Introduction
Colorectal cancer (CRC) is one of the most frequent
cancers in Western countries, accounting for more than 10% of all
cancer deaths (1). Survival of CRC
has been shown to be highly dependent on the stage of the disease
at the time of diagnosis (2,3).
Several methods of colorectal neoplasm screening are currently
available, including colonoscopy (4), barium enema (BE) (5) and fecal occult blood testing (FOBT)
(6,7). These methods have several
disadvantages for population-based screening, which include
invasiveness, relative high cost, frequent false-positive results
or the requirement of expert endoscopists. For example,
colonoscopy, which remains the gold standard for identification of
neoplasia (8), is unsuited for
mass screening due to its invasiveness and requirement of expert
endoscopists. FOBT is widely used as an initial screening method
for colorectal tumors, however, it is not a robust assay because
false-positive results are frequent (9). An ideal CRC-screening method, which
has a high sensitivity and specificity for the target pathology at
a curable stage, is therefore needed.
The Kirsten RAS (KRAS) is the most frequently
mutated proto-oncogene that is critical for tumor progression.
Activating mutations of KRAS in colorectal tumorigenesis are
thought to alter GTPase activity, leading to unregulated cellular
proliferation and malignant transformation (10–12).
KRAS mutations occur early in the colon tumorigenesis
pathway, hence, detection of KRAS mutations would be
beneficial for early diagnosis, prognosis and evaluation of a
therapeutic outcome in cancer treatment (13,14).
KRAS is an effector molecule of epidermal growth factor
receptor (EGFR), which is a key target of therapeutic strategies
designed to treat metastatic CRC (15). Patients harboring KRAS
mutations in codon 12 or 13 usually do not derive benefit from
anti-EGFR treatment (16,17). However, recent studies indicate
that patients who have KRAS condon 12- or KRAS
13-mutated tumors can respond to anti-EGFR treatment, and the
survival of these patients with KRAS mutations correlates
with anti-EGFR therapy in some cases (18–20).
The American Society of Clinical Oncology has recommended recently
that all patients with metastatic colorectal carcinoma should have
their tumor tested for KRAS mutations before anti-EGFR
therapy with cetuximab or panitumumab (21).
Various methods have been devised to identify
KRAS gene mutations, such as direct DNA sequencing (22), mutant enriched polymerase chain
reaction (PCR) (23), peptide
nucleic acid (PNA)-based PCR (24), restriction endonuclease-mediated
selective (REMS)-PCR (25),
PCR-restriction fragment length polymorphism (PCR-RFLP) (26), mutation tube assay (MUTA) test
(27). There are a number of
disadvantages in these methods, such as they are not convenient for
use in clinical laboratories owing to multiple procedural
manipulations that are laborious, time-consuming and
cost-ineffective. A PamChip microarray system was reported
previously by Maekawa et al (28), however, target regions of different
genotypes were amplified separately in this approach, which is
inefficient. Recently, a quantitative method termed allele-specific
competitive blocker PCR (ACB-PCR) has been described for detecting
KRAS gene mutations (29).
Nevertheless, this method may need optimization in order to be
applied in clinical screening, because multiple procedures are
required to use this approach.
To establish a reliable technique suitable for
detection of KRAS mutations for colon cancer screening, in
this study we have developed an oligonucleotide-tagged microarray.
Using this approach, we were able to detect KRAS codon 12
mutations in cancer cell lines and clinical samples. The optimized
operating conditions permitted successful detection of homozygous
as well as heterozygous DNA samples. Our results showed that 10% of
mutant DNA could be detected in the WT background, suggesting that
the tag-microarray-based method is suitable for CRC routine
diagnosis.
Materials and methods
Oligonucleotides and cell lines
Oligonucleotides used in this study are summarized
in Table I. All cell lines listed
in Table II were maintained in
our laboratory (National Veterinary Institute, Technical University
of Denmark) or kindly provided by Mogens Kruhøffer (Aarhus
University Hospital, Denmark). Cell lines were classified into four
groups based on the sequence of codon 12 of KRAS gene:
wild-type sequence (GGT) was designated ‘WT’, GTT was designated
‘M1T1’, GAT was designated ‘M2T2’, AGT was designated ‘M3T3’
(Table II). Cell lines HT29 and
CALU -1 were maintained in McCoy’s medium (HyClone); SW480, SW620
and A549 cell lines were cultured in Dulbecco’s modified Eagle’s
medium (DMEM) (Invitrogen); all other cell lines were grown in
RPMI-1640 medium (Invitrogen). All media used for cell culture were
supplemented with 10% fetal calf serum (Invitrogen). Cells were
cultured at 37°C in a humidified 5% CO2 atmosphere. Four
unique tags were selected based on published data (30) after analysis with regard to
cross-priming, melting temperature, percentage of G + C contain,
and possible secondary structure. Four single nucleotide
polymorphism (SNP)-specific forward primers specific to four
genotypes were designed. One of the four tags was linked to the
5′-end of each of these primers, respectively, to form forward
chimeric tagged-primers (Table I
and Fig. 1A). Four oligonucleotide
probes were modified at the 5′-end with a poly (T) 10 - poly (C) 10
- probe binding tail (TC tail) to facilitate the attachment to the
solid substrate, as previously described (31,32).
One of the four unique tag sequences was introduced into the 3′-end
of each probe (Table I). The
reverse primer (Cy5-K-ras-RP2) was labeled with Cy5 fluorescent dye
to facilitate detection (Table I).
Oligonucleotides were synthesized at DNA Technology A/S (Arhus,
Denmark).
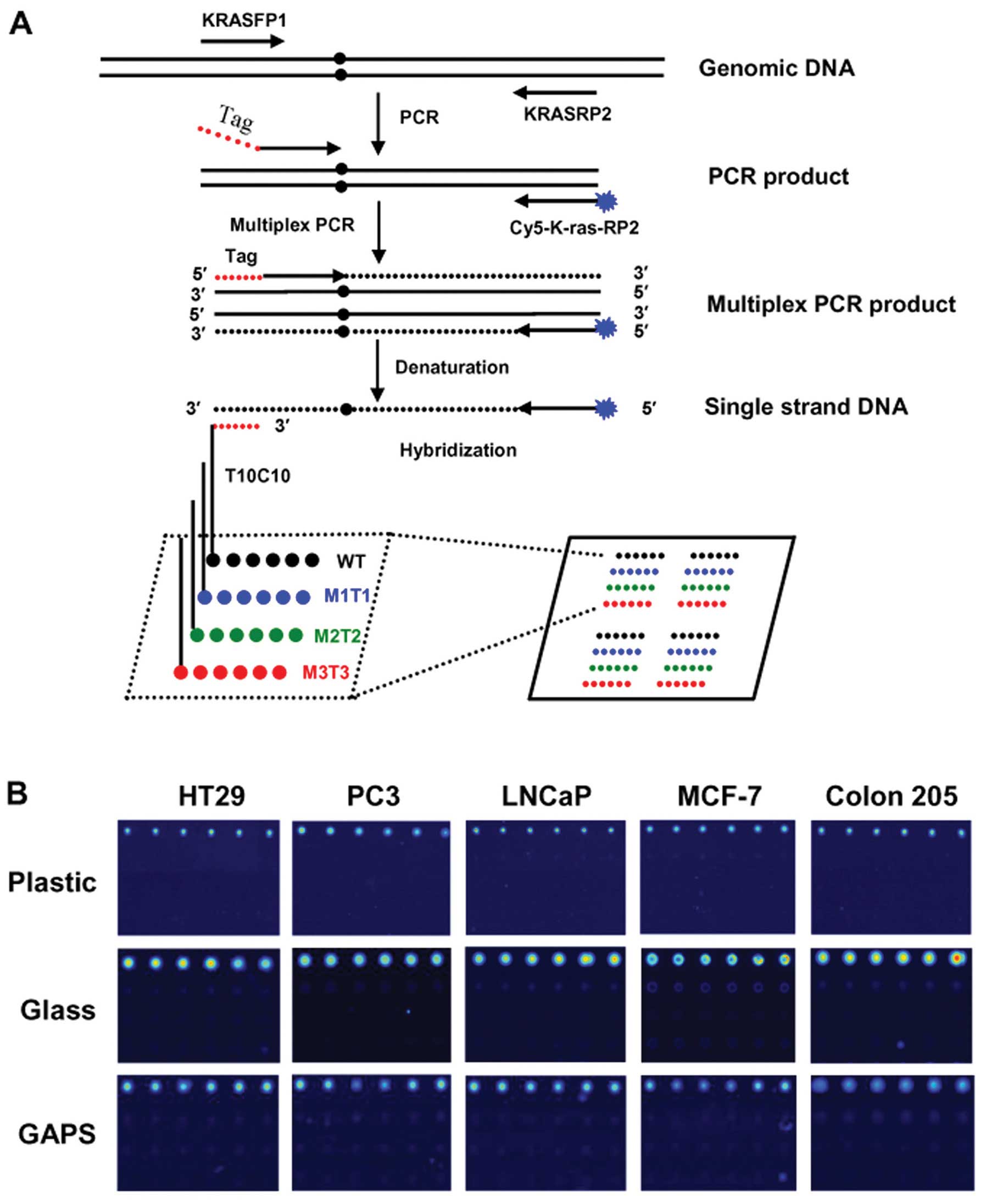 | Figure 1Schematic representation of the
tag-microarray technique and detection of the WT genotypes. (A)
Each slide was spotted with two array areas, each of which was used
for hybridization with one multiplex PCR product. Four kinds of
probes, marked with black, blue, green and red for WT, M1T1, M2T2
and M3T3 genotypes, respectively, were spotted robotically in each
area and each probe was spotted in a row of six identical spots.
Four identical arrays were formed in one area. Fragments spanning
the K-RAS codon 12 ORF were amplified followed by multiplex PCR
using a shared Cy5-labeled reverse primer and forward hybrid
primers which each contains a unique tag sequence and a
locus-specific sequence. The labeled reverse primer was extended to
the 5′-end of the mutation-specific forward primer and was
incorporated into the complementary sequence of the respective tag
(anti-tag). The multiplex PCR product was hybridized to the
immobilized tag probes. To enable immobilization using our simple
UV method, the probes were modified in the 5′-end with poly (T) 10
- poly (C) 10 (31,32). The presence of Cy5 fluorescence
allowed for the detection of specific hybridization. (B)
Representative fluorescence images obtained from hybridization of
WT targets of multiplex PCR product with probes immobilized on
different substrates (plastic, glass, GAPS-II) are shown. |
 | Table IPrimers and probes used in this
study. |
Table I
Primers and probes used in this
study.
| Name | Oligonucleotide
sequences (5′→3′)a | Tm (°C) |
|---|
| KRASFP1 |
ATGACTGAATATAAACTTGT | 35.0 |
| KRASRP2 |
CTCTATTGTTGGATCATATT | 38.1 |
| Tag-K-ras-WT |
ggttctgttcttcgttgacatgaggTGTGGTAGTTGGAGCTGG | 76.7 |
| Tag-K-ras-M1T1 |
gcagaactgatgagcgatccgaata
TGTGGTAGTTGGAGCTGT | 76.2 |
| Tag-K-ras-M2T2 |
aatgatgctctgcgtgatgatgttg
TTGTGGTAGTTGGAGCTGA | 77.6 |
| Tag-K-ras M3T3 |
gcggaacggtcagagagattgatgt
AACTTGTGGTAGTTGGAGCTA | 76.0 |
| Tag-T10C10-WT |
TTTTTTTTTTCCCCCCCCCCggttctgttcttcgttgacatgagg | 81.0 |
|
Tag-T10C10-M1T1 |
TTTTTTTTTTCCCCCCCCCCgcagaactgatgagcgatccgaata | 81.8 |
|
Tag-T10C10-M2T2 |
TTTTTTTTTTCCCCCCCCCCaatgatgctctgcgtgatgatgttg | 81.7 |
|
Tag-T10C10-M3T3 |
TTTTTTTTTTCCCCCCCCCCgcggaacggtcagagagattgatgt | 82.5 |
| Cy5-Tag-WT |
Cy5-ggttctgttcttcgttgacatgagg | 57.8 |
| Cy5-Tag-M1T1 |
Cy5-gcagaactgatgagcgatccgaata | 61.1 |
| Cy5-Tag-M2T2 |
Cy5-aatgatgctctgcgtgatgatgttg | 59.9 |
| Cy5-Tag-M3T3 |
Cy5-gcggaacggtcagagagattgatgt | 61.4 |
|
Anti-Tag-Prob-WT |
TTTTTTTTTTCCCCCCCCCCCCTCATGTCAACGAAGAACAGAACC | 69.4 |
|
Anti-Tag-Prob-M1T1 |
TTTTTTTTTTCCCCCCCCCCCTATTCGGATCGCTCATCAGTTCTGC | 69.1 |
|
Anti-Tag-Prob-M2T2 |
TTTTTTTTTTCCCCCCCCCCCAACATCATCACGCAGAGCATCATT | 69.4 |
|
Anti-Tag-Prob-M3T3 |
TTTTTTTTTTCCCCCCCCCCACATCAATCTCTCTGACCGTTCCGC | 69.5 |
| Anti-P2-T10C10 |
TTTTTTTTTTCCCCCCCCCCTTATTCAGAATCATTTTGTGGACGAA
TATGATCCAACAATAGAG | 81.7 |
| Cy5-K-ras-RP2 |
Cy5-CTCTATTGTTGGATCATATTCGTCCACAAAATGATTCTGAATTAG | 69.6 |
| Table IICell lines used in this study. |
Table II
Cell lines used in this study.
| Cell line | Histology | Mutation | Sequence type | Genotype |
|---|
| HT29 | Human colon
adenocarcinoma | GGT | Homozygous | WT |
| PC3 | Human prostate
adenocarcinoma | GGT | Homozygous | WT |
| LNCaP | Human prostate
carcinoma | GGT | Homozygous | WT |
| Colon205 | Human colon
adenocarcinoma | GGT | Homozygous | WT |
| MCF-7 | Human breast
carcinoma | GGT | Homozygous | WT |
| SW480 | Human colon
adenocarcinoma |
GGT→GTTa | Homozygous | M1T1 |
| SW620 | Human colon
adenocarcinoma |
GGT→GTTa | Homozygous | M1T1 |
| LS174T | Human colon
adenocarcinoma |
GGT→GATa | Heterozygous | M2T2 |
| A427 | Human lung
adenocarcinoma |
GGT→GATa | Heterozygous | M2T2 |
| A549 | Human lung
adenocarcinoma | GGT→AGTa | Homozygous | M3T3 |
| CALU -1 | Human lung
adenocarcinoma | GGT→AGTa | Homozygous | M3T3 |
Polymerase chain reaction (PCR) and
multiplex PCR
Genomic DNA from all cell lines (Table II) was isolated using QIAamp DNA
Mini kit (Qiagen, Hilden, Germany). A 111-bp fragment encompassing
codon 12 of KRAS gene was amplified by PCR using primers
KRASFP1 and KRASRP2 (Table I). PCR
was performed in a 20 μl reaction mixture containing 10 μl
HotStarTaq Plus Master Mix buffer (Qiagen), 2.5 mM
MgCl2, 20 μM forward and reverse primers and 10 ng
extracted DNA. The reaction mixtures were subjected to
amplification on PTC-200 Peltier Thermal Cycler (MJ Research,
Watertown, NY, USA) with initial step at 95°C for 5 min, followed
by 30 cycles with denaturation at 95°C for 40 sec, annealing at
55°C for 40 sec and extension at 72°C for 30 sec. Final extension
was at 72°C for 10 min. The PCR products were purified by QIAquick
PCR purification kit (Qiagen) and were sequenced directly to
determine the altered nucleotide of KRAS gene in codon 12.
Equal amounts of four forward ‘tag-primers’ (Table I) were mixed thoroughly and used
for multiplex PCR. The multiplex PCR was carried out in a total
volume of 20 μl containing the following: 10 μl HotStarTag Plus
Master Mix, 2.5 mM MgCl2, 50 pg purified PCR product,
0.05 pM of mixed forward primer and 2.5 pM of reverse primer
Cy5-K-ras-RP2 (Table II). PCR was
performed with initial step at 95°C for 5 min, followed by 30
cycles with denaturation at 95°C for 40 sec, annealing at 68°C for
40 sec and extension at 72°C for 50 sec. Final extension at 72°C
for 10 min.
Preparation of the tag-microarray and
hybridization
Topas plastic microscope slides (Microfluidic,
Germany) were treated with 95% ethanol for 1 h and then rinsed with
sterile water and dried at room temperature before use. Super Frost
glass microscope slides were purchased from Menzel (Braunschweig,
Germany). GAPS-II coated glass slides were purchased from Corning
(NY, USA). Four DNA probes (Table
I) modified at the 5′-end with TC tail (31) were diluted in 150 mM sodium
phosphate buffer (pH 8.5) to a final concentration of 50 pM, and
deposited in a volume of 10 nl/spot onto the slides using Q-Array
robot spotter (Genetix, UK).
The DNA probes were linked to the solid support
using a simple one-step method previously described (31,32).
After UV irradiation, the slides were washed with agitation in
filtered 0.2% (w/v) sodium dodecyl sulfate (SDS) for 5 min followed
by 0.1X saline-sodium citrate (SSC, 300 mM NaCl, 30 mM sodium
citrate, pH 7.0) for 5 min and then spin-dried.
Cy5-labeled multiplex PCR products (without any
additional post-PCR manipulation steps) were mixed with PerfectHyb
Plus hybridization buffer (Sigma). Single strand DNA was obtained
by boiling the mixture for 5 min followed by incubation in
ice-water for 2 min. The solution was transferred immediately onto
the surface of the slide. The microarrays were hybridized under a
glass coverslip for 1 h at 37°C in a humidified chamber. Slides
were washed at room temperature in filtered 0.5 × SDS + 0.1% SSC
for 10 min with agitation, rinsed in water and spin-dried. The
microarrays were scanned in a LaVision scanner (LaVision BioTech,
Germany) using appropriate laser power and exposure time suggested
by the manufacturer. The fluorescent intensities of the spots were
quantified using Fips BioAnalyzer 4F/4S software.
Sensitivity of the tag-microarray
assay
Genomic DNA extracted from CALU -1 (M3T3) was mixed
with either Colon205 DNA (WT) or DNA extracted from feces of a
healthy donor at the indicated ratios. The mixed genomic DNA was
used as a template for PCR and hybridization.
Genotyping of clinical tumor samples
Twenty-eight DNA samples from patients with tumors
were used as template for PCR and hybridization as described above
to determine the KRAS codon 12 mutations, which were further
verified by direct DNA sequencing.
Thermal cycling stability and re-use of
slide
Hybridization was performed as described above using
WT DNA as a target. To remove the hybridized oligonucleotide DNA
target, the arrays were boiled in distilled water for 5 min. SW480
DNA (M1T1) was used for hybridization utilizing the same array.
Further boiling and hybridization were performed using the same
arrayed slide and A549 DNA (M3T3) as the target. Before and after
hybridization, the DNA arrays were scanned for Cy5
fluorescence.
Results
Tag-microarray design
To develop a robust, affordable, high-throughput
method for the detection of KRAS mutations, a
tag-microarray-based genotyping protocol, as shown schematically in
Fig. 1A, was developed. A 111-bp
fragment encompassing the SNP region of KRAS codon 12 was
amplified. The obtained PCR product was used as a template for
nested multiplex PCR. In the multiplex PCR, the forward primers
were chimeric tag-primers consisting of one of the four unique
tags, linked at 3′-end with allele-specific sequences corresponding
to WT and three mutant genotypes (M1T1, M2T2, M3T3, see Tables I and II). These primers were used in pair with
a 5′-Cy5-labeled reverse primer (Cy5-K-ras-RP2, see Table I). The PCR products were hybridized
to the tag probes immobilized on solid substrates (Fig. 1A).
The selection of the tag is an important step
because the tag sequence significantly affects efficiency and
specificity of hybridization (29,33–35).
To minimize cross-hybridization, we first identified tag sequences
that were predicted to have minimal cross-priming and unlikely to
produce secondary structure. The selected four tags were tested
experimentally for cross-hybridization by labeling all four tags
with Cy5 fluorescent dye and hybridizing them respectively to a
microarray containing four anti-tag probes (Table I). Strong fluorescent signal was
reproducibly observed when Cy5-labeled tag target was hybridized
with its complementary probe, whereas no signal was observed in
random-combination groups (data not shown).
Detection of single-base variations of
the KRAS gene of cancer cells
To assess the usefulness of the tag-microarray
approach for detection of SNPs in genomic DNA, we examined DNA
samples from various cancer cell lines carrying WT or different
mutations in KRAS codon 12 (27,29,36,37).
The specificity of the tag-microarray protocol described here
relies largely on the performance of multiplex PCR, because the
forward ‘tag-primers’ are allele-specific for the mutations of
KRAS. We established an effective PCR amplification system
in which no PCR amplicon was observed in gel-electrophoresis when
one forward ‘tag-primer’ was omitted and its corresponding genotype
DNA template was used in multiplex PCR which was accomplished based
essentially on the same conditions described above (data not
shown). DNA isolated from each cancer cell line (Table II) was used as template to obtain
PCR amplicons containing codon 12. The PCR amplicons were purified
and sequenced. The sequencing results revealed that there were five
WT genotypes (HT29, PC3, LNCaP, MCF-7, Colon205), four homozygous
mutants (SW480, SW620, A549, CALU -1), and two heterozygous mutants
(LS174T, A427) (Table II). These
results were consistent with data previously reported (2,14,22,23,27,38).
A multiplex PCR was subsequently performed and the PCR products
were used directly for the tag-microarray hybridization.
Representative images obtained from WT samples were shown in
Fig. 1B. As expected, specific
signals were readily observed in all WT hybridizations in repeated
experiments (Fig. 1B), indicating
that DNA isolated from WT cell lines was hybridized specifically to
its complementary probe immobilized on all substrates (Topas
plastic, glass and GAPS-II coated slides). No cross-hybridization
was detected in any WT hybridizations (Fig. 1B), demonstrating specificity of the
hybridization. It should be noted that the size of the spot on
plastic surface was slightly smaller than that on the surface of
glass slide or GAPS-II coated slide (Fig. 1B, compare top panel and middle or
bottom panel). This likely resulted from the different physical
characteristics (surface tension) between plastic and glass
substrates, rather than from hybridization. Relative fluorescence
intensity value was calculated by dividing the value of intensity
of fluorescence obtained from each spot to that obtained from
background. The final relative value for each probe was determined
as the average value obtained from six spots in three independent
experiments. The results showed that higher averaged relative
fluorescent signals were detected in normal glass slide, compared
to those obtained in Topas plastic slide or GAPS-II coated slide
(data not shown). However, overall, the intensity of fluorescence
measured on different substrates was not significantly
different.
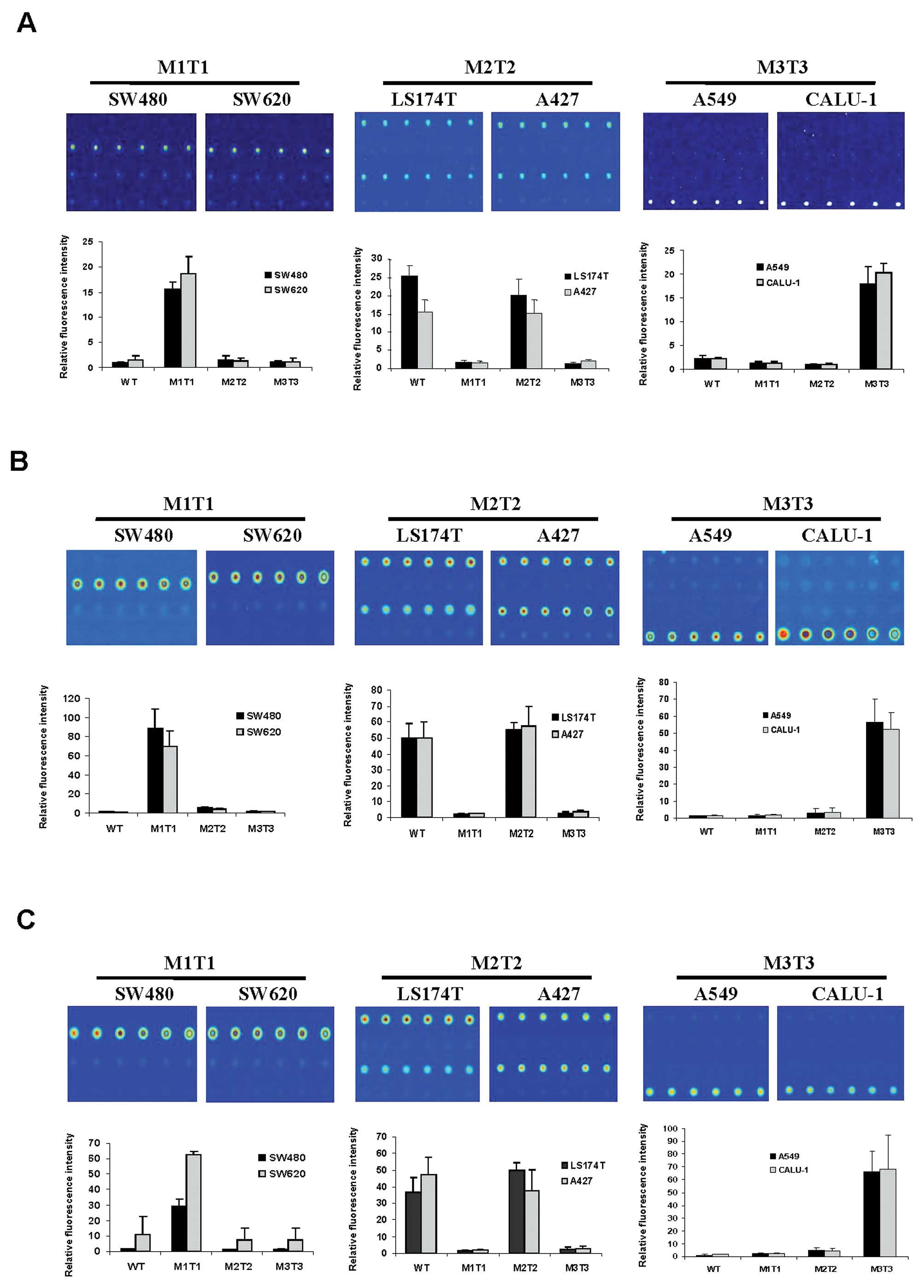
Next, we analyzed all mutant DNAs extracted from
cancer cell lines using procedures as described above. The results
indicated that all mutant genotypes were assigned correctly
(Fig. 2). Fluorescent signal was
consistently detected at the expected genotyped sites in repeated
experiments (Fig. 2), indicating
that mutations in codon 12 could be distinguished from each other
using our tag-microarray approach. The sequencing results showed
that LS174 and A427 cells contain a WT allele in addition to the
mutated genotype in KRAS gene (data not shown).
Consistently, fluorescent signals were detected in both WT and M2T2
probe areas on all slides where the genomic DNAs extracted from
LS174T and A427 cells were examined (Fig. 2, M2T2). The intensity of
fluorescence between WT and M2T2 genotyped sites was equivalent
within the margin of error for LS174T and A427 (Fig. 2), suggesting that the ratio between
M2T2 and WT genotypes is 50% as expected in a heterozygous sample.
The relative fluorescent intensity of all mutant DNAs hybridized to
all immobilized probes was determined, and are shown in Fig. 2. Overall, the calculated values
correlated well with the observed images with respect to the
intensity of signal (Fig. 2). The
presence of WT and mutations in different allele argued that it is
of importance to include four primer sets (WT, M1T1, M2T2 and M3T3)
in a multiplex PCR. Moreover, targeting both WT and all three
mutation possibilities in a multiplex PCR allowed the assay to be
self-controlled on a hybridization slide, as at least one set of
the probes would be detected as positive, demonstrating the assay
was successful. Thus, we did not test other primer mixes, e.g.,
with less primer sets, against a specific template. In sum, our
data indicated that all genotypes of the cells tested were
accurately discriminated by using the tag-microarray hybridization
technique.
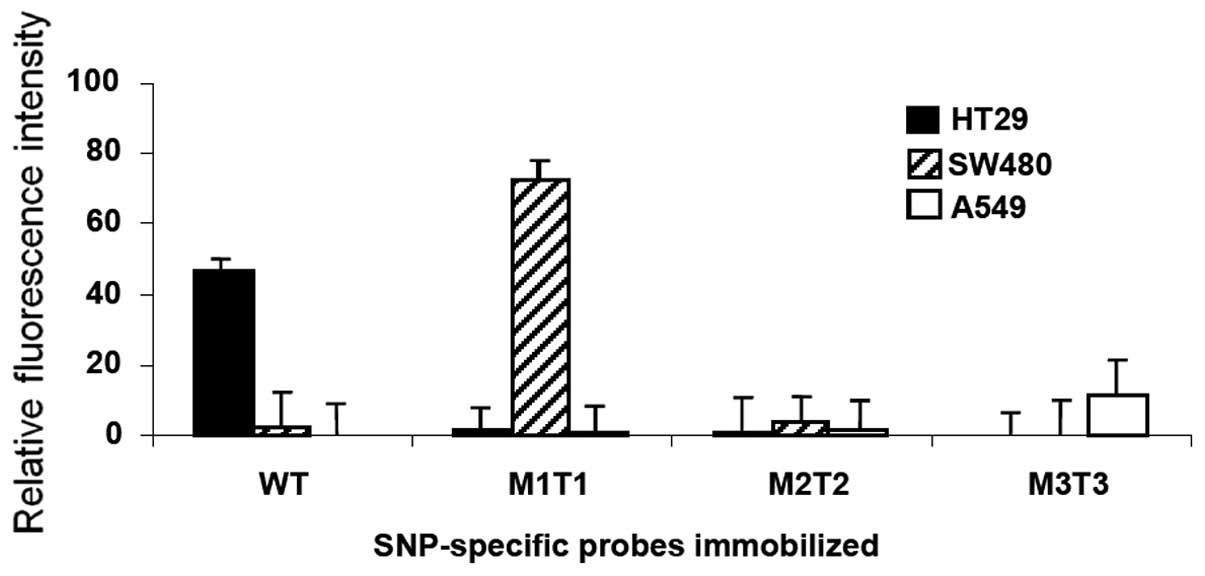
Sensitivity of the method
To quantify the detection limit of the
tag-microarray method in a background of WT DNA, CALU -1 (M3T3)
cell genomic DNA was mixed with varying quantities of WT genomic
DNA from Colon205 cells (Fig. 3A)
or fecal samples (Fig. 3B)
collected from a healthy donor to generate spiked DNA samples.
Fragment containing KRAS codon 12 was amplified using the
mixed DNA sample as template, and hybridization was performed using
normal glass slides, as described above. The results revealed that
the KRAS mutant sequence was detectable when it was present
only in 10% or more of the starting mixed materials (Fig. 3, proportion of DNA 90:10),
demonstrating that the tag-microarray assay was sensitive. Overall,
the intensity of the signal observed in both WT and mutant DNA
samples was dose-dependent (Fig.
3).
Clinical validation
To evaluate the applicability of the tag-microarray
approach, 28 tumor DNAs from clinical patients were analyzed using
this technique. As shown in Table
III, 20 (75%) of 28 samples were detected as WT and seven (25%)
of those samples were detected as mutant. All detections were
confirmed by direct sequencing. Of seven detected KRAS
mutations, two (28.6%) were identified as M1T1 (GGT→GTT); four
(57.1%) were identified as M2T2 (GGT→GAT); one (14.3%) was
determined as M3T3 (GGT→AGT) (Table
III). As verified by a comparison with direct sequencing, all
the WT and mutant samples were correctly identified, suggesting
that the tag-microarray detection technique performed well.
| Table IIIClinical validation of the
method. |
Table III
Clinical validation of the
method.
| | Mutants |
|---|
| |
|
|---|
| Genotype | WT | M1T1 | M2T2 | M3T3 |
|---|
| Percent | (21/28, 75%) | 2/7 (28.6%) | 4/7 (57.1%) | 1/7 (14.3%) |
Stability of the attached probes and
re-use of slide
Finally, we investigated the stability of
immobilization of probes on the solid surfaces and examined whether
the normal glass slides were reusable. We performed
hybridization-boiling-hybridization cycles. Following a successful
hybridization with significant fluorescence read, the 5-min boiling
led to a background signal (data not shown), indicating that the
hybridized targets were completely removed by the boiling.
Re-hybridization of the boiled slides with HT29 target re-gained a
specific signal, indicating the probes are stably attached and
recognized by its target sequence after hybridization-boiling
cycle. Similar results were observed for SW480 and A549 targets,
and that an additional 15 min of boiling did not result in
significant loss of the signal (data not shown). These results
suggested that the immobilized probes were stable on the solid
phase and the slide could be re-used after a thermo treatment. The
corresponding value of signal for each target in repeated
experiments is shown in Fig.
4.
Discussion
Currently, great attention is being paid to the
detection of genetic variation in the development of human diseases
such as cancer. Identification of mutations of host specific
cellular gene is of considerable importance for the diagnosis of
serious diseases such as CRC. DNA microarray has emerged as a
promising tool for large-scale genetic analysis (39,40).
The World Health Organization (WHO) estimates that there are over
940,000 CRC cases annually worldwide, with almost 500,000 deaths
(41). Early diagnosis of CRC
plays an important role in the minimization of death from CRC. We
report here a powerful technique for accurate screening of
mutations of KRAS for the early and fast diagnosis of
CRC.
KRAS codon 12 was selected for this study
since previous investigations revealed that activating mutations of
the KRAS gene occurred in more than 90% of CRC cases, and
most mutations are restricted to codon 12 (2,23,36,37,42).
The codon 12 mutations are therefore an ideal biomarker for early
detection of CRC. Previous study showed that the KRAS codon
12 GGT→GAT mutation was the most common mutation in the mucosa of
colon cancer patients (28). Our
result supported this idea, as 57.1% of clinical patient samples
with KRAS mutations were identified as GGT→GAT mutations.
Cross-hybridization remains one of the significant limitations of
microarray system. We addressed this problem by linking a unique
tag sequence at the 5′-end of forward primers that were used for
multiplex PCR. The Cy5-fluorescence could be detected only in
hybridization in which an entire multiplex
PCR fragment is present (Fig. 1A). Thus, the presence of the unique
tag allows high ability of discrimination upon hybridization on the
microarray and greatly minimizes the risk of false-positive results
due to cross-hybridization, as evidenced by the findings in this
study that all types of mutation in cells and clinical samples
could be unambiguously discriminated. In contrast, attempt of
genotyping by PCR with non-tagged primers was not successful, as
nonspecific signal was readily observed due to cross-hybridization
(data not shown).
Activated KRAS mutants in feces are more
clinically significant than those in other samples (3,25,43).
However, clinical specimens such as feces often contain only a
small percentage of mutant cells in a large background of normal
cells. Thus, assays to detect mutations leading to cancer need to
be sensitive under such conditions (36). Hence, we investigated the detection
limit of the tag-microarray method by mixing mutant cellular DNA
(M3T3) with varying amount of DNA either from WT cell or from feces
of a healthy donor (Fig. 3). To
our knowledge, this attempt has not been reported previously. We
sought to mimic the clinical sample by doing so, as usually patient
sample is of poor quality rather than a sample which is pure like a
cell line. Without any step of mutant DNA enrichment, an accurate
mutation detection limit of 10% mutant DNA in the mixed cell-cell
or cell-fecal DNA materials was reproducibly obtained using our
technique (Fig. 3), indicating
this approach could be used for clinical samples containing a small
fraction of mutant DNA. The successful detection of clinical tumor
samples confirmed our expectations and strongly verified the
applicability of this approach in practice. We are currently
testing additional clinical samples employing this assay to further
validate the performance of this method. Overall, the sensitivity
and accuracy of this method were satisfactory and comparable to the
best results described by other groups (25,27,43).
Using the tag-microarray method, the examination of clinical
samples, from DNA isolation to the eventual result interpretation,
can be finished within one working day, thus, our assay greatly
shortens the hand-on time and time to results, compared to the
method of direct sequencing which normally takes 2–3 working days.
Our technique has additional advantage over other existing methods,
such as it is ideal for large-scale screening of patient samples at
an early stage.
It is desirable to lower the cost of genotyping as
much as possible to obtain more benefits. Toward this direction, we
used different solid substrates for spotting. We found that the
quality of signal and reproducibility of hybridization were higher
when a normal glass slide was used, compared to that of Topas
plastic slide or GAPS-II coated glass slide. Moreover, the regular
glass material is cheaper and easier to use compared to other two
materials tested. Thus, we suggest using the regular glass slide as
a substrate for further research and practical applications. It
should also be noted that the probes immobilized on the solid
surface could undergo treatment of high temperature, indicating
that it is feasible to remove the hybridized target DNA from the
surface of the substrate without disturbing the immobilized probes.
Consequently, new DNA samples can be examined using the same glass
slide as well as the same immobilized probes. The re-use of the
slide and probes makes our approach inexpensive in practice. In
addition, the property of thermo-resistance of the immbolization
allows the possibility of developing a solid PCR platform.
Experiments are underway to establish a solid-PCR system based on
the tag-microarray approach.
Looking solely at the common mutations in a single
codon, as we did, does not rule out other mutations at the same
codon or other codons, such as 13, 59, 61 or 146 in the case of
KRAS (22,28,38,44).
In addition, detection of KRAS codon 12 mutations does not
cover the possibilities of all CRC cases, as it was reported that
KRAS mutations are present in 30–40%, but not 100%, of
colorectal carcinoma cases (45).
Nevertheless, our method is flexible, as in theory multiple
mutations in different codons can be amplified and detected
simultaneously using the technique described here. In addition to
the four mutations in the ‘hotspot’ codon 12 described here,
detection of more mutations in other codons, such as codon 13, 59
and/or 61, is underway using the tag-microarray technique. The
tag-microarray approach described here is highly specific,
noninvasive, cost-effective, and should provide an alternative for
direct sequencing for KRAS mutation detection in colon
cancer patients, particularly if used in combination with FOBT or
other methods for early and mass detecting genetic abnormalities.
We stress the importance of our tag-microarray method as an
innovative technique that can be used for routine diagnosis of CRC
in clinical practice.
Acknowledgements
We thank Dr Mogens Kruhøffer for providing cancer
cell lines and clinical DNA samples, Dr Kurt J. Handberg for
providing cell culture facilities, Jonas Høgberg for expert
technical assistance. This study was supported by the Danish
Council for Independent Research; Technology and Production
Sciences (FTP) (grant no. 274-05-0017). The funders had no role in
study design, data collection and analysis, decision to publish or
preparation of the manuscript.
References
|
1
|
Landis S, Murray T, Bolden S and Wingo P:
Cancer statistics. CA Cancer J Clin. 48:6–29. 1998.
|
|
2
|
Moerkerk P, Arends J, van Driel M, de
Bruine A, de Goeij A and ten Kate J: Type and number of Ki-ras
point mutations relate to stage of human colorectal cancer. Cancer
Res. 54:3376–3378. 1994.PubMed/NCBI
|
|
3
|
Sidransky D, Tokino T, Hamilton S, et al:
Identification of ras oncogene mutations in the stool of patients
with curable colorectal tumors. Science. 256:102–105. 1992.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Winawer S, Fletcher R, Miller L, et al:
Colorectal cancer screening: clinical guidelines and rationale.
Gastroenterology. 112:594–642. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Rex D, Rahmani E, Haseman J, Lemmel G,
Kaster S and Buckley J: Relative sensitivity of colonoscopy and
barium enema for detection of colorectal cancer in clinical
practice. Gastroenterology. 112:17–23. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Hardcastle J, Chamberlain J, Robinson M,
Moss S, Amar S and Balfour T: Randomised controlled trial of
faecal-occult-blood screening for colorectal cancer. Lancet.
348:1472–1477. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lieberman D and Weiss D: One-time
screening for colorectal cancer with combined fecal occult-blood
testing and examination of the distal colon. N Engl J Med.
345:555–560. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Fearon ER: Molecular genetics of
colorectal cancer. Ann NY Acad Sci. 768:101–110. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Burt RW: Colon cancer screening.
Gastroenterology. 119:837–853. 2000. View Article : Google Scholar
|
|
10
|
Hoffman M: Getting a handle on Ras
activity. Science. 255:1591992. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kiaris H and Spandidos DA: Mutations of
ras genes in human tumours (Review). Int J Oncol. 7:413–421.
1995.
|
|
12
|
Shirasawa S, Furuse M, Yokoyama N and
Sasazuki T: Altered growth of human colon cancer cell lines
disrupted at activated Ki-ras. Science. 260:85–88. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Jacobson D and Mills N: A highly sensitive
assay for mutant ras genes and its application to the study of
presentation and relapse genotypes in acute leukemia. Oncogene.
9:553–563. 1994.PubMed/NCBI
|
|
14
|
Nishikawa T, Maemura K, Hirata I, et al: A
simple method of detecting K-ras point mutations in stool samples
for colorectal cancer screening using one-step polymerase chain
reaction/restriction fragment length polymorphism analysis. Clin
Chim Acta. 318:107–112. 2002. View Article : Google Scholar
|
|
15
|
Markman B, Javier F, Capdevila J and
Tabernero J: EGFR and KRAS in colorectal cancer. Adv Clin Chem.
51:71–119. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Siena S, Sartore-Bianchi A, Di
Nicolantonio F, Balfour J and Bardelli A: Biomarkers predicting
clinical outcome of epidermal growth factor receptor-targeted
therapy in metastatic colorectal cancer. J Natl Cancer Inst.
101:1308–1324. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Linardou H, Dahabreh J, Kanaloupiti D, et
al: Assessment of somatic K-RAS mutations as a mechanism associated
with resistance to EGFR-targeted agents: A systematic review and
meta-analysis of studies in advanced non-small-cell lung cancer and
metastatic colorectal cancer. Lancet Oncol. 9:962–972. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Benvenuti S, Sartore-Bianchi A, Di
Nicolantonio F, et al: Oncogenic activation of the RAS/RAF
signaling pathway impairs the response of metastatic colorectal
cancers to anti-epidermal growth factor receptor antibody
therapies. Cancer Res. 67:2643–2648. 2007. View Article : Google Scholar
|
|
19
|
Lee C, Chen H and Liu H: Favorable
response to erlotinib in a lung adenocarcinoma with both epidermal
growth factor receptor exon 19 deletion and K-ras G13D mutations. J
Clin Oncol. 28:e111–e112. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
De Roock W, Jonker D, Nicolantonio F, et
al: Association of KRAS p. G13D mutation with outcome in patients
with chemotherapy-refractory metastatic colorectal cancer treated
with cetuximab. J Am Med Assoc. 304:1812–1820. 2010.PubMed/NCBI
|
|
21
|
Allegra C, Jessup J, Somerfield M, et al:
American Society of Clinical Oncology provisional clinical opinion:
testing for KRAS gene mutations in patients with metastatic
colorectal carcinoma to predict response to anti-epidermal growth
factor receptor monoclonal antibody therapy. J Clin Oncol.
27:2091–2096. 2009. View Article : Google Scholar
|
|
22
|
Toyooka S, Tsukuda K, Ouchida M, et al:
Detection of codon 61 point mutations of the K-ras gene in lung and
colorectal cancers by enriched PCR. Oncol Rep. 10:1455–1459.
2003.PubMed/NCBI
|
|
23
|
Nollau P, Moser C, Weinland G and Wagener
C: Detection of K-ras mutations in stools of patients with
colorectal cancer by mutant-enriched PCR. Int J Cancer. 66:332–336.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Luo J, Chan E, Shih C, et al: Detection of
rare mutant K-ras DNA in a single-tube reaction using peptide
nucleic acid as both PCR clamp and sensor probe. Nucleic Acids Res.
34:e122006. View Article : Google Scholar
|
|
25
|
Mixich F, Ioana M, Voinea F, Saftoiu A and
Ciurea T: Noninvasive detection through REMS-PCR technique of K-ras
mutations in stool DNA of patients with colorectal cancer. J
Gastrointestin Liver Dis. 16:5–10. 2007.PubMed/NCBI
|
|
26
|
Dieterle C, Conzelmann M, Linnemann U and
Berger M: Detection of isolated tumor cells by polymerase chain
reaction-restriction fragment length polymorphism for K-ras
mutations in tissue samples of 199 colorectal cancer patients. Clin
Cancer Res. 10:641–650. 2004. View Article : Google Scholar
|
|
27
|
Lopez-Crapez E, Chypre C, Saavedra J,
Marchand J and Grenier J: Rapid and large-scale method to detect
K-ras gene mutations in tumor samples. Clin Chem. 43:936–942.
1997.PubMed/NCBI
|
|
28
|
Maekawa M, Nagaoka T, Taniguchi T, et al:
Three-dimensional microarray compared with PCR-single-strand
conformation polymorphism analysis/DNA sequencing for mutation
analysis of K-ras codons 12 and 13. Clin Chem. 50:1322–1327. 2004.
View Article : Google Scholar
|
|
29
|
Parsons B, Marchant-Miros K, Delongchamp
R, et al: ACB-PCR quantification of K-RAS codon 12 GAT and GTT
mutant fraction in colon tumor and non-tumor tissue. Cancer Invest.
28:364–375. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Hirschhorn J, Sklar P, Lindblad-Toh K, et
al: SBE-TAGS: an array-based method for efficient single-nucleotide
polymorphism genotyping. Proc Natl Acad Sci USA. 97:12164–12169.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Dufva M, Petersen J, Stoltenborg M,
Birgens H and Christensen C: Detection of mutations using
microarrays of poly(C)10-poly(T)10 modified DNA probes immobilized
on agarose films. Anal Biochem. 352:188–197. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Gudnason H, Dufva M, Bang D and Wolff A:
An inexpensive and simple method for thermal stable immobilization
of DNA on an unmodifed glass surface: UV linking of poly (T)
10-poly (C) 10 tagged DNA probes. Bio Technique. 45:261–271.
2008.PubMed/NCBI
|
|
33
|
Winzeler E, Shoemaker D, Astromoff A, et
al: Functional characterization of the S. cerevisiae genome
by gene deletion and parallel analysis. Science. 285:901–906.
1999.
|
|
34
|
Fan J, Chen X, Halushka M, et al: Parallel
genotyping of human SNPs using generic high-density oligonucleotide
tag arrays. Genome Res. 10:853–860. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Pastinen T, Raitio M, Lindroos K, Tainola
P, Peltonen L and Syvanen AC: A system for specific,
high-throughput genotyping by allele-specific primer extension on
microarrays. Genome Res. 10:1031–1042. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Lleonart M, Ramony Cajal S, Groopman J and
Friesen M: Sensitive and specific detection of K-ras mutations in
colon tumors by short oligonucleotide mass analysis. Nucleic Acids
Res. 32:e532004. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Finkelstein S, Sayegh R, Christensen S and
Swalsky P: Genotypic classification of colorectal adenocarcinoma.
Biologic behavior correlates with K-ras-2 mutation type. Cancer.
71:3827–3838. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Chang Y, Yeh K, Chang T, et al: Fast
simultaneous detection of K-RAS mutations in colorectal cancer. BMC
Cancer. 9:1792009. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Huang J, Mehrens D, Wiese R, et al:
High-throughput genomic and proteomic analysis using microarray
technology. Clin Chem. 47:1912–1916. 2001.PubMed/NCBI
|
|
40
|
Kim I, Kang H, Jang S, et al:
Oligonucleotide microarray analysis of distinct gene expression
patterns in colorectal cancer tissues harboring BRAF and K-ras
mutations. Carcinogenesis. 27:392–404. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
The World Cancer Report - the major
findings. Cent Eur J Public Health. 11:177–179. 2003.
|
|
42
|
Fumagalli D, Gavin P, Taniyama Y, et al: A
rapid, sensitive, reproducible and cost-effective method for
mutation profiling of colon cancer and metastatic lymph nodes. BMC
Cancer. 10:1012010. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Chien C, Chen S, Liu C, Lee C, Yang R and
Huang C: Correlation of K-ras codon 12 mutations in human feces and
ages of patients with colorectal cancer (CRC). Transl Res.
149:96–102. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Cerottini J, Caplin S, Saraga E, Givel J
and Benhattar J: The type of K-ras mutation determines prognosis in
colorectal cancer. Am J Surg. 175:198–202. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Cho K and Vogelstein B: Genetic
alterations in the adenomacarcinoma sequence. Cancer. 70:1727–1731.
1992. View Article : Google Scholar : PubMed/NCBI
|