Introduction
Cytotoxic chemotherapy of most solid cancers rarely
cures the cancer. Molecular targeted therapy has the same
limitation, despite significantly enhanced cancer control and
resulting prolonged survival, especially when cancers with driver
mutations are treated with specific inhibitors, for example, when
non-small cell lung cancer with an epidermal growth factor-receptor
(EGFR) mutation is treated by EGFR-tyrosine kinase
inhibitors (TKIs) (1). Thus, even
after a complete response after a long period of treatment with
gefitinib, the tumors grew again after withdrawal of the agent in
an animal study (2). The secondary
point mutation of T790M in EGFR (3) and establishment of a bypass signal
transduction via cMET amplification (4) are known genetic alterations
responsible for acquired EGFR-TKI resistance. If chemotherapy were
sufficiently effective to kill the entire cell population of the
tumor in a short period, however, the cancer would be cured before
genetic adaptation and chemoresistance. Nevertheless, some cells in
the tumor escape from effective chemotherapy and these residual
cells survive until a chemoresistant phenotype is obtained.
Therefore, complete elimination of residual cells would represent
significant progress in cancer therapy. A recent study showed that
the drug-tolerant phenotype, induced by acute response to
chemotherapeutic agents, is reversible and that the phenotype
maintains viability via engagement of insulin-like growth factor
(IGF)-1 receptor signaling and an altered chromatin state that
requires histone demethylase (5).
This observation importantly provides a vision for a new strategy
to treat cancer by specifically targeting the residual cells after
chemotherapy.
Metformin is a safe biguanide that has been used
worldwide to treat type 2 diabetes mellitus. Metformin activates
AMP-activated protein kinase (AMPK), an enzyme that plays an
important role in insulin signaling, whole body energy balance and
the metabolism of glucose and fats, resulting in lowering of blood
glucose (6). Metformin recently
attracted attention for its potential anticancer effects (7). Epidemiological studies (8–10)
first suggested a link between metformin and cancer prevention by
demonstrating a lower incidence of death from cancer in patients
with diabetes mellitus treated with metformin than those treated
with other antidiabetic agents. These studies were followed by
clinical observations, suggesting a link between metformin and
increased pathologically complete response rate by induction
chemotherapy in patients with breast cancer (10) as well as lower incidence rate of
metastasis and a reduced risk of death in patients with lung cancer
(11). These findings triggered a
number of in vitro and in vivo experiments, revealing
its antiproliferative properties in a variety of cancers (12–20).
Although the precise mechanism is unclear, activation of AMPK might
be crucial. First, liver kinase B1 (LKB1), a well-recognized tumor
suppressor, activates AMPK (21,22)
and metformin requires LKB1 for growth inhibitory action (23). Second, AMPK inhibits the mammalian
target of rapamycin (mTOR) and the S6 kinase I pathways (24,25)
and this inhibition appears to be achieved by phosphorylating
tuberous sclerosis complex-2, another tumor suppressor and upstream
regulator of mTOR (26). Notably,
metformin blocks the growth-promoting effects of both insulin and
IGF-1, deregulates AMPK activity and inhibits mTOR activity, S6
kinase activity and protein synthesis both in transformed and
non-transformed mammary gland cells (14). However, it is unknown whether
metformin causes apoptosis of cancer cells (13,17)
or not (12,16), or whether metformin kills cancer
cells synergistically with other cytotoxic agents (15,18,20,27)
or antagonistically to cisplatin (28,29).
In the present study, in vivo experiments
suggested a unique anticancer action for metformin, specifically on
residual cells after chemotherapy. The mechanism was further
elucidated with a series of in vitro experiments.
Materials and methods
Cell culture and reagents
A human lung adenocarcinoma cell line, PC9,
purchased from Riken Cell Bank (accession no. RCB4455, Tsukuba,
Japan), was used throughout the study. This cell line has an
activating deletion of the EGFR gene (del E746–A750) in exon
19 (30). The cells were cultured
as a monolayer in RPMI-1640 medium supplemented with 10% fetal
bovine serum (FBS), 100 U/ml penicillin and 100 mg/ml streptomycin
in a 37°C humidified atmosphere containing 5% CO2.
Gefitinib (cat no. 3000, Tocris Bioscience, Ellisville, MO, USA)
was dissolved in dimethyl sulfoxide (DMSO) and stored at −20°C
until use. Metformin (1,1-dimethylbiguanide hydrochloride, cat no.
D150959-5G, Sigma-Aldrich, St. Louis, MO, USA) was dissolved in
phosphate buffered saline (PBS) at a concentration of 100 mM and
stored at 4°C. A cisplatin solution at a concentration of 0.5 mg/ml
(pH 2.5–5.5) was purchased from Nihon Kayaku (Tokyo, Japan). Each
drug was diluted in the complete medium for each experiment and the
final concentration of DMSO was <0.1%.
Combined treatment of metformin and
gefitinib in a mouse xenograft model
Five to 6-week-old female severe combined
immunodeficient (SCID) mice were acclimatized to local conditions
for a week before starting the experiments. Aliquots of the cell
suspension (2×106 cells per mouse) were injected
subcutaneously into their flanks. At day 16 (when the tumor volumes
had reached ∼300 mm3), the mice were randomly allocated
into 4 groups (7 mice per group). In every group, administration of
either saline alone or gefitinib suspended in saline (150
mg/kg/day, every day, p.o. with gavage) and either PBS alone or
metformin dissolved in PBS (250 mg/kg/day, every day, i.p.) were
started. Either saline alone or gefitinib suspended in saline was
continued for 14 days and either PBS alone or metformin dissolved
in PBS was continued until terminating observation. In the first
group, only saline (p.o.) and PBS (i.p.) were administered
(control). In the second group, metformin dissolved in PBS was
administered. In the third group, gefitinib suspended in saline was
administered. In the fourth group, both gefitinib and metformin
were administered (Fig. 1). The
administration route of metformin was selected because a previous
study showed that i.p. was better tolerated and more effective than
p.o. in vivo (31). The
dose of metformin was selected according to a preliminary
experiment that showed that this dose was near maximal without
causing death or body-weight loss in the animals (data not shown).
The dose of gefitinib was previously published (32). In each animal, the tumor was
allowed to grow until reaching ∼2,000 mm3, or until day
66, after which the animal was sacrificed. The tumor size was
estimated by 2-dimensional caliper measurements and calculation
with the formula π/6 × (A × B)3/2, where A and B
represent the larger and smaller diameters of the tumor,
respectively. The tumor size was measured twice a week during the
observation period. The animal experiments were approved by the
animal ethics review board of Chiba University (protocol no.
A22-186) and were conducted in an animal facility at Chiba
University under the strict SPF conditions in accordance with the
established institutional guidelines.
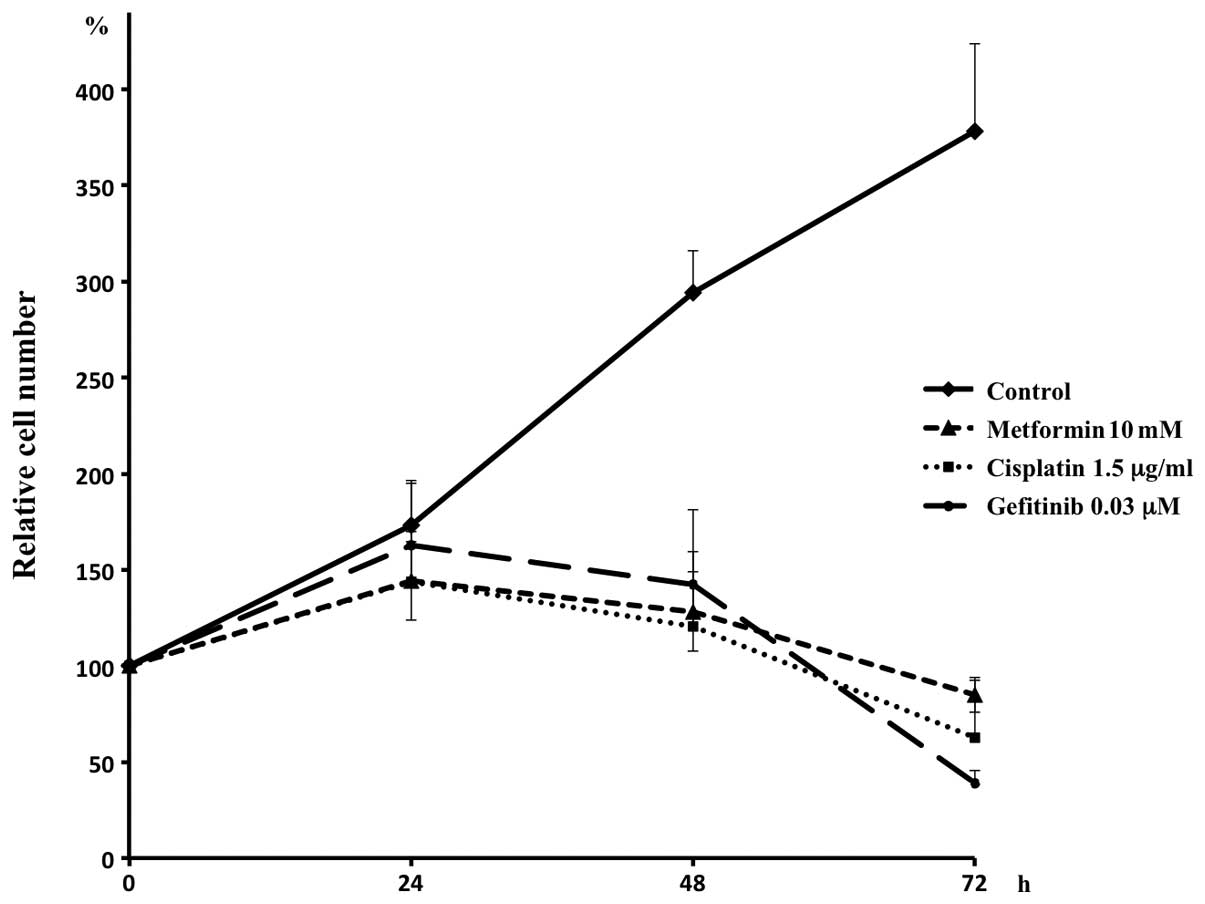
 | Figure 1.Effects of treatment with metformin,
gefitinib and a combination of metformin and gefitinib, on the
growth of PC9 xenograft tumors in SCID mice. After growing tumors
for 16 days, the animals were randomly divided into 4 groups. In
group 1 (fine broken line), saline (p.o.) was administered daily
for 14 days (until day 30) and PBS (i.p.) was then administered
daily until terminating the observation (day 66). In group 2 (fine,
solid line), gefitinib (p.o., 150 mg/kg/day) suspended in saline
was administered daily for 14 days and PBS (i.p.) was then
administered daily until day 66. In group 3 (thick, broken line),
saline (p.o.) was administered daily for 14 days and metformin
(i.p., 250 mg/kg/day) dissolved in PBS was then administered daily
until day 66. In group 4 (thick, solid line), both gefitinib and
metformin were administered. The regrowth of the tumors after
withdrawing gefitinib was significant, with each given p-value in
the figure, suppressed by metformin (compare groups 3 and 4),
whereas metformin exerted no effects on tumor growth (compare
groups 1 and 2) and tumor shrinkage by gefitinib (compare groups 3
and 4). Each point represents the mean and the bars represent the
SE (n=7). |
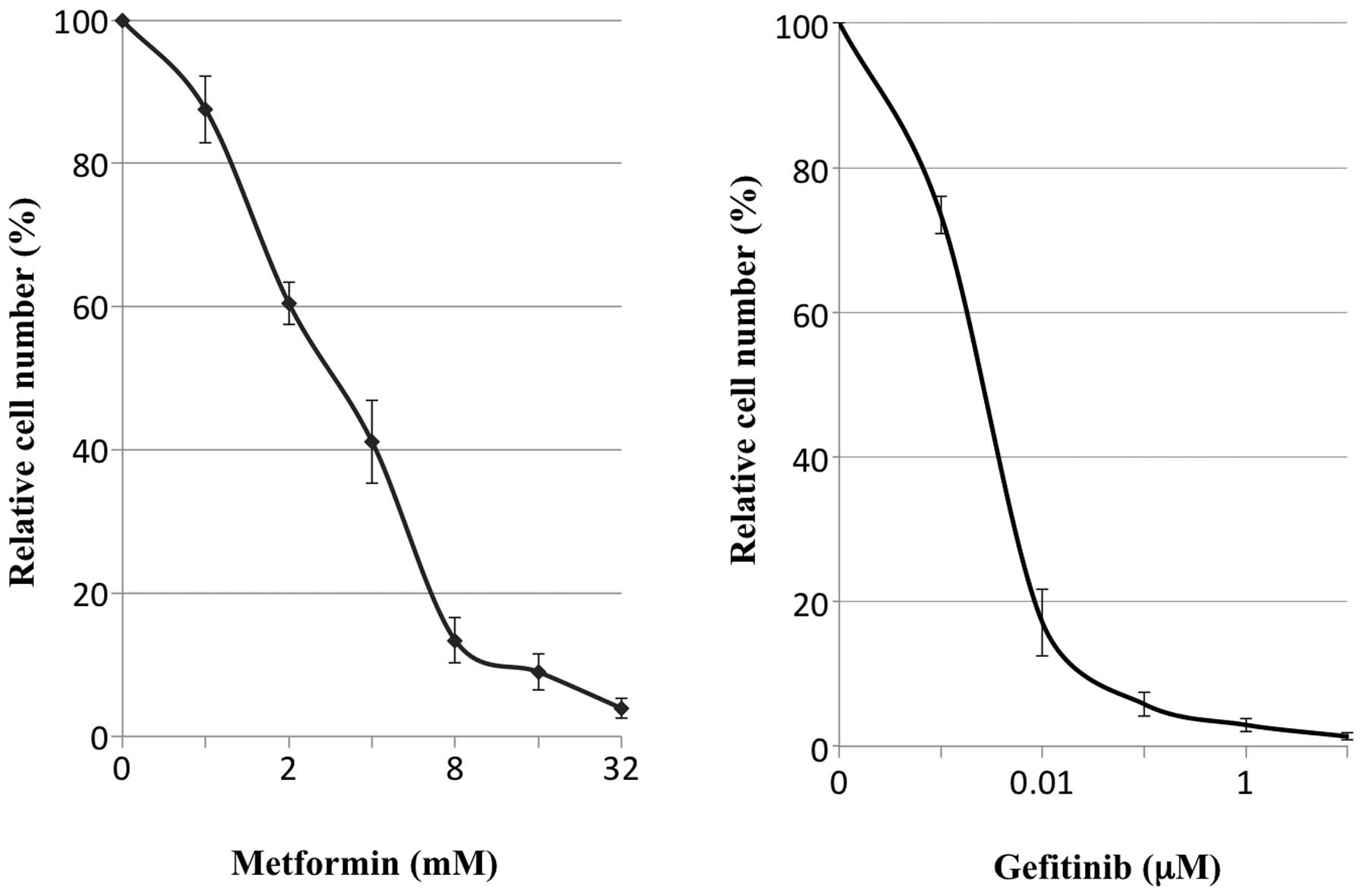
Cell proliferation in vitro
For the in vitro chemosensitivity assay,
5×105 cells per 6-cm-diameter culture dish were plated
and cultured for 24 h until adding the agents metformin, gefitinib,
cisplatin, or their combinations, to the medium for further
culture. The cells were harvested, counted and the survival at
defined time points (24, 48 and 72 h after adding the agents) was
calculated. A series of preliminary experiments indicated that
metformin at 10 mM, gefitinib at 0.03 μM and cisplatin at
1.5 μg/ml were nearly equivalent in reducing cell numbers to
10% of the cell number obtained without adding the agents (control)
at 72 h (Fig. 2). Therefore, these
concentrations were used for further experiments.
Apoptosis
Apoptosis was evaluated by Hoechst staining and
caspase activity determination. For Hoechst staining, cells treated
with agents for 48 h were trypsinized and harvested together with
the floating cells, fixed with 70% ethanol, stained with 2
μg/ml bisbenzimide H33342 trihydrochloride (cat no.
B2261-25MG; Sigma-Aldrich) and examined by fluorescence microscopy
according to the manufacturer's instructions. Cells with aggregated
or fragmented chromatin were regarded as apoptotic cells and 500
cells in each experiment were counted to calculate the apoptotic
cell ratio. To determine the caspase activity in the cell extracts,
a colorimetric assay was used to monitor the absorbance at 405 nm
of p-nitroanilide (pNA) released from the synthetic
substrates. Caspase 3 and 8 activities were evaluated with
synthetic substrates DEVD-pNA and IETD-pNA, respectively, using the
colorimetric assay kit APOPCYTO (Medical & Biological
Laboratories, Nagoya, Japan) according to the manufacturer's
instructions. In each assay, ∼200 μg of protein was
extracted from the cells treated with each agent for 24 h.
Cell cycle
Cell cycle distributions were determined by a
propidium iodide single-color flow cytometric method (FACSCanto II;
BD Biosciences, San Jose, CA, USA), according to the manufacturer's
instructions. Briefly, the cells were trypsinized, washed twice
with ice-cold PBS, fixed with 70% ethanol and then stored at −20°C
until analysis. Before analysis by FACS and CellQuest software (BD
Biosciences), the cell suspensions were washed twice with PBS,
suspended in 500 μl of PI/RNase staining buffer (BD
Biosciences) and incubated for 15 min at room temperature.
Immunofluorescent staining for CD133
The cells were cultured in a chamber slide for 24 h,
followed by treatment with the agents for 24 h. After removal of
the medium containing the agents, the cells were fixed in a 1:1
mixture of methanol and acetone for 2 min, followed by blocking
with normal goat serum for 30 min. The cells were incubated with
primary anti-human CD133 antibodies (cat no. 130-090-422, Miltenyi
Biotec, Bergisch Gladbach, Germany) overnight at 4°C. The cells
were then washed 3 times in PBS and incubated with the secondary
antibody (anti-mouse IgG) conjugated to the Alexa488 fluorescent
dye for 1 h at room temperature. The stained cells were embedded in
VectaShield mounting medium with DAPI (Vector Laboratories,
Burlingame, CA, USA) and were examined with a Nikon Eclipse 80i
microscope (Nikon, Tokyo, Japan) using the VB-7210 imaging system
(Keyence, Tokyo, Japan). Staining results were directly observed at
a magnification of ×200 using a fluorescence microscope.
CD24 and CD44 expression determined by
FACS
For analysis of cell-surface marker expression by
FACS, anti-human CD24 antibodies conjugated to phycoerythrin (cat
no. 311105, BioLegend, San Diego, CA, USA) and anti-human CD44
antibodies conjugated with allophycocyanin (cat no. 103011,
BioLegend) were used. The cells treated with the agents for 24 h
were trypsinized and washed 3 times with PBS. The cells
(1×106) in a single-cell suspension were resuspended in
the staining buffer (PBS containing 2% FBS) and labeled with the
antibodies, followed by washing and resuspension in 500 μl
of staining buffer according to the manufacturer's instructions.
The cells were also labeled with propidium iodide to enrich viable
cells and analyzed with a JSAN cell sorter and AppSan software (Bay
Bioscience, Kobe, Japan).
Enrichment of CD24-positive cells
To obtain a cell population enriched in
CD24-positive cells, a magnetic cell-sorting system with the
Miltenyi Biotec MACS Cell Separation kit was used according to the
manufacturer's instructions. The sorted cells were analyzed with a
JSAN cell sorter and the AppSan software as described above. The
cell sorting and subsequent brief culture for propagation were
repeated up to 4 times to obtain a cell population consisting of
∼80% CD24-positive cells.
Statistical analysis
Data are presented as the means ± SEs as indicated
and were analyzed by Student's t-test. A p-value of <0.05
(2-tailed) was considered statistically significant.
Results
Effect of gefitinib and metformin on
xenograft tumors in vivo
When the administration was initiated day 16 or when
the tumor size reached ∼300 mm3, metformin did not
reduce tumor growth (compare groups 2 to 1, Fig. 1). In addition, no additional effect
was observed with metformin in tumor shrinkage with gefitinib (see
the curves from days 16 to 31 in groups 3 and 4). Metformin,
however, significantly reduced tumor regrowth after withdrawal of
gefitinib treatment (see the curves from days 35 to 66 in groups 3
and 4).
Suppression of in vitro cell
proliferation
In vitro administration of metformin
suppressed proliferation of PC9 cells in a dose-dependent manner,
similarly to gefitinib (Fig. 2)
and the concentrations that resulted in 90% reduction of cell
numbers compared with controls at 72 h after the administration
were 11.6±1.87 mM with metformin, 0.042±0.024 μM with
gefitinib and 1.48±0.18 μg/ml with cisplatin. In subsequent
experiments, concentrations of 10 mM for metformin, 0.03 μM
for gefitinib and 1.5 μg/ml for cisplatin were employed
(Fig. 3).
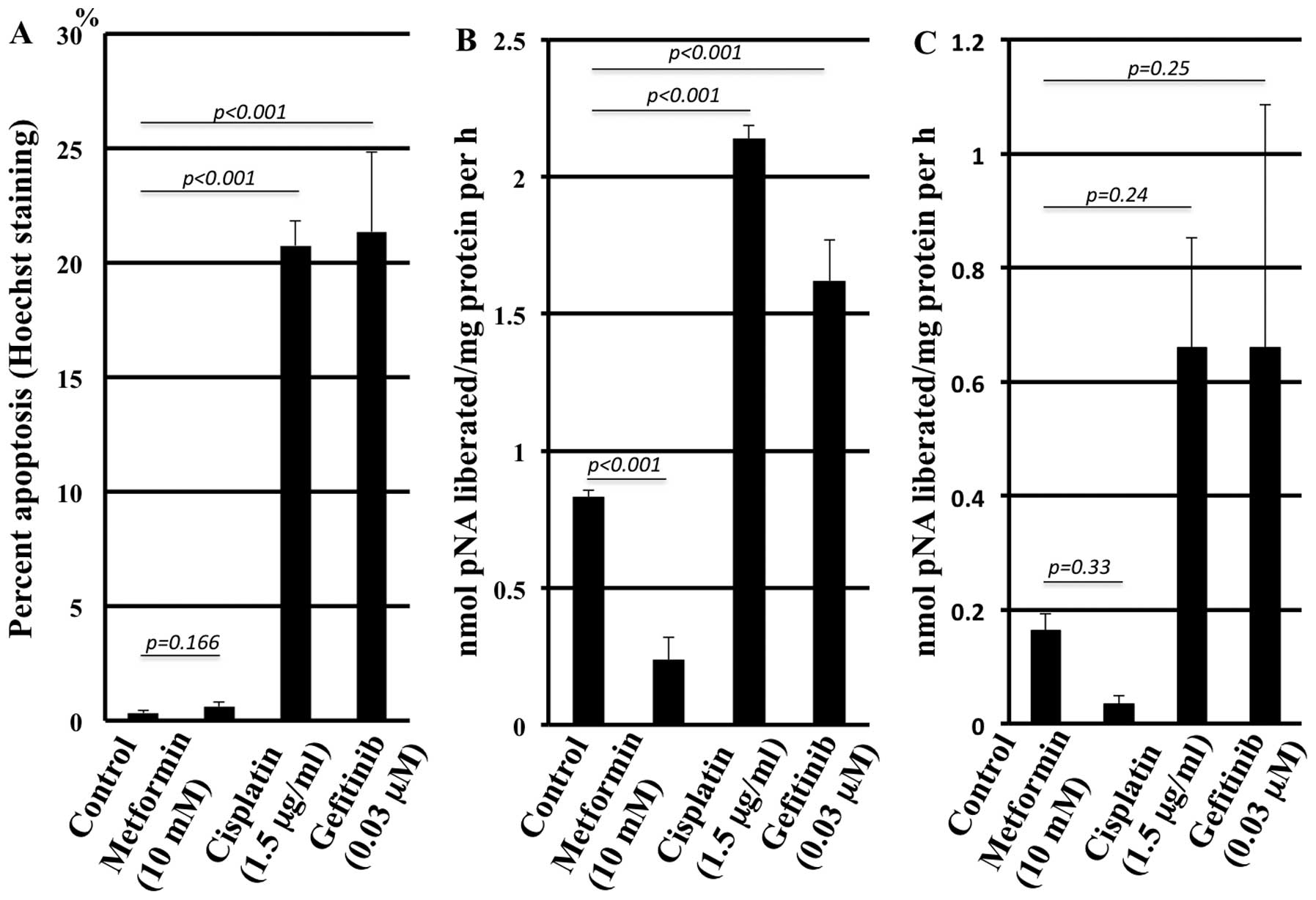
Apoptosis induction by the agents
As assessed by Hoechst staining, metformin did not
induce more apoptotic cells compared with the control in contrast
to cisplatin and gefitinib (Fig.
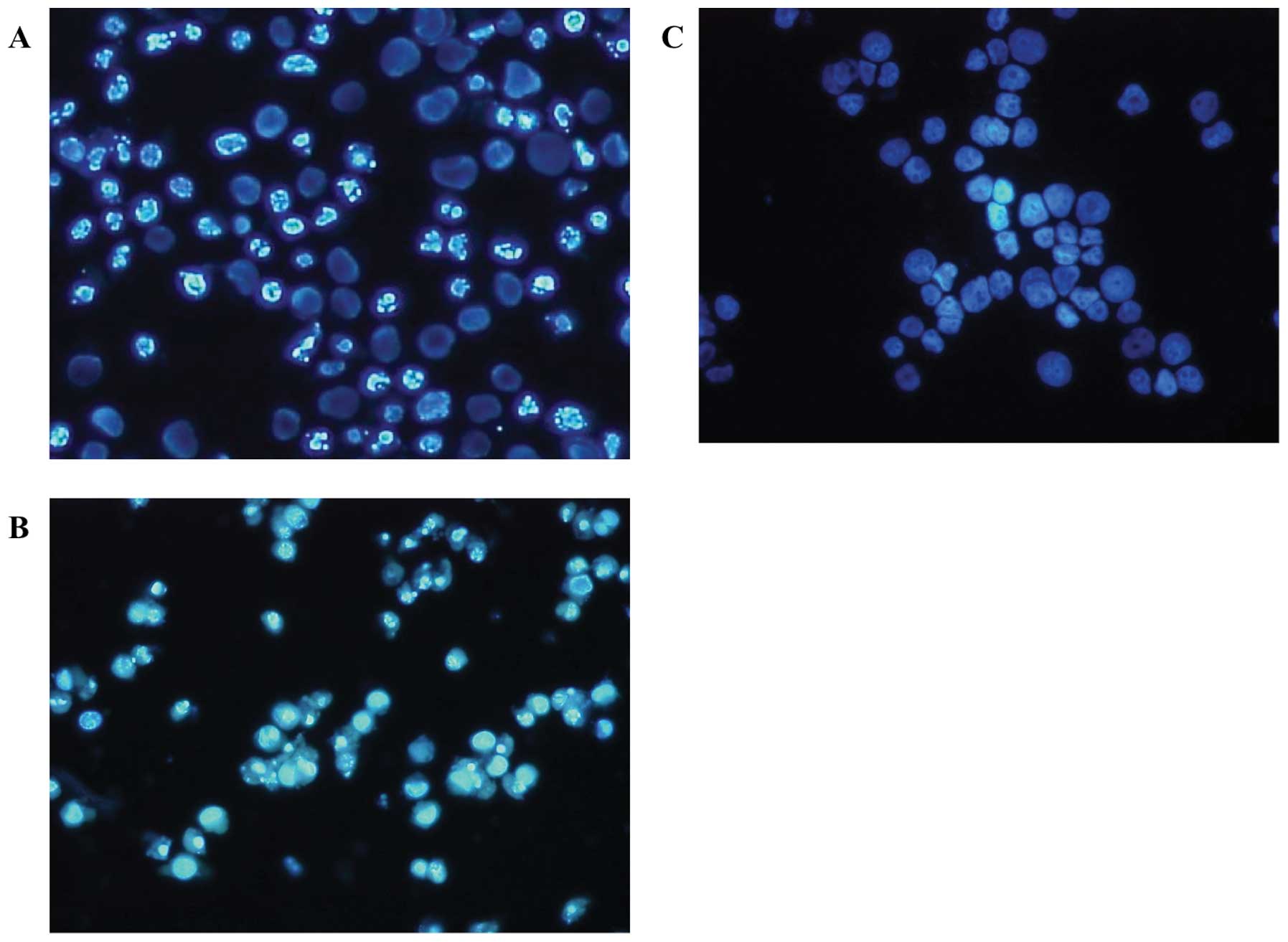
4A). The representative morphology in the presence of the
agents is shown in Fig. 5. The
caspase 3 assay confirmed the results (Fig. 4B) with statistically significant
differences, although the caspase 8 assay solely revealed a trend
without statistical significance (Fig.
4C).
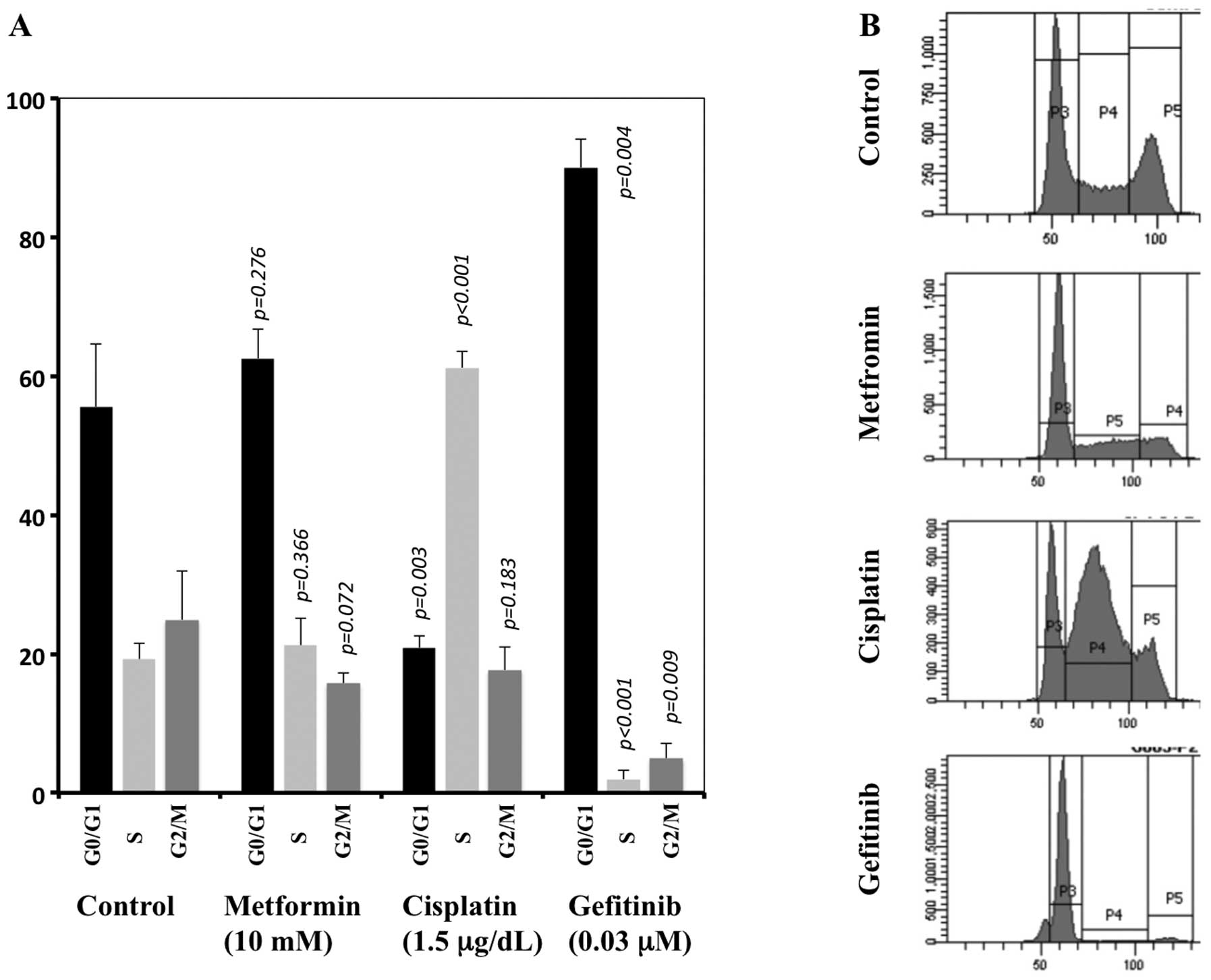
Cell cycle alteration
FACS analysis showed that metformin did not
significantly alter the cell cycle distribution compared to the
control. In contrast, cisplatin caused a marked cell accumulation
at the S phase together with a marked decrease of cells at the
G0/G1 phases. Gefitinib caused marked accumulation at the G0/G1
phases together with a marked decrease at the S and G2/M phases
compared with the control (Fig.
6).
Expression of CD24, CD44 and CD133 in
surviving cells after exposure to gefitinib
Because we failed to detect reproducible CD133
expression by FACS analysis, CD133 expression was assessed solely
by immunofluorescence staining. The CD133-positive cells were
significantly enriched after exposure to gefitinib for 24 h. In
contrast, exposure to metformin alone or to gefitinib combined with
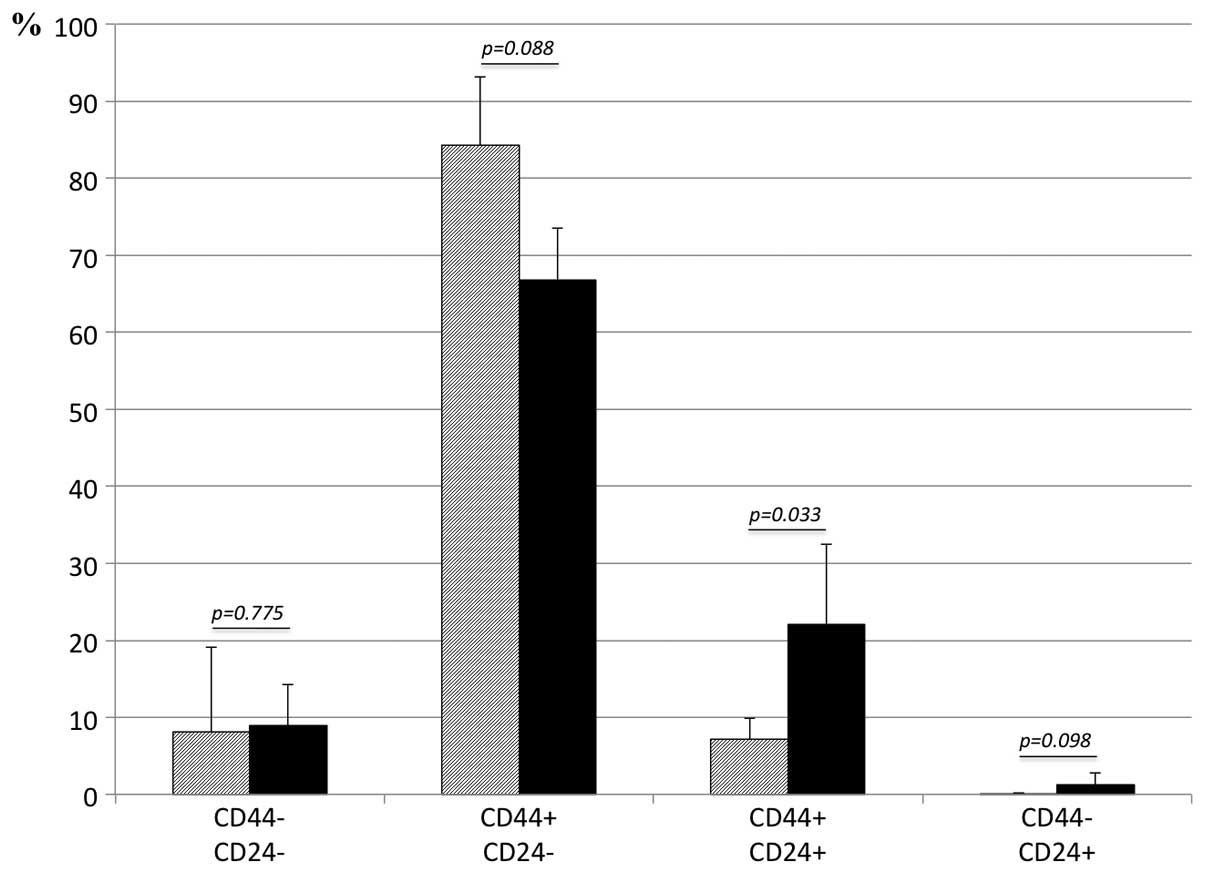
metformin did not augment CD133 expression (Fig. 7). Altered expression of CD24 and
CD44, assessed by FACS, are shown in Fig. 8. The cell population with negative
staining for CD44, either with or without CD24 expression, was not
significantly altered by exposure to gefitinib for 24 h. The cell
population with positive staining for CD24 increased significantly
after exposure to gefitinib for 24 h.
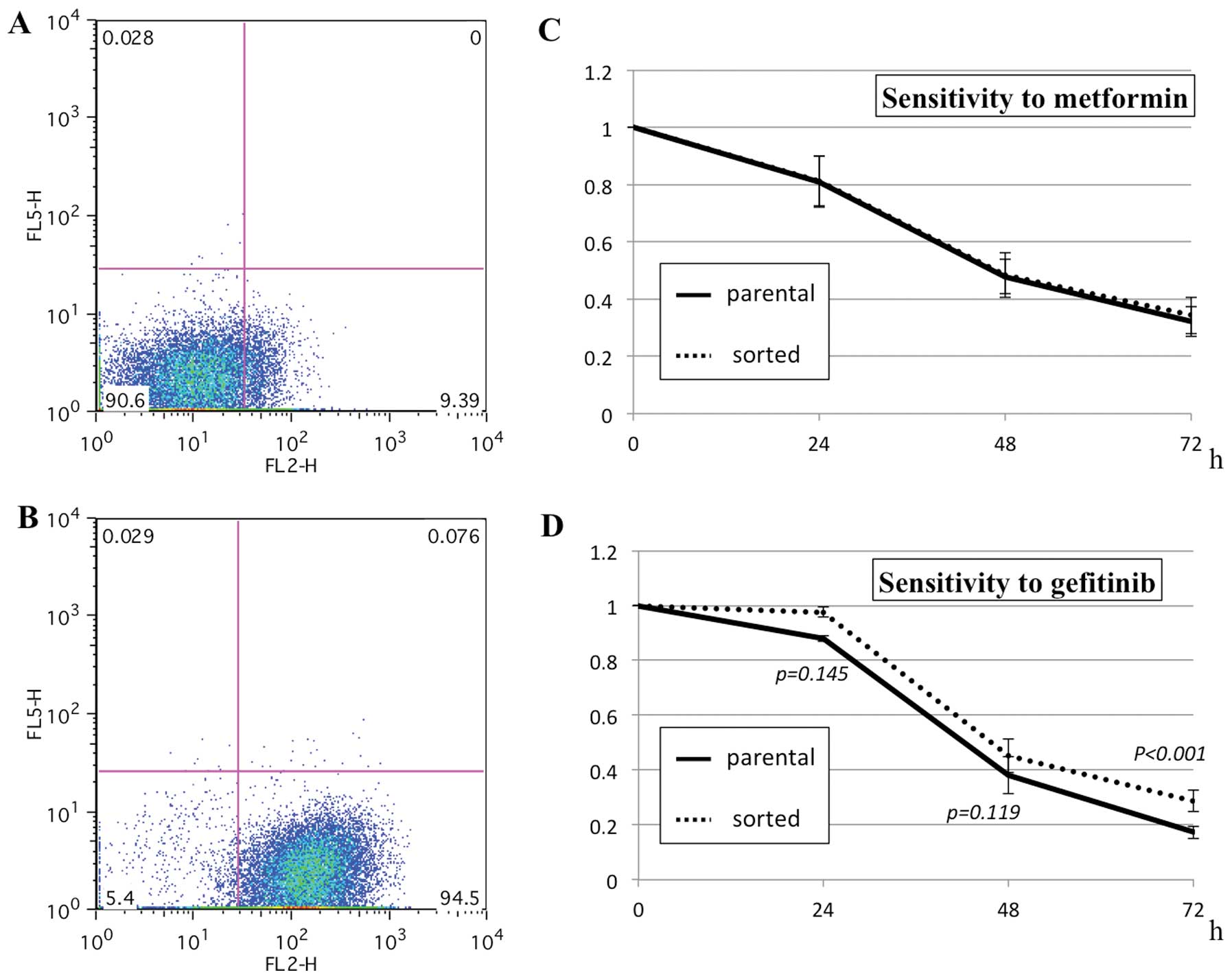
Sensitivity to metformin and gefitinib in
CD24-positive cells
To obtain a cell population enriched in
CD24-positive cells, the parental cells were sorted by FACS
magnetic separation. Because a single sorting was not sufficient to
enrich CD24-positive cells, the sorting was repeated up to 4 times.
The proportion of CD24-positive cells in the parental cells
(7.1±4.2%, n=3) was enriched to 81.8±12.1% (n=3). The in
vitro sensitivity assay revealed that the parental and sorted
cells showed nearly identical sensitivity to metformin. The sorted
cells, however, were slightly but significantly more resistant to
gefitinib than the parental cells (Fig. 9).
Discussion
The present study employed the human lung
adenocarcinoma cell line PC9, which possesses an EGFR exon
19 deletion mutation that renders EGFR sensitive to the TKIs. In
vivo experiments with xenografts derived from these cells
resulted in the following observations: i) metformin exerted no
effect on already grown tumors (>300 mm3), ii)
metformin had no additional effect on tumor shrinkage by gefitinib,
iii) the tumors regrew after withdrawing gefitinib even after the
treatment had resulted in complete regression of the tumors and iv)
metformin significantly suppressed the regrowth of the tumors after
withdrawing gefitinib treatment. These observations suggest that
metformin is effective specifically on residual cells after
gefitinib treatment; however, metformin is not sufficiently
effective to suppress growth of already established tumors.
To test our hypothesis, a series of in vitro
experiments were conducted. Cisplatin was included as a positive
control in some of the experiments. A dose that resulted in an
in vitro cell number reduction to 10% of the original cell
number was chosen for each agent. Metformin did not induce
apoptosis when assessed by Hoechst staining and caspase 3 and 8
activity determination, in contrast to gefitinib and cisplatin.
Moreover, apoptosis induction by metformin treatment was
significantly lower, even lower than what was observed in the
control experiments, suggesting an apoptosis-protective property of
metformin. This is consistent with a previous report that
demonstrated a preventive effect of AMPK on apoptosis (33,34).
Although metformin decreased the cells at the G2/M phases, cell
cycle alteration with metformin was not dramatic in contrast with
cisplatin and gefitinib, which induced significant accumulation at
the S and G0/G1 phases, respectively. Although the results obtained
with cisplatin (35) and gefitinib
(36,37) are consistent with previously
reported data, the results obtained with metformin are rather
complicated. Some studies reported an absence of apoptosis
induction with metformin (17),
whereas other reports described a significant apoptosis induction
(12). Similarly, although some
have reported a significant cell cycle shift with metformin
(12,16), others have reported only mild cell
cycle arrest at the G0-G1 phases in the presence of metformin
(38,39). The effects of metformin on
apoptosis and cell cycle arrest may vary depending on the cell line
examined, as previously reported with human lung cancers of a
variety of histological types (40). Nevertheless, the minimum effects of
metformin on apoptosis induction and cell cycle alteration in
vitro in the present experimental system may explain the
absence of effects on tumor growth inhibition in vivo.
Based on the reported drug resistance of cancer stem
cells (41) and the information
presented in a previous study (5),
the expression of 3 putative cancer stem cell markers, CD133
(42), CD44 (43) and CD24 (44), was examined. After gefitinib
treatment of the cells in vitro, cells with CD133 expression
were enriched as assessed by immunofluorescence staining. FACS
analysis revealed an enrichment of cells with CD24 expression after
gefitinib treatment in vitro. The population of cells with
CD44 expression was unaltered. These observations are similar to
those in a previous report (5),
suggesting that cells with CD133 or CD24 expression may be
resistant to gefitinib. In fact, metformin treatment in
vitro did not enrich cells with CD133 expression, in contrast
to what was observed with gefitinib. In addition, combined
treatment with metformin and gefitinib canceled the enrichment
observed in the treatment with gefitinib alone. These results
strongly suggest that metformin is effective against residual cells
after in vitro treatment with gefitinib, consistent with the
in vivo experiment. A cell population consisting of ∼80%
cells expressing CD24 (∼10-fold enrichment compared with the
parental cells) was obtained by FACS sorting to directly examine
chemosensitivity. These cells were slightly but significantly more
resistant to gefitinib than the parental cells, whereas their
sensitivity to metformin was identical to the parental cells,
suggesting that metformin was effective against residual cells
after gefitinib treatment. Nevertheless, the degree of augmented
resistance to gefitinib in cells with CD24 expression was
unexpectedly small. This can be explained if the resistant cells
express CD24 and if only a part of the cell population with CD24
expression is resistant to gefitinib.
The nature and properties of the residual cells
after treatment with gefitinib are unclear. Although CD133 and CD24
are putative markers for cancer stem cells in human brain and colon
cancers, respectively, the present results do not indicate that the
residual cells are cancer stem cells because cancer stem cells in
human lung cancer have not yet been identified. Considering their
prompt emergence in a short period, non-mutational mechanisms,
including epigenetic change and selection of resistant cells from a
heterogeneous cell population, seem to be the most likely routes of
chemo-resistance. Cancer stem cells and epithelial-mesenchymal
transitions might be possible candidates for selection. Specific
targeting of residual cells after chemotherapy may be a suitable
approach to cure cancers. The present study highlighted metformin
as a candidate for targeting residual cells and we envision that
further elucidation of the detailed molecular mechanism of the
cytotoxicity of metformin represents progress in cancer
therapy.
Acknowledgements
Yuichi Takiguchi has received
honoraria for lectures not related to this study and unrestricted
research and educational grants from AstraZeneca, K.K., Japan, and
Chugai Pharmaceutical Co., Japan. Ikuo Sekine and Koichiro Tatsumi
have received honoraria for lectures not related to this study-from
AstraZeneca, K.K., Japan, and Chugai Pharmaceutical Co., Japan.
Osamu Yokosuka has received research grants not related to this
study from Chugai Pharmaceutical Co., Japan. This study was
supported by a Grant-in-Aid for Scientific Research (grant no.
23591136) from the Japan Society for the Promotion of Science and
the Ministry of Education, Culture, Sports, Science and Technology
of Japan.
References
|
1.
|
Inoue A, Kobayashi K, Maemondo M, et al:
Updated overall survival results from a randomized phase III trial
comparing gefitinib with carboplatin-paclitaxel for chemo-naive
non-small cell lung cancer with sensitive EGFR gene mutations
(NEJ002). Ann Oncol. 24:54–59. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2.
|
Wakeling AE, Guy SP, Woodburn JR, et al:
ZD1839 (Iressa): an orally active inhibitor of epidermal growth
factor signaling with potential for cancer therapy. Cancer Res.
62:5749–5754. 2002.PubMed/NCBI
|
|
3.
|
Kosaka T, Yatabe Y, Endoh H, et al:
Analysis of epidermal growth factor receptor gene mutation in
patients with non-small cell lung cancer and acquired resistance to
gefitinib. Clin Cancer Res. 12:5764–5769. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Engelman JA, Zejnullahu K, Mitsudomi T, et
al: MET amplification leads to gefitinib resistance in lung cancer
by activating ERBB3 signaling. Science. 316:1039–1043. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Sharma SV, Lee DY, Li B, et al: A
chromatin-mediated reversible drug-tolerant state in cancer cell
subpopulations. Cell. 141:69–80. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Towler MC and Hardie DG: AMP-activated
protein kinase in metabolic control and insulin signaling. Circ
Res. 100:328–341. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Pollak MN: Investigating metformin for
cancer prevention and treatment: the end of the beginning. Cancer
Discov. 2:778–790. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Evans JM, Donnelly LA, Emslie-Smith AM,
Alessi DR and Morris AD: Metformin and reduced risk of cancer in
diabetic patients. BMJ. 330:1304–1305. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Bowker SL, Majumdar SR, Veugelers P and
Johnson JA: Increased cancer-related mortality for patients with
type 2 diabetes who use sulfonylureas or insulin. Diabetes Care.
29:254–258. 2006. View Article : Google Scholar
|
|
10.
|
Jiralerspong S, Palla SL, Giordano SH, et
al: Metformin and pathologic complete responses to neoadjuvant
chemotherapy in diabetic patients with breast cancer. J Clin Oncol.
27:3297–3302. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Mazzone PJ, Rai H, Beukemann M, Xu M, Jain
A and Sasidhar M: The effect of metformin and thiazolidinedione use
on lung cancer in diabetics. BMC Cancer. 12:4102012. View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Alimova IN, Liu B, Fan Z, et al: Metformin
inhibits breast cancer cell growth, colony formation and induces
cell cycle arrest in vitro. Cell Cycle. 8:909–915. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Liu B, Fan Z, Edgerton SM, et al:
Metformin induces unique biological and molecular responses in
triple negative breast cancer cells. Cell Cycle. 8:2031–2040. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Zakikhani M, Dowling R, Fantus IG,
Sonenberg N and Pollak M: Metformin is an AMP kinase-dependent
growth inhibitor for breast cancer cells. Cancer Res.
66:10269–10273. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Rocha GZ, Dias MM, Ropelle ER, et al:
Metformin amplifies chemotherapy-induced AMPK activation and
antitumoral growth. Clin Cancer Res. 17:3993–4005. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Ben Sahra I, Laurent K, Loubat A, et al:
The antidiabetic drug metformin exerts an antitumoral effect in
vitro and in vivo through a decrease of cyclin D1 level. Oncogene.
27:3576–3586. 2008.PubMed/NCBI
|
|
17.
|
Wang LW, Li ZS, Zou DW, Jin ZD, Gao J and
Xu GM: Metformin induces apoptosis of pancreatic cancer cells.
World J Gastroenterol. 14:7192–7198. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Gotlieb WH, Saumet J, Beauchamp MC, et al:
In vitro metformin anti-neoplastic activity in epithelial ovarian
cancer. Gynecol Oncol. 110:246–250. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Rattan R, Giri S, Hartmann LC and Shridhar
V: Metformin attenuates ovarian cancer cell growth in an AMP-kinase
dispensable manner. J Cell Mol Med. 15:166–178. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Rattan R, Graham RP, Maguire JL, Giri S
and Shridhar V: Metformin suppresses ovarian cancer growth and
metastasis with enhancement of cisplatin cytotoxicity in vivo.
Neoplasia. 13:483–491. 2011.PubMed/NCBI
|
|
21.
|
Hawley SA, Boudeau J, Reid JL, et al:
Complexes between the LKB1 tumor suppressor, STRAD alpha/beta and
MO25 alpha/beta are upstream kinases in the AMP-activated protein
kinase cascade. J Biol. 2:282003. View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Lizcano JM, Goransson O, Toth R, et al:
LKB1 is a master kinase that activates 13 kinases of the AMPK
subfamily, including MARK/PAR-1. EMBO J. 23:833–843. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Dowling RJ, Zakikhani M, Fantus IG, Pollak
M and Sonenberg N: Metformin inhibits mammalian target of
rapamycin-dependent translation initiation in breast cancer cells.
Cancer Res. 67:10804–10812. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
24.
|
Bolster DR, Crozier SJ, Kimball SR and
Jefferson LS: AMP-activated protein kinase suppresses protein
synthesis in rat skeletal muscle through down-regulated mammalian
target of rapamycin (mTOR) signaling. J Biol Chem. 277:23977–23980.
2002. View Article : Google Scholar
|
|
25.
|
Kimura N, Tokunaga C, Dalal S, et al: A
possible linkage between AMP-activated protein kinase (AMPK) and
mammalian target of rapamycin (mTOR) signalling pathway. Genes
Cells. 8:65–79. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
26.
|
Inoki K, Zhu T and Guan KL: TSC2 mediates
cellular energy response to control cell growth and survival. Cell.
115:577–590. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
27.
|
Iliopoulos D, Hirsch HA and Struhl K:
Metformin decreases the dose of chemotherapy for prolonging tumor
remission in mouse xenografts involving multiple cancer cell types.
Cancer Res. 71:3196–3201. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
28.
|
Janjetovic K, Vucicevic L, Misirkic M, et
al: Metformin reduces cisplatin-mediated apoptotic death of cancer
cells through AMPK-independent activation of Akt. Eur J Pharmacol.
651:41–50. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
29.
|
Harhaji-Trajkovic L, Vilimanovich U,
Kravic-Stevovic T, Bumbasirevic V and Trajkovic V: AMPK-mediated
autophagy inhibits apoptosis in cisplatin-treated tumour cells. J
Cell Mol Med. 13:3644–3654. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
30.
|
Costa DB, Halmos B, Kumar A, et al: BIM
mediates EGFR tyrosine kinase inhibitor-induced apoptosis in lung
cancers with oncogenic EGFR mutations. PLoS Med. 4:1669–1679. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
31.
|
Memmott RM, Mercado JR, Maier CR, Kawabata
S, Fox SD and Dennis PA: Metformin prevents tobacco
carcinogen-induced lung tumorigenesis. Cancer Prev Res.
3:1066–1076. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
32.
|
Sirotnak FM, Zakowski MF, Miller VA, Scher
HI and Kris MG: Efficacy of cytotoxic agents against human tumor
xenografts is markedly enhanced by coadministration of ZD1839
(Iressa), an inhibitor of EGFR tyrosine kinase. Clin Cancer Res.
6:4885–4892. 2000.PubMed/NCBI
|
|
33.
|
Blazquez C, Geelen MJ, Velasco G and
Guzman M: The AMP-activated protein kinase prevents ceramide
synthesis de novo and apoptosis in astrocytes. FEBS Lett.
489:149–153. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
34.
|
Shaw RJ, Kosmatka M, Bardeesy N, et al:
The tumor suppressor LKB1 kinase directly activates AMP-activated
kinase and regulates apoptosis in response to energy stress. Proc
Natl Acad Sci USA. 101:3329–3335. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
35.
|
Ormerod MG, Orr RM and Peacock JH: The
role of apoptosis in cell killing by cisplatin: a flow cytometric
study. Br J Cancer. 69:93–100. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
36.
|
Janmaat ML, Kruyt FA, Rodriguez JA and
Giaccone G: Response to epidermal growth factor receptor inhibitors
in non-small cell lung cancer cells: limited antiproliferative
effects and absence of apoptosis associated with persistent
activity of extracellular signal-regulated kinase or Akt kinase
pathways. Clin Cancer Res. 9:2316–2326. 2003.
|
|
37.
|
Tracy S, Mukohara T, Hansen M, Meyerson M,
Johnson BE and Janne PA: Gefitinib induces apoptosis in the
EGFRL858R non-small-cell lung cancer cell line H3255. Cancer Res.
64:7241–7244. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
38.
|
Cantrell LA, Zhou C, Mendivil A, Malloy
KM, Gehrig PA and Bae-Jump VL: Metformin is a potent inhibitor of
endometrial cancer cell proliferation - implications for a novel
treatment strategy. Gynecol Oncol. 116:92–98. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
39.
|
Kato K, Gong J, Iwama H, et al: The
antidiabetic drug metformin inhibits gastric cancer cell
proliferation in vitro and in vivo. Mol Cancer Ther. 11:549–560.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
40.
|
Ashinuma H, Takiguchi Y, Kitazono S, et
al: Antiproliferative action of metformin in human lung cancer cell
lines. Oncol Rep. 28:8–14. 2012.
|
|
41.
|
Matsui W, Wang Q, Barber JP, et al:
Clonogenic multiple myeloma progenitors, stem cell properties, and
drug resistance. Cancer Res. 68:190–197. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
42.
|
Hemmati HD, Nakano I, Lazareff JA, et al:
Cancerous stem cells can arise from pediatric brain tumors. Proc
Natl Acad Sci USA. 100:15178–15183. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
43.
|
Al-Hajj M, Wicha MS, Benito-Hernandez A,
Morrison SJ and Clarke MF: Prospective identification of
tumorigenic breast cancer cells. Proc Natl Acad Sci USA.
100:3983–3988. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
44.
|
Vermeulen L, Todaro M, de Sousa Mello F,
et al: Single-cell cloning of colon cancer stem cells reveals a
multi-lineage differentiation capacity. Proc Natl Acad Sci USA.
105:13427–13432. 2008. View Article : Google Scholar : PubMed/NCBI
|