Introduction
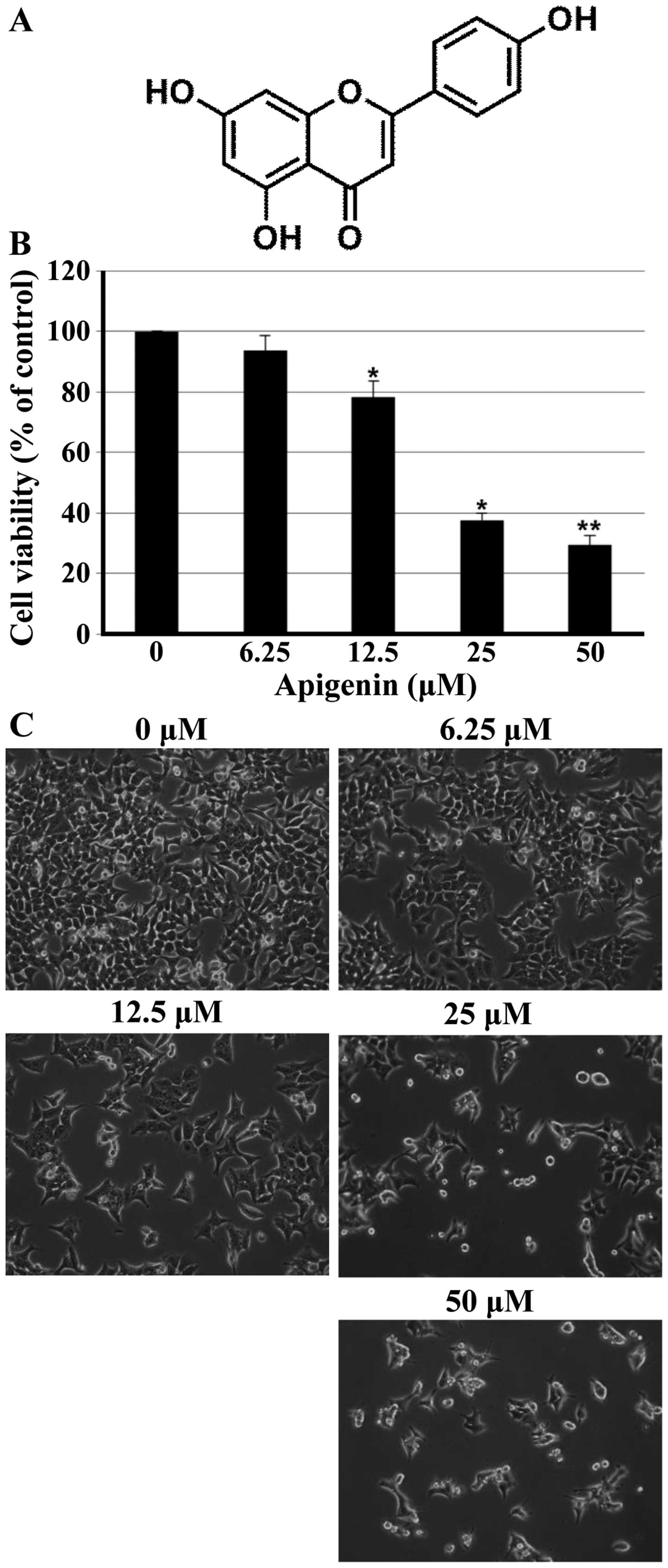
Apigenin (4’,5,7-trihydroxyflavone; Fig. 1A), a naturally occurring flavone,
it is widely distributed in many fruits and vegetables such as
parsley, onions, apples, tea and chamomile. In recent years,
apigenin has been increasingly recognized as a cancer
chemopreventive agent. The chemopreventive aspects of apigenin have
been evaluated both in vitro and in vivo. Apigenin
has been shown to be growth inhibitory in a variety of human cancer
cell lines including colon, pancreatic, oral squamous, lung and
leukemia cells (1–5). An important effect of apigenin is to
increase the stability of the tumor suppressor p53 gene in normal
cells. It has been shown that apigenin induced G2/M cell cycle
arrest in colon cancer cells (1),
and in vivo it is involved in
p21CIP1/WAF1-independent pathway for inhibitory
phosphorylation of p34 (Cdc2) and concomitant G2/M arrest in mouse
keratinocytes (6). Apigenin was
shown to induce apoptosis in a variety of cancer cells (3,4,7,8).
Apigenin has shown to inhibit tumor cell invasion and metastases by
regulating the hypoxia-inducible factor 1-α protein level and to
inhibit transforming growth factor β 1-induced vascular endothelial
growth factor expression in human prostate cancer cells (9). Moreover, apigenin has been reported
to potentiate the effect of tumor necrosis factor-related
apoptosis-inducing ligand, paclitaxel, ABT-263, 5-fluorouracil
(5-FU) and cisplatin against various human cancers (10–13).
Autophagy, an evolutionarily conserved process,
sequesters and degrades long-lived cellular proteins and organelles
through the lysosomal machinery (14,15).
The purpose of autophagy is the recycling of cellular components to
sustain metabolism under stress conditions such as nutrient
deprivation and to prevent accumulation of damaged proteins and
organelles (16). The first
evidence for a role of autophagy in cancer was found by Liang et
al (17). The
autophagy-promoting activity of beclin-1 in human breast cancer
cells is associated with inhibition of MCF7 cellular proliferation
(17). It is reported that
beclin-1, a phylo-genetically conserved protein that is essential
for autophagy, can inhibit tumorigenesis and is expressed at
decreased levels in human breast carcinoma. Recent evidence
indicates that the phosphoinositide 3-kinase (PI3K), Akt and
mammalian target of rapamycin (mTOR) pathway, which is activated in
many types of cancer (18) is
important in autophagy regulation, especially through activating
mTOR kinase, leading to suppression of autophagy (15). Recently, increasing evidence
indicates that autophagy is closely associated with tumors. It
participates as a tumor suppressor in tumor development in the
early stages and as a proto-oncogene in advanced stages (19). Autophagy has been show to increase
as a result of chemotherapy, leading the cancer cells to autophagic
cell death (programmed cell death) (20). However, although autophagy is a
potential therapeutic target in adjuvant chemotherapy, the exact
role and the relationship of autophagy with cancer development and
progression, autophagic cell death, and apoptosis in cancer are
still unclear.
Colorectal cancer (CRC) is the third most common
incident cancer among men and the second most frequent among women
in worldwide (21). Although
specific causes of colon cancer are not known, nutritional and
environmental factors have been associated with the development of
colon cancer. In Korea, incidence of CRC has significantly
increased. Annual percentage changes in age-standardized incident
rates were 6.2% in men and 6.8% in women between 1999 and 2009
using the world standard population as a reference population. It
is the second common cancer after stomach among men and the third
most common after cancers of thyroid and breast among women in
Korea (22,23). Surgical resection currently remains
the only curative treatment for CRC, however, it is unsatisfactory.
This is because only 70% of colorectal tumors are resectable, 75%
of which are curable, and many patients have to receive adjuvant
chemotherapy (24). Due to the
incomplete therapeutic options for colon cancer, there is a need to
develop preventive treatment approaches for this malignancy. This
study was conducted to investigate the ability of apigenin to
induce apoptotic cell death and the ability of apigenin to induce
autophagy in HCT116 human colon cancer cells, and whether
inhibition of autophagy could potentiate the proapoptotic effect of
apigenin.
Materials and methods
Reagents
Apigenin, acridine orange, 3-methyladenine (3-MA),
and monoclonal antibody against β-actin were purchased from
Sigma-Aldrich (St. Louis, MO, USA). Apigenin was dissolved in
dimethylsulfoxide (DMSO) and stored at −20°C before the experiments
and dilutions were made in culture medium.
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT)
was obtained from Amresco (Solon, OH, USA). Antibodies against
beclin-1, Cdc2, Cdc25c, cyclin B1, p21, p53, poly (ADP-ribose)
polymerase (PARP), caspase-3, caspase-8 and caspase-9 were
purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA).
Polyclonal antibody against LC3B was obtained from Cell Signaling
Technology (Danvers, MA, USA). RPMI-1640, fetal bovine serum (FBS),
and penicillin-streptomycin were purchased from HyClone (Logan, UT,
USA).
Cell culture and apigenin treatment
The human colorectal cancer cell line HCT116 was
purchased from the American Type Culture Collection (Manassas, VA,
USA) and maintained at 37°C in a humidified 95% air and 5%
CO2 in RPMI-1640 supplemented with 10% FBS, 100 U/ml
penicillin and 100 μg/ml streptomycin.
Assays for cell viability
The cell growth was determined by MTT assay. Cells
were seeded in 6-well culture plate and incubated at 37°C for 24 h.
Then, cells were treated with different concentration of apigenin
(0–50 μM) for 24 h. The cells were incubated in the dark
with MTT reagent (0.5 mg/ml) at 37°C for 2 h. Medium was removed,
formazan was dissolved in DMSO and the absorbance at 540 nm was
measured by using an ELISA plate reader.
Hoechst staining and observation of
nuclear structure
Cells were washed twice with phosphate-buffered
saline (PBS) and fixed with 3.7% paraformaldehyde (Sigma-Aldrich)
in PBS for 10 min at room temperature. Fixed cells were washed with
PBS, and stained with 4 μg/ml Hoechst 33342 for 20 min at
room temperature. The stained cells were washed twice with PBS and
analyzed by a fluorescent microscope.
Cell cycle analyses by flow
cytometry
Cells were harvested with trypsinization and washed
once with PBS. After centrifugation, the cells were fixed in 70%
ethanol at −20°C overnight. Fixed cells were prepared for flow
cytometry analysis by washing twice with PBS and then stained in
propidium iodide (PI) (Sigma-Aldrich) solution (50 μg/ml in
PBS) for 30 min at room temperature in the dark. Flow cytometry
analysis was performed on a Cytomic FC500 (Beckman Coulter,
Istanbul, Turkey).
Western blot analysis
After apigenin treatment, cells were harvested and
washed with cold PBS. Total cells were lysed in lysis buffer [40 mM
Tris (pH 8.0), 120 mMNaCl, 0.5% NP-40, 2 μg/ml aprotinin, 2
μg/ml leupeptine and 100 μg/ml phenymethylsulfonyl
fluoride (PMSF)]. After centrifugation, the supernatant was
collected and protein concentration was determined by protein assay
reagents (Bio-Rad, Hercules, CA, USA). Equal amount of protein
extracts were denatured by boiling at 100°C for 5 min in sample
buffer (Bio-Rad). The proteins were separated on subject to 6–15%
SDS-PAGE and transferred to polyvinylidenedi fluoride (PVDF)
membrane. The membranes were blocked with 5% non-fat dry milk in
Tris-buffered saline with Tween-20 buffer (TBS-T) (20 mM Tris, 100
mM NaCl, pH 7.5 and 0.1% Tween-20) for 1 h at room temperature. The
membranes were washed 10 min, 4 times with TBS-T buffer each for 10
min and incubated for 1 h with horseradish peroxidase-conjugated
secondary antibodies (Santa Cruz Biotechnology). The membranes were
washed once for 10 min, 4 times with TBS-T buffer. Antigen-antibody
complex were detected by the enhanced chemiluminescence (ECL)
detection system (GE Healthcare, Piscataway, NJ, USA).
Annexin V staining
The Annexin V-FITC is used to quantitatively
determine the percentage of cells within a population that are
actively undergoing apoptosis. Cells harvested were washed twice
with cold PBS, suspended the cells in 1X binding buffer (BD
Biosciences, San Diego, CA, USA). The counted cells were stained in
PI and Annexin V-FITC solution (BD Pharmingen FITC Annexin V
Apoptosis Detection kit, BD Biosciences) for 15 min at room
temperature in the dark, and then, the stained cells were analyzed
by flow cytometry.
Detection of autophagy with acridine
orange staining
The formation of acidic vesicular organelles (AVOs)
is a well-known feature of autophagy. Cells were seeded in 6-well
culture plate and incubated at 37°C for 24 h. Then, cells were
treated with different concentration of apigenin (0–50 μM)
for 24 h, and stained with acridine orange (1 μg/ml) for 15
min. Subsequently, cells were washed with PBS and examined under a
fluorescent microscope. Depending on intracellular acidity,
autophagic lysosomes appeared as orange/red fluorescent cytoplasmic
vesicles, while the nuclei were displayed bright green.
Alternatively, to quantify the development of AVOs,
apigenin-treated cells were stained with acridine orange (1
μg/ml) for 15 min, trypsinized, and then washed with PBS.
The stained cells were then analyzed using a flow cytometer.
GFP-LC3 assay
HCT116 cells were transfected with LC3-GFP plasmid
using Lipofectamine 2000 reagent (Invitrogen, Grand Island, NY,
USA) according to the manufacturer’s instructions. Cells were
treated with 3-MA (5 mM), apigenin (25 μM) or apigenin +
3-MA for 24 h and the formation of punctuate LC3-positive
structures was observed by confocal microscopy.
Statistical analysis
Results are expressed as the mean ± SD of three
separate experiments and analyzed by Student’s t-test, and
considered significantly different at p<0.05 or p<0.01.
Results
Apigenin suppresses the growth of HCT116
cells
To investigate the growth inhibitory effect of
apigenin (Fig. 1A), the MTT assay
was performed in HCT116 cells. As shown in Fig. 1B, apigenin inhibited effectively
cell proliferation of HCT116 cells in a concentration-dependent
manner. The lower concentrations of apigenin (6.25 μM) did
not affect cell viability; however, the higher concentrations (25
and 50 μM) of apigenin significantly reduced the cell
viability of HCT116 cells. Additional experiment was done to
confirm the growth inhibitory activity of apigenin on HCT116 cells
when analyzed by microscopic observations. As shown in Fig. 1C, cells treated with apigenin
displayed distinct morphological changes compared with untreated
cells. In particular, cells were rounded and cell number was
decreased in a concentration-dependent manner after apigenin
treatment.
Apigenin modulates cell cycle progression
in HCT116 cells
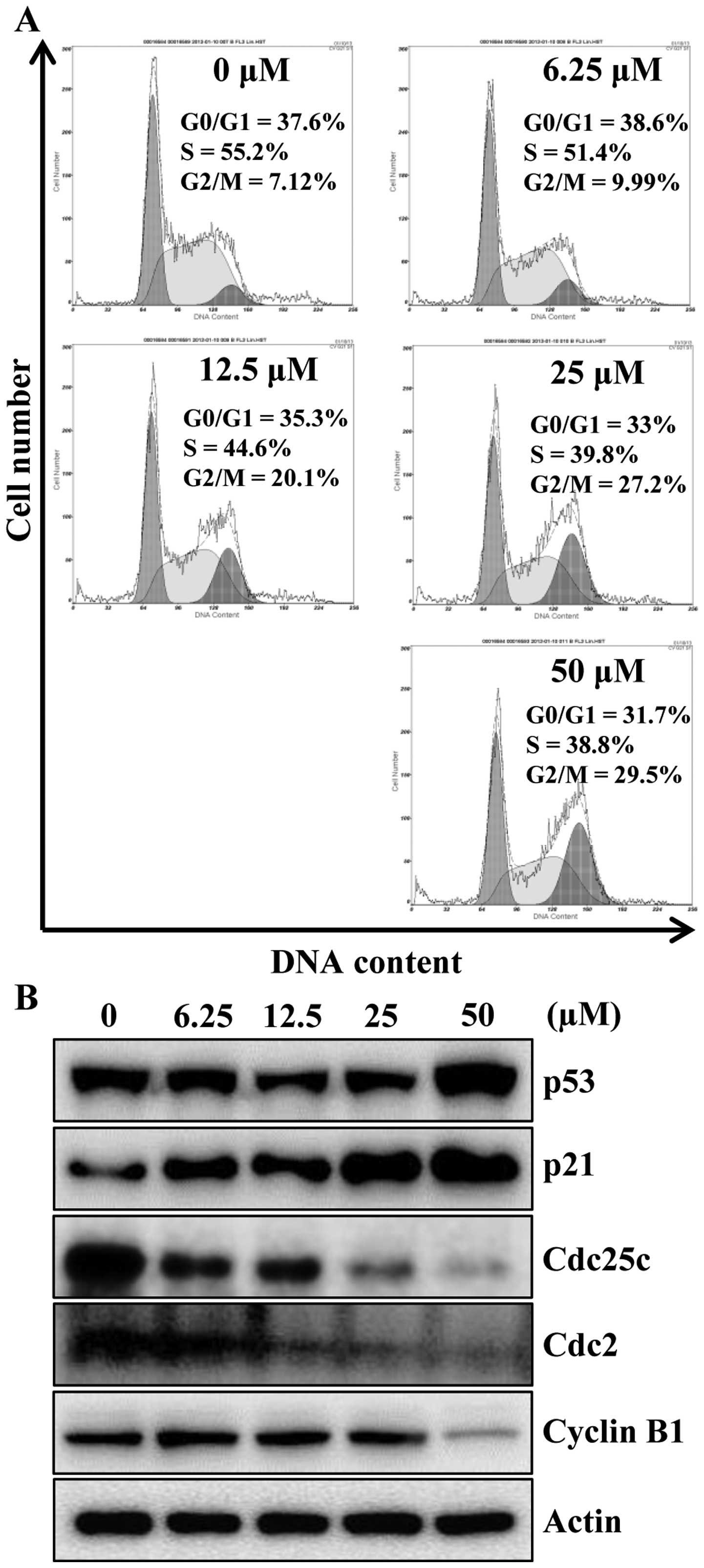
To identify the mechanism responsible for
apigenin-induced cell growth inhibition, cell cycle progression was
evaluated by flow cytometry analysis, as shown in Fig. 2A, apigenin treatment caused a
significant cell cycle arrest in the G2/M phase
concentration-dependently. An accumulation of cells in G2/M phase
of 9.99, 20.1, 27.2 and 29.5% was observed for 6.25, 12.5, 25 and
50 μM, respectively, when compared with untreated cells
(7.12%). Next, we examined whether apigenin can modulate the
expression of G2/M cell cycle regulators. Cells were treated with
various concentrations of apigenin for 24 h and then the level of
G2/M cell cycle regulatory proteins were examined by western blot
analysis. As indicated in Fig. 2B,
after the cells were exposed to apigenin, the levels of p53 and its
downstream protein p21CIP1/WAF1, a potent
cyclin-dependent kinase (CDK) inhibitor in G1 and G2/M phases, were
increased in a concentration-dependent manner. Therefore, in
agreement with previous findings (Fig.
2A), apigenin treatment decreased Cdc25c and its regulatory
protein Cdc2 as well as cyclin B1 (Fig. 2B). These data suggest the
possibility that apigenin-induced growth inhibition of HCT116 cells
was the result of G2/M arrest.
Apigenin induces apoptosis in HCT116
cells
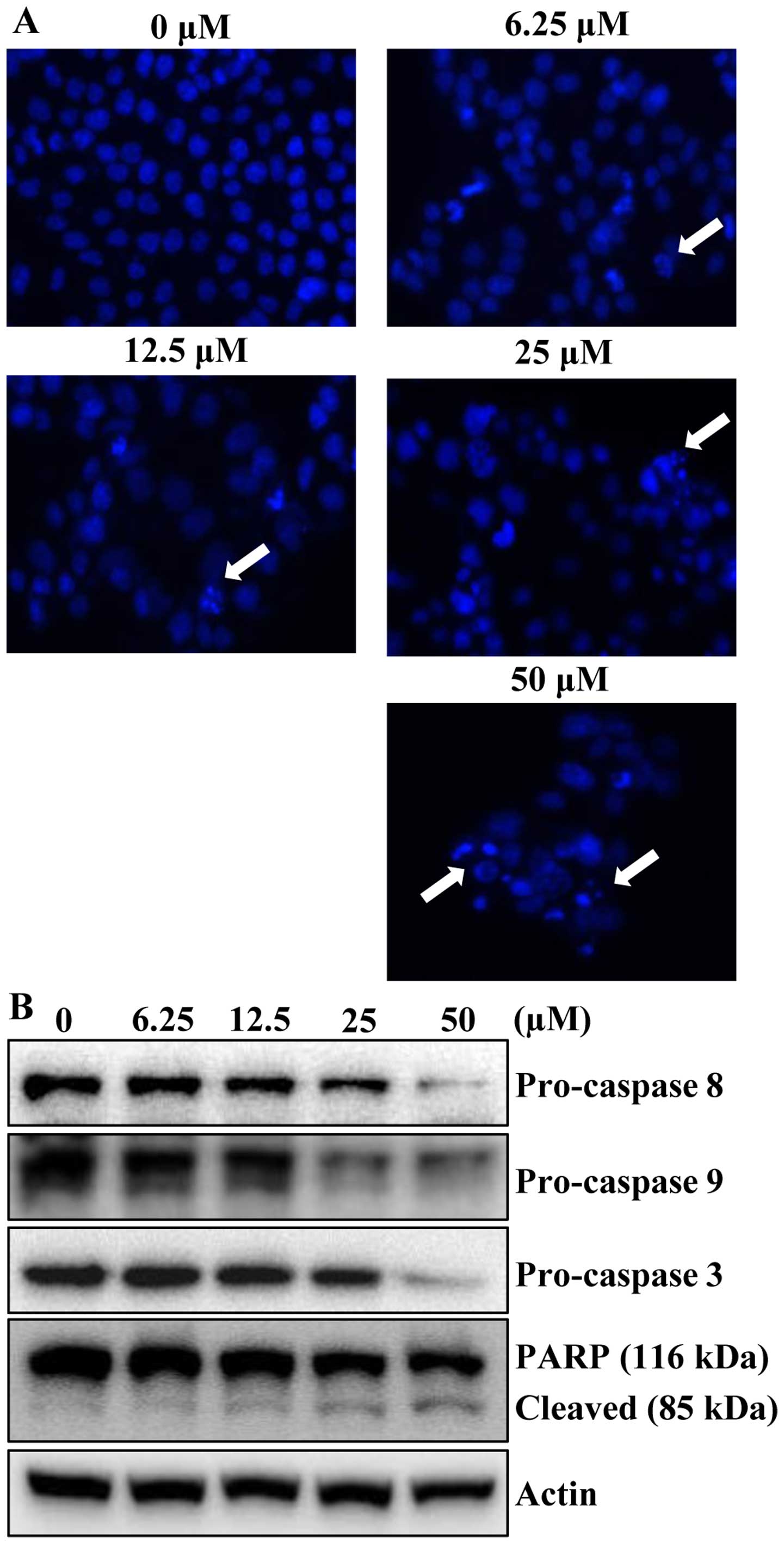
To determine whether the growth inhibitory effects
of apigenin were due to apoptotic cell death, the morphological
changes of cellular structures were assessed with Hoechst 33342
staining. The untreated control cells displayed intact nuclear
structure, while cells were treated with apigenin resulted in
chromosomal condensation and formation of apoptotic bodies, which
are characteristics of programmed cell death (Fig. 3A, arrows). The effect of apigenin
on apoptosis was further verified by western blot analysis.
Apigenin treatment decreased the protein levels of procaspase-8, -9
and -3 in concentration-dependently. In accordance with the
decrease of procaspases with consequent increase of the levels of
active forms of caspases, the cleavage of PARP was increased in a
concentration-dependent manner (Fig.
3B). These results suggested that apigenin induced apoptotic
cell death in HCT116 cells.
Apigenin induces autophagy in HCT116
cells
Autophagy, an evolutionally conserved self-digestive
process, is known as a cell survival mechanism during nutrient
depletion and is essential to cellular homeostasis maintenance by
facilitating the disposal of unfolded proteins and cellular
constitutes (25). It has been
reported that inhibition of autophagy has potential to promote
apoptosis induced by several anticancer agents, which suggested
that autophagy may play a protective role in cancer cells (26,27).
Recently, apigenin has been reported to induce autophagy in human
breast cancer cells (28). To
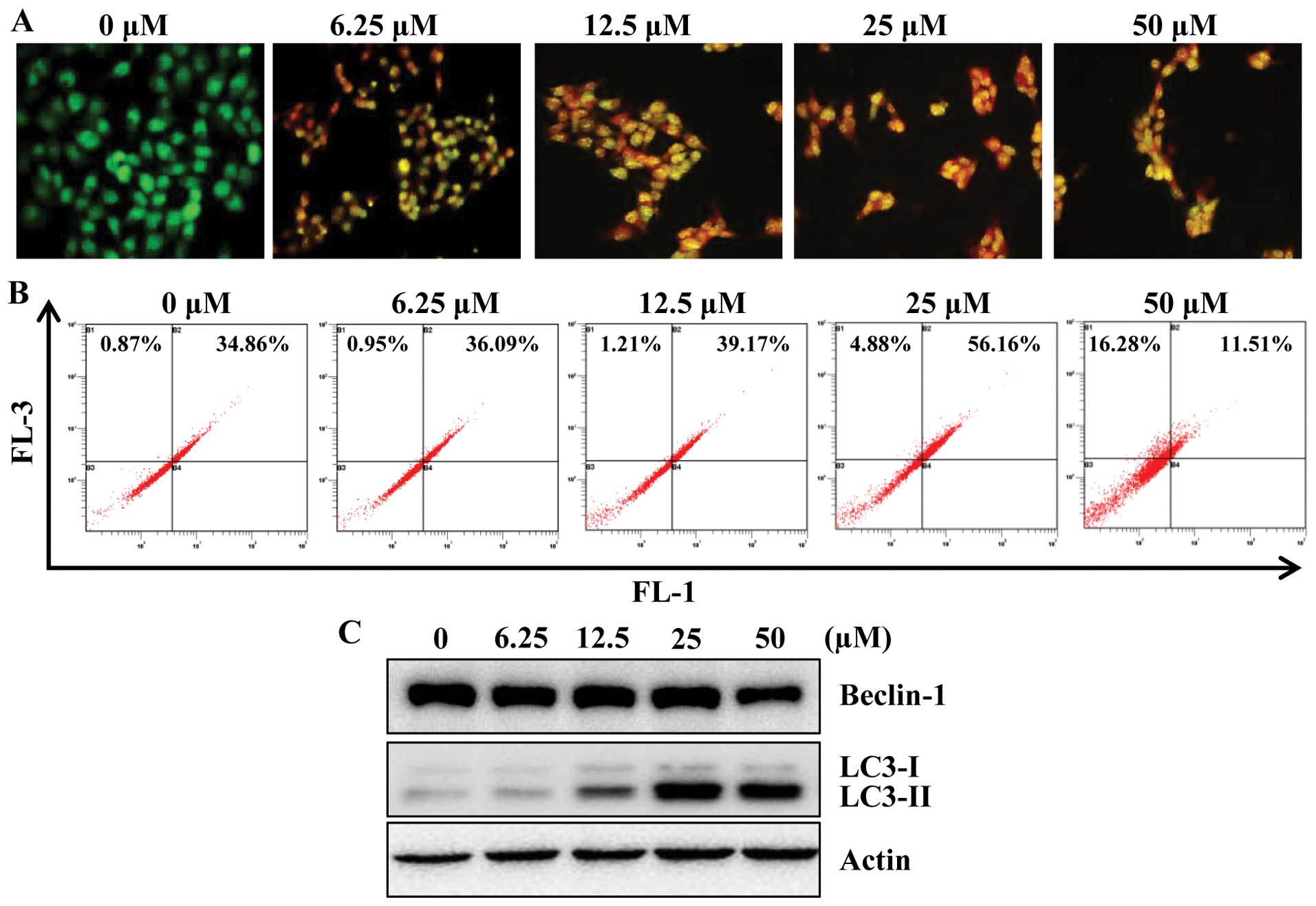
determine whether apigenin induces autophagy in HCT116 cells, we
examined the effect of apigenin on the formation of acidic
vesicular organelles (AVOs) in HCT116 cells. For this, we used
acridine orange (AO), which accumulates in acidic cell compartments
and displays bright red fluorescence (29). The formation of AVOs can be
detected by fluorescence microscopy and quantified by flow
cytometry. As shown in Fig. 4A,
apigenin treatment caused progressive increase of AVO formation in
HCT116 cells. In agreement with microscopic observation, flow
cytometry analysis also showed a concentration-responsive increase
in AVOs in apigenin-treated cells as compared with their respective
untreated control cells (Fig.
4B).
It is known that the beclin-1 level and LC3
conversion (LC3-I to LC3-II) are selective markers of autophagy. As
shown in Fig. 4C, apigenin
treatment markedly increased the expression of LC3-II, suggestive
of autophagy induction. However, apigenin did not affect the
expression of beclin-1 significantly. To further confirm the
induction of autophagy by apigenin, we utilized fluorescence
microscopy to examine autophagosome formation in HCT116 cells
transiently expressing GFP-LC3 protein. Due to the conversion of
LC3-I (a soluble form) to the LC3-II (a lipidized form), which
associates with the membranes of autophagosomes (30), this conversion can be detected by
observing the formation of punctate structures. Treatment of these
cells for 24 h with apigenin resulted in relocalization of the
GFP-LC3 protein to punctate cytoplasmic dots, indicators of
autophagosome formation. Furthermore, co-treatment with apigenin
and 3-MA, inhibits autophagy at initiation of double membrane
encapsulation, significantly inhibiting GFP-LC3 dot formation in
HCT116 cells (Fig. 4D).
Autophagy inhibition enhances
apigenin-induced apoptosis in HCT116 cells
Accumulating data suggest that inhibition of
autophagy may enhance chemosensitization in human cancer cells
(31). Thus, we determined whether
inhibition of autophagy could potentiate apigenin-induced apoptotic
cell death. For this, we first performed Annexin V/PI staining to
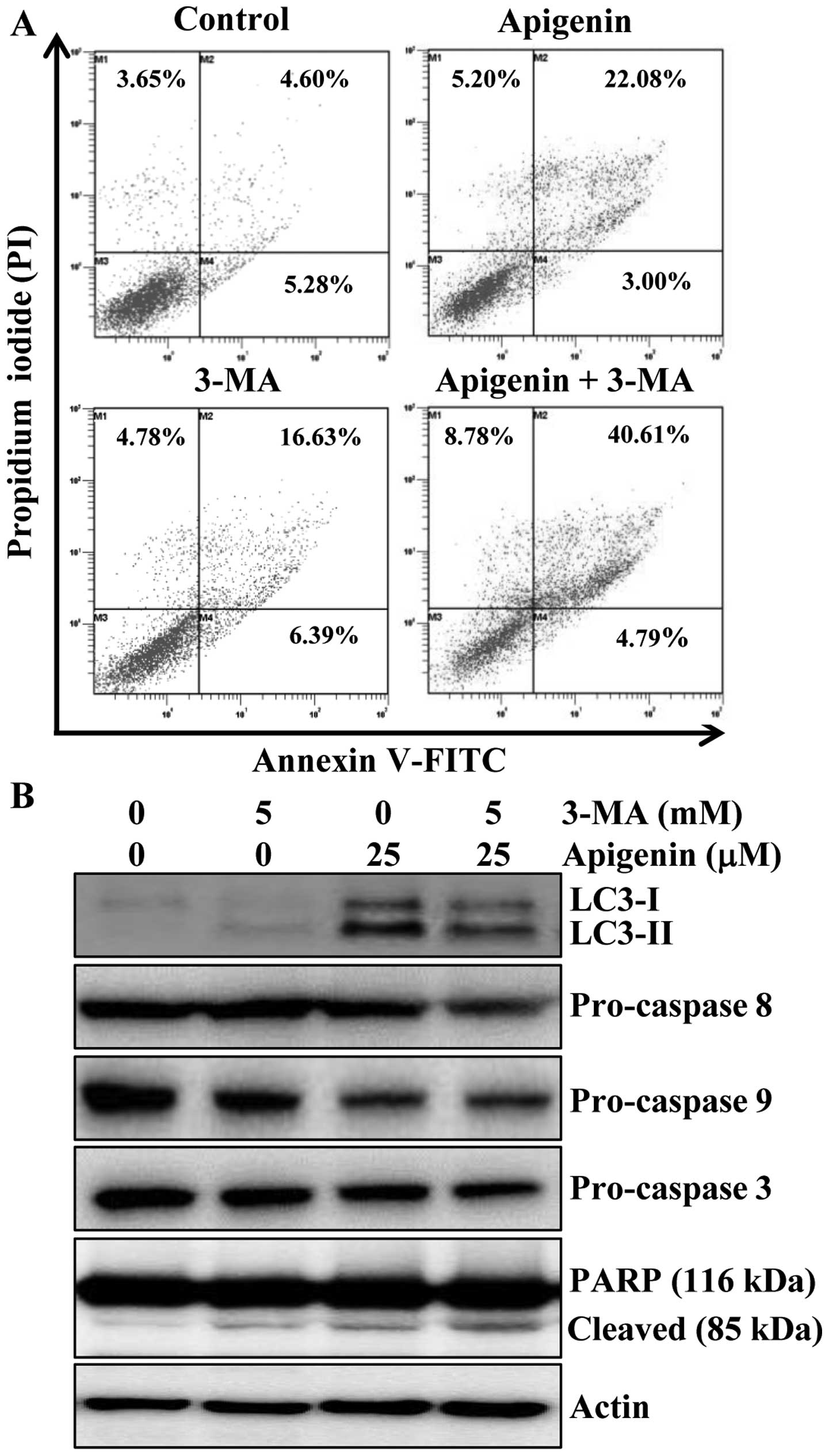
evaluate the effects of 3-MA on apigenin-induced apoptosis. Results
indicated that apoptosis induction was 25% by apigenin, 23% by
3-MA, and 45.3% by the combination of the two agents (Fig. 5A). Furthermore, the alteration in
the protein levels of LC3 and apoptotic proteins procaspase-8, -9
and -3 and PARP cleavage were also observed by western blot
analysis. The protein level of LC3-II was increased by apigenin,
however, addition of 3-MA abrogated the effect of apigenin on
LC3-II induction (Fig. 5B). We
also found that the combination of 3-MA and apigenin resulted in a
significant increase in the level of PARP cleavage and decrease in
procaspase-3, -8 and -9 compared to apigenin treatment alone
(Fig. 5B). Altogether, these
results indicated that inhibition of autophagy by 3-MA enhanced
apigenin-induced cell death in HCT116 cells.
Discussion
Many studies have demonstrated that apigenin
exhibits chemo-preventive effects on various cancer cells. Apigenin
has been reported to act via several mechanisms, including
promotion of cell cycle arrest (1,32),
apoptosis and suppression of signal transduction (33). One of most obvious mechanisms of
apigenin is to induce p53 tumor suppressor protein at the
translational level, followed by p21 induction (34). In the present study, apigenin
effectively suppressed the growth of HCT116 cells by cell cycle
arrest and this growth inhibition was attributed to apoptotic cell
death. Furthermore, an alteration in the ratio of Bax to Bcl-2 has
been proposed to play an important role in anticancer agent-induced
cell death (35). The effect of
apigenin on Bcl-2 family protein is controversial. Several reports
indicated that apigenin could upregulate Bax and down-modulate
Bcl-2 expression in numerous cancer cell lines including lung and
breast cancer (4,28). However, in agreement with the
report of Shao et al (12),
we could not find any significant changes in levels of
pro-apoptotic protein, Bax and anti-apoptotic protein, Bcl-2, in
HCT116 cells (data not shown). The precise reason for this
difference is not clear but cell type and conditions for experiment
used may count for this discrepancy.
We found that apigenin induces autophagy in colon
cancer cells. In the autophagic pathways, there are two molecular
regulation mechanisms, PI3K/protein kinase B (Akt)/mTOR and class
III PI3K signal transduction pathways. In the process of autophagy,
the complex of beclin-1 and class III PI3K-dependent autophagy has
an important function in mediating the localization of
autophagy-related proteins to the preautophagosomal membrane
(15). Autophagy has been shown by
several antineoplastic agents in a great number of cancer cells,
such as cervical carcinoma (36),
glioma (37) or colon cancer cells
(38). Our result showed
progressive increase of LC3-II protein levels compared with the
control cells by apigenin. At 25 μM concentration, apigenin
dramatically increased LC3-II, but higher concentration of apigenin
(50 μM) decreased LC3-II and beclin-1 levels. It is likely
50 μM apigenin induced more cell death than 25 μM.
The knockdown of beclin-1 caused slow cell growth compared with
cells with normal expression of beclin-1 (39). Thus, we observed that apigenin
induced both apoptosis and autophagy. This phenomenon may be
explained by the effect of apigenin on the PI3K/Akt/mTOR pathway.
It has been indicated that the anticancer effect of apigenin is
related to the inhibition of the PI3K/Akt/mTOR pathway (32,40),
which is also an essential pathway that negatively regulates
autophagy (41).
Furthermore, we found that autophagy inhibition
using 3-MA pronounced apigenin-induced cell death. Adjusting
autophagy appropriately may increase the cytotoxicity of anticancer
drugs in tumor cells (42). A
recent report indicated that 3-MA worked well with chemotherapeutic
drugs by triggering apoptosis in some cancer cells (39). For example, autophagy inhibition by
using 3-MA augmented chemotherapeutic effect of 5-FU in several
in vitro models of human colon cancer (43). Therefore, we hypothesized that
treatment of autophagy inhibitors might sensitize HCT116 cells to
apigenin chemotherapy by improving the rate of apoptosis cell death
induced by apigenin or by converting the autophagy process to an
apoptotic process. It has been indicated that, inhibition of
autophagy by 3-MA enhances the apoptosis in many cancer cells. The
mitochondrial apoptotic pathway is a relatively important death
receptor pathway for the induction of apoptosis by chemotherapeutic
drugs (24).
In conclusion, apigenin suppressed growth of HCT116
cells in a concentration-dependent manner by causing G2/M cell
cycle arrest. In addition, apigenin-treated cells exhibited
inductions of apoptosis and autophagy. The inhibition of autophagy
by 3-MA enhanced the apoptosis induced by apigenin through
activations of pro-caspases-8, -9 and -3 and cleavage of PARP.
These results provide evidence that inhibition of autophagy may be
an effective way to improve the chemotherapy of anti-cancer agents
against human colon cancer.
Acknowledgements
This research was supported by Basic
Science Research Program through the National Research Foundation
of Korea (NRF) funded by the Korea government (MSIP)
(NRF-2009-0071649). This study was also financially supported by
the National Research Foundation of Korea (NRF) grant funded by the
Korea government (MSIP) (no. 2009-0083538). We thank Aging Tissue
Bank for providing research information. The authors thank Mr.
Kyoung-Pil Lee (Pusan National University) for helping with
confocal microscopy.
References
|
1.
|
Wang W, Heideman L, Chung CS, Pelling JC,
Koehler KJ and Birt DF: Cell-cycle arrest at G2/M and growth
inhibition by apigenin in human colon carcinoma cell lines. Mol
Carcinog. 28:102–110. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
2.
|
Wu DG, Yu P, Li JW, et al: Apigenin
potentiates the growth inhibitory effects by IKK-beta-mediated
NF-kappaB activation in pancreatic cancer cells. Toxicol Lett.
224:157–164. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Maggioni D, Garavello W, Rigolio R,
Pignataro L, Gaini R and Nicolini G: Apigenin impairs oral squamous
cell carcinoma growth in vitro inducing cell cycle arrest
and apoptosis. Int J Oncol. 43:1675–1682. 2013.PubMed/NCBI
|
|
4.
|
Das S, Das J, Samadder A, Boujedaini N and
Khuda-Bukhsh AR: Apigenin-induced apoptosis in A375 and A549 cells
through selective action and dysfunction of mitochondria. Exp Biol
Med (Maywood). 237:1433–1448. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Jayasooriya RG, Kang SH, Kang CH, et al:
Apigenin decreases cell viability and telomerase activity in human
leukemia cell lines. Food Chem Toxicol. 50:2605–2611. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
McVean M, Weinberg WC and Pelling JC: A
p21(waf1)-independent pathway for inhibitory phosphorylation of
cyclin-dependent kinase p34(cdc2) and concomitant G(2)/M arrest by
the chemopreventive flavonoid apigenin. Mol Carcinog. 33:36–43.
2002. View
Article : Google Scholar
|
|
7.
|
Zhu Y, Mao Y, Chen H, et al: Apigenin
promotes apoptosis, inhibits invasion and induces cell cycle arrest
of T24 human bladder cancer cells. Cancer Cell Int. 13:542013.
View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Kim SH, Kang JG, Kim CS, et al: Apigenin
induces c-Myc-mediated apoptosis in FRO anaplastic thyroid
carcinoma cells. Mol Cell Endocrinol. 369:130–139. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Mirzoeva S, Kim ND, Chiu K, Franzen CA,
Bergan RC and Pelling JC: Inhibition of HIF-1 alpha and VEGF
expression by the chemopreventive bioflavonoid apigenin is
accompanied by Akt inhibition in human prostate carcinoma PC3-M
cells. Mol Carcinog. 47:686–700. 2008. View
Article : Google Scholar
|
|
10.
|
Horinaka M, Yoshida T, Shiraishi T, Nakata
S, Wakada M and Sakai T: The dietary flavonoid apigenin sensitizes
malignant tumor cells to tumor necrosis factor-related
apoptosis-inducing ligand. Mol Cancer Ther. 5:945–951. 2006.
View Article : Google Scholar
|
|
11.
|
Xu Y, Xin Y, Diao Y, et al: Synergistic
effects of apigenin and paclitaxel on apoptosis of cancer cells.
PLoS One. 6:e291692011. View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Shao H, Jing K, Mahmoud E, Huang H, Fang X
and Yu C: Apigenin sensitizes colon cancer cells to antitumor
activity of ABT-263. Mol Cancer Ther. 12:2640–2650. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Chan LP, Chou TH, Ding HY, et al: Apigenin
induces apoptosis via tumor necrosis factor receptor- and
Bcl-2-mediated pathway and enhances susceptibility of head and neck
squamous cell carcinoma to 5-fluorouracil and cisplatin. Biochim
Biophys Acta. 1820:1081–1091. 2012. View Article : Google Scholar
|
|
14.
|
Mizushima N: Autophagy: process and
function. Genes Dev. 21:2861–2873. 2007. View Article : Google Scholar
|
|
15.
|
Shintani T and Klionsky DJ: Autophagy in
health and disease: a double-edged sword. Science. 306:990–995.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Xie Z and Klionsky DJ: Autophagosome
formation: core machinery and adaptations. Nat Cell Biol.
9:1102–1109. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Liang XH, Jackson S, Seaman M, et al:
Induction of autophagy and inhibition of tumorigenesis by beclin 1.
Nature. 402:672–676. 1999. View
Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Vivanco I and Sawyers CL: The
phosphatidylinositol 3-kinase AKT pathway in human cancer. Nat Rev
Cancer. 2:489–501. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Ng G and Huang J: The significance of
autophagy in cancer. Mol Carcinog. 43:183–187. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Gozuacik D and Kimchi A: Autophagy as a
cell death and tumor suppressor mechanism. Oncogene. 23:2891–2906.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
21.
|
Ferlay J, Shin HR, Bray F, Forman D,
Mathers C and Parkin DM: Estimates of worldwide burden of cancer in
2008: GLOBOCAN 2008. Int J Cancer. 127:2893–2917. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Jung KW, Park S, Kong HJ, et al: Cancer
statistics in Korea: incidence, mortality, survival, and prevalence
in 2009. Cancer Res Treat. 44:11–24. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Shin A, Kim KZ, Jung KW, et al: Increasing
trend of colorectal cancer incidence in Korea, 1999–2009. Cancer
Res Treat. 44:219–226. 2012.PubMed/NCBI
|
|
24.
|
Huerta S, Goulet EJ and Livingston EH:
Colon cancer and apoptosis. Am J Surg. 191:517–526. 2006.
View Article : Google Scholar
|
|
25.
|
Kundu M and Thompson CB: Autophagy: basic
principles and relevance to disease. Annu Rev Pathol. 3:427–455.
2008. View Article : Google Scholar
|
|
26.
|
Lin JF, Tsai TF, Liao PC, et al: Benzyl
isothiocyanate induces protective autophagy in human prostate
cancer cells via inhibition of mTOR signaling. Carcinogenesis.
34:406–414. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
27.
|
Cheng P, Ni Z, Dai X, et al: The novel
BH-3 mimetic apogossypolone induces Beclin-1- and ROS-mediated
autophagy in human hepatocellular carcinoma cells. Cell Death Dis.
4:e4892013. View Article : Google Scholar : PubMed/NCBI
|
|
28.
|
Cao X, Liu B, Cao W, et al: Autophagy
inhibition enhances apigenin-induced apoptosis in human breast
cancer cells. Chin J Cancer Res. 25:212–222. 2013.PubMed/NCBI
|
|
29.
|
Paglin S, Hollister T, Delohery T, et al:
A novel response of cancer cells to radiation involves autophagy
and formation of acidic vesicles. Cancer Res. 61:439–444.
2001.PubMed/NCBI
|
|
30.
|
Klionsky DJ, Abeliovich H, Agostinis P, et
al: Guidelines for the use and interpretation of assays for
monitoring autophagy in higher eukaryotes. Autophagy. 4:151–175.
2008. View Article : Google Scholar
|
|
31.
|
Carew JS, Nawrocki ST and Cleveland JL:
Modulating autophagy for therapeutic benefit. Autophagy. 3:464–467.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
32.
|
Shukla S and Gupta S: Apigenin-induced
cell cycle arrest is mediated by modulation of MAPK, PI3K-Akt, and
loss of cyclin D1 associated retinoblastoma dephosphorylation in
human prostate cancer cells. Cell Cycle. 6:1102–1114. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
33.
|
Van Dross RT, Hong X and Pelling JC:
Inhibition of TPA-induced cyclooxygenase-2 (COX-2) expression by
apigenin through downregulation of Akt signal transduction in human
keratinocytes. Mol Carcinog. 44:83–91. 2005.PubMed/NCBI
|
|
34.
|
King JC, Lu QY, Li G, et al: Evidence for
activation of mutated p53 by apigenin in human pancreatic cancer.
Biochim Biophys Acta. 1823:593–604. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
35.
|
Adams JM and Cory S: The Bcl-2 apoptotic
switch in cancer development and therapy. Oncogene. 26:1324–1337.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
36.
|
Harhaji-Trajkovic L, Vilimanovich U,
Kravic-Stevovic T, Bumbasirevic V and Trajkovic V: AMPK-mediated
autophagy inhibits apoptosis in cisplatin-treated tumour cells. J
Cell Mol Med. 13:3644–3654. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
37.
|
Nishikawa T, Tsuno NH, Okaji Y, et al:
Inhibition of autophagy potentiates sulforaphane-induced apoptosis
in human colon cancer cells. Ann Surg Oncol. 17:592–602. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
38.
|
Galluzzi L, Aaronson SA, Abrams J, et al:
Guidelines for the use and interpretation of assays for monitoring
cell death in higher eukaryotes. Cell Death Differ. 16:1093–1107.
2009. View Article : Google Scholar
|
|
39.
|
Fang J, Xia C, Cao Z, Zheng JZ, Reed E and
Jiang BH: Apigenin inhibits VEGF and HIF-1 expression via
PI3K/AKT/p70S6K1 and HDM2/p53 pathways. FASEB J. 19:342–353. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
40.
|
Kim J, Kundu M, Viollet B and Guan KL:
AMPK and mTOR regulate autophagy through direct phosphorylation of
Ulk1. Nat Cell Biol. 13:132–141. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
41.
|
Shingu T, Fujiwara K, Bogler O, et al:
Inhibition of autophagy at a late stage enhances imatinib-induced
cytotoxicity in human malignant glioma cells. Int J Cancer.
124:1060–1071. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
42.
|
Li J, Hou N, Faried A, Tsutsumi S and
Kuwano H: Inhibition of autophagy augments 5-fluorouracil
chemotherapy in human colon cancer in vitro and in vivo model. Eur
J Cancer. 46:1900–1909. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
43.
|
Li J, Hou N, Faried A, Tsutsumi S,
Takeuchi T and Kuwano H: Inhibition of autophagy by 3-MA enhances
the effect of 5-FU-induced apoptosis in colon cancer cells. Ann
Surg Oncol. 16:761–771. 2009. View Article : Google Scholar : PubMed/NCBI
|