Introduction
Bladder cancer is one of the most common urologic
malignances with over 300,000 new cases worldwide diagnosed
annually (1,2). Despite the advances of
chemotherapeutics, no improvement in survival of advanced or
metastatic bladder cancer has been reported (3–7).
Intense studies on molecular mechanisms have led to the
identification of numerous signaling pathways involving bladder
carcinogenesis, which provide new hope for bladder cancer
treatment. Among them, the following features of TGF-β1 signaling
pathway has attracted much attention: i) the association of changes
affecting either the level of TGF-β1 or the expression of its
receptors with both aggressive bladder carcinoma and poor outcome
(8); ii) importance of
interactions among molecules in the TGF-β1 signaling for the
progression of bladder cancer (9);
iii) the potential therapeutic use of TGF-β1 in bladder carcinoma
(10); and iv) the role of TGF-β1
and its receptor in immune escape in bladder cancer (11).
TGF-β1 is a pleiotropic cytokine, physiologically
involved in the proliferation and differentiation of cells,
embryonic development, angiogenesis, wound healing and immune
regulation (12). Under malignant
conditions, TGF-β1 however is considered as a major modulator of
tumor behavior. During the initiation and early stage of tumor
development, TGF-β1 may serve as a tumor suppressor by inhibiting
proliferation and accelerating apoptosis; later on, TGF-β1 becomes
a protumor factor by favoring tumor migration, invasion,
angiogenesis and immune evasion (13,14).
In the latter mode, tumor cells organize strategies to overcome
TGF-β1-mediated growth arrest by downregulating or mutating
receptors or other means so that they cannot be targeted (15–22).
There are three TGF-β receptors: TGF-βR type I, II
and III, respectively. TGF-βRIII has a very short cytoplasmic tail
and lacks any signaling motif, whereas TGF-βRI and II are
serine/threonine kinases that are essential part of TGF-β1 signal
transduction (12,13). TGF-β receptor downregulation and/or
mutation in various tumor types, including human bladder cancer,
are well covered and discussed in the literature (14–19,23).
Therefore, the role of abnormal TGF-β receptors appears to render
malignant cells resistant to TGF-β-mediated adverse effects.
In contrast to the loss or mutation of its
receptors, TGF-β1 usually is highly expressed by tumor cells
(14). Mechanisms of tumor immune
evasion, including those summarized below, might be helpful to
explain this phenomenon: i) TGF-β1 is capable of inducing
regulatory T cells (24); ii)
TGF-β1 impairs tumor antigen presentation by inhibiting the
maturation of dendritic cells (24); and iii) TGF-β1 ‘educates’
macrophages and fibroblasts to become tumor-associated macrophages
or fibroblasts (23,24). None of these mechanisms precludes
tumor cells from directly utilizing TGF-β1 signaling through
certain alternative mechanism, to promote tumor cell invasion and
metastasis, without inhibiting tumor cell proliferation (14,23).
We provide evidence that bladder cancer cells exploit mutated TGF-β
receptor for TGF-β signal transduction, leading to their enhanced
migration and invasion as well as avoidance of growth arrest.
Materials and methods
Ethics statement
The study protocol was approved by the Medical
Ethics Committee of Tongji Medical College and performed according
to the declaration of Helsinki. All patients gave their written
informed consent before participating in this study. The University
of Padova and the Thomas Jefferson University Institutes’ ethics
regulations on research conducted on human tissues were
followed.
Human cell line
Cell lines T24, ScaBER and BIU-87 (bladder cancer);
PC-3 and DU145 (prostate cancer); A549 (lung cancer); HeLa
(cervical cancer); AGS (gastric cancer); A375 and A875 (melanoma);
HepG2 and SMMC-7721 (hepatocarcinoma); MDA-MB-435, MDA-MB-231 and
MCF-7 (breast cancer); Raji and K562 (leukemia); and L02 (embryo
hepatocyte derived) were involved in this study. BIU-87, A875,
MDA-MB-435, SMMC-7721, Raji, K562 and L02 were purchased from China
Center for Type Culture Collection (CCTCC, Wuhan, China). T24,
ScaBER, PC-3, DU145, A549, HeLa, AGS, A375, HepG2, MDA-MB-231 and
MCF-7 were purchased from the American Type Culture Collection
(ATCC, Manassas, VA, USA). Cells were cultured in Dulbecco’s
modified Eagle’s medium (Gibco-BRL, Carlsbad, CA, USA) supplemented
with 100 ml/l fetal bovine serum (HyClone, Logan, UT, USA) at 37°C
in a humidified atmosphere containing 5% CO2.
Plasmids and transfections
Plasmids used in this study included
pIRES2-EGFP-TβRII-Fc (TβRII-Fc) and pIRES2-EGFP (control). Those
interested in further details are kindly referred to the following
citation (25). Plasmids were
transfected into L02, T24 or ScaBER cell line using Lipofectamine
Plus reagent, according to the manufacturer’s (Invitrogen,
Carlsbad, CA, USA) instruction.
siRNAs, siRNA of Smad2 and Smad3 and their controls,
on the other hand, were purchased from RiboBio Company (Guangzhou,
China), and transfected into cells also using Lipofectamine Plus
reagent, according to the manufacturer’s instruction. Forty-eight
hours after the transfections, the cells were sorted for the
detection of GFP expression or other experiments.
Real-time PCR analysis
Total RNA was extracted from cells with TRIzol
reagent (Invitrogen) according to the manufacturer’s instructions.
For real-time RT-PCR assays, the cDNA sequences of genes were
retrieved from NCBI database. The primers were designed with the
Oligo Primer Analysis 4.0 software and the sequences were blasted
(http://www.ncbi.nlm.nih.gov/BLAST/).
Real-time RT-PCR was done as described previously (26). The mRNA level of the detected gene
was expressed as the relative level to that of β-actin. The
sequences of the primers were as follows: β-actin, 5′-CCTAGAAGCATTT
GCGGTGG-3′ (sense) and 5′-GAGCTACGAGCTGCCT GACG-3′ (antisense);
TGF-β1, 5′-ACTACTACGCCAAGGA GGTCAC-3′ (sense) and
5′-GAGCAACACGGGTTCAGGT-3′ (antisense); TGF-βRII,
5′-AGACGGCTCCCTAAACAC TAC-3′ (sense) and
5′-GAATGCTCTATGTCACCCACTC-3′ (antisense); TGF-βRI,
5′-GACAACGTCAGGTTCTGG CTCA-3′ (sense) and 5′-ATCGACCTTTGCCAATGCT
TTC-3′ (antisense); p15, 5′-GGCAGACAGGTTTAGCTGTTT CATG-3′ (sense)
and 5′-CCACAATGGAGCTAGAAGCA GGA-3′ (antisense); CUTL1,
5′-AAAGACCAGCCTGAAAGT CGG-3′ (sense) and
5′-CCAGGGATGAGCTGAAAAAGT-3′ (antisense); and Cdc25A,
5′-CTCCTCCGAGTCAACAGAT TCA-3′ (sense) and
5′-CAGCCACGAGATACAGGTCTTA-3′ (antisense).
Patient samples
A total of 46 clinical bladder cancer specimens were
consecutively acquired during the period of October 2009 to October
2010, with approval by the Ethics Committee of the Medical Faculty
of Tongji Medical College, by transurethral resection or radical
cystectomy from untreated cancer patients (Table I). Informed consent was obtained in
accordance with the Declaration of Helsinki from all subjects. Data
on the patients’ clinical and pathologic states were collected,
including sex, age, tumor size, pathologic T stage, tumor grade and
multiplicity. The pathologic stage of bladder cancer was assessed
according to the 2002 UICC TNM tumor stage classification by: i)
the superficial bladder cancer (T1) includes pTa and pT1 tumors;
and ii) the muscle invasive bladder cancer (T2) includes pT2, pT3
and pT4. Tumor grade was assessed according to the 2004
WHO/International Society of Urologic Pathology grading
classification by: i) the well differentiated papillary urothelial
neoplasm includes low malignant potential and low grade tumor
(low); and ii) the poorly differentiated papillary urothelial
neoplasm which includes high grade bladder cancer (high).
 | Table I.Patient characteristics. |
Table I.
Patient characteristics.
| Characteristic | Patients (n) |
|---|
| Sex | |
| Male | 40 (87.0) |
| Female | 6 (13.0) |
| Age (year) | |
| <45 | 7 (15.2) |
| ≥45 | 39 (84.8) |
| Tumor size
(cm) | |
| <3 | 35 (76.1) |
| ≥3 | 11 (23.9) |
| Multiplicity | |
| Single | 32 (69.6) |
| Multiple | 14 (30.4) |
| T stage | |
| T1 | 19 (41.3) |
| T2 | 27 (58.7) |
| Tumor grade | |
| Low | 23 (50.0) |
| High | 23 (50.0) |
| Expression type of
TGF-βRII | |
| Non-point
mutation | 28 (61.9) |
|
Mutation-free | 8 (17.4) |
|
Undetectable | 9 (19.6) |
|
Frame-shift | 11 (24.0) |
| Point
mutation | 18 (39.1) |
Isolation of human primary bladder cancer
cells
Fresh bladder cancer specimens were used for tumor
cell isolation as described previously (27). Briefly, tumor tissue was washed
three times in cold DMEM medium containing 1% FBS and digested with
hyaluronidase (Sigma-Aldrich, St. Louis, MO, USA), collagenase and
DNase for 1 h at 37°C. After grinding with semifrosted slides and
lysis of RBC, the dissociated cells were incubated on ice for 20
min and then spun down at 500 rpm for 1 min. This process was
repeated twice and the cells were first incubated for 2 h to get
rid of adhesive cells. Tumor cells were then cultured in DMEM
supplemented with 10% FBS, 2 mmol/l L-glutamine, 1.0 mmol/l sodium
pyruvate, 100 U/ml penicillin G sodium, and 100 μg/ml
streptomycin sulfate in 6-well plate in a humidified incubator at
37°C with 5% CO2.
DNA sequencing
Total RNA, extracted from cell lines or primary
bladder cancer cells, was reversely transcribed to cDNA. The latter
was used to amplify the whole coding sequence of TGF-βRII by PCR
with primers (5′-CTGGAAGATGCTGCTT CTC-3′ and
5′-ACTGCTCTGAAGTGTTCTGC-3′). PCR products were sequenced by Sangon
Biotech (Shanghai, China).
Flow cytometry analysis
Cells were incubated with 1 μg of human
IgG/105 cells for 15 min at room temperature prior to
staining, and then stained with phycoerythrin-conjugated anti-human
TGF-β1 or TGF-βRII antibodies and their isotypes (eBioscience, San
Diego, CA, USA). The stained cells were used for flow cytometric
analysis (BD Biosciences, LSR II).
Immunohistochemistry
To detect TGF-βRII protein, cells were grown in
glass slides overnight. After washing twice, slides were fixed in
dry acetone for 10 min at room temperature and air-dried for
another 10 min. Rabbit anti-human TGF-βRII primary antibody
(Millipore), biotinylated goat anti-rabbit IgG, and
streptavidin-conjugated horseradish peroxidase (eBioscience) were
used for immunohistochemical staining.
Cell proliferation assays
Cells were incubated with PKH-26 dye for membrane
staining, and then cultured in the presence or absence of TGF-β1 (2
ng/ml, PeproTech) in 24-well plate, and 24 to 48 h later, the
proliferation was analyzed by flow cytometry and expressed with
stimulation index (SI).
Cell migration and invasion
Bladder cancer cell motility and invasion were
evaluated by Transwell assay as described previously (28). In migration or invasion experiment,
cells were allowed to reach confluence in serum-containing complete
growth medium and then incubated for 16 h in serum-free medium
before treatment of TGF-β1 or TβRII-Fc and smad2+3 siRNA
transfection. Matrigel invasion assay was performed in Transwell
plates with polycarbonate membrane filters (Corning, Corning, NY,
USA). Precoated filters (6.5 mm in diameter, 8 μm pore size,
Matrigel 100 μg/cm2) were rehydrated with 0.1 ml
medium. Then, 2×105 pretreated cells in 0.2 ml DMEM were
added to the top chamber. Medium (0.6 ml) supplemented with 20%
fetal bovine serum was added to each well of the plate to act as a
chemoattractant in the lower chamber. Following incubation for 18 h
at 37°C, non-invading cells at the upper surface of the filter were
wiped off with a cotton swab, and the invading cells at the lower
surface of the filter were fixed for 5 min in 100% methanol and
stained with hematoxylin and eosin. Cells that moved through the
insert were counted in five random fields and expressed as the
average number of cells per field. Experiments were repeated in
triplicate. Transwell migration assays were done using the same
procedure but without coating filters with Matrigel.
Co-immunoprecipitation assay
Co-immunoprecipitation was performed as described
previously (29). Briefly, cells
were harvested at 0, 5, 10 and 20 min following TGF-β1 (2 ng/ml)
treatment. Cell extracts were first precleared with 25 μl of
protein A-agarose (Sigma-Aldrich). The supernatants were
immunoprecipitated with anti-TGF-βRII antibody for 1 h at 4°C,
followed by incubation with protein A-agarose overnight at 4°C. The
complexes were collected by centrifugation for western blot
analysis.
Western blot analysis
Cell lysates and prestained m.w. markers were
separated by SDS-PAGE followed by transfer onto nitrocellulose
membranes. The membranes were blocked in TBST (Tris-buffered saline
with 0.5% of Triton X-100) containing 5% non-fat milk or BSA, and
probed with primary antibodies (R&D Systems). After incubation
with the secondary Ab conjugated with HRP, membranes were
extensively washed, and the immunoreactivity was visualized by ECL
according to the manufacturer’s protocol (ECL kit, Santa Cruz
Biotechnology, Santa Cruz, CA, USA). Antibodies smad2/3, psmad2/3,
β-actin, p15, CUTL1 and Cdc25A were purchased from Santa Cruz
Biotechnology.
Statistical analysis
Results were expressed as mean values ± SD and
interpreted by ANOVA or χ2 test (in statistics of
patient sample, we used χ2 continuity correction when
1≤T<5). SPSS version 12.0 (Chicago, IL, USA) was used for
statistical analysis. Differences were considered to be
statistically significant when P<0.05.
Results
TGF-βRII is highly expressed in bladder
cancer cell line T24
This study first examined TGF-β1 expression and its
receptors TGF-βRI and TGF-βRII by real-time RT-PCR in human tumor
cell lines derived from a variety of tissues, including breast,
liver, lung, gastric, prostate, skin, bladder and bone marrow. As
expected, all the tumor cell lines were shown to highly express
TGF-β1 but to have low expression of TGF-βRI (Fig. 1A), which was consistent with
previous reports (14–16,20–23).
Unexpectedly however, we found that although TGF-βRII was weakly
expressed in most tumor cell lines but strongly expressed in T24
bladder cancer cells (Fig. 1A).
Such abnormal high expression of TGF-βRII in T24 cells was further
confirmed by flow cytomety and cellular immunohistochemical
staining (Fig. 1B and C). Thus,
bladder cancer cell line T24 was identified as highly expressing
TGF-βRII.
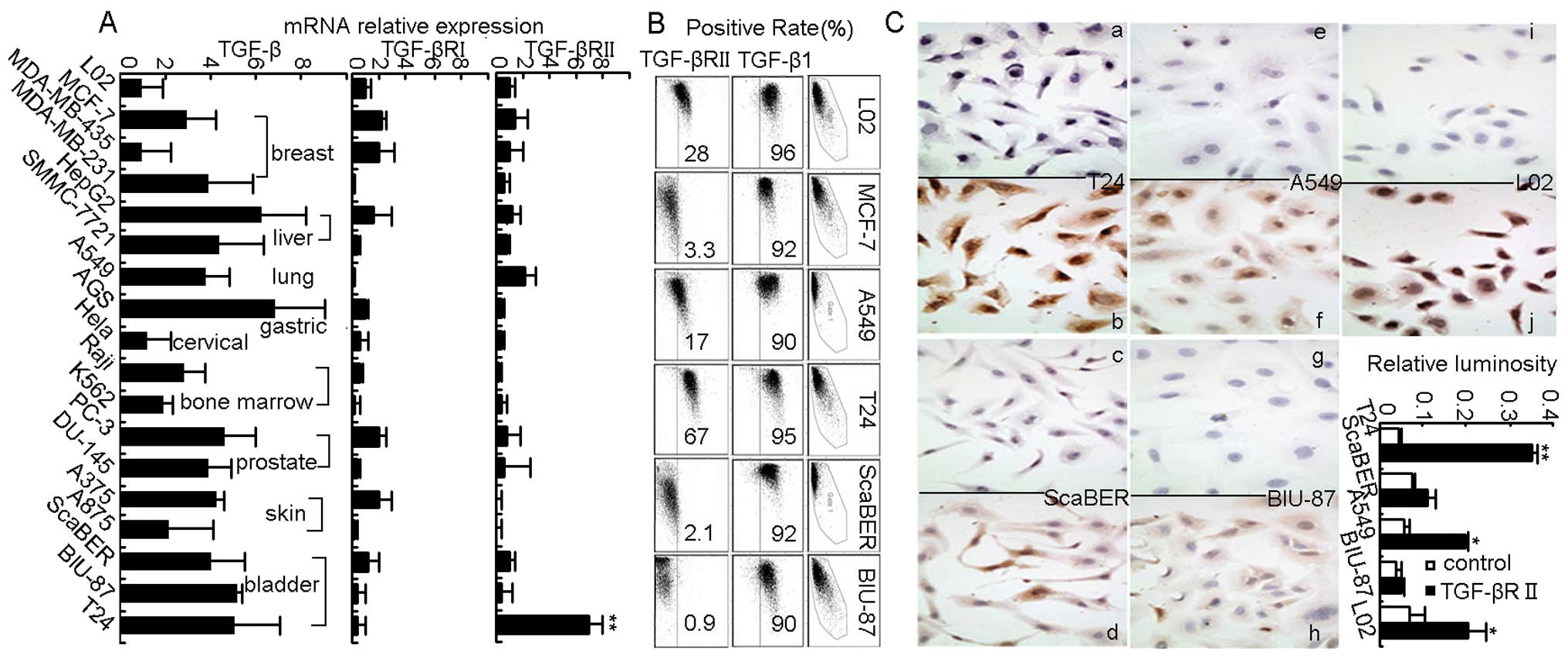 | Figure 1.TGF-βRII is highly expressed in human
bladder cancer cell line T24. (A) qRT-PCR showed expression of
TGF-β1, TGF-βRI and TGF-βRII in different kinds of human cancer
cell lines, **P<0.01 for each cancer cell line
compared to embryo hepatocyte derived L02 cells. (B) Flow cytometry
showed expression of cytoplasmic TGF-β1 and membraned TGF-βRII in
human cancer cell lines. (C) Cell immunohistochemistry (a, c, e, g
and i, isotype control; and b, d, f, h and j, experimental group)
also showed strong staining of TGF-βRII in T24 cell membrane,
*P<0.05, **P<0.01 compared to control
group. Magnification, ×400. Similar results were obtained at least
in three separate experiments. |
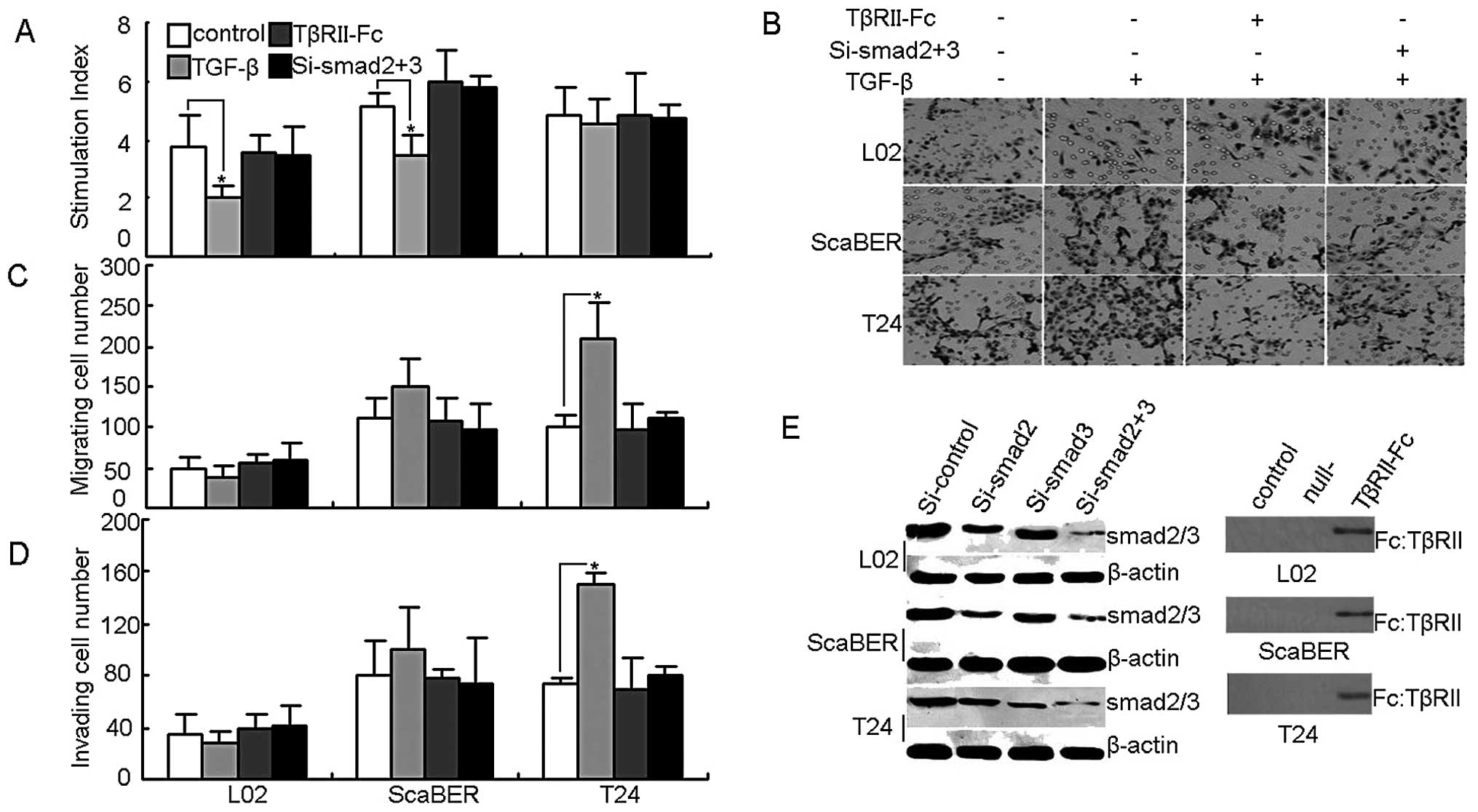
TGF-βRII engagement does not induce
growth arrest but strengthens TGF-β1-mediated cell invasion in T24
bladder cancer cells
Next, this study turned to the question of whether
TGF-βRII in T24 bladder cancer cells was functional. TGF-β1 impacts
on cell growth arrest are well known in the literature (12–14).
First we performed the in vitro proliferation assay, and
found that the addition of TGF-β1 effectively inhibited the growth
of normal liver cell line L02 and the bladder cancer cell line
ScaBER, but not T24 cell growth (Fig.
2A). On the other hand, an in vitro Transwell assay
found that the addition of TGF-β1 markedly promoted the migration
and invasion of T24 cells (Fig.
2B, C and D). To test and confirm these initial results, a
comparable approach was used to transfect TGF-βRII-Fc vector or
smad2/3 siRNA, resulting in the blocking of TGF-βRII or silencing
smad2/3, the downstream molecules of TGF-βR signaling (Fig. 2E). Under such conditions, we found
that the above effects of TGF-β on T24 cells were relieved
(Fig. 2A, B, C and D). These
findings suggest that TGF-βRII does not mediate T24 cell growth
arrest but is capable of promoting T24 cell migration and invasion
through the TGF-β signal pathway.
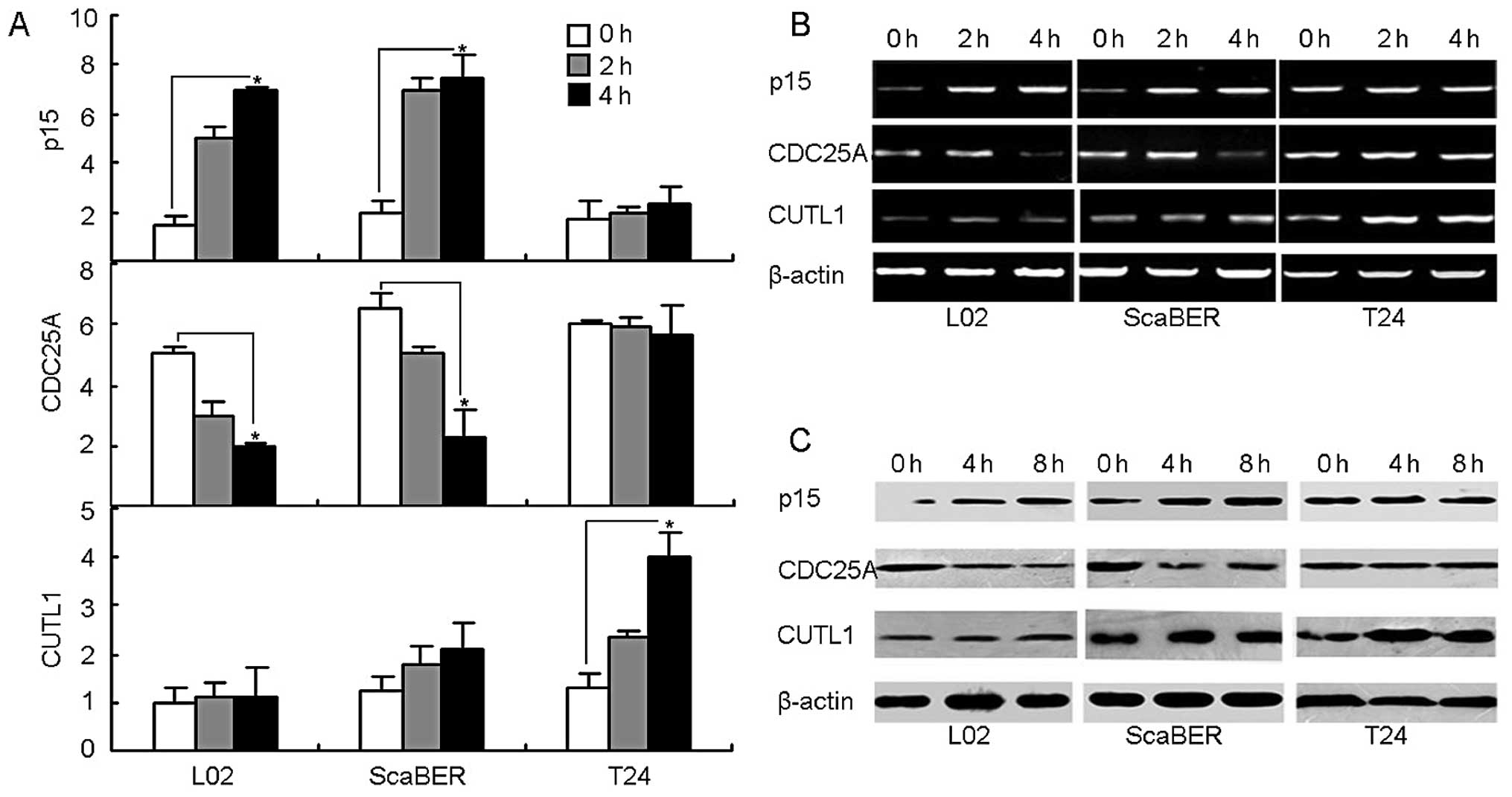
TGF-βRII signaling fails to regulate
proliferation-associated p15 and Cdc25A expression but enhances
invasiveness-associated CUTL1 expression in T24 cells
Having identified a vital role for TGF-βRII in cell
migration and invasiveness, this study turned next to explore the
possible molecular mechanism through which TGF-β1 binding TGF-βRII
resulted in the failure of growth arrest but the enhancement of
motility in T24 bladder cancer cells. The literature has already
identified the mechanisms of TGF-β1-mediated growth arrest which
involve upregulating cyclin-dependent kinase (CDK) inhibitor p15
and downregulating CDK4/6-activating phosphatase Cdc25A (30–32).
Looking further, this study found that TGF-β1 stimulation was
capable of upregulating p15 and downregulating Cdc25A in both
ScaBER and L02 cell lines without any undue effect on the
expression of p15 and Cdc25A in T24 cell line, evaluated by both
RT-PCR and real-time PCR (Fig. 3A and
B). Consistently, p15 and Cdc25A proteins in T24 cells were not
affected by TGF-β stimulation, evaluated by a western blot analysis
(Fig. 3C). On the other hand,
homeobox transcription factor CUTL1 has been well demonstrated as a
critical target of TGF-β1 signaling to mediate the promotion of
cancer cell motility and invasiveness (33–35).
Therefore, besides p15 and Cdc25A, this study was able to also
determine the expression of CUTL1. Interestingly, the expression of
CUTL1 was significantly upregulated by the addition of TGF-β1 in
T24 cells but not in L02 or ScaBER cells (Fig. 3A, B and C). Therefore, in T24
bladder cancer cells, the TGF-βRII signaling pathway is ineffective
in the regulation of p15 and Cdc25A expression but highly
efficacious in the regulation of CUTL1 expression, leading to the
evasion of growth arrest and the enhancement of motility and
invasion.
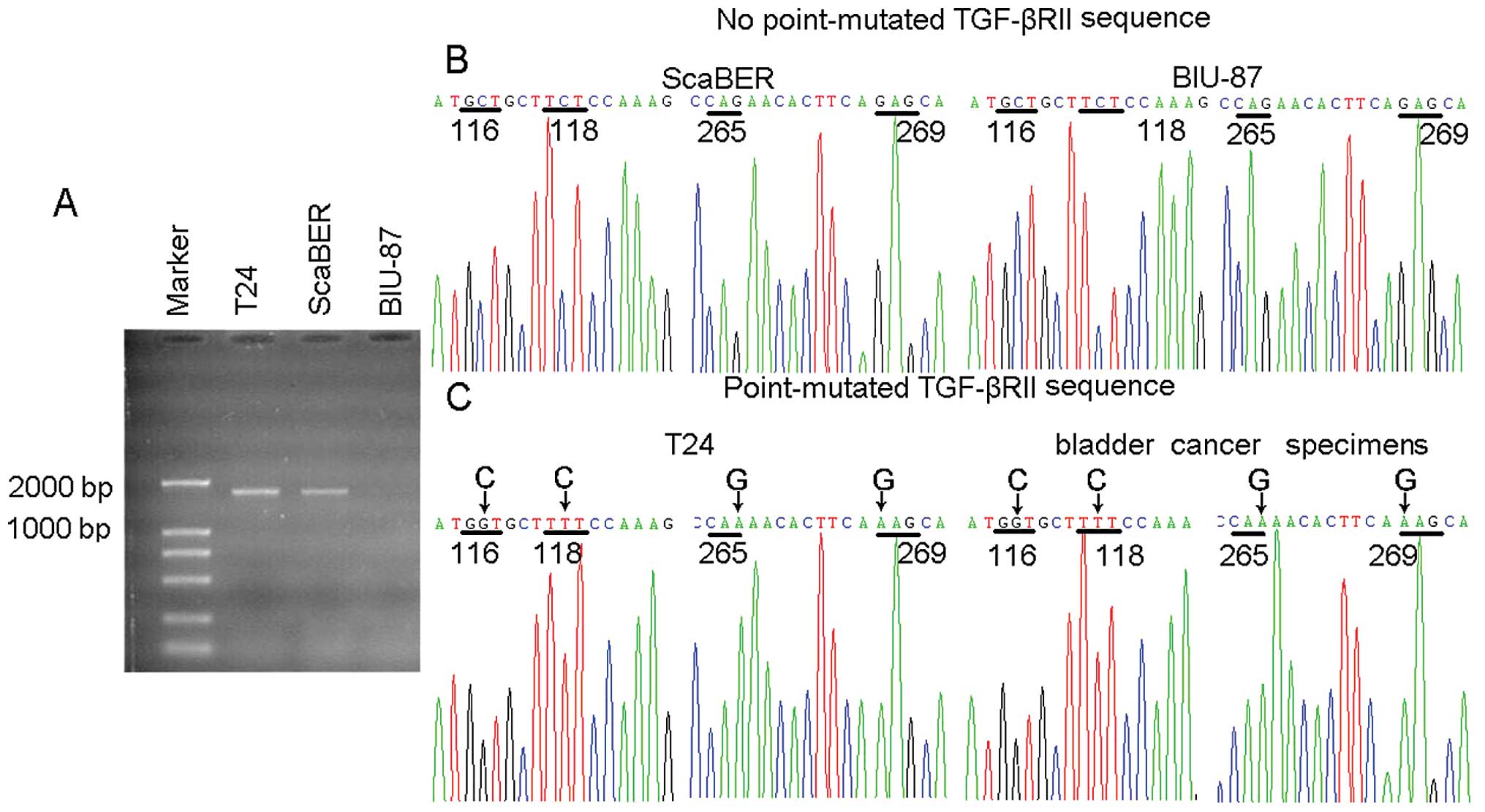
Sequencing analysis of TGF-βRII mutation
in T24 cell line
Further investigation was necessary into the
molecular basis by which TGF-β1 signaling had no effect on p15 and
Cdc25A expression but was efficacious in the upregulation of CUTL1
in T24 cells. In bulk tumor cell population the growth of
TGF-βRhigh tumor cells is declined due to TGF-β1
signaling-mediated growth arrest, leading, in turn, to the
domination of TGF-βRlow/neg tumor cells, depending on
the type of cultured cells involved. Paradoxically, T24 bladder
cancer cell line maintains the high expression of TGF-βRII. To
explain this, it was reasonable to speculate that T24 cells
employed a mutation strategy to encrouch TGF-β signaling. In this
regard, a pair of primers was designed to amplify 1707 bp cDNA
fragment covering the coding region of TGF-βRII. By 25 cycles, the
visible PCR products from T24 and ScaBER cell lines were confirmed
in an agarose gel (Fig. 4A).
Further sequencing analysis indicated that TGF-βRII was not mutated
in either ScaBER or BIU-87, but mutated with several single
nucleotides in T24 (Fig. 4B and
C). Notably, one point mutation was the GAG to AAG transition,
leading to corresponding amino acid transition of
Glu269→Lys in the cytoplasmic domain of TGF-βRII. Glu
and Lys belong to acidic and basic amino acids, respectively. Thus,
this Glu269→Lys mutation might profoundly change the
electric property of TGF-βRII, leading to altered conformations and
abnormal signaling pathways.
Mutated TGF-βRII-transduced signaling is
Smad2/3 dependent in T24 cells
The fact, that Smad2/3 are the classical downstream
molecules for TGF-β signaling transduction (23,24),
begged the question of whether Smad2/3 were also required for the
mutated TGF-βRII signaling pathway in T24 cells. This, in turn,
first called for the measurement of the active form of Smad2/3 by
measuring its phosphorylation state by western blot analysis. It
was found that the phosphorylation was induced 30 min after the
stimulation of 2 ng/ml TGF-β1 in L02 and ScaBER cells as well as
T24 cells (Fig. 5A), suggesting
that the mutated TGF-βRII signaling transduction might be through
the classical Smad2/3 pathway. Moreover, by pulling down the
TGF-βRII complex with anti-TGF-βRII antibody, it was observed that
Smad2/3 proteins were bound to the complex (Fig. 5B), suggesting the interaction of
Smad2/3 and TGF-βRII in T24 cells. When silencing Smad2 and Smad3
in T24, the binding complex can not be detected with additional
TGF-β1, expression of CUTL1 then having almost no change (Fig. 5C), indicating that elevated level
of CUTL1 was Smad2/3 dependent in T24 cells. Taken together, these
findings suggested that the transduction of TGF-β signaling by
mutated TGF-βRII is Smad2/3-dependent in bladder cancer T24 cell
line.
GAG→AAG mutation of TGF-βRII occurs in
bladder cancer patients and is correlated to high
aggressiveness
To translate the implications of the above in
vitro data in vivo, the sequence of TGF-βRII cDNA in
bladder cancer patients was further analyzed. Primary bladder
cancer cells were isolated from fresh specimens of bladder cancer
patients (n=46, Table I), and then
used for RT-PCR to amplify the TGF-βRII cDNA (25 cycles) for
sequencing. As expected, similar mutation patterns were found in
primary bladder cancer cells as in T24 cells. A total of 18
specimens showed GAG→AAG mutation (39.1%), which was also companied
by other mutations observed in T24 cells (Fig. 4C). Therefore, it is safe to say
that GAG→AAG mutation of TGF-βRII also occurs in bladder cancer
patients. To explore the possible clinical significance of such
point mutations, patients were subdivided into 2 groups:
non-mutated TGF-βRII (group 1, n=28) and point-mutated TGF-βRII
(group 2, n=18). In cases of bladder cancer, tumor grade is used to
reflect the relapse and metastasis, and pathologic T stage,
however, may reflect the degree of infiltration and invasion by
tumor cells. By comparing the clinical and pathologic features of 2
groups, significant differences were found in both pathologic T
stage and tumor grade stages (P<0.01; Table II). Together, these findings
suggested that GAG→AAG mutation of TGF-βRII may also occur in
patients with bladder cancer and such TGF-βRII mutations seem to be
correlated with worse malignant phenotypes and poor prognosis.
| Table II.Comparison of clinical and pathologic
characteristics. |
Table II.
Comparison of clinical and pathologic
characteristics.
| Variable | Patients (n)
| χ2
trend | P-value |
| Non-point-mutated
TGF-βRII | Point-mutated
TGF-βRII |
| Pathologic T
stage | | | | |
| T1 | 16 (57.1) | 3 (16.7) | 7.404 | 0.007 |
| T2 | 12 (42.9) | 15 (83.3) | | |
| Tumor grade | | | | |
| Low | 19 (67.9) | 4 (22.2) | 9.127 | 0.003 |
| High | 9 (32.1) | 14 (77.8) | | |
| Tumor size
(cm) | | | | |
| <3 | 23 (82.1) | 12 (66.7) |
Δ0.717 | 0.397 |
| ≥3 | 5 (17.9) | 6 (33.3) | | |
| Age | | | | |
| <45 | 4 (14.3) | 3 (16.7) |
Δ0.003 | 0.956 |
| ≥45 | 24 (85.7) | 15 (83.3) | | |
| Gender | | | | |
| Female | 5 (17.9) | 1 (0.6) |
Δ0.578 | 0.447 |
| Male | 23 (82.1) | 17 (94.4) | | |
| Multiplicity | | | | |
| Single | 22 (78.6) | 10 (55.6) | 2.741 | 0.098 |
| Multiplicity | 6 (21.4) | 8 (44.4) | | |
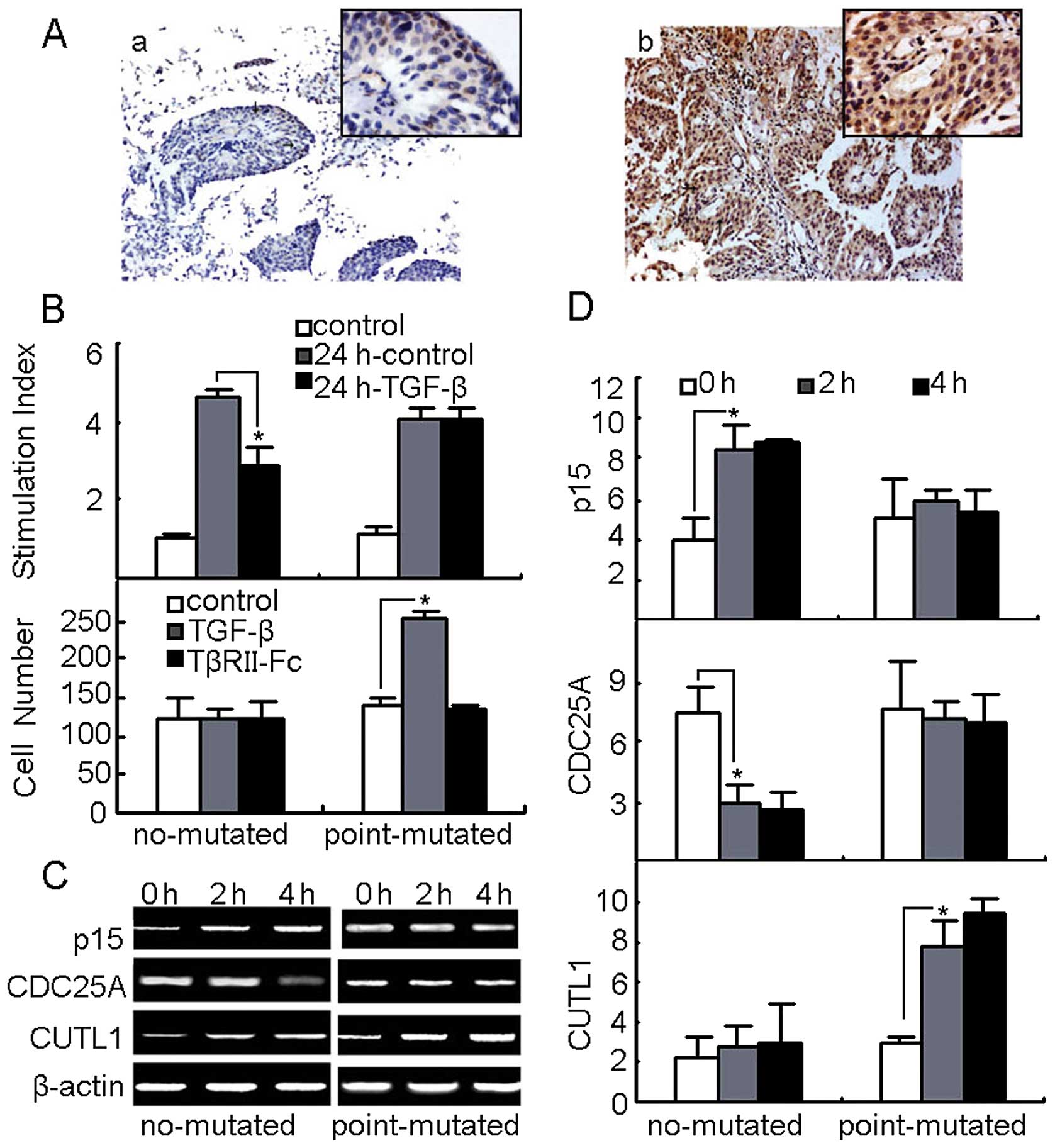
Mutated TGF-βRII transduces TGF-β1
signaling to modulate the behavior of primary bladder cancer
cells
The mutation-induced relief from growth arrest and
strengthened motility in T24 bladder cancer cell line TGF-βRII were
found to be similarly at work in primary bladder cancer cells. On
the basis of the sequencing results, the primary bladder cancer
cells from 12 patients were classified into two groups: no TGF-βRII
mutation (n=6) and mutation with GAG→AAG (n=6). The expression of
TGF-βRII between these two groups was then compared. The
immunohistochemical staining showed that TGF-βRII protein was
strongly expressed in the mutation with GAG→AAG group but not in
the no mutation group (Fig. 6A).
As a parallel observation, the primary bladder cancer cell growth
was not affected by TGF-β treatments in the mutation group
(Fig. 6B). On the other hand,
TGF-β treatment enhanced the invasion of these cells from GAG→AAG
group, which were impaired by TGF-βRII blockade (Fig. 6B). Measurements, furthermore, of
the expression of p15, Cdc25A and CUTL1 in the cultured primary
bladder cancer cells in the presence or absence of TGF-β, as
expected, showed that p15 and Cdc25A were not affected, whilst
CUTL1 was upregulated by TGF-β in GAG→AAG group (Fig. 6C and D). Therefore, TGF-βRII
GAG→AAG mutation in bladder cancer patients might favor cancer cell
migration and survival.
Discussion
Tumor cells evolve multiple strategies, including
the mutation of TGF-βRII, to overcome TGF-β signaling-mediated
growth arrest. Previous studies have reported that TGF-βRII
mutations might abolish TGF-β-induced growth inhibition in breast,
head and neck, colon and endometrial cancers (16). This study has provided further
evidence that bladder cancer cells evolve point mutations in the
extracellular and cytoplasmic regions of TGF-βRII, which
incapacitate the TGF-β-mediated growth arrest but enhance the
tumor-promoting effect of TGF-β in the migration and invasion of
bladder cancer cells.
Overexpression of TGF-β in human cancers is a
general biological phenomenon and switching the role of TGF-β from
a tumor suppressor to a tumor promoter is an important step in
malignant development. To accomplish this goal, tumor cells employ
a variety of molecular mechanisms to downregulate the expression of
TGF-β receptors or simply disable their function by using mutation
strategies. In the case of TGF-βRII, for example, truncation,
deletion, or decreased expression of TGF-βRII has been detected in
a variety of primary tumors and tumor cell lines (15–22).
Moreover, mutations in TGF-βRII frequently occur at the coding
region of exon 3 with a special sequence called microsatellite-like
repeats consisting of a 10-base pair (bp) poly-adenine per repeat.
Such mutations are characterized by an insertion/deletion of one or
two adenines, leading to a truncated protein that lacks the
transmembrane domain and the intracellular serine/threonine kinase
domain, and found in a variety of malignancies including colon,
gastric, non-small cell lung and biliary tract cancers and glioma
(36–40). In addition, point mutations in the
kinase domain of TGF-βRII causing defective autophosphorylation
have been reported in human head and neck carcinoma cell lines
(41). In this study, we further
showed that point mutations of TGF-βRII occurred in bladder cancer
cell line and patients. Two point mutations were detected in the
extracellular region and another two were found in the cytoplasmic
serine/threonine kinase domain. For the latter, one is synonymous
mutation and the other is missense mutation by
Glu269→Lys, thus profoundly changing the proximal charge
and subsequently influencing the phosphorylation of TGF-βRI. The
TGF-β signaling can still be transduced through classical Smad2/3
pathway, regardless of the potent effect of Glu269→Lys
mutation on TGF-βRI, whose activation recruits and phosphorylates
Smad2/3. In the present study, although we did not investigate how
Glu269→Lys mutation affects the phosphorylation of
TGF-βRI, elucidating the underlying molecular mechanism undoubtedly
has been useful for our understanding of the significance of
TGF-βRII in bladder tumorigenesis.
Bladder cancer is the most and second most common
genitourinary neoplasia in China and the USA, respectively, which
causes up to 12,000 or more annual deaths. However, to date, the
progression of bladder cancer is still not well understood.
Previous studies have showed the serum levels of TGF-β1 were
significantly elevated in invasive bladder cancer patients and
TGF-β1, rather than TGF-β2 or 3 was the predominant isoform in
bladder cancer cells at protein as well as mRNA levels (42,43),
suggesting that TGF-β1 signaling is involved in the progression of
bladder cancer. Hung et al explored the molecular profile
involving TGF-β signaling pathway in bladder cancer, which
emphasized the importance of TGF-β signaling in bladder cancer
progression (9). Given the known
growth-inhibiting effect induced by TGF-β signaling, how bladder
cancer cells escape TGF-β-mediated growth arrest still remains
elusive. Early studies indicated that TGF-βRII is necessary for
TGF-β-mediated growth inhibitory response and TGF-βRII is
downregulated in invasive bladder cancer lesions (44). Nevertheless, the increase of
TGF-βRII expression was also reported in muscle invasive bladder
cancer (45,46). Our present study suggests a
reconciliation of this paradox by the mutation of TGF-βRII.
Although TGF-βRII seems to be downregulated in most human bladder
cancer cell lines as well as other human cancer cell lines, the
bladder cancer cell line T24 was found to be capable of
overexpressing TGF-βRII (Fig. 1),
attributable perhaps to the point mutations of TGF-βRII (Fig. 4). In line with these in
vitro data, we also found that TGF-βRII was highly expressed in
bladder cancer lesions in patients with such point mutations. Thus,
by adapting the mutation strategy, bladder cancer cells keep the
high expression of TGF-βRII but evade the growth inhibition by
TGF-β signaling. Interestingly, this mutation strategy may enhance
the intrinsic promoting effect of TGF-β on migration and invasion.
This phenomenon was observed in both the bladder cancer cell line
and primary tumor cells. In line with our findings, previous
studies have showed that a point mutation in TGF-βRII may have
failed to restore TGF-β-induced growth arrest but was still able to
induce TGF-β-induced migration (47,48).
On the basis of the findings here and in other studies, it appears
that the mutation of TGF-βRII abrogates the tumor suppressor
functions of TGF-β but strengthens other pro-oncogenic effects of
TGF-β in bladder cancer.
CUTL1, also known as CDP, Cut, or Cux-1, a
homeodomain transcriptional regulator of development and cell cycle
progression, and has been identified as a key downstream effector
of TGF-β signaling in modulating tumor cell motility and invasion.
Michl et al found that TGF-β induces CUTL1 expression via
Smad4 and p38MAPK and CUTL1 may stabilize Src protein, leading to
the activation of Src-regulated downstream signaling molecules such
as RhoA, Rac1, Cdc42 and ROCK and subsequent cell mobility and
invasion (33,35). This study found that the addition
of TGF-β increased the expression of CUTL1 and the point mutations
of TGF-βRII, albeit, further augmented such upregulation in bladder
cancer cell line T24 and some primary bladder cancer cells.
Although the underlying mechanism was not elucidated here, some
clues may be drawn from other studies. In addition to the canonical
Smad-mediated signaling pathway, other signal molecules may also be
integrated and execute TGF-β signaling. It is known that
cytoskeleton reorganization is prerequisite for cell mobility and
invasion and governed by small guanosine triphosphatases (GTPases)
of the Rho/Rac/Cdc42 family. Coincidently, recent studies found the
crosstalk between the classical TGF-β/Smad pathway and Rho GTPases
and through TGF-β signaling pathway small GTPase molecules could be
transcriptionally upregulated and functionally activated (30). Activation of Rho GTPases by binding
GTP instead of GDP leads to the interaction with multiple effector
proteins, most of which are serine-threonine kinases, such as Rho
coiled-coiled kinase (ROCK1), thereby resulting in actin
polymerization via the ROCK1/LIMK2/cofilin pathway. In the present
study, although the Rho pathway was not determined per se,
it is logical to assume that the point mutations of TGF-βRII were
useful for the activation Rho GTPases such as RhoA. Besides
TGF-βRI, TGF-βRII has also been shown to interact with other
molecules including cyclin B2, Hsp90, endoglin and AP2B1. Whether
TGF-βRII, especially in its mutated form, is capable of interacting
with Rho GTPases, promoting bladder cancer cell motility, is
nevertheless worthy of verification.
In summary, this study showed that TGF-βRII, by
virtue of its extracellular and cytoplasmic point mutations,
confers on bladder cancer cells a desensitivity to TGF-β-mediated
growth arrest but, at the same time, more ability for
TGF-β-promoted motility and migration. These point mutations have
potential clinical significance in both prognosis and treatment of
patients with bladder cancer.
Acknowledgements
We are grateful to Xiaoping Zhao
(Huazhong University of Science and Technology, P.R. China) for
vector construction, Xing Zeng, Haiyang Lu (Department of Urology,
Tongji Hospital Affiliated Tongji Medical College, Huazhong
University of Science and Technology, Wuhan, P.R. China) for
collection of bladder cancer specimens. This study was supported by
the National Key and Basic Research Development Program of China
Grant 2013CB530505, the Natural Science Foundation of China (nos.
81102219 and 81101944).
References
|
1.
|
Scelo G and Brennan P: The epidemiology of
bladder and kidney cancer. Nat Clin Pract Urol. 4:205–217. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
2.
|
Niu HT, Shao SX, Zhang ZL, et al:
Quantitative risk stratification and individual comprehensive
therapy for invasive bladder cancers in China. Int Urol Nephrol.
41:571–577. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Carthon BC, Wolchok JD, Yuan J, et al:
Preoperative CTLA-4 blockade: tolerability and immune monitoring in
the setting of a presurgical clinical trial. Clin Cancer Res.
16:2861–2871. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Havaleshko DM, Smith SC, Cho H, et al:
Comparison of global versus epidermal growth factor receptor
pathway profiling for prediction of lapatinib sensitivity in
bladder cancer. Neoplasia. 11:1185–1193. 2009.PubMed/NCBI
|
|
5.
|
Vaughn DJ, Srinivas S, Stadler WM, et al:
Vinflunine in platinum-pretreated patients with locally advanced or
metastatic urothelial carcinoma: results of a large phase 2 study.
Cancer. 115:4110–4117. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Calabro F, Lorusso V, Rosati G, et al:
Gemcitabine and paclitaxel every 2 weeks in patients with
previously untreated urothelial carcinoma. Cancer. 115:2652–2659.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Milowsky MI, Nanus DM, Maluf FC, et al:
Final results of sequential doxorubicin plus gemcitabine and
ifosfamide, paclitaxel, and cisplatin chemotherapy in patients with
metastatic or locally advanced transitional cell carcinoma of the
urothelium. J Clin Oncol. 27:4062–4067. 2009. View Article : Google Scholar
|
|
8.
|
Kim JH, Shariat SF, Kim IY, Menesses-Diaz
A, Tokunaga H, Wheeler TM and Lerner SP: Predictive value of
expression of transforming growth factor-beta(1) and its receptors
in transitional cell carcinoma of the urinary bladder. Cancer.
92:1475–1483. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Hung TT, Wang H, Kingsley EA, Risbridger
GP and Russell PJ: Molecular profiling of bladder cancer:
involvement of the TGF-beta pathway in bladder cancer progression.
Cancer Lett. 265:27–38. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Champelovier P, El Atifi M, Mantel F,
Rostaing B, Simon A, Berger F and Seigneurin D: In vitro tumoral
progression of human bladder carcinoma: role for TGFbeta. Eur Urol.
48:846–851. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Helmy A, Hammam OA, El Lithy TR and El
Deen Wishahi MM: The role of TGF-beta-1 protein and TGF-beta-R-1
receptor in immune escape mechanism in bladder cancer. Med Gen Med.
9:342007.PubMed/NCBI
|
|
12.
|
Shi Y and Massague J: Mechanisms of
TGF-beta signaling from cell membrane to the nucleus. Cell.
113:685–700. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Rahimi RA and Leof EB: TGF-beta signaling:
a tale of two responses. J Cell Biochem. 102:593–608. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Derynck R, Akhurst RJ and Balmain A:
TGF-beta signaling in tumor suppression and cancer progression. Nat
Genet. 29:117–129. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Fukai Y, Fukuchi M, Masuda N, Osawa H,
Kato H, Nakajima T and Kuwano H: Reduced expression of transforming
growth factor-beta receptors is an unfavorable prognostic factor in
human esophageal squamous cell carcinoma. Int J Cancer.
104:161–166. 2003. View Article : Google Scholar
|
|
16.
|
Levy L and Hill CS: Alterations in
components of the TGF-beta superfamily signaling pathways in human
cancer. Cytokine Growth Factor Rev. 17:41–58. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Lynch MA, Nakashima R, Song H, DeGroff VL,
Wang D, Enomoto T and Weghorst CM: Mutational analysis of the
transforming growth factor beta receptor type II gene in human
ovarian carcinoma. Cancer Res. 58:4227–4232. 1998.PubMed/NCBI
|
|
18.
|
Garrigue-Antar L, Souza RF, Vellucci VF,
Meltzer SJ and Reiss M: Loss of transforming growth factor-beta
type II receptor gene expression in primary human esophageal
cancer. Lab Invest. 75:263–272. 1996.PubMed/NCBI
|
|
19.
|
Zhang HT, Chen XF, Wang MH, et al:
Defective expression of transforming growth factor beta receptor
type II is associated with CpG methylated promoter in primary
non-small cell lung cancer. Clin Cancer Res. 10:2359–2367. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Gobbi H, Arteaga CL, Jensen RA, et al:
Loss of expression of transforming growth factor beta type II
receptor correlates with high tumour grade in human breast in-situ
and invasive carcinomas. Histopathology. 36:168–177. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
21.
|
Kim IY, Ahn HJ, Lang S, Oefelein MG, Oyasu
R, Kozlowski JM and Lee C: Loss of expression of transforming
growth factor-beta receptors is associated with poor prognosis in
prostate cancer patients. Clin Cancer Res. 4:1625–1630.
1998.PubMed/NCBI
|
|
22.
|
Kim IY, Ahn HJ, Zelner DJ, et al: Loss of
expression of transforming growth factor beta type I and type II
receptors correlates with tumor grade in human prostate cancer
tissues. Clin Cancer Res. 2:1255–1261. 1996.PubMed/NCBI
|
|
23.
|
Massague J: TGFbeta in cancer. Cell.
134:215–230. 2008. View Article : Google Scholar
|
|
24.
|
Bierie B and Moses HL: Tumour
microenvironment: TGFbeta: the molecular Jekyll and Hyde of cancer.
Nat Rev Cancer. 6:506–520. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
25.
|
Zhao XP, Huang YY, Huang Y, et al:
Transforming growth factor-beta1 upregulates the expression of CXC
chemokine receptor 4 (CXCR4) in human breast cancer MCF-7 cells.
Acta Pharmacol Sin. 31:347–354. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26.
|
Zhou Y, Wang S, Ma JW, et al: Hepatitis B
virus protein X induced expression of the CXC chemokine IP-10 is
mediated through activation of NF-kappaB and increases migration of
leukocytes. J Biol Chem. 285:12159–12168. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
27.
|
Huang B, Zhao J, Shen S, et al: Listeria
monocytogenes promotes tumor growth via tumor cell toll-like
receptor 2 signaling. Cancer Res. 67:4346–4352. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
28.
|
Huang B, Lei Z, Zhang GM, et al:
SCF-mediated mast cell infiltration and activation exacerbate the
inflammation and immunosuppression in tumor microenvironment.
Blood. 112:1269–1279. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
29.
|
Han M, Li AY, Meng F, et al: Synergistic
co-operation of signal transducer and activator of transcription 5B
with activator protein 1 in angiotensin II-induced angiotensinogen
gene activation in vascular smooth muscle cells. FEBS J.
276:1720–1728. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
30.
|
Brown K and Bhowmick NA: Linking
TGF-beta-mediated Cdc25A inhibition and cytoskeletal regulation
through RhoA/p160(ROCK) signaling. Cell Cycle. 3:408–410. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
31.
|
Feng XH, Lin X and Derynck R: Smad2, Smad3
and Smad4 cooperate with Sp1 to induce p15(Ink4B) transcription in
response to TGF-beta. EMBO J. 19:5178–5193. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
32.
|
Seoane J, Pouponnot C, Staller P, Schader
M, Eilers M and Massague J: TGFbeta influences Myc, Miz-1 and Smad
to control the CDK inhibitor p15INK4b. Nat Cell Biol. 3:400–408.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
33.
|
Aleksic T, Bechtel M, Krndija D, et al:
CUTL1 promotes tumor cell migration by decreasing
proteasome-mediated Src degradation. Oncogene. 26:5939–5949. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
34.
|
Michl P and Downward J: CUTL1: a key
mediator of TGFbeta-induced tumor invasion. Cell Cycle. 5:132–134.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
35.
|
Michl P, Ramjaun AR, Pardo OE, et al:
CUTL1 is a target of TGF(beta) signaling that enhances cancer cell
motility and invasiveness. Cancer Cell. 7:521–532. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
36.
|
Grady WM, Myeroff LL, Swinler SE, et al:
Mutational inactivation of transforming growth factor beta receptor
type II in microsatellite stable colon cancers. Cancer Res.
59:320–324. 1999.PubMed/NCBI
|
|
37.
|
Izumoto S, Arita N, Ohnishi T, et al:
Microsatellite instability and mutated type II transforming growth
factor-beta receptor gene in gliomas. Cancer Lett. 112:251–256.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
38.
|
Nagai M, Kawarada Y, Watanabe M, et al:
Analysis of micro-satellite instability, TGF-beta type II receptor
gene mutations and hMSH2 and hMLH1 allele losses in
pancreaticobiliary maljunction-associated biliary tract tumors.
Anticancer Res. 19:1765–1768. 1999.
|
|
39.
|
Tani M, Takenoshita S, Kohno T, Hagiwara
K, Nagamachi Y, Harris CC and Yokota J: Infrequent mutations of the
transforming growth factor beta-type II receptor gene at chromosome
3p22 in human lung cancers with chromosome 3p deletions.
Carcinogenesis. 18:1119–1121. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
40.
|
Wu MS, Lee CW, Shun CT, Wang HP, Lee WJ,
Sheu JC and Lin JT: Clinicopathological significance of altered
loci of replication error and microsatellite instability-associated
mutations in gastric cancer. Cancer Res. 58:1494–1497. 1998.
|
|
41.
|
Bharathy S, Xie W, Yingling JM and Reiss
M: Cancer-associated transforming growth factor beta type II
receptor gene mutant causes activation of bone morphogenic
protein-Smads and invasive phenotype. Cancer Res. 68:1656–1666.
2008. View Article : Google Scholar
|
|
42.
|
Eder IE, Stenzl A, Hobisch A, Cronauer MV,
Bartsch G and Klocker H: Transforming growth factors-beta 1 and
beta 2 in serum and urine from patients with bladder carcinoma. J
Urol. 156:953–957. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
43.
|
Eder IE, Stenzl A, Hobisch A, Cronauer MV,
Bartsch G and Klocker H: Expression of transforming growth factors
beta 1, beta 2 and beta 3 in human bladder carcinomas. Br J Cancer.
75:1753–1760. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
44.
|
Geiser AG, Burmester JK, Webbink R,
Roberts AB and Sporn MB: Inhibition of growth by transforming
growth factor-beta following fusion of two nonresponsive human
carcinoma cell lines. Implication of the type II receptor in growth
inhibitory responses. J Biol Chem. 267:2588–2593. 1992.
|
|
45.
|
Izadifar V, de Boer WI, Muscatelli-Groux
B, Maille P, van der Kwast TH and Chopin DK: Expression of
transforming growth factor beta1 and its receptors in normal human
urothelium and human transitional cell carcinomas. Hum Pathol.
30:372–377. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
46.
|
McGarvey TW, Tait E, Tomaszewski JE and
Malkowicz SB: Expression of transforming growth factor-beta
receptors and related cell-cycle components in transitional-cell
carcinoma of the bladder. Mol Urol. 3:371–380. 1999.PubMed/NCBI
|
|
47.
|
Li F, Goncalves J, Faughnan K, et al:
Targeted inhibition of wound-induced PAI-1 expression alters
migration and differentiation in human epidermal keratinocytes. Exp
Cell Res. 258:245–253. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
48.
|
Providence KM and Higgins PJ: PAI-1
expression is required for epithelial cell migration in two
distinct phases of in vitro wound repair. J Cell Physiol.
200:297–308. 2004. View Article : Google Scholar : PubMed/NCBI
|