Introduction
Historically, replicative DNA was the main target of
anti-cancer agents for many years, but because of the selectivity
or occurrence of resistance to the drugs, agents which have new
strategy such as molecular targeted therapy show promise for the
treatment of cancer. Efforts to improve cancer therapy have focused
on the development of more selective, biological mechanism-based
agents that can overcome tumor resistance, as well as minimize
toxic effects to normal cells (1).
Ribonucleases are enzymes which catalyze the
degradation of RNA. RNases display various biological roles,
including nutritional function, remobilization of phosphate,
senescence, self-incompatibility, defensin-like activity and the
conspicuous function in RNA metabolism (2,3).
Some members of RNases are reported to exhibit angiogenic,
neurotoxic, antitumor, or immunosuppressive activities (4). Bovine pancreatic ribonuclease A
(RNase A), EC 3.1.27.5 (5), was
the first RNase tested for a possible anticancer activity in
vitro (6–8) and in vivo (9–12).
While RNase A needed high amounts to observe the anticancer
activity, more effective RNases have been reported in recent years.
The proposed mechanism of ribonuclease-induced cytotoxicity is: i)
cell surface binding and internalization, ii) translocation to the
cytosol, iii) evasion of the cytosolic ribonuclease inhibitor
protein (RI) and iv) degradation of cellular RNA. Differences in
the efficiency of any of these steps could affect the cell
susceptibility (13). One
promising RNase for cancer therapeutic drug is onconase, a
ribonuclease isolated from Rana pipiens oocytes. Onconase,
manifests cytotoxic and cytostatic effects (14), presents synergism with several
kinds of anti-cancer drugs (15–22)
and at present is in phase II/III clinical trials as an anticancer
drug (1,23). Onconase has demonstrated some
advantages for potential clinical applications, including: a)
evading human RNase inhibitors in cytosol, b) inhibitory activity
against broad types of human tumors, c) without any untoward immune
response and exerting only weak and reversible renal toxicity
(24). The phase III clinical
trial of onconase has prompted the genetic engineering of known
RNases as well as a search for new medicinal RNases (3,12,24,25).
Sialic acid binding lectin (SBL) isolated from R.
catesbeiana oocytes was found as a lectin, because SBL
agglutinates various kinds of tumor cells and the agglutination was
inhibited by sialoglycoprotein or ganglioside (26–28).
Agglutination induced by SBL was observed in tumor cells, but not
in normal red blood cells or fibroblasts (28). Amino acid sequence of SBL shows
that it has homology to the member of RNase A superfamily and it
has been revealed that SBL practically has pyrimidine base-specific
ribonuclease activity (29–32).
The antitumor effect of SBL was reported using P388 and L1210
murine leukemia cells in vitro and sarcoma 180, Ehrlich and
Mep 2 ascites cells in vivo (33–35).
RC-RNase isolated from R. catesbeiana is identical to SBL
(36,37). It was also reported that RC-RNase
seems to harbor a more specific anticancer activity compared with
onconase (38).
However, the mechanism of antitumor effect of SBL is
unclear and the validity for human leukemia cells has not been
fully studied. We studied the antitumor effect of SBL using some
human leukemia cell lines. We found that SBL shows cytotoxicity to
some cell lines, including multiple drug resistant (MDR) cells. The
mechanism of SBL-induced cytotoxicity is analyzed in detail by
combinational usage of specific caspase inhibitors and
mitochondrial membrane depolarization detector JC-1 and we clearly
show that cytotoxicity is induced through caspase-dependent
apoptosis in which mitochondrial perturbation occurs as upstream
events. It is extrapolated that the novel mechanistic apoptosis
inducing activity toward various human leukemia cells regardless of
P-glycoprotein (P-gp) expression indicating that SBL is a new
candidate as an alternative to conventional DNA-damaging anticancer
drugs.
Materials and methods
Materials
SBL was isolated in sequential chromatography on
Sephadex G-75, DEAE-cellulose, hydroxyapatite and SP-Sepharose as
described previously (28).
Etoposide (ETO), doxorubicin (DOX) and anti-β-actin antibody were
purchased from Sigma-Aldrich (Tokyo, Japan). Tumor necrosis
factor-related apoptosis inducing ligand (TRAIL) was purchased from
R&D Systems (Minneapolis, MN, USA). Caspase inhibitors
(zVAD-fmk, zIETD-fmk, zLEHD-fmk) and anti-caspase-9 antibody were
purchased from Medical & Biological Laboratories Co., Ltd.
(MBL, Nagoya, Japan). Anti-caspase-8 antibody, anti-caspase-3
antibody and anti-Bid antibody were purchased from Cell Signaling
Technology (Beverly, MA, USA). Anti-cytochrome c antibody
was purchased from Becton-Dickinson (Franklin Lakes, NJ, USA).
Horseradish peroxidase (HRP)-conjugated anti-mouse IgG actibody and
HRP-conjugated anti-rabbit IgG andibody was purchased from Zymed
(South San Francisco, CA, USA) and Cedarlane Lab. Ltd. (Hornby,
Ontario, Canada), respectively.
Cell culture
Human leukemia Jurkat T-cells, erythroleukemia K562
cells, Adriamycin-resistant and P-gp-overexpressing K562 cells
(K562/ADR), Burkitt’s lymphoma Raji cells and promyelocytic
leukemia U937 cells were obtained from the Cell Resource Center of
the Biomedical Research, Institute of Development, Ageing and
Cancer, Tohoku University (Sendai, Japan). Cells were routinely
kept in RPMI-1640 medium (Nissui Pharmaceutical Co. Ltd., Tokyo,
Japan) supplemented with 10% fetal calf serum (FCS), penicillin
(100 U/ml) and streptomycin (100 μg/ml) at 37°C in a 95% air
and 5% CO2 atmosphere.
RNA extraction and analysis
The cells were treated with SBL (2 μM) for
indicated time. Total RNA of the cells was extracted with TRIzol
reagent (Invitrogen, Carlsbad, CA, USA) according to the
manufacturer’s instructions. RNA (1 μg) was electrophoresed
on 2% agarose gel containing formaldehyde (18%). The gels were
visualized by ethidium bromide staining.
Measurement of cell viability
To determine the cytotoxicity, WST-8 assays
(39), were done in accordance
with the manufacturer’s instructions. Briefly, the cells
(2×104 cells/well) were plated into 96-well plates.
Various concentration of reagents were added in triplicate to the
cultures and incubated for indicated times before adding the WST-8
solution. The absorbance of the resulting product was measured 4 h
later at a wavelength of 450 nm with back ground subtraction at 650
nm. The IC50 which shows the compound concentration
required for 50% inhibition of the cell growth was calculated
employing GraphPad Prism 3.0 software. Cell viability was
determined by trypan blue dye exclusion assay. The cells
(2×105 cells/ml) were cultured in 100 μl in
96-well plates. After treatment with SBL, the cells were stained
with 0.25% trypan blue and both viable and non-viable cells were
counted.
Observation of nuclear morphology
The cells (2×105 cells/ ml) were cultured
in 5 ml in 6-well plates. After treatment with SBL, the cells were
collected by centrifugation and washed with PBS. Then the cells
were fixed with 1% paraformaldehyde (100 μl) for 15 min at
4°C, and stained with Hoechst 33258 (50 μl, 1 mg/ml) for 15
min at 4°C. After three washes with PBS, the cells were mounted on
slide glass using Prolong gold antifade reagent (Molecular Probes).
The fluorescence was visualized with a fluorescence microscope,
Zeiss Axioscope 2 (Carl Zeiss, Jena GmbH, Jena, Germany).
Detection of DNA fragmentation
The cells (2×105 cells/ml) were cultured
in 100 μl in 96-well plates. After treatment with SBL, the
cells were collected by centrifugation, washed with PBS, then lysed
with cell lysis buffer [50 mM Tris-HCl (pH 6.8), 10 mM EDTA, 0.5%
w/v sodium-N-lauroylsarcosinate]. The samples were incubated for 30
min with RNase A (final concentration, 500 μg/ml) at 50°C,
before being digested for 30 min with proteinase K (final
concentration, 500 μg/ml) at 50°C. Then the samples were
electrophoresed on 1.8% agarose gel, DNA bands were visualized by
ethidium bromide staining.
Flow cytometric analysis of Annexin V
binding and propidium iodide (PI) incorporation
Annexin V binding and PI incorporation were detected
with a MEBCYTO apoptosis kit (MBL) according to the manufacturer’s
instructions. The cells (2×105 cells/ml) were cultured
in 1 ml in 24-well plates. Fluorescence intensity of fluorescein
isothiocyanate (FITC)-Annexin V and PI was determined using a
FACSCalibur flow cytometer (Becton-Dickinson).
Detection of caspase activity
Caspase activity was measured with caspase
colorimetric protease assay kit (MBL) in accordance with the
manufacturer’s instructions. After treatment with SBL (2 μM)
for indicated time, cells were lysed with cell lysis buffer and
incubated for 10 min at 4°C. Then samples were centrifuged at
12,400 rpm and supernatant was collected. Samples (50 μl, 1
μg/μl) were mixed with equal amount of 2X reaction
buffer and substrates (DEVD-pNA for caspase-3, LEHD-pNA for
caspase-9, IETD-pNA for caspase-8) were added at final
concentration 200 μM. After incubation for 2 h at 37°C, the
absorbance of the resulting product was measured at a wavelength of
405 nm.
Treatment of caspase inhibitors
The role of caspase activation in the process was
studied by the addition of zVAD-fmk (pancaspase inhibitor),
zIETD-fmk (caspase-8 specific inhibitor) and zLEHD-fmk (caspase-9
specific inhibitor). Each caspase inhibitor (50 μM) was
added to culture medium 30 min before the addition of reagents.
Western blotting
Whole cell lysate was prepared by lysing the cells
with extraction buffer [150 mM NaCl, 1% Triton X-100, 10 mM
Tris-HCl (pH 7.5), 5 mM EDTA (pH 8.0), 1 mM phenylmethylsulfonyl
fluoride (PMSF), 1 tablet/10 ml protease inhibitor cocktail (Roche
Applied Science, Indianapolis, IN, USA)]. Lysates of organelle
(mitochondria) fraction or cytosol fraction were prepared by
ProteoExtract Subcellular Proteome Extraction Kit (Merck Millipore,
Billerica, MA, USA). Soluble proteins were collected and
concentrations were measured by DC protein assay kit (Bio-Rad,
Richmond, CA, USA) in accordance with instructions. Proteins were
separated by SDS-PAGE and transferred to polyvinylidene difluoride
(PVDF) membrane (GE Healthcare, Little Chalfont, UK). The membrane
was blocked by 5% fat-free skim milk for 1 h. After the membrane
was washed with TBST [20 mM Tris-HCl (pH 7.6), 137 mM NaCl, 0.05%
Tween-20], primary and secondary antibodies were added to the
membrane, respectively. The proteins on membrane were detected
using ECL western blotting detection regents (GE Healthcare).
Detection of mitochondrial membrane
potential (MMP) reduction
MMP was assessed using a fluorescent probe
5,50,6,60-tetrachloro-1,10,3,30-tetraethyl-benzamidazolocarbocyanin
iodide (JC-1, AnaSpec, Fremont, CA, USA). Red emission from the dye
is attributed to a potential-dependent aggregation of JC-1 in the
mitochondria. Green fluorescence reflects the monomeric form of
JC-1, appearing in the cytoplasm after mitochondrial membrane
depolarization. Cells were cultured in condition of each experiment
and then incubated with JC-1 (2 μM) dye diluted in culture
medium at 37°C for 15 min. The cells were washed three times with
PBS and analyzed immediately using FACSCalibur
(Becton-Dickinson).
Statistical analysis
Results were collected from three independent
experiments, each performed in triplicate and data are expressed as
mean ± SD. Statistical analysis was performed using GraphPad Prism
3.0 and comparisons were made using one-way or two-way analysis of
variance (ANOVA), followed by Bonferroni’s post hoc
tests.
Results
SBL shows cytotoxicity to some human
leukemia cell lines including MDR cells
We have previously shown that SBL has antitumor
activity against mouse leukemia cells in vitro and in
vivo (33,34,40).
In this study, antitumor activity of SBL for human leukemia cell
line and the signaling mechanism was evaluated. To determine the
effect of SBL on cell viability of some human leukemia cell lines,
WST assay was performed. As shown in Table I, SBL shows cytotoxic effect for
all cell lines tested at low concentration (0.15–1.39 μM)
and the lowest IC50 value was observed in Jurkat cells
(0.15 μM). The cytotoxic effect of SBL was observed
regardless of the P-gp expression level, while ETO and DOX which
are used clinically for leukemia as DNA damaging agent did not
cause cytotoxic effect on P-gp-overexpressing K562 cells.
 | Table I.Inhibitory effect of SBL, ETO and DOX
on the viability of human leukemia cell lines. |
Table I.
Inhibitory effect of SBL, ETO and DOX
on the viability of human leukemia cell lines.
| | IC50
(mM)
|
|---|
| Cell name |
Characteristics | SBL | ETO | DOX |
|---|
| Jurkat | T-cell
leukemia | 0.15±0.07 | 2.09±0.78 | 1.10±0.71 |
| K562 |
Erythroleukemia | 1.39±0.92 | 13.23±3.86 | 3.51±1.84 |
| K562/ADM |
P-glycoprotein-overexpressing K562
cells | 0.36±0.18 | N/A | N/A |
| U937 | Promyelocytic
leukemia | 0.81±0.24 | 0.46±0.09 | 0.34±0.27 |
| Raji | Burkitt’s
lymphoma | 0.88±0.54 | 0.49±0.04 | 0.28±0.19 |
SBL degrades cellular RNA and inhibits
cell proliferation of Jurkat cells
Because SBL has pyrimidine base-specific
ribonuclease activity (31,32),
we analyzed whether the cellular RNA is degraded by SBL or not.
Total RNA extracted from SBL-treated Jurkat cells was analyzed by
agarose gel electrophoresis and partial RNA degradation was found
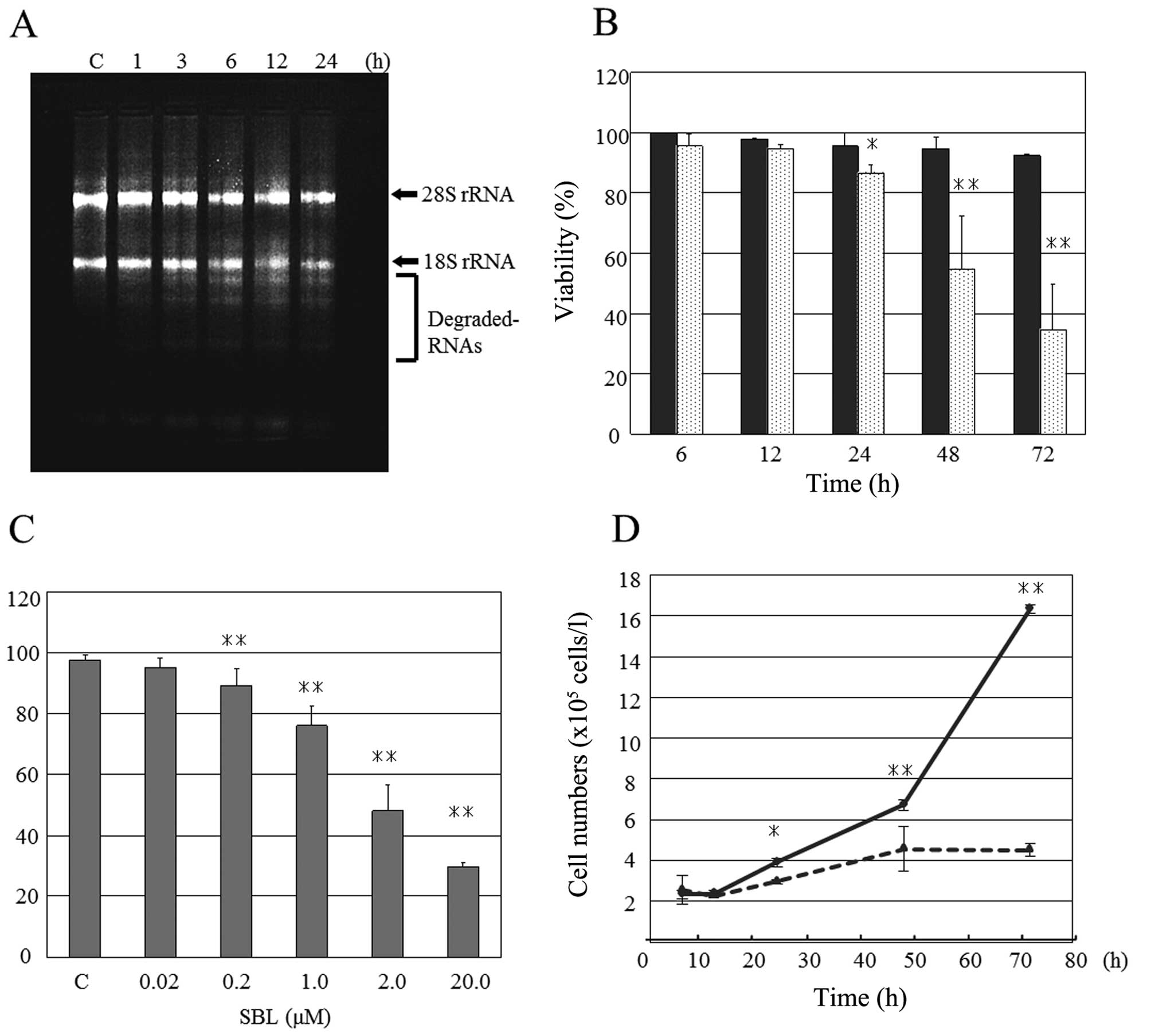
from 3-h treatment, then increased time-dependently (Fig. 1A). Next, the effects of SBL on
Jurkat cell proliferation were analyzed in detail. Trypan blue dye
exclusion assay showed that SBL (2 μM) exhibited
cytotoxicity toward Jurkat cells from 24 h time-dependently and
that SBL (0.2 μM or above) exhibited cytotoxicity in 48-h
treatment concentration-dependently (Fig. 1B and C). Furthermore, significant
inhibition of Jurkat cell proliferation was observed from 24-h
treatment (Fig. 1D).
SBL induces apoptosis in Jurkat
cells
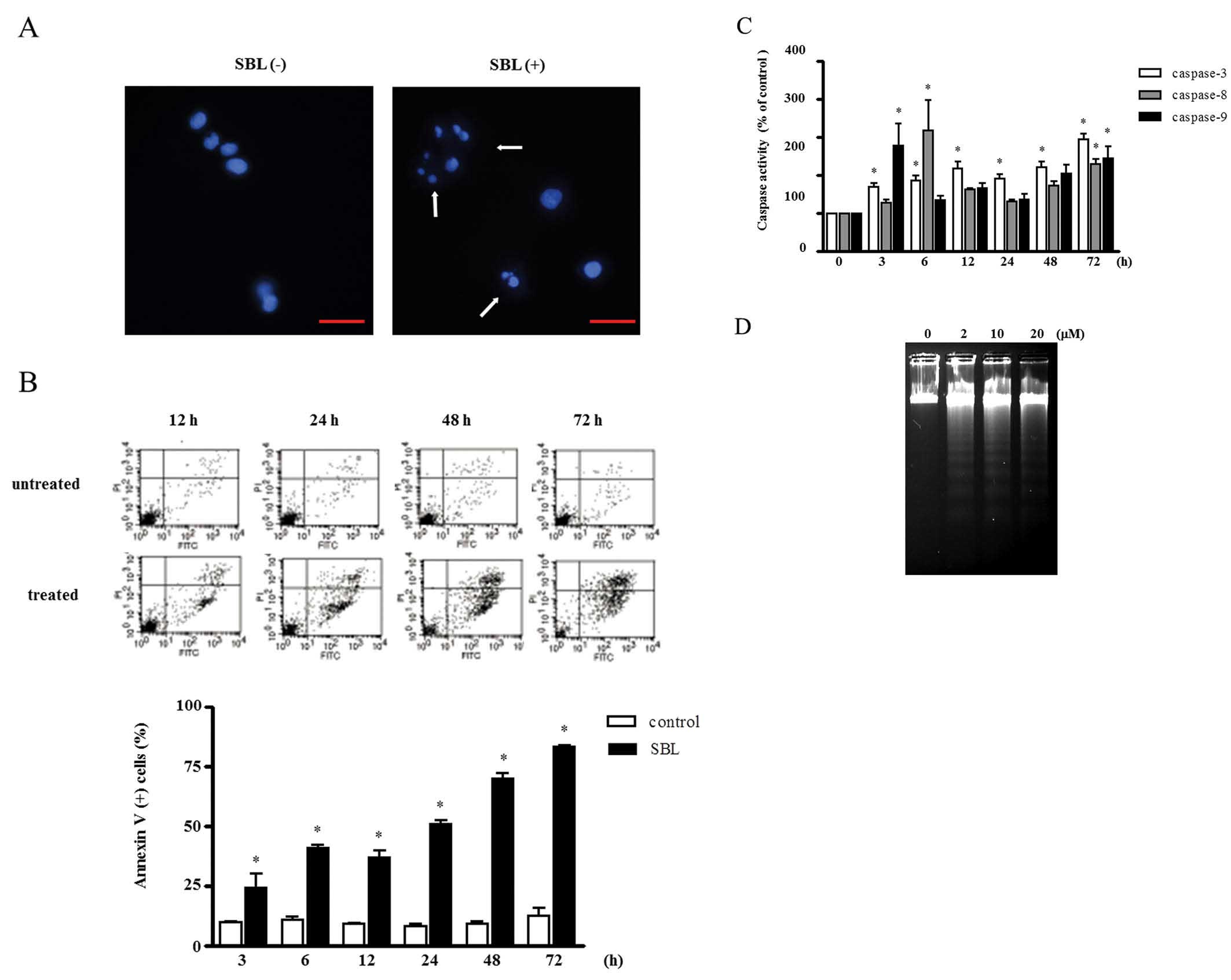
To study the mechanism involved in the cytotoxicity
of SBL, first we investigated the morphological changes in
SBL-treated Jurkat cells using Hoechst 33258. Exposure of SBL
resulted in typical apoptotic morphological alterations, such as
karyorrhexis, nuclear condensation and nuclear fragmentation
(Fig. 2A). We further observed the
apoptotic biological changes in SBL-treated Jurkat cells. Annexin-V
binding which is attributed to externalization of
phosphatidylserine (PS) was observed from 3-h treatment of SBL
(Fig. 2B). Simultaneously, the
activation of initiator caspase (−8 and −9) and effector caspase
(−3) was observed from 3 or 6 h (Fig.
2C). Similarly, DNA fragmentation was observed in a
dose-dependent manner (Fig. 2D).
These data indicate that SBL induces apoptosis in Jurkat cells.
SBL-induced apoptosis is dependent on
caspases and caspase-9 is activated more strongly than
caspase-8
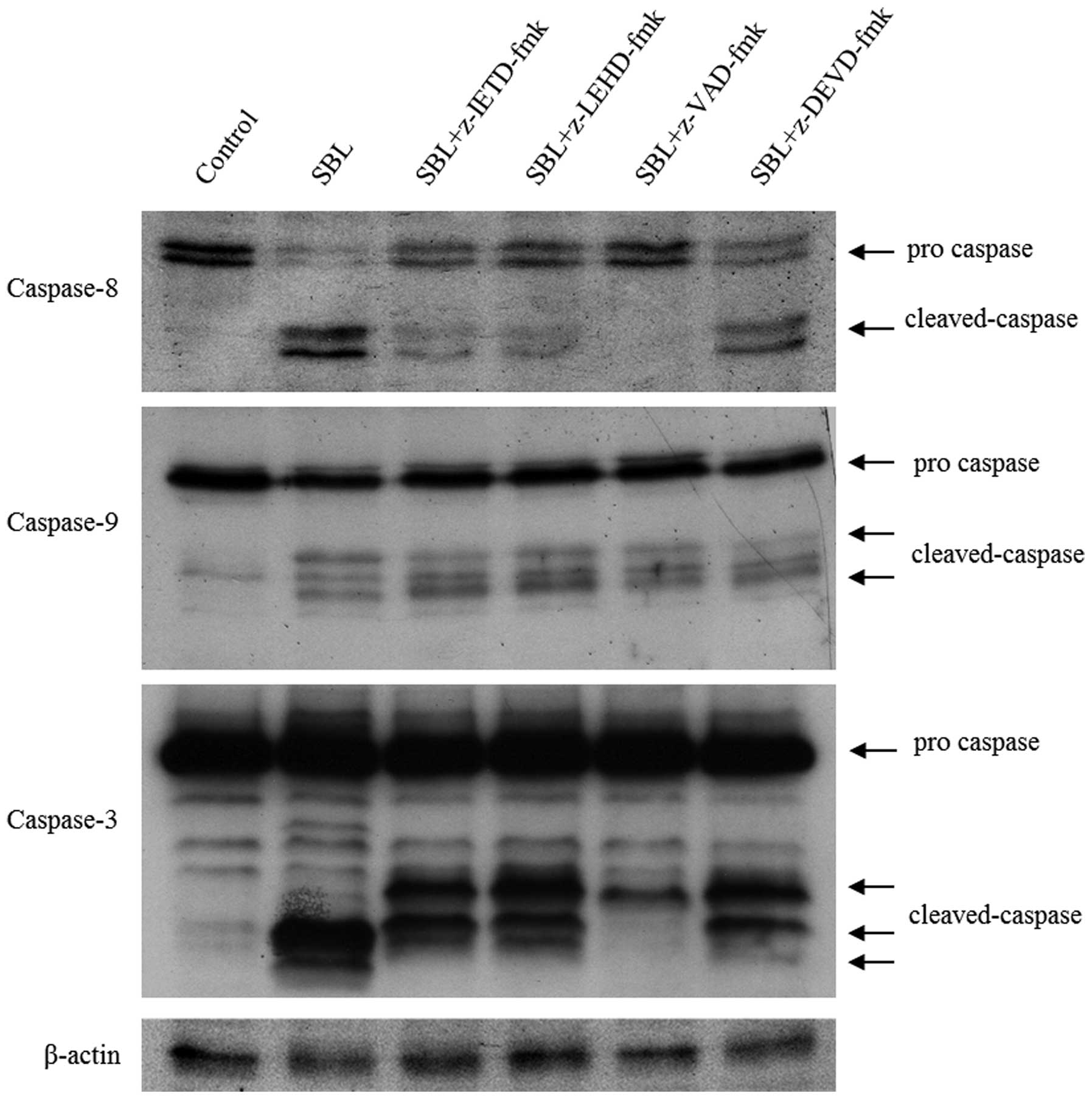
To analyze the detail of SBL-induced caspase
activation, we performed experiments using caspase inhibitors.
Pretreatment of z-VAD inhibited SBL-induced cell death (Fig. 3A) and completely blocked
SBL-induced DNA fragmentation (Fig.
3B). Next, we analyzed activation pattern of caspase-8, -9 and
-3 under the presence of specific inhibitors for each of the
caspases (Fig. 4). Pretreatment of
caspase-9 inhibitor, z-LEHD, inhibited activation of caspase-8. On
the other hand, pretreatment of caspase-8 inhibitor, z-IETD, could
not inhibit caspase-9 activation and the activation of caspase-9
was not affected even by the treatment of z-VAD or z-LEHD.
Pretreatment of caspase-3 inhibitor, z-DEVD did not have an effect
on activation of caspase-8 or -9. These data indicate that SBL
induces caspase-dependent apoptosis and caspase-9 is activated
strongly in SBL-induced apoptosis.
SBL induces activation of p38 and JNK,
but not ERK
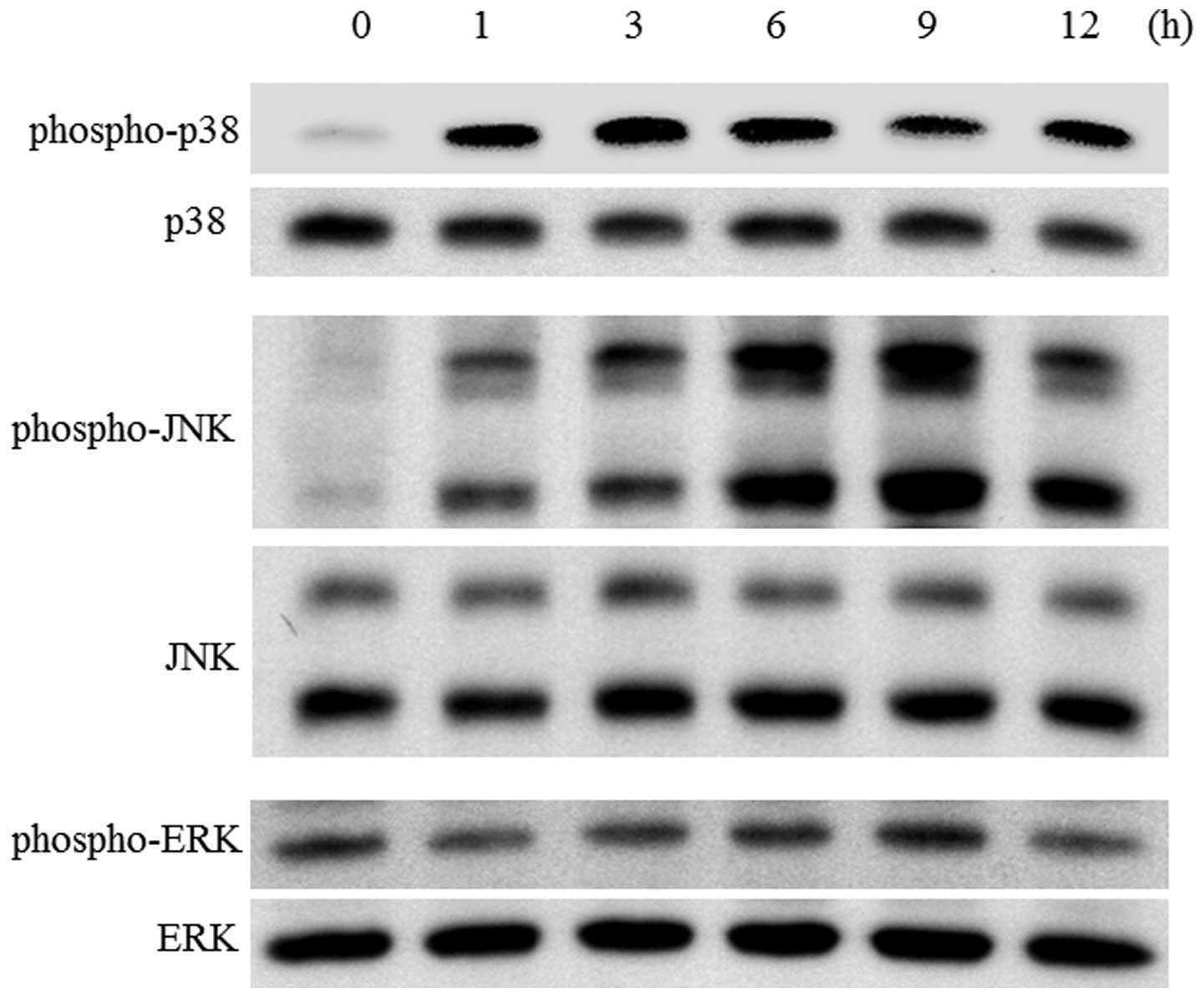
We monitored the activation of mitogen-activated
protein kinases (MAPKs), extracellular signal-regulated kinases
(ERKs), c-jun N-terminal kinase (JNKs)/stress-activated protein
kinases and p38 in SBL-treated cells. Fig. 5 shows p38 kinase was activated as
early as 1 h and sustained to 12 h. Furthermore, activation of
JNK1/2 was observed from 1-h treatment and maximal at 6–9 h,
whereas, activation of ERK was not observed in this condition.
These results suggest that p38 and JNK may be involved in
SBL-induced apoptotic signaling.
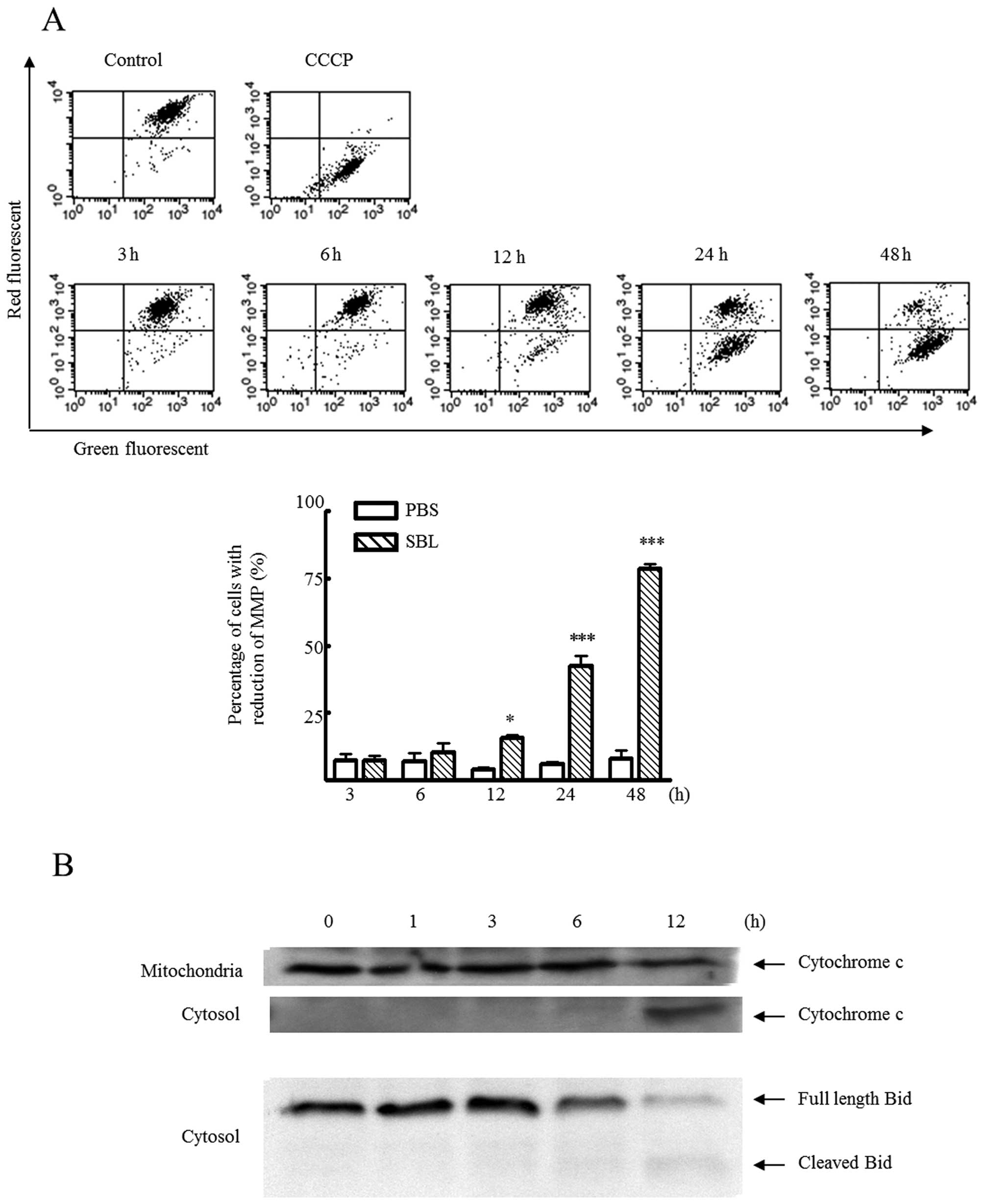
Mitochondrial perturbation occurs before
activation of caspases
Because caspase-9 is known as initiator caspase in
apoptosis through mitochondria pathway, we analyzed the
mitochondrial perturbation in SBL-induced apoptosis. During
apoptosis, loss of mitochondrial membrane potential (MMP) is
observed. We detected mitochondrial membrane depolarization using
JC-1 fluorescent dye in SBL-treated Jurkat cells and found that SBL
caused mitochondrial membrane depolarization in a time-dependent
manner (Fig. 6A). At the same
time, release of cytochrome c from mitochondria to cytosol
was also observed (Fig. 6B) and
the cleavage of Bid, which causes an efflux of cytochrome c
from the mitochondria, was observed from 6 h. These results show
occurrence of mitochondrial perturbation in SBL-treated Jurkat
cells.
To study the importance of mitochondrial
perturbation in SBL-induced apoptosis, we analyzed cell viability
and the mitochondrial depolarization under the presence of caspase
inhibitors comparing with TRAIL and ETO known as inducer of
apoptosis through death receptor pathway and mitochondrial pathway,
respectively. At 48-h treatment with each of the reagents,
pretreatment of z-VAD completely inhibited TRAIL-induced
cytotoxicity, but did not or only partially inhibited SBL- or
ETO-induced cytotoxicity (Fig.
7A). Similarly, mitochondrial depolarization caused by TRAIL
was completely inhibited by z-VAD, while SBL or ETO-induced
mitochondrial depolarization was not affected by z-VAD (Fig. 7B). Furthermore, z-IETD inhibited
TRAIL-induced mitochondrial depolarization to similar extent with
z-VAD, whereas, z-LEHD was less effective. On SBL- or ETO-induced
mitochondrial depolarization, neither IETD nor LEHD showed
inhibitory effect like z-VAD (Fig.
7C). These results indicate that SBL-induced mitochondrial
perturbation is not dependent on caspase activation. Thus, SBL
invokes mitochondrial perturbation first and this process is
followed by caspase activation and the amplification of death
signal executes apoptotic cell death.
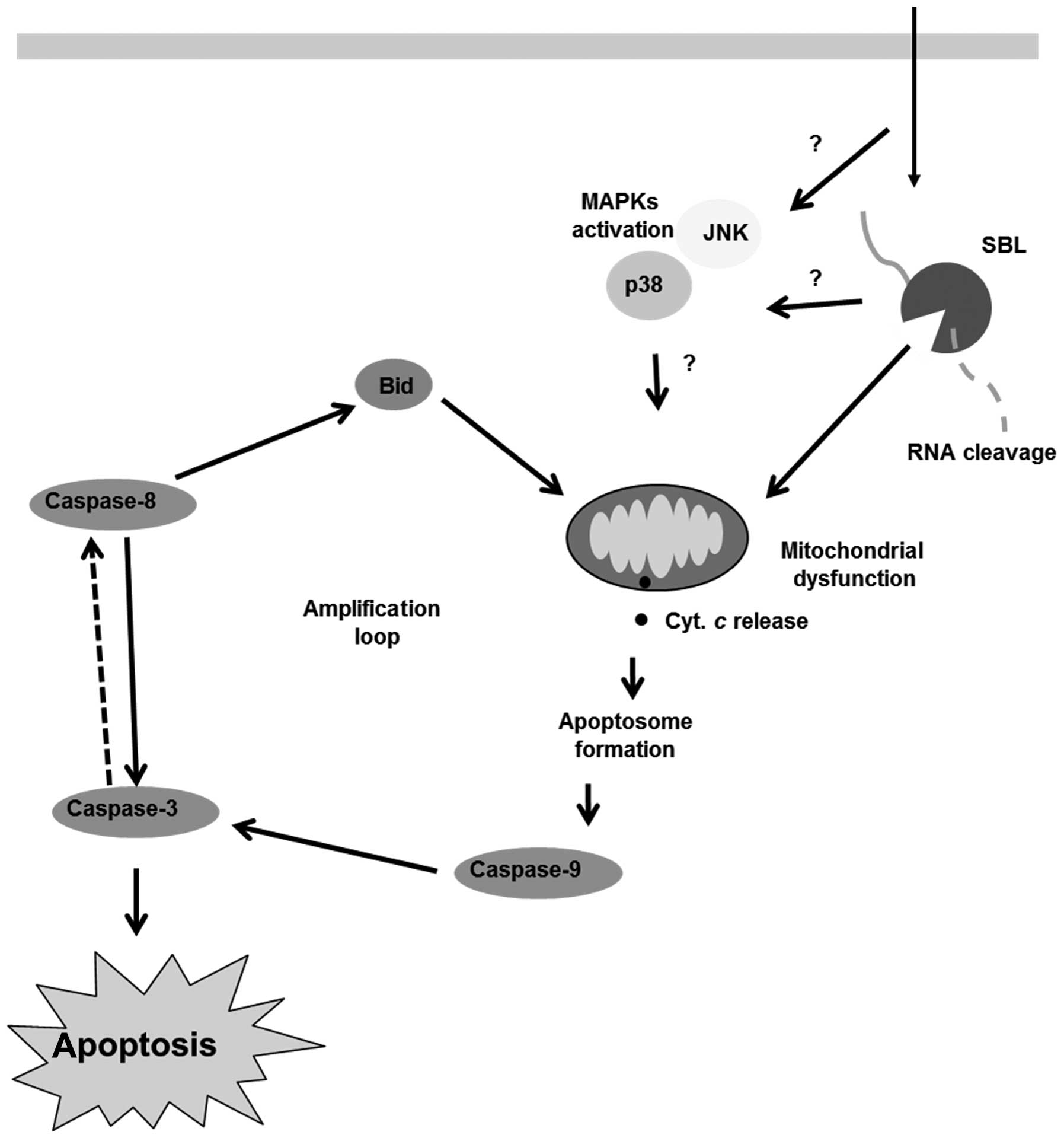
Discussion
SBL is a multifunctional protein which has lectin
activity, ribonuclease activity and antitumor activity. The
proposed mechanism of SBL-induced cell death is shown in Fig. 8. SBL binds to cell surface,
internalizes into tumor cells and degrades cellular RNA and this
ribotoxic stress triggers mitochondrial perturbation. The
activation of p38 and JNK may be involved in the above process.
Then, apoptotic signal is amplified by activation of caspase and
leads to cell death.
SBL selectively agglutinates tumor cells, but not
erythrocytes or fibroblasts (28).
SBL shows cytotoxity to various tumor cells, but not to human
primary WI-38 lung fibroblasts, normal mesothelial Met-5A cells
(data not shown), human primary HFW fibroblasts, immortalized
murine NIH- 3T3/3 cells (40),
human primary HS-68 foreskin fibroblasts (37) or hamster kidney BHK-21 cells
(41). It seems that the selective
effect of SBL on cancer cells is due to its selective binding to
tumor cells, because sialidase treatment of cells abolished the
tumor cell agglutination and also the antiproliferative effect
induced by SBL (33).
In this study, we showed that SBL manifests
cytotoxicity to some human leukemia cell lines including MDR cells,
while conventional DNA-damaging agents, ETO and DOX which have been
used clinically were not able to show cytotoxicity to MDR cells
(Table I). The resistance of
tumors occurs as a cross-resistance to a whole range of drugs with
different structures and this phenomenon is called MDR. The
cytotoxic drugs that are most frequently associated with MDR are
hydrophobic and amphipathic natural products, such as the taxanes
(paclitaxel and docetaxel), vinca alkaloids (vinorelbine,
vincristine and vinblastine), anthracyclines (DOX, daunorubicin and
epirubicin), epipodophyllotoxins (ETO and teniposide),
antimetabolites (methorexate, fluorouracil, cytosar, 5-azacytosine,
6-mercaptopurine and gemcitabine), topotecan, dactinomycin and
mitomycin c (42–46). Overexpression of ATP-binding
cassette (ABC) transporters such as P-gp is known to be responsible
for MDR (46). Cytotoxic RNase,
PE5 (a variant of human pancreatic ribonuclease carrying a nuclear
localization signal) reduced the expression level of the P-gp in
MDR cell lines (47). It is
believed that SBL displays novel mechanistic and tumor-selective
cytotoxic effects regardless of P-gp expression and SBL is
favorable as a new candidate anticancer drug.
Apoptosis, also known as programmed cell death,
plays a critical role in various biological phenomena, such as
development, immunity and also cell death induced by
chemotherapeutic drugs (48).
Apoptosis may occur through death receptor-dependent (extrinsic) or
independent (intrinsic or mitochondrial) pathways and the pathways
finally activate the effector caspase-3, which leads to finally
execution of apoptosis (49).
During the execution phase of apoptosis, typical apoptotic changes
such as chromatin condensation, nuclear collapse, internucleosomal
DNA fragmentation are observed. A variety of studies have
demonstrated that in cancer therapy, induction of apoptosis is a
frequent outcome of effective therapy. In this study, we showed
SBL-treated Jurkat cells present typical apoptotic morphological
alterations, such as karyorrhexis, nuclear condensation and
fragmentation (Fig. 2A) and
apoptotic biological changes such as PS externalization, activation
of caspases, and DNA fragmentation (Fig. 2B–D). This SBL-induced DNA
fragmentation was completely blocked by z-VAD indicating that the
cytotoxicity of SBL is induced through caspase-dependent
apoptosis.
It has been reported that some chemotherapeutics and
natural toxins which induce ribotoxic stress response activates
MAPK (50,51). Regarding ribotoxic stress, He et
al demonstrated ribotoxins, such as deoxynivalenol (DON),
anisomycin, satratoxin G (SG) and ricin activate p38, JNKs and ERK
in RAW264 mouse macrophage cell line (52). It was reported that activation of
JNK is important for cytotoxicity of onconase using
jnk1−/− jnk2−/− mouse embryo
fibroblast (MEF) cells (53). Fang
et al reported that RNase MC2 induces phosphorylation of
p38, JNK and ERK in MCF-7 cells (3) and this RNase-mediated apoptotic
signaling is contributed by dual phosphorylation of ERK and JNK in
Hep G2 cells (54). Although it
has been implicated largely that activation of p38 and JNK are
proapoptotic (50,55) and that phosphorylation of ERK is
linked with both antitumor activity (56) and tumor progression (57), some complicated results have been
reported. Costro et al reported that PE5 kills
adriamycin-resitant MCF-7 (MCF-7/ADR) cells through apoptosis
associated with the inactivation of JNK, while onconase did not
change the phosphorylation level of JNK in the cells (25). We showed that SBL is capable of
inducing activation of p38 and JNK, but not ERK. The activation of
p38 and JNK were observed as early as 1-h treatment with SBL
suggesting that p38 and JNK may be activated upstream of
mitochondrial perturbation. Although we tested the effects of p38
inhibitor (SP600125) and JNK inhibitor (SB203580), they did not
affect the cytotoxicity induced by SBL (data not shown). There are
some possible explanations for this phenomenon: i.e., binding to
cell surface or the internalization into cytosol is able to display
RNase activity of SBL, or the first cleavage of RNA, which is a
non-detectable amount by electrophoresis can activate p38 and JNK.
In addition, the activation of p38 and JNK may not be related to
SBL cytotoxicity, or the inhibition of their activation may induce
alternative death signals. The contribution of p38- and
JNK-activation in SBL-induced cytotoxicity remains to be
elucidated.
Wolf and Green reported that caspase-3 is capable of
eliciting cleavage and activation of caspase-8 (58). Activation of caspase-8 results in
the cleavage of Bid to produce a truncated form of the protein.
Truncated Bid translocates from the cytoplasm to the mitochondria,
where it appears to interact with and antagonize the actions of
anti-apoptotic members of the Bcl-2 family, thereby causing an
efflux of cytochrome c from the mitochondria (59–63).
This, in turn, can result in the activation of caspase-9.
Therefore, caspase-8 could amplify apoptotic signals through the
continued release of cytochrome c and subsequent activation
of caspase-9 and -3 (64). Once
activating signals of apoptotic caspase are induced, the
amplification signal can activate caspase-8, -9 and -3. In
addition, the determination of exact time course of the sequential
events is limited by detection sensitivity of experiments. These
facts disturb discrimination of which pathway is involved in the
stimuli. We utilized a combination of specific caspase inhibitors
and mitochondrial membrane depolarization detector JC-1 to
distinguish the SBL-induced signaling pathway comparatively with
TRAIL and ETO. It was clearly shown that SBL-induced mitochondrial
depolarization was not diminished by z-VAD, while TRAIL-induced
mitochondrial depolarization was completely inhibited by z-VAD
(Fig. 7A and B). These results
indicate that cytotoxicity of SBL is induced through
caspase-dependent apoptosis in which mitochondrial perturbation
occurs as upstream events.
In conclusion, we report that SBL, a multifunctional
protein shows cytotoxicity for some human leukemia cell lines
including MDR cells (Fig. 8). The
details of apoptotic signal induced by SBL was analyzed by
combinational usage of specific caspase inhibitors and the
mitochondrial membrane depolarization detector JC-1. The use of
this combination was shown in detail to distinguish the apoptotic
pathway. SBL displays novel mechanistic and tumor-selective
cytotoxic effects regardless of P-gp expression and SBL has
potential as an alternative molecule to conventional DNA-damaging
anticancer drugs.
Acknowledgements
This study was supported by the
‘Academic Frontier’ Project for Private Universities from the
Ministry of Education, Culture, Sports, Science and Technology of
Japan.
References
|
1.
|
Costanzi J, Sidransky D, Navon A and
Goldsweig H: Ribonucleases as a novel pro-apoptotic anticancer
strategy: review of the preclinical and clinical data for
ranpirnase. Cancer Invest. 23:643–650. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
2.
|
Deshpande RA and Shankar V: Ribonucleases
from T2 family. Crit Rev Microbiol. 28:79–122. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Fang EF, Zhang CZ, Fong WP and Ng TB:
RNase MC2: a new Momordica charantia ribonuclease that
induces apoptosis in breast cancer cells associated with activation
of MAPKs and induction of caspase pathways. Apoptosis. 17:377–387.
2012.
|
|
4.
|
D’Alessio G: New and cryptic biological
messages from RNases. Trends Cell Biol. 3:106–109. 1993.PubMed/NCBI
|
|
5.
|
Raines RT: Ribonuclease A. Chem Rev.
98:1045–1066. 1998. View Article : Google Scholar
|
|
6.
|
Ita M, Halicka HD, Tanaka T, Kurose A,
Ardelt B, Shogen K and Darzynkiewicz Z: Remarkable enhancement of
cytotoxicity of onconase and cepharanthine when used in combination
on various tumor cell lines. Cancer Biol Ther. 7:1104–1108. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Rybak SM, Pearson JW, Fogler WE, Volker K,
Spence SE, Newton DL, Mikulski SM, Ardelt W, Riggs CW, Kung HF and
Longo DL: Enhancement of vincristine cytotoxicity in drug-resistant
cells by simultaneous treatment with onconase, an antitumor
ribonuclease. J Natl Cancer Inst. 88:747–753. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Kim DH, Kim EJ, Kalota A, Gewirtz AM,
Glickson J, Shogen K and Lee I: Possible mechanisms of improved
radiation response by cytotoxic RNase, Onconase, on A549 human lung
cancer xenografts of nude mice. Adv Exp Med Biol. 599:53–59. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Halicka HD, Murakami T, Papageorgio CN,
Mittelman A, Mikulski SM, Shogen K and Darzynkiewicz Z: Induction
of differentiation of leukaemic (HL-60) or prostate cancer (LNCaP,
JCA-1) cells potentiates apoptosis triggered by onconase. Cell
Prolif. 33:407–417. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Tsai SY, Hsieh TC, Ardelt B, Darzynkiewicz
Z and Wu JM: Combined effects of onconase and IFN-beta on
proliferation, macromolecular syntheses and expression of STAT-1 in
JCA-1 cancer cells. Int J Oncol. 20:891–896. 2002.PubMed/NCBI
|
|
11.
|
Mikulski SM, Viera A, Darzynkiewicz Z and
Shogen K: Synergism between a novel amphibian oocyte ribonuclease
And lovastatin in inducing cytostatic and cytotoxic effects in
human lung and pancreatic carcinoma cell lines. Br J Cancer.
66:304–310. 1992. View Article : Google Scholar
|
|
12.
|
Rutkoski TJ, Kink JA, Strong LE, Schilling
CI and Raines RT: Antitumor activity of ribonuclease multimers
created by site-specific covalent tethering. Bioconjug Chem.
21:1691–1702. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Haigis MC, Kurten EL and Raines RT:
Ribonuclease inhibitor as an intracellular sentry. Nucleic Acids
Res. 31:1024–1032. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Ledoux L and Brachet J: Remarks on
preparations of ribonuclease from different manufacturing sources.
Biochim Biophys Acta. 16:2901955.PubMed/NCBI
|
|
15.
|
Ledoux L: Action of ribonuclease on
neoplastic growth. II. Action on Landschutz ascites cells in vitro.
Biochim Biophys Acta. 20:369–377. 1956. View Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Darzynkiewicz Z, Carter SP, Mikulski SM,
Ardelt WJ and Shogen K: Cytostatic and cytotoxic effects of Pannon
(P-30 Protein), a novel anticancer agent. Cell Tissue Kinet.
21:169–182. 1988.PubMed/NCBI
|
|
17.
|
Easty DM, Ledoux L and Ambrose EJ: The
action of ribonuclease on neoplastic growth. III. Studies by
interference microscopy. Biochim Biophys Acta. 20:528–537. 1956.
View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Telford IR, Kemp JF, Taylor EF and Yeaman
MW: Effect of ribonuclease on survival of ascites tumor bearing
mice. Proc Soc Exp Biol Med. 100:829–831. 1959. View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Ledoux L: Action of ribonuclease on two
solid tumours in vivo. Nature. 176:36–37. 1955. View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Ardelt W, Mikulski SM and Shogen K: Amino
acid sequence of an anti-tumor protein from Rana pipiens
oocytes and early embryos. Homology to pancreatic ribonucleases. J
Biol Chem. 266:245–251. 1991.PubMed/NCBI
|
|
21.
|
Ledoux L and Revell SH: Action of
ribonuclease on neoplastic growth. I. Chemical aspects of normal
tumour growth: the Landschutz ascites tumour. Biochim Biophys Acta.
18:416–426. 1955.PubMed/NCBI
|
|
22.
|
Aleksandrowicz J, Urbanczyk J, Ostrowska A
and Sierko J: Further research on the activity of ribonucleases in
the blood and urine of patients suffering from proliferative
hemocytopathia. Blood. 13:652–664. 1958.PubMed/NCBI
|
|
23.
|
Vert A, Castro J, Ruiz-Martinez S, Tubert
P, Escribano D, Ribo M, Vilanova M and Benito A: Generation of new
cytotoxic human ribonuclease variants directed to the nucleus. Mol
Pharm. 9:2894–2902. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24.
|
Fang EF and Ng TB: Ribonucleases of
different origins with a wide spectrum of medicinal applications.
Biochim Biophys Acta. 1815:65–74. 2011.PubMed/NCBI
|
|
25.
|
Castro J, Ribo M, Navarro S, Nogues MV,
Vilanova M and Benito A: A human ribonuclease induces apoptosis
associated with p21WAF1/CIP1 induction and JNK inactivation. BMC
Cancer. 11:92011. View Article : Google Scholar : PubMed/NCBI
|
|
26.
|
Kawauchi H, Sakakibara F and Watanabe K:
Agglutinins of frog eggs: a new class of proteins causing
preferential agglutination of tumor cells. Experientia. 31:364–365.
1975. View Article : Google Scholar : PubMed/NCBI
|
|
27.
|
Sakakibara F, Kawauchi H, Takayanagi G and
Ise H: Egg lectin of Rana japonica and its receptor
glycoprotein of Ehrlich tumor cells. Cancer Res. 39:1347–1352.
1979.
|
|
28.
|
Nitta K, Takayanagi G, Kawauchi H and
Hakomori S: Isolation and characterization of Rana
catesbeiana lectin and demonstration of the lectin-binding
glycoprotein of rodent and human tumor cell membranes. Cancer Res.
47:4877–4883. 1987.PubMed/NCBI
|
|
29.
|
Titani K, Takio K, Kuwada M, Nitta K,
Sakakibara F, Kawauchi H, Takayanagi G and Hakomori S: Amino acid
sequence of sialic acid binding lectin from frog (Rana
catesbeiana) eggs. Biochemistry. 26:2189–2194. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
30.
|
Kamiya Y, Oyama F, Oyama R, Sakakibara F,
Nitta K, Kawauchi H, Takayanagi Y and Titani K: Amino acid sequence
of a lectin from Japanese frog (Rana japonica) eggs. J
Biochem. 108:139–143. 1990.PubMed/NCBI
|
|
31.
|
Nitta K, Oyama F, Oyama R, Sekiguchi K,
Kawauchi H, Takayanagi Y, Hakomori S and Titani K: Ribonuclease
activity of sialic acid-binding lectin from Rana catesbeiana
eggs. Glycobiology. 3:37–45. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
32.
|
Okabe Y, Katayama N, Iwama M, Watanabe H,
Ohgi K, Irie M, Nitta K, Kawauchi H, Takayanagi Y, Oyama F, et al:
Comparative base specificity, stability, and lectin activity of two
lectins from eggs of Rana catesbeiana and R. japonica
and liver ribonuclease from R catesbeiana. J Biochem.
109:786–790. 1991.PubMed/NCBI
|
|
33.
|
Nitta K, Ozaki K, Ishikawa M, Furusawa S,
Hosono M, Kawauchi H, Sasaki K, Takayanagi Y, Tsuiki S and Hakomori
S: Inhibition of cell proliferation by Rana catesbeiana and
Rana japonica lectins belonging to the ribonuclease
superfamily. Cancer Res. 54:920–927. 1994.PubMed/NCBI
|
|
34.
|
Nitta K, Ozaki K, Tsukamoto Y, Furusawa S,
Ohkubo Y, Takimoto H, Murata R, Hosono M, Hikichi N, Sasaki K, et
al: Characterization of a Rana catesbeiana lectin-resistant
mutant of leukemia P388 cells. Cancer Res. 54:928–934. 1994.
|
|
35.
|
Nitta K, Ozaki K, Tsukamoto Y, Hosono M,
Ogawakonno Y, Kawauchi H, Takayanagi Y, Tsuiki S and Hakomori S:
Catalytic lectin (leczyme) from bullfrog (Rana catesbeiana)
eggs. Int J Oncol. 9:19–23. 1996.PubMed/NCBI
|
|
36.
|
Liao YD: A pyrimidine-guanine
sequence-specific ribonuclease from Rana catesbeiana
(bullfrog) oocytes. Nucleic Acids Res. 20:1371–1377. 1992.
View Article : Google Scholar : PubMed/NCBI
|
|
37.
|
Liao YD, Huang HC, Leu YJ, Wei CW, Tang PC
and Wang SC: Purification and cloning of cytotoxic ribonucleases
from Rana catesbeiana (bullfrog). Nucleic Acids Res.
28:4097–4104. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
38.
|
Tang CH, Hu CC, Wei CW and Wang JJ:
Synergism of Rana catesbeiana ribonuclease And IFN-gamma
triggers distinct death machineries in different human cancer
cells. FEBS Lett. 579:265–270. 2005.PubMed/NCBI
|
|
39.
|
Ishiyama M, Miyazono Y, Sasamoto K, Ohkura
Y and Ueno K: A highly water-soluble disulfonated tetrazolium salt
as a chromogenic indicator for NADH as well as cell viability.
Talanta. 44:1299–1305. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
40.
|
Liao YD, Huang HC, Chan HJ and Kuo SJ:
Large-scale preparation of a ribonuclease from Rana
catesbeiana (bullfrog) oocytes and characterization of its
specific cytotoxic activity against tumor cells. Protein Expr
Purif. 7:194–202. 1996.PubMed/NCBI
|
|
41.
|
Hu CC, Lee YH, Tang CH, Cheng JT and Wang
JJ: Synergistic cytotoxicity of Rana catesbeiana
ribonuclease And IFN-gamma on hepatoma cells. Biochem Biophys Res
Commun. 280:1229–1236. 2001.PubMed/NCBI
|
|
42.
|
Thomas H and Coley HM: Overcoming
multidrug resistance in cancer: an update on the clinical strategy
of inhibiting P-glycoprotein. Cancer Control. 10:159–165.
2003.PubMed/NCBI
|
|
43.
|
Ambudkar SV, Dey S, Hrycyna CA,
Ramachandra M, Pastan I and Gottesman MM: Biochemical, cellular,
and pharmacological aspects of the multidrug transporter. Annu Rev
Pharmacol Toxicol. 39:361–398. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
44.
|
Krishna R and Mayer LD: Multidrug
resistance (MDR) in cancer. Mechanisms, reversal using modulators
of MDR and the role of MDR modulators in influencing the
pharmacokinetics of anticancer drugs. Eur J Pharm Sci. 11:265–283.
2000.PubMed/NCBI
|
|
45.
|
Stavrovskaya AA: Cellular mechanisms of
multidrug resistance of tumor cells. Biochemistry (Mosc).
65:95–106. 2000.PubMed/NCBI
|
|
46.
|
Ozben T: Mechanisms and strategies to
overcome multiple drug resistance in cancer. FEBS Lett.
580:2903–2909. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
47.
|
Castro J, Ribo M, Puig T, Colomer R,
Vilanova M and Benito A: A cytotoxic ribonuclease reduces the
expression level of P-glycoprotein in multidrug-resistant cell
lines. Invest New Drugs. 30:880–888. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
48.
|
Elmore S: Apoptosis: a review of
programmed cell death. Toxicol Pathol. 35:495–516. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
49.
|
Grutter MG: Caspases: key players in
programmed cell death. Curr Opin Struct Biol. 10:649–655. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
50.
|
Mansouri A, Ridgway LD, Korapati AL, Zhang
Q, Tian L, Wang Y, Siddik ZH, Mills GB and Claret FX: Sustained
activation of JNK/p38 MAPK pathways in response to cisplatin leads
to Fas ligand induction and cell death in ovarian carcinoma cells.
J Biol Chem. 278:19245–19256. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
51.
|
Bunyard P, Handley M, Pollara G, Rutault
K, Wood I, Chaudry M, Alderman C, Foreman J, Katz DR and Chain BM:
Ribotoxic stress activates p38 and JNK kinases and modulates the
antigen-presenting activity of dendritic cells. Mol Immunol.
39:815–827. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
52.
|
He K, Zhou HR and Pestka JJ: Mechanisms
for ribotoxin-induced ribosomal RNA cleavage. Toxicol Appl
Pharmacol. 265:10–18. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
53.
|
Iordanov MS, Wong J, Newton DL, Rybak SM,
Bright RK, Flavell RA, Davis RJ and Magun BE: Differential
requirement for the stress-activated protein kinase/c-Jun
NH(2)-terminal kinase in RNA damage-induced apoptosis in primary
and in immortalized fibroblasts. Mol Cell Biol Res Commun.
4:122–128. 2000. View Article : Google Scholar
|
|
54.
|
Fang EF, Zhang CZ, Zhang L, Fong WP and Ng
TB: In vitro and in vivo anticarcinogenic effects of RNase MC2, a
ribonuclease isolated from dietary bitter gourd, toward human liver
cancer cells. Int J Biochem Cell Biol. 44:1351–1360. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
55.
|
Dasmahapatra G, Lembersky D, Kramer L,
Fisher RI, Friedberg J, Dent P and Grant S: The pan-HDAC inhibitor
vorinostat potentiates the activity of the proteasome inhibitor
carfilzomib in human DLBCL cells in vitro and in vivo. Blood.
115:4478–4487. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
56.
|
Chen J, Rusnak M, Luedtke RR and Sidhu A:
D1 dopamine receptor mediates dopamine-induced cytotoxicity via the
ERK signal cascade. J Biol Chem. 279:39317–39330. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
57.
|
Wilhelm SM, Carter C, Tang L, Wilkie D,
McNabola A, Rong H, Chen C, Zhang X, Vincent P, McHugh M, Cao Y,
Shujath J, Gawlak S, Eveleigh D, Rowley B, Liu L, Adnane L, Lynch
M, Auclair D, Taylor I, Gedrich R, Voznesensky A, Riedl B, Post LE,
Bollag G and Trail PA: BAY 43-9006 exhibits broad spectrum oral
antitumor activity and targets the RAF/MEK/ERK pathway and receptor
tyrosine kinases involved in tumor progression and angiogenesis.
Cancer Res. 64:7099–7109. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
58.
|
Wolf BB and Green DR: Suicidal tendencies:
apoptotic cell death by caspase family proteinases. J Biol Chem.
274:20049–20052. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
59.
|
Kuwana T, Smith JJ, Muzio M, Dixit V,
Newmeyer DD and Kornbluth S: Apoptosis induction by caspase-8 is
amplified through the mitochondrial release of cytochrome c.
J Biol Chem. 273:16589–16594. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
60.
|
Li H, Zhu H, Xu CJ and Yuan J: Cleavage of
BID by caspase 8 mediates the mitochondrial damage in the Fas
pathway of apoptosis. Cell. 94:491–501. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
61.
|
Luo X, Budihardjo I, Zou H, Slaughter C
and Wang X: Bid, a Bcl2 interacting protein, mediates cytochrome
c release from mitochondria in response to activation of
cell surface death receptors. Cell. 94:481–490. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
62.
|
Schendel SL, Azimov R, Pawlowski K, Godzik
A, Kagan BL and Reed JC: Ion channel activity of the BH3 only Bcl-2
family member, BID. J Biol Chem. 274:21932–21936. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
63.
|
Wei MC, Zong WX, Cheng EH, Lindsten T,
Panoutsakopoulou V, Ross AJ, Roth KA, MacGregor GR, Thompson CB and
Korsmeyer SJ: Proapoptotic BAX and BAK: a requisite gateway to
mitochondrial dysfunction and death. Science. 292:727–730. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
64.
|
Viswanath V, Wu Y, Boonplueang R, Chen S,
Stevenson FF, Yantiri F, Yang L, Beal MF and Andersen JK: Caspase-9
activation results in downstream caspase-8 activation and bid
cleavage in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced
Parkinson’s disease. J Neurosci. 21:9519–9528. 2001.PubMed/NCBI
|