Introduction
The apoptosome-driven cell death pathway plays an
important role not only in embryonic development and homeostasis of
postnatal tissues (1,2), but it can be also involved in
pathogenesis and treatment response of cancer (3,4). The
apoptosome pathway is launched upon cytochrome c
(cyt-c) release from mitochondria into the cytosol (5). Cyt-c molecules bind to
cytosolic Apaf-1 monomers containing 13 WD repeats (6,7) and
induce, together with (d)ATP binding via nucleotide exchange, a
conformation change of Apaf-1 monomers allowing them to oligomerize
into a heptameric complex called apoptosome (8,9).
Subsequent binding of procaspase-9 (PC-9) molecules to apoptosome
leads to their activation via autoproteolytic processing, yielding
the active apoptosome-bound cleaved caspase-9 (CS-9) (8,10–12).
The active CS-9 in the holo-apoptosome then cleaves and activates
the zymogens of the executioner caspase-3 (CS-3) and caspase-7
(CS-7) (8,10–14).
The processes of assembly and function of apoptosome
complexes can be positively or negatively regulated by numerous
factors (15,16). There is evidence that not only
dysfunction of apoptosome (17–20),
but also its hyperactivity (21–24)
can contribute to development and progression of malignant tumours
and their susceptibility to therapy. It has been reported that
although numerous non-small cell lung carcinoma (NSCLC) cell lines
and tumours express Apaf-1, PC-9 and procaspase-3 (PC-3) proteins
in levels sufficient to launch the apoptosome pathway, their
capability of the apoptosome-dependent caspase activation may be
low or absent (25–28). Despite the studies of several
possible regulators of apoptosome in NSCLC cells, including the
inhibitor of apoptosis proteins XIAP, cIAP-1 and cIAP-2, TUCAN,
Smac, and PHAPI (28–32), and the evidence of
high-Mr apoptosome complexes incapable of PC-9
processing (33–35), the regulation of apoptosome
assembly and signalling in NSCLC is still elusive.
We demonstrated previously that although the levels
of PC-9 protein were comparable in NSCLC tumours and matched lungs,
the expression of both Apaf-1 and PC-3 proteins was frequently
upregulated and the induced activity of apoptosome apparatus tended
to be higher in the tumours as compared to lungs (27). To explore further the functionality
of apoptosome apparatus in NSCLC, we studied its sensitivity to
activation in the cell-free cytosol from NSCLC cells and NSCLC
tumours and matched lungs, the assembly of apoptosome complexes and
functional stability apoptosome precursors, the impact of
clinico-pathological parameters of NSCLC tumours on the level of
apoptosome-generated CS-3-like activity, and the involvement of
XIAP in the regulation of apoptosome activity in NSCLC tumours.
Materials and methods
Reagents
Most reagents used in this study were obtained from
suppliers as described previously (27). Sephacryl S300HR, Gel Filtration
Molecular Weight Markers (cat. no. MW-GF-1000), bovine serum
albumin (BSA; cat. no. A7030), the affinity purified rabbit
anti-caspase-3 and rabbit anti-Apaf-1 antibodies (cat. nos. C9598
and A8469, respectively), and the goat anti-rabbit IgG horseradish
peroxidase (HRP) conjugate (cat. no. A4914), used as a secondary
antibody, were from Sigma (St. Louis, MO, USA). The rabbit
anti-caspase-9 antibody was from Cell Signaling (cat. no. 9502,
Danvers, MA, USA). The pre-stained Precision Plus Protein Standards
and Blotting-Grade Blocker (BGB) were from Bio-Rad Laboratories
(Hercules, CA, USA). The peptides AVPIAQK (P1) and ATPFQEG (P2)
were custom synthesized by Clonestar Peptide Services (Brno, Czech
Republic).
Cell cultures
NSCLC cell lines used in the present study were
squamous cell lung carcinoma-derived cell lines CALU-1, NCI-H520
and SKMES-1, and lung adenocarcinoma-derived cell lines A549,
SKLU-1, LXF-289 and COLO-699. They were obtained from the following
depositories: CALU-1, SKMES-1, A549 and SKLU-1 were from the
European Collection of Cell Cultures (Salisbury, UK), LXF-289 and
COLO-699 were from the German Collection of Microorganisms and Cell
Cultures (Braunschweig, Germany), and NCI-H520 was from the
American Type Culture Collection (ATCC, Rockville, MD, USA). The
cells were cultured in humidified atmosphere of 5% CO2
and air at 37°C in the Eagle’s minimum essential medium
supplemented with 2 mM L-glutamine, 26.2 mM NaHCO3, 1%
of a stock solution of non-essential amino acids, 10 mg/l
apo-transferrin, 0.25 μM Fe(NO3)3, 5%
foetal bovine serum, 105 IU/l of penicillin-G and 100 mg/l of
streptomycin. After reaching confluence, the cells were harvested
for preparation of cell-free cytosols.
Patients and tissues
Specimens of NSCLC tumours and matched lung
parenchyma were obtained from 62 surgically treated patients
(median age of 63 years, age range of 47–78 years; 41 men and 21
women; 48 smokers, 10 non-smokers and 4 ex-smokers), who did not
received radiotherapy nor chemotherapy before surgery for lung
cancer. The study was approved by the local institutional ethics
committee and was conducted in accordance with the Declaration of
Helsinki. Signed written informed consent was obtained from each
patient before entry to the study. Tissue samples (∼1 g, wet mass)
from non-necrotic part of the tumour and from the lung parenchyma
at a site located as distantly as possible from the tumour, were
excised from the resected lung lobe or lung immediately after
surgery. All tissues were snap-frozen in liquid nitrogen and stored
at −78°C until cell-free cytosols preparation. The
histopathological classification of the tumours was done according
to the World Health Organization criteria (36) and included squamous cell lung
carcinoma (SQCLC; n=30, including 1, 15 and 14 tumours of grade 1,
2 and 3, respectively), lung adenocarcinoma (LAC; n=25, including
3, 8 and 11 tumours of grade 1, 2 and 3, respectively), mixed type
SQCLC and LAC (n=1), large-cell lung carcinoma (LCLC; n=1),
sarcomatoid lung carcinoma (SLC; n=3) and undifferentiated lung
carcinoma (UNDIF; n=2). Tumour staging was performed according to
the new international pTNM system (37), and included 9 and 21 tumours of
stage IA and IB, respectively, 2 and 13 tumours of stage IIA and
IIB, respectively, 15 and 1 tumours of stage IIIA and IIIB,
respectively, and 1 tumour of stage IV.
Preparation of cell-free cytosol
samples
Unless otherwise indicated, the preparation of
cell-free cytosols was performed at 0–4°C. Cells at confluence were
washed and gently scraped into the Hank’s balanced salt solution
containing 4.16 mM NaHCO3. Subsequently, cells were
centrifuged at 300 × g for 10 min and the pellets were resuspended
in 1/4-1/2 volume of 25 mM HEPES/NaOH, 4 mM Na2EDTA, 1.5
mM MgCl2, 10 mM KCl buffer, pH 7.4 (HEMK buffer). After
gentle shaking for 30 min, the cells were disrupted using whole
glass Dounce homogenizer equipped with the pestle B. To prepare
tissue cytosols, samples of NSCLC tumours and lung parenchyma were
disrupted using three cycles of liquid nitrogen pre-chilling in a
Teflon container and pulverizing for 30 sec on Mixer Mill MM 200
(Retsch, Haan, Germany) at a speed of 30 sec−1. The
powders were resuspended in HEMK buffer (1.5 ml per 1 g of wet
tissue) and shaken on ice for 30 min. The suspensions were
homogenized three times for 5 sec on a homogenizer Ultra-Turrax T25
(IKA, Staufen, Germany) at 24,000 rpm. The cell and tissue
homogenates were centrifuged at 200,000 × g for 60 min and the
supernatants (i.e. cytosols) were stored in small aliquots at −78°C
until analysis.
Apoptosome apparatus activation and assay
of caspase-3- like activity
In order to activate the apoptosome apparatus,
cytosol samples (150 μl), containing 2.5 mg of total
protein/ml in HEMK buffer with 5 mM D,L-dithiothreitol (DTT), were
incubated with 10 μM of cyt-c and 1 mM of dATP at
37°C for 30 min. When analysing the effect of the XIAP-neutralizing
peptides AVPIAQK (P1) and ATPFQEG (P2), the peptides were added to
the activation reactions in a concentration of 500 μM, which
was previously shown to be very effective in relieving the
(cyt-c + dATP)-induced apoptosome activity from the
XIAP-mediated inhibition (38,52).
Cytosol samples incubated without cyt-c and dATP and without
the peptides P1 and P2 served as controls. Subsequently, aliquots
of the incubated cytosol samples (20 μl) were used for
measurement of the endogenous (E, in the controls) and the total
(cyt-c + dATP)-induced (T) CS-3-like activity, using a
kinetic enzyme assay. The incremental induced (I) CS-3-like
activity was then calculated as the difference between the T and E
activities (I=T-E). The enzyme reactions, carried out in a total
volume of 200 μl at 37°C and in duplicate, were done with
100 μM of Ac-DEVD-AFC as a substrate in an assay buffer
containing 50 mM HEPES/NaOH, 1.63 mM CHAPS, 1 mM
Na2EDTA, 292 mM sucrose, 100 mM NaCl, and 5 mM DTT, pH
7.2. Further control reactions, run in parallel, were performed in
the presence of a caspase inhibitor Ac-DEVD-CHO (10 μM). The
fluorescence of the enzymatically released
7-amino-4-trifluoromethylcoumarin (AFC) was measured on a
microplate fluorometer SpectraFluor (Tecan, Salzburg, Austria). The
caspase activity was calculated from the progress curves in steady
state. The specific caspase activity was expressed in pkat
(pmol/sec) per 1 mg of total protein.
Gel filtration chromatography
For the study of the assembly of apoptosome
complexes, samples of cell-free cytosol from COLO-699, CALU-1 and
A549 cells (5 mg of total protein/ml) were incubated in HEMK buffer
with 5 mM DTT in the presence or absence of cyt-c (10
μmol/l) and dATP (1 mmol/l) at 37°C for 30 min.
Subsequently, the samples were fractionated by gel filtration
chromatography (GFC) on a column (70×1.6 cm) of Sephacryl S300HR at
2–4°C. The column was eluted with a buffer containing 25 mM
HEPES/NaOH, 1 mM Na2EDTA, 1.5 mM MgCl2, 10 mM
KCl, 1 mM DTT, and 100 mM NaCl, pH 7.2. The elution rate was 0.5
ml/min and 0.5-ml fractions were collected. Aliquots of the
fractions were denatured in a sample buffer and were subjected to
sodium dodecyl sulphate-polyacrylamide gel electrophoresis
(SDS-PAGE) followed by immunoblotting analysis. In some
experiments, the samples of cell-free cytosol without exogenous
cyt-c and dATP, serving as a control, were kept on ice
before loading onto the chromatography column. The column was
calibrated with Blue Dextran 2000 and 6 different protein
Mr-markers.
SDS-PAGE and immunoblotting
The assembly of apoptosome complexes and the
proteolytic processing of PC-9 and PC-3 in the control and the
(cyt-c + dATP)-treated cell-free cytosols were investigated
by SDS-PAGE and immunoblotting using specific antibodies. Prior
loading onto SDS-polyacrylamide gels, cytosol samples (2.5 or 5 mg
of total protein/ml) were incubated at 37°C for the indicated times
without or with cyt-c (10 μM) and dATP (1 mM) in HEMK
buffer, pH 7.4, with 5 mM DTT. Where indicated, the caspase
inhibitor Ac-DEVD-CHO (1 μM) was added into the incubated
samples. SDS-PAGE was carried out in 16.5% T/3% C and 8% T/3% C
gels, for analysis of caspases and Apaf-1, respectively, using the
Tris-Tricine-SDS buffer system (39). The samples were denaturated by 5
min boiling in a sample buffer, pH 7.4, containing 2% SDS, 0.01%
Serva Blue G, 50 mM Tris/HCl, 100 mM DTT, and 10% glycerol, and
were gel-loaded (50 μg of total protein per lane) in
parallel with pre-stained protein Mr markers. The
separated proteins were electrotransferred to Hybond-P PVDF
membrane sheets (GE Healthcare, Little Chalfont, UK) with using a
transfer buffer containing 48 mM Tris, 39 mM glycine, 1.3 mM SDS,
20% methanol, pH 9.2. The following immunoblotting procedure,
coupled with an enhanced chemiluminescence detection system, was
carried out at room temperature as follows. The membrane sheets
were blocked with 5% BGB and 1% BSA in TBST buffer (20 mM Tris/HCl,
100 mM NaCl, 0.1 v/v% Tween-20, pH 7.6) for 1 h, and incubated with
anti-Apaf-1 antibody (1 μg/ml) or anti-caspase-3 antibody
(0.5 μg/ml) for 2 h or with anti-caspase-9 antibody (at a
dilution of 1:1,500 or 1:500) overnight at 4°C. The primary
antibodies were diluted in TBST buffer. The membranes were then
extensively washed in TBST buffer, incubated with the secondary
HRP-conjugated antibody (at a dilution of 1:10,000 in 5% BSA in
TBST buffer) for 1 h, extensively washed in TBST buffer, and
incubated with the ECL Plus Reagent (GE Healthcare) for 5 min. The
chemiluminiscence signal of specific immunocomplexes was captured
onto BioMax Light-1 film (Eastman Kodak, Rochester, NY, USA).
Determination of XIAP and total
protein
The levels of XIAP protein in cell-free cytosol
samples were determined by sandwich ELISA as described previously
(40). Total protein concentration
in cell-free cytosol samples was determined by the bicinchoninic
acid assay using BSA as a standard (41).
Statistical analysis
The statistical analysis was performed with
SigmaStat software (Systat Software, Point Richmond, CA, USA) and
Stat200 (Biosoft, Cambridge, UK). A two-sided p-value lower than
0.05 was accepted as statistically significant difference.
Results
It has been recently demonstrated that the
activation of PC-9 at the Apaf-1-apoptosome requires its
proteolytic processing to CS-9 (12). To assess the functionality of
apoptosome apparatus in NSCLC cell lines, we studied the
proteolytic processing of PC-9 to CS-9 and the subsequent
proteolytic activation of PC-3 to CS-3 and generation of CS-3-like
activity upon incubation of cell-free cytosol samples with
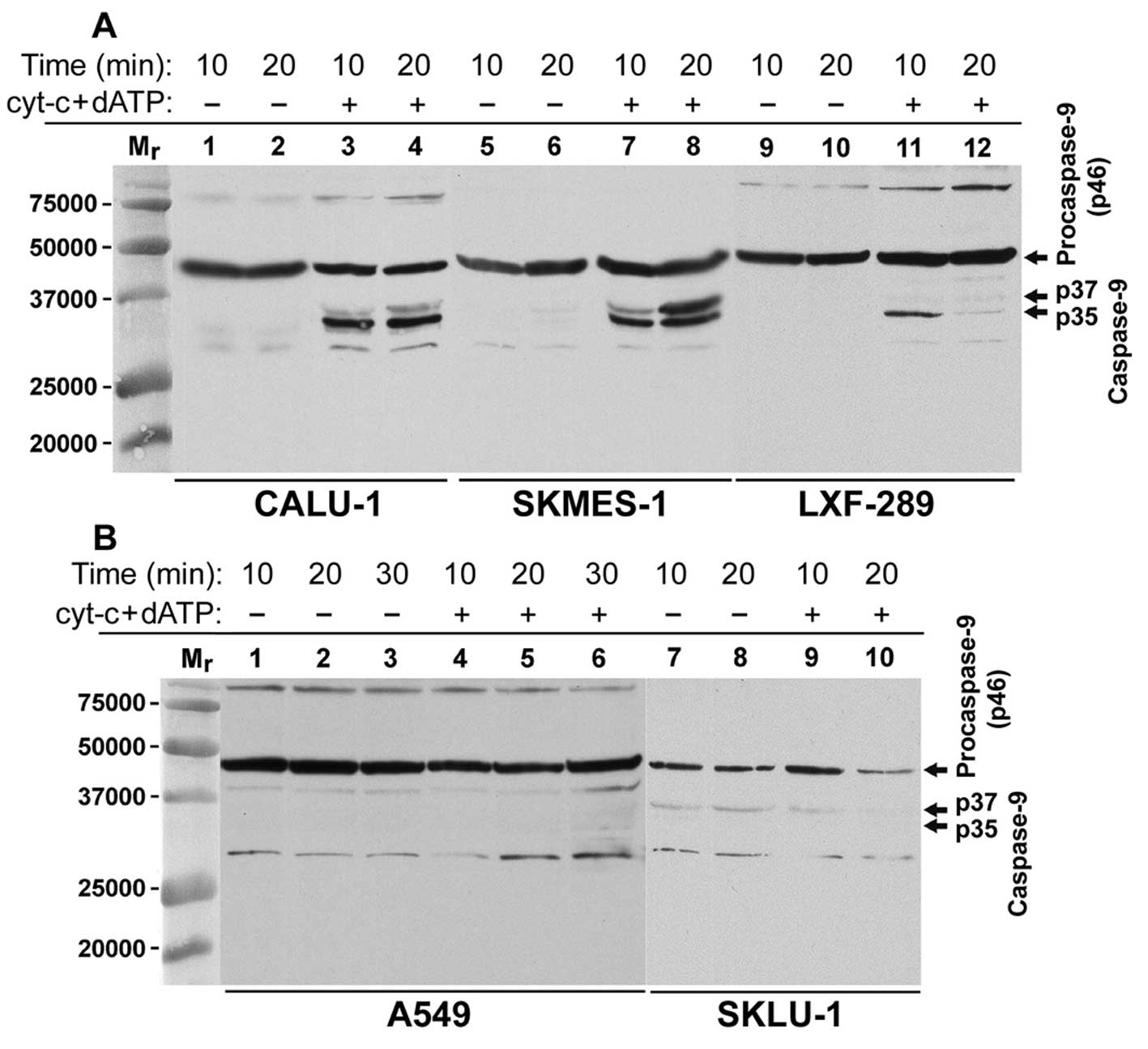
exogenous cyt-c and dATP. In CALU-1, SKMES-1, LXF-289 and
NCI-H520 cells, we detected the generation of the p35 CS-9 cleaved
form already within 5–10 min after the addition of cyt-c and
dATP, while the p37 CS-9 cleaved form appeared later (Figs. 1A and 2A, left part). Surprisingly, the p35 CS-9
form in the (cyt-c + dATP)-treated cell-free cytosol from
LXF-289 cells, detected in 10 min of activation, disappeared later
(Fig. 1). This may be due to the
time-dependent and yet unexplained loss of an antigenic epitope
from the C-terminus of the p35 caspase-9 subunit, which was
recognised by the used anti-caspase-9 antibody. The (cyt-c +
dATP)-treated cell-free cytosol from A549 cells showed PC-9 protein
(p46), but the p35 and p37 cleaved CS-9 forms were not detected,
indicating inhibition of apoptosome activation (Fig. 1B). Finally, the cell-free cytosol
from SKLU-1 cells also did not respond with the p35 CS-9 form
generation to the cyt-c and dATP treatment (Fig. 1B). However, we regularly detected a
faint band of the p37 CS-9 form in the untreated as well as the
(cyt-c + dATP)-treated cytosol samples from SKLU-1 cells
(Fig. 1B), indicating the presence
of an endogenous CS-3-like activity in these cells (Fig. 3).
To study the involvement of CS-3 in the proteolytic
processing of PC-9, we incubated cell-free cytosol from NCI-H520
cells for different times with cyt-c and dATP and in the
absence and the presence of a typical CS-3 inhibitor Ac-DEVD-CHO (1
μM). The western blot (WB) analysis showed that in the
absence of the inhibitor, PC-9 was processed into two forms, one
containing the p35 subunit and the other one the p37 subunit
(Fig. 2A, lanes 2–4). The
formation of the CS-9 p35 form preceded the generation of the CS-9
p37 form (Fig. 2A, lane 1 vs. lane
2). The time-dependent disappearance of PC-9 (p46) was faster in
samples without Ac-DEVD-CHO compared to the samples with
Ac-DEVD-CHO (Fig. 2A, lanes 1–4
vs. lanes 5–8). In addition, the formation of the CS-9 p37 form
correlated with the generation of active CS-3 forms containing the
p20 or p17 subunit (Fig. 2A and B,
left parts). On the contrary, the generation of both the CS-9 p37
form and the active CS-3 forms was completely blocked in the
presence of Ac-DEVD-CHO (Fig. 2A and
B, right parts). These results clearly indicate that
Ac-DEVD-CHO can inhibit the activity of CS-9 p35 form in the
holo-apoptosome, leading to the absence of PC-3 activation, but it
does not inhibit the apoptosome-driven PC-9 activation and
autoprocessing to CS-9 p35 form.
To further confirm functionality or suppression of
apoptosome apparatus in the studied NSCLC cell lines, we analysed
and compared the levels of the endogenous and the (cyt-c +
dATP)-induced CS-3-like activity in their cell-free cytosol, using
a fluorimetric CS-3-like activity assay with Ac-DEVD-AFC as the
substrate. The (cyt-c + dATP)-treated cytosol samples showed
a significant increase of CS-3-like activity in 5 (CALU-1,
NCI-H520, SKMES-1, LXF-289 and COLO-699) of 7 studied NSCLC cell
lines (Fig. 3), confirming the
results of WB analysis of the (cyt-c + dATP)-induced
processing of PC-9 and PC-3 proteins (Figs. 1, 2A and 4C). The cell-free cytosol samples from
A549 and SKLU-1 cells did not show any significant increase of the
endogenous CS-3-like activity after the treatment with cyt-c
and dATP (Fig. 3). Interestingly,
we regularly found a high level of endogenous CS-3-like activity in
the (cyt-c + dATP)-untreated cell-free cytosol from SKLU-1
cells (Fig. 3). This unusual
finding was accompanied with the expression of a p24 protein
co-detected by WB with the unprocessed PC-3 (unpublished data).
Both, the endogenous and the (cyt-c + dATP)-induced
CS-3-like activities were completely inhibited by the caspase
inhibitor Ac-DEVD-CHO at 10 μM (data not shown).
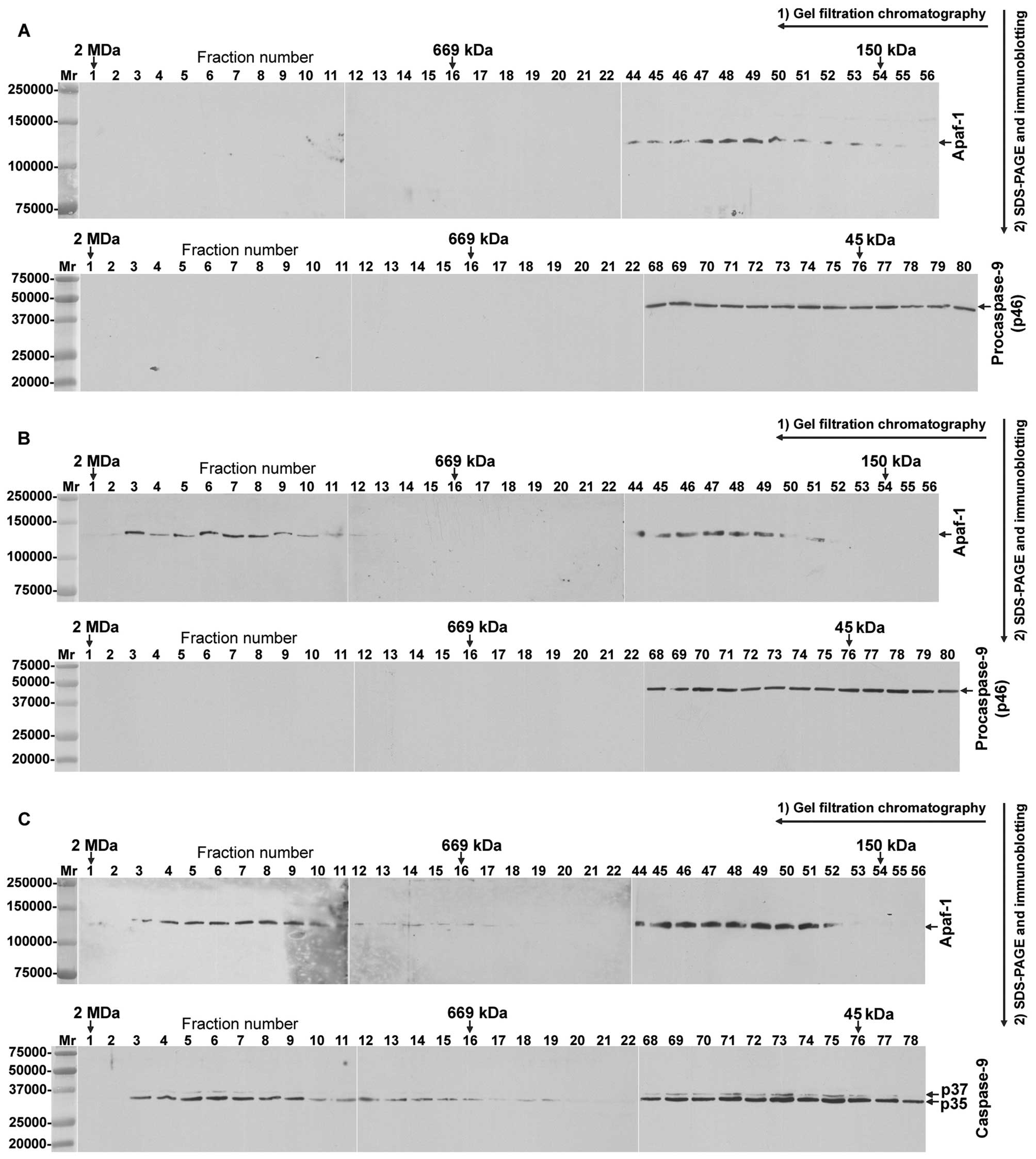
To find out the possible assembly difference between
the functional and dysfunctional apoptosomes in the studied NSCLC
cell lines, we analysed the formation of apoptosome complexes in
cell-free cytosol, using GFC and WB techniques. As the primary
control, we used cell-free cytosol from NSCLC cell lines which was
kept on ice, instead of being incubated prior to GFC. Using this
material from COLO-699 cells, Apaf-1 and PC-9 eluted in
Mr-region corresponding to their unoligomerized forms
(Fig. 4A). Surprisingly, if the
cytosol from COLO-669 cells was incubated alone at 37°C for 30 min,
Apaf-1, but not PC-9, eluted in two Mr-regions, one
corresponding to its high-Mr aggregate (∼1,400 kDa) and
the other one to its unoligomerized forms (Fig. 4B, upper panel). On the contrary,
when the cytosol from COLO-669 cells was incubated with
cyt-c and dATP at 37°C for 30 min, PC-9 was completely
converted to the CS-9 p35 and p37 cleaved forms, which co-eluted
with Apaf-1 in two Mr-regions, one corresponding to
their high-Mr complexes (∼1,400 and ∼700 kDa) and the
other one to their unoligomerized forms (Fig. 4C). Similar results were obtained in
the apoptosome assembly experiments carried out with the cell-free
cytosol from CALU-1 cells (data not shown). By contrast, the
incubation of A549 cell-free cytosol with cyt-c and dATP
induced a shift of a portion of both Apaf-1 (Fig. 5A) and PC-9 (Fig. 5B) proteins into the
high-Mr fractions corresponding to complexes of ∼1,400
and ∼700 kDa. These fractions contained predominantly PC-9 while
the CS-9 p35 and p37 forms were only barely detectable when using
three times higher concentration of the anti-caspase-9 antibody.
However, the majority of PC-9 and some CS-9 eluted in the
low-Mr fractions (Fig.
5B).
To asses the effect of thermal pre-treatment of the
cell-free cytosol on the (cyt-c + dATP)-induced CS-3-like
activity, we pre-incubated aliquots of the cytosol from NCI-H520
cells without cyt-c and dATP at different temperatures and
for different times. Subsequently, the samples were incubated with
exogenous cyt-c and dATP at 37°C for 30 min and then
assayed, along with controls, for CS-3-like activity. As shown in
Fig. 6A and B, the (cyt-c +
dATP)-mediated induction of CS-3-like activity was rapidly lost in
cytosol samples which were exposed to thermal pre-treatment during
the period of apoptosome activation. Similar results were obtained
in the thermal pre-treatment experiments carried out at different
temperatures and for different times with the cell-free cytosol
from CALU-1 cells (data not shown). Importantly, the pre-incubation
at 37°C for 10 min leads to irreversible loss of apoptosome
activation ability in all cell-free cytosol samples tested,
including cytosols from seven cell lines (Fig. 6C), four NSCLC tumours of different
histopathological types and lung parenchyma (Fig. 6D). These results indicate that
thermal pre-treatment of cell-free cytosol samples should be
avoided in apoptosome activation analysis in order to prevent loss
of apoptosome activation ability.
We next studied the sensitivity of apoptosome
apparatus to activation in NSCLC tumours and matched lungs, using
analysis of CS-3-like activity in cell-free cytosols. The
endogenous (E) as well as the (cyt-c + dATP)-induced total
(T) and the (cyt-c + dATP)-induced incremental (I; I=T-E)
CS-3-like activities were significantly higher in NSCLC tumours as
compared to matched lungs (Table
I). When the robust (cyt-c + dATP)-induced CS-3-like
activity generation (i.e. I/E ≥2) was considered, it was found in
19 (31%) of 62 examined tumours and only in 5 (8%) of 62 examined
lungs (p=0.003; χ2 test). When all (cyt-c +
dATP)-dependent increases of CS-3-like activity were considered
(i.e. I/E >0), they were found in 47 (76%) of 62 examined
tumours and 29 (47%) of 62 examined lungs (p=0.0017; χ2
test). Moreover, the tumour/lung CS-3-like activity ratio was much
higher than 2 for the (cyt-c + dATP)-induced activities (T
and I) and was the predominant finding in the majority of examined
NSCLC patients (Table I). These
results indicate that the apoptosome apparatus can be more robustly
and more frequently activated in NSCLC tumours as compared to
matched lungs. Nevertheless, the tumour histopathological type,
grade and stage or the patient’s gender and smoking habit had no
statistically significant impact on the levels of endogenous and
the (cyt-c + dATP)-induced CS-3-like activity in the tumours
(p>0.08, Mann-Whitney test; data not shown).
 | Table I.Analysis of the (cytochrome c
+ dATP)-induced activation of apoptosome apparatus in cytosols from
non-small cell lung carcinoma tissues and matched lung
parenchyma. |
Table I.
Analysis of the (cytochrome c
+ dATP)-induced activation of apoptosome apparatus in cytosols from
non-small cell lung carcinoma tissues and matched lung
parenchyma.
| Category of
caspase-3-like activity | Caspase-3-like
activity a,b (pkat/mg of total protein) | Statistical
difference (p-value) of caspase-3-like activity in Tu vs.
Luc | Tu/Lu ratio of
caspase-3-like activityb,d | No. of patients
with Tu/Lu activity ratio >2 |
|---|
|
|---|
| Tumours (Tu) | Lungs (Lu) |
|---|
| Endogenous (E) | 0.717
(0–7.710) | 0.019
(0–1.351) |
9.2×10−11 | 13.8
(0.3–107.1) | 27 (79%) |
| Total induced
(T) | 1.428
(0–88.706) | 0.064
(0–5.645) |
9.8×10−11 | 17.1
(0.6–773.3) | 38 (88%) |
| Incremental induced
(I)e | 0.289
(0–88.429) | 0.004
(0–5.245) |
1.3×10−5 | 9.1
(0.3–2,364.9) | 19 (70%) |
|
| | | |
| Activity ratio
I/Ef | 1.07
(0.03–536.3) | 0.49
(0.001–28.4) | | | |
We further tested whether XIAP, the only direct and
simultaneous inhibitor of CS-9 and CS-3 or CS-7 (42–45),
inhibits the apoptosome activity in cytosol from NSCLC tumours. The
levels of XIAP protein assayed in the cytosols from NSCLC tumours
(n=28) ranged from 17.6 to 126.6 nmol per mg of total protein. The
(cyt-c + dATP)-untreated cytosol samples from these tumours
showed negative linear correlation between the level of XIAP
protein and the low endogenous CS-3-like activity (r=−0.480,
p=0.009). However, once treated with cyt-c and dATP, the
cytosol samples displayed positive linear correlation between the
level of XIAP protein and the (cyt-c + dATP)-induced
CS-3-like activity (r=0.414, p=0.028). This indicates that the
apoptosome-generated CS-3 activity can overcome the potential
caspase inhibitory effect of XIAP in NSCLC tumours. To test whether
XIAP is involved in the inhibition of endogenous and
apoptosome-generated caspase activity in cytosol from NSCLC tumours
we used the heptapeptides AVPIAQK (P1) and ATPFQEG (P2) to
neutralize the XIAP inhibitory action against the caspases
(38,46). In the (cyt-c +
dATP)-untreated cytosol samples from NSCLC tumours (n=21), the
respective endogenous CS-3-like activities (in pkat/mg of total
protein, median and range) in the absence (E) and the presence of
500 μM of peptide P1 (E-P1) or peptide P2 (EP-2) were: E,
1.36 and 0.27–7.16; EP-1, 1.39 and 0.28–8.30; and EP-2, 1.39 and
0.29–7.45. The slight increase of the endogenous CS-3-like activity
in the presence of peptide P1 (Fig. 7A
and C) was statistically significant (p=7.0×10−4,
Wilcoxon matched pair test). When the cytosols samples from NSCLC
tumours were treated with cyt-c and dATP in the presence of
500 μM of peptide P1 or P2 some of them showed markedly
higher CS-3-like activity as compared with the cytosol samples
treated with cyt-c and dATP in the absence of the peptides
(Fig. 7A, B and D). The respective
(cyt-c + dATP)-induced incremental CS-3-like activities (in
pkat/mg of total protein, median and range) in the absence (I) and
the presence of 500 μM of peptide P1 (I-P1) or peptide P2
(IP-2) were: I, 6.2 and 0.2–99.3; I-P1, 7.6 and 0.1–124.7; and
I-P2, 9.9 and 0.4–130.5. The moderate increase of the (cyt-c
+ dATP)-induced incremental CS-3-like activity in the presence of
peptide P2 was statistically significant (p= 0.012, Wilcoxon
matched pair test). In addition, the CS-3-like activity ratios
I-P1/I and I-P2/I higher than 1.5 were found in 4 (21%) and 9 (47%)
of 19 evaluated NSCLC tumours, respectively (Fig. 7D), further indicating somewhat
higher potency of peptide P2 to derepress the inhibition of
apoptosome-generated CS-3-like activity.
 | Figure 7.Effect of XIAP-neutralizing peptides
on the endogenous and the (cyt-c + dATP)-induced
caspase-3-like activity in cell-free cytosols from NSCLC tumours.
(A and B) Caspase-3-like reactions in cell-free cytosol sample
aliquots from an SQCLC tumour and a UNDIF tumour, respectively,
that had been preincubated without (a control, C) or with
cyt-c + dATP (CydA) and without or with 500 μM of the
XIAP-neutralizing peptide AVPIAQK (P1) or ATPFQEG (P2). In (A), the
rates of the caspase-3-like reactions labelled C, P1, CydA and CydA
+ P1 were, respectively, 0.23, 0.29, 2.4 and 4.7 nkat. In (B), the
rates of the caspase-3-like reactions labelled C, P2, CydA and CydA
+ P2 were, respectively, 0.28, 0.28, 7.6 and 13.0 nkat. (C and D)
depict the responses of the endogenous and the (cyt-c +
dATP)-induced caspase-3-like activities, respectively, in cell-free
cytosols from 21 NSCLC tumours to the peptides AVPIAQK (P1, 500
μM) and ATPFQEG (P2, 500 μM). The incremental
(cyt-c + dATP)-induced caspase-3-like activities in the
absence of the peptides (a control, I) and in their presence (I-P1
and I-P2) were calculated as described in Materials and methods.
Data indicated as mean ± SEM from three independent experiments.
The caspase-3-like activity ratios I-P1/I and I-P2/I higher than 2
(+) and higher than 1.5 (*) but lower than 2 are indicated. |
Discussion
The results of the present study showed that
apoptosome apparatus was functional in the majority of examined
NSCLC cell lines and NSCLC tumours. This evidence was obtained in
experiments demonstrating the (cyt-c + dATP)-induced
proteolytic processing of PC-9 to CS-9 leading to the proteolytic
activation of PC-3 to CS-3 and to generation of CS-3-like activity
in cell-free cytosol samples. Recent studies provided clear
evidence that the activation of PC-9 at the Apaf-1-apoptosome
requires its proteolytic processing to CS-9 (12). The lack of the (cyt-c +
dATP)-mediated induction of CS-3-like activity we observed in
cell-free cytosols from A549 and SKLU-1 cell lines was associated
with the absence of PC-9 processing to the p35 CS-9 form.
It was previously reported that the ∼1,400- and
∼700-kDa apoptosome complexes, both containing the p35 and p37
forms of CS-9, were formed in the dATP-activated lysates from human
tumour cells lines THP.1 and MCF-7 (13,14,33).
In this study, we confirmed abundant formation of the
high-Mr apoptosome complex of ∼1,400 kDa in the
(cyt-c + dATP)-responsive COLO-699 and CALU-1 cytosols, but
the ∼700 kDa complex was also detected. Interestingly, after
treatment of the cytosol samples with cyt-c and dATP for 30
min, PC-9 was completely processed to CS-9 p35 and p37 forms. These
CS-9 forms co-eluted with both apoptosome complexes, but were
mostly present in the low-Mr fractions corresponding to
CS-9 monomers. This can be explained by the recruitment-dependent
PC-9 processing function of apoptosome, where PC-9 has higher
binding affinity to apoptosome and displaces the processed CS-9
(10). Thus, in COLO-699 and
CALU-1 cell-free cytosols, apparently all PC-9 molecules can be
processed to CS-9 after the activation of apoptosome apparatus. By
contrast, in the (cyt-c + dATP)-treated A549 cell-free
cytosol, the formed high-Mr apoptosome complexes were
able to bind PC-9, but the proteolytic processing of the
apoptosome-associated PC-9 was blocked and the majority of PC-9 was
present outside the apoptosomes. The mechanism underlying the
failure of proteolytic processing of the apoptosome-bound PC-9 is
not known at present. It might involve phosphorylations of PC-9
molecules that do not interfere with PC-9 recruitment to
apoptosome, but can prevent the apoptosome-mediated PC-9 activation
(48). The trace amounts of CS-9
p35 and p37 forms were probably extra-apoptosome generated CS-9
forms, since they could be immunodetected in A549 cell-free cytosol
regardless of the treatment with cyt-c and dATP, using three
times higher concentration of the anti-caspase-9 antibody (data not
shown). Although the origin of the cleaved CS-9 forms in A549 cells
cytosol is not known, for instance, calpains are capable of PC-9
cleavage to CS-9 p35 and p37 forms (47).
It was previously observed that incubation of
recombinant Apaf-1 with cyt-c in the absence of dATP
resulted in the formation of non-functional high-Mr
Apaf-1 aggregates (34). In the
present study, we found that incubating cell-free cytosol from all
tested cell lines and tissues without addition of cyt-c and
dATP at temperatures higher than 4°C leads to loss of the
(cyt-c + dATP)-mediated induction of CS-3-like activity.
This thermal-induced loss of apoptosome activation is likely due to
not only the formation of high-Mr Apaf-1 aggregates
incapable of recruitment and activation of PC-9 but also by yet
unexplained inactivation of Apaf-1 monomers (Fig. 4B and C, upper panels). The Apaf-1
aggregates were not present in cell-free cytosols which were not
exposed to thermal treatment prior to GFC and WB analysis (Fig. 4A upper panel). Moreover, contrary
to a previous report (34), the
addition of exogenous cyt-c during the thermal treatment of
cell-free cytosols had a protective role on the subsequent
(cyt-c + dATP)-induction of CS-3-like activity (data not
shown). Our results indicate that the propensity of cytosolic
Apaf-1 to irreversible thermal aggregation and inactivation
complicates the attempts to reactivate non-functional apoptosomes,
for instance through enzymatic dephosphorylation of phosphorylated
PC-9 (48) or Apaf-1 (49,50).
Previous studies showed increased activation ability
of apoptosomes in breast cancer cells (21) and brain tumours (22), because of PHAPI and Apaf-1
overexpression, respectively. In the present study, we found
differential sensitivity to apoptosome apparatus activation in
NSCLC tumours and matched lungs. The heightened sensitivity to
apoptosome activation in these tumours may be caused by
overexpression of both Apaf-1 and PC-3 proteins (27). Although the activation of
apoptosome apparatus was much more robust and frequent in NSCLC
tumours as compared to lungs, it was not associated with tumour
histology, grade or stage. The low sensitivity to apoptosome
activation of lung parenchyma cells, which are exposed to various
environmental stressors, is important for their long-term survival
and restriction from unwanted apoptosis. It has been already
reported that compared to inflammatory cells airway epithelial
cells have some innate resistance to apoptosis stimuli (51). Thus, the differential sensitivity
of NSCLC tumours and lung parenchyma to apoptosome activation might
be exploited for the apoptosis-based therapies. On the other hand,
however, the apoptosome-generated CS-3 activity in tumour cells can
significantly increase the rate of tumour recurrence and death of
patients (23,24). The mechanism of this cancer cell
repopulation involves the CS-3-mediated proteolytic activation of
calcium independent phospholipase-A2 leading to
increased production of arachidonic acid and
prostaglandin-E2, which stimulates proliferation of
tumour cells (23,24).
XIAP acts in the initiation as well as the execution
phases of apoptosome pathway due to inhibition of the
apoptosome-associated CS-9 p35/p12 form, CS-3 and CS-7 (42,44,46,52).
Since XIAP is highly expressed in NSCLC tumours (40), it might be involved in suppression
of apoptosome activity in these tumours. However, the inhibition of
the apoptosome-associated CS-9 by XIAP can be reverted by Smac
protein, which sequesters XIAP and relieves CS-9 to activate PC-3
to CS-3 (46). The active CS-3,
but not CS-7, in turn removes the IAP binding motif from the
N-terminus of CS-9 p12 subunit making the CS-9 resistant to XIAP
inhibition (46). To test whether
the inactivation of XIAP can enhance the activity of apoptosome
apparatus in cytosol from NSCLC tumours, we used the
XIAP-neutralizing peptides AVPIAQK (P1) and ATPFQEG (P2). These
peptides displayed similar binding affinities toward XIAP as the
target (46). The former peptide
(38) as well as their shorter
analogues (52) were previously
shown to be very effective derepressors of the apoptosome aparatus.
We found, that the (cyt-c + dATP)-induced CS-3-like activity
was moderately increased by the CS-9 p12 subunit-mimetic peptide
ATPFQEG in some tested tumours. These results indicate that XIAP
does not effectively suppress the activity of the apoptosome
apparatus in NSCLCs.
Acknowledgements
This study was supported by the
research project RVO-NNB/2013 from the Ministry of Health, Czech
Republic. We would like to thank Dr Jan Cermak, Department of
Pneumology and Thoracic Surgery, Hospital Bulovka, Prague, for
cooperation in obtaining tissue samples and clinico-pathological
data from the patients entering the study.
References
|
1.
|
Cecconi F, Alvarez-Bolado G, Meyer BI,
Roth KA and Gruss P: Apaf1 (CED-4 homolog) regulates programmed
cell death in mammalian development. Cell. 94:727–737. 1998.
View Article : Google Scholar
|
|
2.
|
Hao Z, Duncan GS, Chang CC, Elia A, Fang
M, Wakeham A, Okada H, Calzascia T, Jang Y, You-Ten A, Yeh WC,
Ohashi P, Wang X and Mak TW: Specific ablation of the apoptotic
functions of cytochrome C reveals a differential requirement for
cytochrome C and Apaf-1 in apoptosis. Cell. 121:579–591. 2005.
View Article : Google Scholar
|
|
3.
|
Soengas MS, Capodieci P, Polsky D, Mora J,
Esteller M, Opitz-Araya X, McCombie R, Herman JG, Gerald WL,
Lazebnik YA, Cordon-Cardo C and Lowe SW: Inactivation of the
apoptosis effector Apaf-1 in malignant melanoma. Nature.
409:207–211. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Nguyen JT and Wells JA: Direct activation
of the apoptosis machinery as a mechanism to target cancer cells.
Proc Natl Acad Sci USA. 100:7533–7538. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Jiang X and Wang X: Cytochrome C-mediated
apoptosis. Annu Rev Biochem. 73:87–106. 2004. View Article : Google Scholar
|
|
6.
|
Hausmann G, O’Reilly LA, van Driel R,
Beaumont JG, Strasser A, Adams JM and Huang DC: Pro-apoptotic
apoptosis protease-activating factor 1 (Apaf-1) has a cytoplasmic
localization distinct from Bcl-2 or Bcl-x(L). J Cell Biol.
149:623–634. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Benedict MA, Hu Y, Inohara N and Nunez G:
Expression and functional analysis of Apaf-1 isoforms. Extra Wd-40
repeat is required for cytochrome c binding and regulated
activation of procaspase-9. J Biol Chem. 275:8461–8468. 2000.
View Article : Google Scholar
|
|
8.
|
Yuan S and Akey CW: Apoptosome structure,
assembly, and procaspase activation. Structure. 21:501–515. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Yuan S, Topf M, Reubold TF, Eschenburg S
and Akey CW: Changes in Apaf-1 conformation that drive apoptosome
assembly. Biochemistry. 52:2319–2327. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Malladi S, Challa-Malladi M, Fearnhead HO
and Bratton SB: The Apaf-1•procaspase-9 apoptosome complex
functions as a proteolytic-based molecular timer. EMBO J.
28:1916–1925. 2009.
|
|
11.
|
Yuan S, Yu X, Asara JM, Heuser JE, Ludtke
SJ and Akey CW: The holo-apoptosome: activation of procaspase-9 and
interactions with caspase-3. Structure. 19:1084–1096. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Hu Q, Wu D, Chen W, Yan Z and Shi Y:
Proteolytic processing of caspase-9 zymogen is required for
apoptosome-mediated activation of caspase-9. J Biol Chem.
288:15142–15147. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Twiddy D, Brown DG, Adrain C, Jukes R,
Martin SJ, Cohen GM, Macfarlane M and Cain K: Pro-apoptotic
proteins released from the mitochondria regulate the protein
composition and caspase-processing activity of the native
Apaf-1/caspase-9 apoptosome complex. J Biol Chem. 279:19665–19682.
2004. View Article : Google Scholar
|
|
14.
|
Twiddy D, Cohen GM, Macfarlane M and Cain
K: Caspase-7 is directly activated by the approximately 700-kDa
apoptosome complex and is released as a stable XIAP-caspase-7
approximately 200-kDa complex. J Biol Chem. 281:3876–3888. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Schafer ZT and Kornbluth S: The
apoptosome: physiological, developmental, and pathological modes of
regulation. Dev Cell. 10:549–561. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Bratton SB and Salvesen GS: Regulation of
the Apaf-1-caspase-9 apoptosome. J Cell Sci. 123:3209–3214. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Liu JR, Opipari AW, Tan L, Jiang Y, Zhang
Y, Tang H and Nunez G: Dysfunctional apoptosome activation in
ovarian cancer: implications for chemoresistance. Cancer Res.
62:924–931. 2002.PubMed/NCBI
|
|
18.
|
Corvaro M, Fuoco C, Wagner M and Cecconi
F: Analysis of apoptosome dysregulation in pancreatic cancer and of
its role in chemoresistance. Cancer Biol Ther. 6:209–217. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Christoph F, Hinz S, Weikert S,
Kempkensteffen C, Schostak M, Miller K and Schrader M: Comparative
promoter methylation analysis of p53 target genes in urogenital
cancers. Urol Int. 80:398–404. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Tan L, Kwok RP, Shukla A, Kshirsagar M,
Zhao L, Opipari AW Jr and Liu JR: Trichostatin A restores Apaf-1
function in chemoresistant ovarian cancer cells. Cancer.
117:784–794. 2011. View Article : Google Scholar
|
|
21.
|
Schafer ZT, Parrish AB, Wright KM,
Margolis SS, Marks JR, Deshmukh M and Kornbluth S: Enhanced
sensitivity to cytochrome c-induced apoptosis mediated by PHAPI in
breast cancer cells. Cancer Res. 66:2210–2218. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Johnson CE, Huang YY, Parrish AB, Smith
MI, Vaughn AE, Zhang Q, Wright KM, Van Dyke T, Wechsler-Reya RJ,
Kornbluth S and Deshmukh M: Differential Apaf-1 levels allow
cytochrome c to induce apoptosis in brain tumors but not in normal
neural tissues. Proc Natl Acad Sci USA. 104:20820–20825. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Huang Q, Li F, Liu X, Li W, Shi W, Liu FF,
O’Sullivan B, He Z, Peng Y, Tan AC, Zhou L, Shen J, Han G, Wang XJ,
Thorburn J, Thorburn A, Jimeno A, Raben D, Bedford JS and Li CY:
Caspase 3-mediated stimulation of tumor cell repopulation during
cancer radiotherapy. Nat Med. 17:860–866. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24.
|
Zimmerman MA, Huang Q, Li F, Liu X and Li
CY: Cell death-stimulated cell proliferation: a tissue regeneration
mechanism usurped by tumors during radiotherapy. Semin Radiat
Oncol. 23:288–295. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
25.
|
Joseph B, Marchetti P, Formstecher P,
Kroemer G, Lewensohn R and Zhivotovsky B: Mitochondrial dysfunction
is an essential step for killing of non-small cell lung carcinomas
resistant to conventional treatment. Oncogene. 21:65–77. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
26.
|
Gomyo Y, Sasaki J, Branch C, Roth JA and
Mukhopadhyay T: 5-Aza-2′-deoxycytidine upregulates caspase-9
expression cooperating with p53-induced apoptosis in human lung
cancer cells. Oncogene. 23:6779–6787. 2004.
|
|
27.
|
Krepela E, Prochazka J, Liu X, Fiala P and
Kinkor Z: Increased expression of Apaf-1 and procaspase-3 and the
functionality of intrinsic apoptosis apparatus in non-small cell
lung carcinoma. Biol Chem. 385:153–168. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
28.
|
Hoffarth S, Zitzer A, Wiewrodt R, Hahnel
PS, Beyer V, Kreft A, Biesterfeld S and Schuler M: pp32/PHAPI
determines the apoptosis response of non-small-cell lung cancer.
Cell Death Differ. 15:161–170. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
29.
|
Ekedahl J, Joseph B, Grigoriev MY, Muller
M, Magnusson C, Lewensohn R and Zhivotovsky B: Expression of
inhibitor of apoptosis proteins in small- and non-small-cell lung
carcinoma cells. Exp Cell Res. 279:277–290. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
30.
|
Bartling B, Lewensohn R and Zhivotovsky B:
Endogenously released Smac is insufficient to mediate cell death of
human lung carcinoma in response to etoposide. Exp Cell Res.
298:83–95. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
31.
|
Checinska A, Giaccone G, Hoogeland BS,
Ferreira CG, Rodriguez JA and Kruyt FA: TUCAN/CARDINAL/CARD8 and
apoptosis resistance in non-small cell lung cancer cells. BMC
Cancer. 6:1662006. View Article : Google Scholar : PubMed/NCBI
|
|
32.
|
Checinska A, Hoogeland BS, Rodriguez JA,
Giaccone G and Kruyt FA: Role of XIAP in inhibiting
cisplatin-induced caspase activation in non-small cell lung cancer
cells: a small molecule Smac mimic sensitizes for
chemotherapy-induced apoptosis by enhancing caspase-3 activation.
Exp Cell Res. 313:1215–1224. 2007. View Article : Google Scholar
|
|
33.
|
Cain K, Bratton SB, Langlais C, Walker G,
Brown DG, Sun XM and Cohen GM: Apaf-1 oligomerizes into
biologically active approximately 700-kDa and inactive
approximately 1.4-MDa apoptosome complexes. J Biol Chem.
275:6067–6070. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
34.
|
Kim HE, Du F, Fang M and Wang X: Formation
of apoptosome is initiated by cytochrome c-induced dATP hydrolysis
and subsequent nucleotide exchange on Apaf-1. Proc Natl Acad Sci
USA. 102:17545–17550. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
35.
|
Kim HE, Jiang X, Du F and Wang X: PHAPI,
CAS, and Hsp70 promote apoptosome formation by preventing Apaf-1
aggregation and enhancing nucleotide exchange on Apaf-1. Mol Cell.
30:239–247. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
36.
|
Travis WD, Brambilla E, Müller-Hermelink
HK and Harris CC: Pathology & Genetics: Tumours of the Lung,
Pleura, Thymus and Heart. IARC Press; Lyon: 2004
|
|
37.
|
Detterbeck FC, Boffa DJ and Tanoue LT: The
new lung cancer staging system. Chest. 136:260–271. 2009.
View Article : Google Scholar
|
|
38.
|
Srinivasula SM, Datta P, Fan XJ,
Fernandes-Alnemri T, Huang Z and Alnemri ES: Molecular determinants
of the caspase-promoting activity of Smac/DIABLO and its role in
the death receptor pathway. J Biol Chem. 275:36152–36157. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
39.
|
Schägger H and von Jagow G: Tricine-sodium
dodecyl sulfatepolyacrylamide gel electrophoresis for the
separation of proteins in the range from 1 to 100 kDa. Anal
Biochem. 166:368–379. 1987.
|
|
40.
|
Krepela E, Dankova P, Moravcikova E,
Krepelova A, Prochazka J, Cermak J, Schützner J, Zatloukal P and
Benkova K: Increased expression of inhibitor of apoptosis proteins,
survivin and XIAP, in non-small cell lung carcinoma. Int J Oncol.
35:1449–1462. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
41.
|
Smith PK, Krohn RI, Hermanson GT, Mallia
AK, Gartner FH, Provenzano MD, Fujimoto EK, Goeke NM, Olson BJ and
Klenk DC: Measurement of protein using bicinchoninic acid. Anal
Biochem. 150:76–85. 1985. View Article : Google Scholar
|
|
42.
|
Bratton SB, Lewis J, Butterworth M,
Duckett CS and Cohen GM: XIAP inhibition of caspase-3 preserves its
association with the Apaf-1 apoptosome and prevents CD95- and
Bax-induced apoptosis. Cell Death Differ. 9:881–892. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
43.
|
Shiozaki EN, Chai J, Rigotti DJ, Riedl SJ,
Li P, Srinivasula SM, Alnemri ES, Fairman R and Shi Y: Mechanism of
XIAP-mediated inhibition of caspase-9. Mol Cell. 11:519–527. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
44.
|
Scott FL, Denault JB, Riedl SJ, Shin H,
Renatus M and Salvesen GS: XIAP inhibits caspase-3 and -7 using two
binding sites: evolutionarily conserved mechanism of IAPs. EMBO J.
24:645–655. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
45.
|
Eckelman BP, Salvesen GS and Scott FL:
Human inhibitor of apoptosis proteins: why XIAP is the black sheep
of the family. EMBO Rep. 7:988–994. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
46.
|
Denault JB, Eckelman BP, Shin H, Pop C and
Salvesen GS: Caspase 3 attenuates XIAP (X-linked inhibitor of
apoptosis protein)-mediated inhibition of caspase 9. Biochem J.
405:11–19. 2007.PubMed/NCBI
|
|
47.
|
Chua BT, Guo K and Li P: Direct cleavage
by the calcium-activated protease calpain can lead to inactivation
of caspases. J Biol Chem. 275:5131–5135. 2000. View Article : Google Scholar
|
|
48.
|
Allan LA and Clarke PR: Apoptosis and
autophagy: regulation of caspase-9 by phosphorylation. FEBS J.
276:6063–6073. 2009. View Article : Google Scholar
|
|
49.
|
Kim J, Parrish AB, Kurokawa M, Matsuura K,
Freel CD, Andersen JL, Johnson CE and Kornbluth S: Rsk-mediated
phosphorylation and 14-3-3varepsilon binding of Apaf-1 suppresses
cytochrome c-induced apoptosis. EMBO J. 31:1279–1292. 2012.
View Article : Google Scholar
|
|
50.
|
Martin MC, Allan LA, Lickrish M, Sampson
C, Morrice N and Clarke PR: Protein kinase A regulates caspase-9
activation by Apaf-1 downstream of cytochrome c. J Biol Chem.
280:15449–15455. 2005. View Article : Google Scholar
|
|
51.
|
White SR: Apoptosis and the airway
epithelium. J Allergy (Cairo). 2011.948406:2011.PubMed/NCBI
|
|
52.
|
Srinivasula SM, Hegde R, Saleh A, Datta P,
Shiozaki E, Chai J, Lee RA, Robbins PD, Fernandes-Alnemri T, Shi Y
and Alnemri ES: A conserved XIAP-interaction motif in caspase-9 and
Smac/DIABLO regulates caspase activity and apoptosis. Nature.
410:112–116. 2001. View Article : Google Scholar : PubMed/NCBI
|