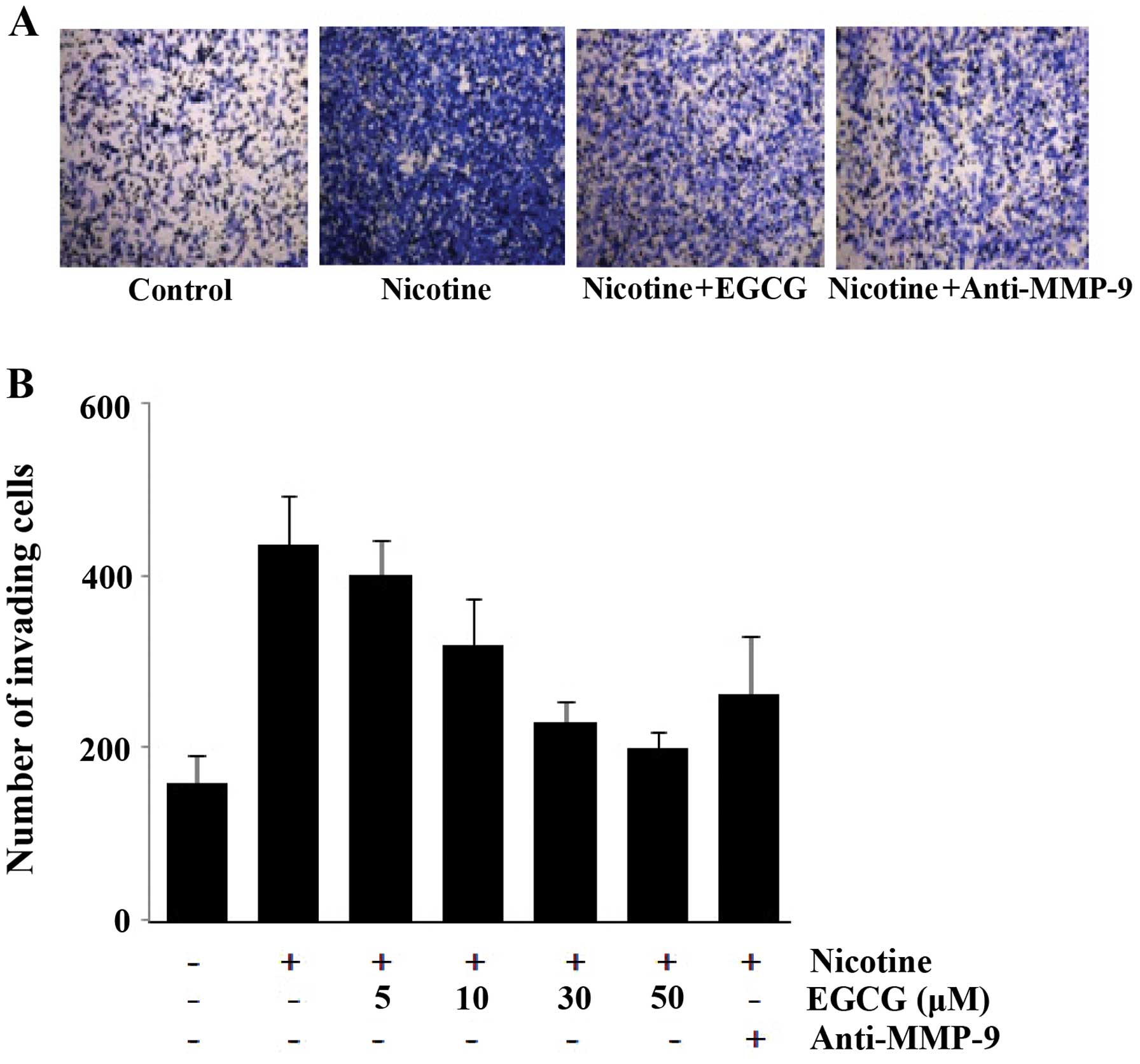
Effect of EGCG and anti-MMP-9 antibody on
nicotine-induced cell invasion
To examine if EGCG and anti-MMP-9 antibody inhibit
nicotine-induced cell invasion, modified Boyden chamber systems
were employed. As shown in Fig. 6,
EGCG and anti-MMP-9 antibody inhibited the invasiveness of ECV304
cells stimulated by nicotine. These results show that EGCG was able
to suppress cell invasion via blocking MMP-9 expression.
Discussion
In endothelial cells, a potential role of the tea
polyphenol (−)-epigallocatechin-3-gallate (EGCG), a major component
of green tea, on nicotine-induced MMP-9 and cell invasion were
investigated. To achieve our goal, we employed human ECV304
endothelial cells that had undergone spontaneous immortalization
from human umbilical vein endothelial cell (HUVEC). HUVEC has been
used for many studies of endothelial cell function. However, as
primary isolates, these cells exhibit a limited life span in
culture and do not survive in serum-deprivation conditions used in
our systems. ECV304 cells have been shown to respond to
extracellular growth factors, are contact inhibited and
differentiate when grown on extracellular matrix (32).
The present study suggests that EGCG inhibited
nicotine-induced MMP-9 and cell invasion in ECV304 endothelial
cells. Interest in green tea as a cancer chemopreventive agent in
humans has intensified for several reasons (33). First, epidemiological evidence
suggests that people who consume a large amount of green tea have a
lower risk of various cancers. Second, green tea has been shown in
animal models to protect against the development and progression of
skin, lung, mammary gland and gastrointestinal tract tumors. Third,
green tea extracts have been shown in vitro to stimulate
apoptosis of various cancer cell lines, including prostate,
lymphoma, colon and lung. Finally, green tea consumption is
associated with few adverse events and it is readily available at
low cost.
Nicotine alone or in combination with other
substances present in the cigarette smoking is recognized as an
agent for the modulation of key cellular processes involved in the
pathobiological effects of tobacco (34). Nicotine has been shown to enhance
cell migration, invasion and subsequent metastasis in many cancer
cells (35). In addition,
Jacob-Ferreira et al(36)
has suggested that nicotine produces cardiovascular effects
involving MMP-9. MMP-9 plays a key role in the degradation of the
extracellular matrix, which is required for various
pathophysiologic responses (37).
Several studies have reported convincing evidence that suppression
of MMPs by different inhibitors markedly reduced tumor cell
invasiveness and metastasis (38,39).
Elevated levels and activities of MMP-9 are found in cancer tissues
and tumor cells. Due to the significant role that MMPs play in
cancer as well as additional human pathologies, considerable
interest has focused on investigating of plant-derived compounds
that can inhibit MMP activities.
Reactive oxygen species (ROS) are important
messenger molecules in downstream signaling pathways and can
ultimately lead to the induction of invasion-related genes
including those for the MMPs (40). Our results show that nicotine
stimulates the intracellular ROS production and EGCG suppress the
ROS production, as determined with
H2O2-sensitive fluorophore DCFDA. The
molecular mechanism for the ROS production by nicotine remains to
be elucidated. Asano et al(41) reported that nicotine activates PKC,
which stimulates NADPH oxidase to generate ROS in C6 glioma cells.
However, non-NADPH oxidase-dependent sources, including
mitochondrial electron transport, may also be importantly involved
in the nicotine-induced ROS generation. EGCG has strong
antioxidative capacity, high affinity for the lipid bilayers of the
cell membrane and can easily enter the nuclei of cancer cells
(42). EGCG is water soluble and
readily oxidizable (43). Its
catechol structure also makes EGCG a strong chelator of metal ions.
EGCG can bind the transition metal ions, prevent formation of
hydroxyl radicals and thus inhibit exogenous ROS-potentiated tumor
invasion (44). Therefore, EGCG
could inhibit tumor cell invasion by scavenging oxygen
radicals.
This study demonstrated that the suppression of
nicotine-induced MMP-9 expression by EGCG occurred at the
transcriptional level as revealed by a transient transfection study
using the MMP-9 promoter luciferase reporter construct. A portion
of the 5′-flanking region of the MMP-9 gene has been cloned. The
promoter of the MMP-9 gene contains AP-1 (−533, −79), NF-κB (−600),
PEA3 (−540) and Sp1 (−558) sites for stimuli that can induce MMP-9
expression (45). EGCG inhibited
the level of active NF-κB and AP-1 induced by nicotine, indicating
that NF-κB and AP-1 inhibition is crucial in the suppression of
MMP-9 expression by EGCG in human ECV304 endothelial cells.
The following observations suggest that the
inhibition of NF-κB by EGCG is involved in MMP-9 gene regulation in
ECV304 cells: i) the nicotine treatment increased I-κB
phosphorylation and NF-κB-dependent transcriptional activity; ii)
EGCG inhibits nicotine-induced I-κB phosphorylation and
NF-κB-dependent transcriptional activity; and iii) MMP-9
transcriptional activity was inhibited by transfection with mutated
NIK, I-κBα or I-κBβ. The potent antioxidative capacity of EGCG may
contribute to the prevention of NF-κB activation. Since the ROS was
known to activate NF-κB through the phosphorylation of I-κB, EGCG
may prevent the ROS induction of NF-κB activation in MMP-9 gene
expression. The results that ROS production was increased within 15
min after exposing the cells to nicotine and the level of ROS were
attenuated by EGCG supported the above assumption.
In the subsequent experiments, the role of AP-1 in
the inhibitory effects of EGCG on nicotine-induced MMP-9 expression
was also investigated. The active AP-1 complex may comprise a
homodimer of c-jun or heterodimers between c-fos, c-jun and ATF2
(46). The c-jun is activated by
N-terminal phosphorylation of specific serine residues (ser63/73)
and it appears to be exclusively activated by JNK. The c-fos
activation can be regulated by JNK and Erk signal pathways
(47). Gum et al(48) suggested that regulation of MMP-9
expression in UMSCC-1 cells was regulated by JNK- and Erk-dependent
signaling pathways. Simon et al(49) demonstrated that phorbol
ester-enhanced MMP-9 secretion and in vitro cell
invasiveness were associated with activation of p38 MAPK. In this
study, EGCG suppressed nicotine-induced c-fos and c-jun expression
and AP-1 activity. Since EGCG inhibited phosphorylation of receptor
tyrosine kinase, it would be logical to assume that the reduction
in activity of mitogen-activated protein kinase (MAPK) and AP-1
were due to the reduction in receptor activities. Previously, we
showed that EGCG could directly inhibit MAPK activities in a
cell-free system, although the mechanism by which EGCG inhibited
the MAPK activities in the cell-free system remained to be
clarified. Thus, the nicotine-induced AP-1 might be inhibited by
dual effects of EGCG in the cells: the suppression of receptor
phosphorylation and direct inhibition of MAPK activities.
Our results suggest new insight into how EGCG may be
involved in nicotine-induced MMP-9 expression and cell invasion in
endothelial cells. Further studies are needed to elucidate the
detailed mechanisms by which EGCG inhibits the MMP-9 expression and
to examine whether EGCG, in fact, exerts the same effects in
vivo.
Acknowledgements
This study was supported by a research grant
(0720570) from the National Cancer Center, by the National Research
Foundation of Korea (NRF) grant (Basic Research Program,
2010-0009910 and MRC for Gene Regulation, 2011-0030132) funded by
the Korea government (MSIP).
References
|
1
|
Brunnemann KD and Hoffmann D: Analytical
studies on tobacco-specific N-nitrosamines in tobacco and tobacco
smoke. Crit Rev Toxicol. 21:235–240. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Davis R, Rizwani W, Banerjee S, Kovacs M,
Haura E, Coppola D and Chellappan S: Nicotine promotes tumor growth
and metastasis in mouse models of lung cancer. PLoS One.
4:e75242009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ji BT, Chow WH, Hsing AW, McLaughlin JK,
Dai Q, Gao YT, Blot WJ and Fraumeni JF Jr: Green tea consumption
and the risk of pancreatic and colorectal cancers. Int J Cancer.
70:255–258. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Stoner GD and Mukhtar H: Polyphenols as
cancer chemopreventive agents. J Cell Biochem (Suppl). 22:169–180.
1995. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Paschka AG, Butler R and Young CY:
Induction of apoptosis in prostate cancer cell lines by the green
tea component, (−)-epigallocatechin-3-gallate. Cancer Lett.
130:1–7. 1998.
|
|
6
|
Jung YD, Kim MS, Shin BA, Chay KO, Ahn BW,
Liu W, Bucana CD, Gallick GE and Ellis LM: EGCG, a major component
of green tea, inhibits tumour growth by inhibiting VEGF induction
in human colon carcinoma cells. Br J Cancer. 84:844–850. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Sen T, Dutta A and Chatterjee A:
Epigallocatechin-3-gallate (EGCG) downregulates gelatinase-B
(MMP-9) by involvement of FAK/ERK/NFkappaB and AP-1 in the human
breast cancer cell line MDA-MB-231. Anticancer Drugs. 21:632–644.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kim MH, Jung MA, Hwang YS, Jeong M, Kim
SM, Ahn SJ, Shin BA, Ahn BW and Jung YD: Regulation of urokinase
plasminogen activator by epigallocatechin-3-gallate in human
fibrosarcoma cells. Eur J Pharmacol. 487:1–6. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Shen JZ, Zheng XF, Wei EQ and Kwan CY:
Green tea catechins evoke a phasic contraction in rat aorta via
H2O2-mediated multiple-signalling pathways.
Clin Exp Pharmacol Physiol. 30:88–95. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Jung YD and Ellis LM: Inhibition of tumour
invasion and angiogenesis by epigallocatechin gallate (EGCG), a
major component of green tea. Int J Exp Pathol. 82:309–316. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Parsons SL, Watson SA, Brown PD, Collins
HM and Steele RJ: Matrix metalloproteinases. Br J Surg. 84:160–166.
1997. View Article : Google Scholar
|
|
12
|
Chandler S, Miller KM, Clements JM, Lury
J, Corkill D, Anthony DC, Adams SE and Gearing AJ: Matrix
metalloproteinases, tumor necrosis factor and multiple sclerosis:
an overview. J Neuroimmunol. 72:155–161. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ogata Y, Pratta MA, Nagase H and Arner EC:
Matrix metalloproteinase 9 (92-kDa gelatinase/type IV collagenase)
is induced in rabbit articular chondrocytes by cotreatment with
interleukin 1 beta and a protein kinase C activator. Exp Cell Res.
201:245–249. 1992. View Article : Google Scholar
|
|
14
|
Ma Z, Qin H and Benveniste EN:
Transcriptional suppression of matrix metalloproteinase-9 gene
expression by IFN-gamma and IFN-beta: critical role of STAT-1alpha.
J Immunol. 167:5150–5159. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Vartak DG and Gemeinhart RA: Matrix
metalloproteases: underutilized targets for drug delivery. J Drug
Target. 15:1–20. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Konstantinopoulos PA, Karamouzis MV,
Papatsoris AG and Papavassiliou AG: Matrix metalloproteinase
inhibitors as anticancer agents. Int J Biochem Cell Biol.
40:1156–1168. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Woodhouse EC, Chuaqui RF and Liotta LA:
General mechanisms of metastasis. Cancer. 80:1529–1537. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Mehlen P and Puisieux A: Metastasis: a
question of life or death. Nat Rev Cancer. 6:449–458. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Arii S, Mise M, Harada T, Furutani M,
Ishigami S, Niwano M, Mizumoto M, Fukumoto M and Imamura M:
Overexpression of matrix metalloproteinase 9 gene in hepatocellular
carcinoma with invasive potential. Hepatology. 24:316–322. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Klein G, Vellenga E, Fraaije MW, Kamps WA
and de Bont ES: The possible role of matrix metalloproteinase
(MMP)-2 and MMP-9 in cancer, e.g acute leukemia. Crit Rev Oncol
Hematol. 50:87–100. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Shin SY, Kim JH, Baker A, Lim Y and Lee
YH: Transcription factor Egr-1 is essential for maximal matrix
metalloproteinase-9 transcription by tumor necrosis factor alpha.
Mol Cancer Res. 8:507–519. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
McKinsey TA, Brockman JA, Scherer DC,
Al-Murrani SW, Green PL and Ballard DW: Inactivation of IkappaBbeta
by the tax protein of human T-cell leukemia virus type 1: a
potential mechanism for constitutive induction of NF-kappaB. Mol
Cell Biol. 16:2083–2090. 1996.PubMed/NCBI
|
|
23
|
Geleziunas R, Ferrell S, Lin X, Mu Y,
Cunningham ET Jr, Grant M, Connelly MA, Hambor JE, Marcu KB and
Greene WC: Human T-cell leukemia virus type 1 Tax induction of
NF-kappaB involves activation of the IkappaB kinase alpha
(IKKalpha) and IKKbeta cellular kinases. Mol Cell Biol.
18:5157–5165. 1998.PubMed/NCBI
|
|
24
|
Hwang YS, Jeong M, Park JS, Kim MH, Lee
DB, Shin BA, Mukaida N, Ellis LM, Kim HR, Ahn BW and Jung YD:
Interleukin-1beta stimulates IL-8 expression through MAP kinase and
ROS signaling in human gastric carcinoma cells. Oncogene.
23:6603–6611. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Mizuma M, Katayose Y, Yamamoto K, Shiraso
S, Sasaki T, Yabuuchi S, Oda A, Masuda K, Rikiyama T, Onogawa T,
Ohtsuka H, Motoi F, Egawa S and Unno M: Up-regulated
p27Kip1 reduces matrix metalloproteinase-9 and inhibits
invasion of human breast cancer cells. Anticancer Res.
28:2669–2677. 2008.
|
|
26
|
Poulsen HE, Jensen BR, Weimann A, Jensen
SA, Sørensen M and Loft S: Antioxidants, DNA damage and gene
expression. Free Radic Res. 33:33–39. 2000.
|
|
27
|
Park MJ, Lee JY, Kwak HJ, Park CM, Lee HC,
Woo SH, Jin HO, Han CJ, An S, Lee SH, Chung HY, Park IC, Hong SI
and Rhee CH: Arsenic trioxide (As2O3)
inhibits invasion of HT1080 human fibrosarcoma cells: role of
nuclear factor-kappaB and reactive oxygen species. J Cell Bochem.
95:955–969. 2005.PubMed/NCBI
|
|
28
|
Tobar N, Villar V and Santibanez JF:
ROS-NFkappaB mediates TGF-beta1-induced expression of
urokinase-type plasminogen activator, matrix metalloproteinase-9
and cell invasion. Mol Cell Biochem. 340:195–202. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kim HS, Kim MH, Jeong M, Hwang YS, Lim SH,
Shin BA, Ahn BW and Jung YD: EGCG blocks tumor promoter-induced
MMP-9 expression via suppression of MAPK and AP-1 activation in
human gastric AGS cells. Anticancer Res. 24:747–753.
2004.PubMed/NCBI
|
|
30
|
Hipskind RA and Bilbe G: MAP kinase
signaling cascades and gene expression in osteoblasts. Front
Biosci. 3:804–816. 1998.PubMed/NCBI
|
|
31
|
Guedea AL, Schrick C, Guzman YF,
Leaderbrand K, Jovasevic V, Corcoran KA, Tronson NC and Radulovic
J: ERK-associated changes of AP-1 proteins during fear extinction.
Mol Cell Neurosci. 47:137–144. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Iwasaka C, Tanaka K, Abe M and Sato Y:
Ets-1 regulates angiogenesis by inducing the expression of
urokinase-type plasminogen activator and matrix metalloproteinase-1
and the migration of vascular endothelial cells. J Cell Physiol.
169:522–531. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Hong MH, Kim MH, Chang HJ, Kim NH, Shin
BA, Ahn BW and Jung YD: (−)-Epigallocatechin-3-gallate inhibits
monocyte chemotactic protein-1 expression in endothelial cells via
blocking NF-kappaB signaling. Life Sci. 80:1957–1965. 2007.
|
|
34
|
Wittel UA, Momi N, Seifert G, Wiech T,
Hopt UT and Batra SK: The pathobiological impact of cigarette smoke
on pancreatic cancer development (Review). Int J Oncol. 41:5–14.
2012.PubMed/NCBI
|
|
35
|
Chen RJ, Chang LW, Lin P and Wang YJ:
Epigenetic effects and molecular mechanisms of tumorigenesis
induced by cigarette smoke: an overview. J Oncol.
2011:6549312011.PubMed/NCBI
|
|
36
|
Jacob-Ferreira AL, Palei AC, Cau SB,
Moreno H Jr, Martinez ML, Izidoro-Toledo TC, Gerlach RF and
Tanus-Santos JE: Evidence for the involvement of matrix
metalloproteinases in the cardiovascular effects produced by
nicotine. Eur J Pharmacol. 627:216–222. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Gialeli C, Theocharis AD and Karamanos NK:
Roles of matrix metalloproteinases in cancer progression and their
pharmacological targeting. FEBS J. 278:16–27. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Noda M, Oh J, Takahashi R, Kondo S,
Kitayama H and Takahashi C: RECK: a novel suppressor of malignancy
linking oncogenic signaling to extracellular matrix remodeling.
Cancer Metastasis Rev. 22:167–175. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Lee HY, Park KS, Kim MK, Lee T, Ryu SH,
Woo KJ, Kwon TK and Bae YS: A small compound that inhibits tumor
necrosis factor-alpha-induced matrix metalloproteinase-9
upregulation. Biochem Biophys Res Commun. 336:716–722. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Brenneisen P, Briviba K, Wlaschek M, Wenk
J and Scharffetter-Kochanek K: Hydrogen peroxide
(H2O2) increases the steady-state mRNA levels
of collagenase/MMP-1 in human dermal fibroblasts. Free Radic Biol
Med. 22:515–524. 1997.
|
|
41
|
Asano H, Horinouchi T, Mai Y, Sawada O,
Fujii S, Nishiya T, Minami M, Katayama T, Iwanaga T, Terada K and
Miwa S: Nicotine- and tar-free cigarette smoke induces cell damage
through reactive oxygen species newly generated by PKC-dependent
activation of NADPH oxidase. J Pharmacol Sci. 118:275–287. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Okabe S, Suganuma M, Hayashi M, Sueoka E,
Komori A and Fujiki H: Mechanisms of growth inhibition of human
lung cancer cell line, PC-9, by tea polyphenols. Jpn J Cancer Res.
88:639–643. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Graham HN: Green tea composition,
consumption and polyphenol chemistry. Prev Med. 21:334–350. 1992.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Zhang G, Miura Y and Yagasaki K:
Suppression of adhesion and invasion of hepatoma cells in culture
by tea compounds through antioxidative activity. Cancer Lett.
159:169–173. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Sato H and Seiki M: Regulatory mechanism
of 92 kDa type IV collagenase gene expression which is associated
with invasiveness of tumor cells. Oncogene. 8:395–405.
1993.PubMed/NCBI
|
|
46
|
van Dam H and Castellazzi M: Distinct
roles of Jun: Fos and Jun: ATF dimers in oncogenesis. Oncogene.
20:2453–2464. 2001.PubMed/NCBI
|
|
47
|
Besirli CG, Wagner EF and Johnson EM Jr:
The limited role of NH2-terminal c-Jun phosphorylation
in neuronal apoptosis: identification of the nuclear pore complex
as a potential target of the JNK pathway. J Cell Biol. 170:401–411.
2005.PubMed/NCBI
|
|
48
|
Gum R, Wang H, Lengyel E, Juarez J and
Boyd D: Regulation of 92 kDa type IV collagenase expression by the
jun aminoterminal kinase- and the extracellular signal-regulated
kinase-dependent signaling cascades. Oncogene. 14:1481–1493. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Simon C, Simon M, Vucelic G, Hicks MJ,
Plinkert PK, Koitschev A and Zenner HP: The p38 SAPK pathway
regulates the expression of the MMP-9 collagenase via
AP-1-dependent promoter activation. Exp Cell Res. 271:344–355.
2001. View Article : Google Scholar : PubMed/NCBI
|